
A strain-reinforcing elastomer adhesive with superior adhesive strength and toughness†
Chuanlong
Li
a,
Wenbo
Dong
a,
Longyu
Li
 a,
Zhengli
Dou
a,
Yuhan
Li
b,
Liuhe
Wei
b,
Qin
Zhang
*a,
Qiang
Fu
a and
Kai
Wu
*a
a,
Zhengli
Dou
a,
Yuhan
Li
b,
Liuhe
Wei
b,
Qin
Zhang
*a,
Qiang
Fu
a and
Kai
Wu
*a
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, P. R. China. E-mail: kaiwu@scu.edu.cn; qinzhang@scu.edu.cn
bCollege of Chemistry and Green Catalysis Center, Zhengzhou Key Laboratory of Elastic Sealing Materials, Zhengzhou University, Zhengzhou 450001, P. R. China
First published on 28th July 2023
Abstract
Strong and ductile adhesives often undergo both interfacial and cohesive failure during the debonding process. Herein, we report a rare self-reinforcing polyurethane adhesive that shows the different phenomenon of only interfacial failure yet still exhibiting superior adhesive strength and toughness. It is synthesized by designing a hanging adhesive moiety, hierarchical H-bond moieties, and a crystallizable soft segment into one macromolecular polyurethane. The former hanging adhesive moiety allows the hot-melt adhesive to effectively associate with the target substrate, providing sufficient adhesion energy; the latter hierarchical H-bond moieties and a crystallizable soft segment cooperate to enable the adhesive to undergo large lap-shear deformations through sacrificing weak bonds and mechano-responsive strength through the fundamental mechanism of strain-induced crystallization. As a result, this polyurethane adhesive can keep itself intact during the debonding process while still withstanding a high lap-shear strength and dissipating tremendous stress energy. Its adhesive strength and work of debonding are as high as 11.37 MPa and 10.32 kN m−1, respectively, outperforming most reported tough adhesives. This self-reinforcing adhesive is regarded as a new member of the family of strong and ductile adhesives, which will provide innovative chemical and structural inspirations for future conveniently detachable yet high-performance adhesives.
New conceptsThe majority of tough adhesives via the incorporation of weak components undergo cohesive failure, mostly along with interfacial failure. This mixed failure behavior is often considered to be the optimal way to improve their adhesive strength and toughness. This work reports a different self-reinforcing polyurethane adhesive to show the phenomenon of only interfacial failure yet still exhibiting superior adhesive strength and toughness. Its material design guideline is also on the basis of the sacrificial phase (weak hydrogen bonds) to provide it with large-deformation and energy-dissipation abilities, while its additional advantage is the presence of mechano-responsive characteristics that enhance the material cohesion and prevent it from any destruction through strain-induced crystallization during the debonding process. As a result, this polyurethane adhesive can keep itself intact during the debonding process while still withstanding a high lap-shear strength and dissipating tremendous stress energy. It can show superb mechanical tensile and adhesive performance, for example that its tensile strength (24.13 MPa) can be comparable to those of some universal polyolefin plastics, while its stretchability (≈800%) is like that of rubbers; its lap-shear strength and work of debonding are as high as 11.37 MPa and 10.32 kN m−1, respectively, outperforming those of most tough adhesives in previous literature. This strong and ductile PUD20 elastomer is also found to be an on-demand adhesive, that is toughly adhesive when applied while being easily detachable when someone needs to peel it off, which is very convenient for the actual use and disassembly. |
1. Introduction
Adhesion is an important and ubiquitous requirement in modern life. It successfully aids in connecting the complex structures of buildings, electronic devices, industrial vehicles, or aircraft into a whole.1,2 Nowadays, people's demand for adhesive materials is increasing and developing constantly, since the adhesion scene has gradually changed from simple to complex, and the bonded object is more and more diverse.3–8 The present variety of adhesives is usually either ductile or strong but seldom both.9–15 Ductility is effective to withstand a large extension and dissipate mechanical stress, which is useful to prevent abrupt bond failure. However, ductile adhesives suffer from cohesive weakness and hardly endure a large external force.16,17 Strong adhesives with intrinsic high cohesion are mechanically robust, which can support large loads while unfortunately suffering from the characteristic of brittleness. When an external force is loaded, their adhesion strength rises and then abruptly decreases with a sudden breakage.18 An ideal adhesive is expected to combine both high adhesive strength and good ductility, enabling the bonded structure to adapt to more complex situations and be more reliable.Ductility is often found in most low-modulus materials that exhibit weakly bonded molecular networks and favorable chain mobility. In contrast, adhesion strength depends on high covalent binding and secure molecular networks. These conflicts in the structural design make the fabrication of strong and ductile adhesives a great challenge. Nature has always been a source of inspiration to put forward ingenious ways to break through the seeming contradictions and achieve balanced performance in one biological material.19–23 One famous example is the seashell nacre, whose rigid component, aragonite platelets, is linked together by soft protein. This typical soft–hard cooperation is effective to dissipate the stress energy and maintain high strength, in which the soft protein allows large deformations to exhibit a mechanical hysteresis feature and the rigid aragonite platelets withstand large external force and stop crack propagation.24–26 Other tough biological examples, such as spider silk and vascular smooth muscle, exhibit similar structural characteristics, that is the incorporation of weakness makes toughness.27–29 Insights from nature, present strong and ductile adhesives were designed with this idea of sacrificial phase (i.e., weak bonding or soft phase) in mind. For example, sacrificial hydrogen bonding was reported to be designed in a copolymer adhesive.18 The formation of the interpolymeric network of breakable bonds was found efficient to dissipate mechanical stresses while leaving the whole material deformable but intact. The tough adhesive exhibits a lap shear adhesion strength of 2.6 MPa and a work of debonding of 0.54 kN m−1. Another example is the incorporation of dynamic covalent linkage.30 Boronicester was added to a triblock polystyrene-b-poly(ethylene-co-butylene)-b-polystyrene polymer, in which the embedded inorganic silica nanoparticles will dynamically crosslink through the reaction between the boronic ester groups and the hydroxyl groups on the silica nanoparticles. During the debonding process, these dynamic covalent bonds will break and cooperate with the large deformation of the soft ethylene butylene block, dissipating large quantities of mechanical stress energy. As a result, this tough adhesive exhibits a lap shear adhesion strength of 10.42 MPa and a work of debonding of 5.43 kN m−1.
The majority of the above tough adhesives via the incorporation of weak components undergo cohesive failure, mostly along with interfacial failure.15,30–32 This mixed failure behavior is often considered to be the optimal way to improve their adhesive strength and toughness. In contrast, herein, a different self-reinforcing polyurethane adhesive is synthesized to show the phenomenon of only interfacial failure yet still exhibiting superior adhesive strength and toughness. Its material design guideline is also on the basis of the sacrificial phase (weak hydrogen bonds) to provide it with large-deformation and energy-dissipation abilities, while its additional advantage is the presence of mechano-responsive characteristics that enhance the material cohesion and prevent it from any destruction through strain-induced crystallization during the debonding process. As a result, the lap shear strength and work of debonding of the polyurethane adhesive are found to be as high as 11.37 MPa and 10.32 kN m−1, respectively, outperforming those of most tough adhesives in previous literature. This mechano-responsive adhesive with only interfacial failure is regarded as a new member of the family of strong and ductile adhesives. It can be on-demand detachable with no residual adhesive on the target substrate, which is very convenient for the actual use and disassembly. Such structural features and debonding fundamentals in this distinctive adhesive will provide new inspirations for future advanced and high-performance adhesives.
2. Results and discussion
Polyurethane adhesives were prepared according to the experimental procedures involving the ordinal synthesis of the T-shaped chain extender with a dopamine component (DMPA-DA) and a three-step copolymerization reaction. These polyurethanes are abbreviated as PUDx, where x refers to the percentage value of DMPA-DA in the total chain extender. The synthetic route of DMPA-DA can be found in Fig. S1, ESI,† and the chemical structures of the intermediate products were confirmed using 1H nuclear magnetic resonance (1H NMR) spectra (Fig. S2–S5, ESI†). Afterward, the PUDx adhesives were synthesized via the random copolymerization of the soft segment poly(tetrahydrofuran) (PTMEG), two chain extenders DMPA-DA and 2,6-pyridinedimethanol (PDM), and isophorone diisocyanate (IPDI). The synthetic route, chemical structure, and molecular weight information are shown in Fig. S6–S8 and Tables S1 and S2, ESI.†According to Fig. 1a, the chemical structure of the PUDx adhesive contains four characteristic segments with different functionalities. Its soft segment is PTMEG which is a linear structure and can be crystallizable. It is designed to offer adhesives with mechano-responsive possibility (strain-induced crystallization), especially at a large lap-shear displacement.33–35 The hydrogen-bond (H-bond) moieties of the adhesives are designed with hierarchical binding energy. As confirmed by atomic force microscopy (Fig. S9, ESI†) and the small angle X-ray scattering test (Fig. S10, ESI†), these H-bond moieties, including carbamate, pyridine, and catechol groups, will assemble into nano-scale hard domains. For the H bonds with low binding energy (i.e., carbamate–carbamate and catechol–catechol), they act as the sacrificial weakness, which is designed to allow the large deformation of the elastomer and dissipate the stress energy. For other H-bonds with high binding energy (i.e., carbamate–pyridine and pyridine–catechol), they are designed to prevent the stretched soft segment from relaxation under a large strain and guarantee the result of strain-induced crystallization.36 The hanging adhesive moiety (DMPA-DA) is designed to offer the PUDx with better ability to associate with the target substrate through secondary relaxations because side-chain relaxation is proved more favorable than the counterpart in the macromolecular main chain.37 Through the above molecular designs, we expect that during the debonding process, the PUDx adhesives can allow large deformations due to the sacrificial H-bonds and withstand high lap-shear forces because of the mechano-responsive merit and the effective interfacial interactions with the substrates. According to the lap-shear adhesive performance in Fig. 1c and Table S3, ESI,† the amorphous PUDx hot melt (differential scanning calorimetry (DSC) curves in Fig. S11, ESI†) exhibit the best lap-shear strength as high as 11.37 MPa and a work of debonding up to 10.32 kN m−1. Although being compared to other adhesives in previous literature, including polymer gels, elastomers, and resins, these PUDx adhesives manifest outperforming lap-shear properties. Taking the PUD20 elastomer adhesive as an example, only a 0.25 g sample can lift an 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-times-weight bucket (4.5 kg), indicating its remarkable tensile strength (Fig. 1d). After being sandwiched by two stainless steel plates, just 300 mm2 PUD20 adhesive can bear a 60 kg adult (Fig. 1e).
000-times-weight bucket (4.5 kg), indicating its remarkable tensile strength (Fig. 1d). After being sandwiched by two stainless steel plates, just 300 mm2 PUD20 adhesive can bear a 60 kg adult (Fig. 1e).
 | ||
| Fig. 1 Molecular design of the polyurethane elastomer adhesive. (a) Molecular structure of the polyurethane elastomer adhesive containing four typical functional segments. (b) Schematic illustration of the assembled dynamic hard domains within the polyurethane elastomer adhesive, which consists of the hierarchical H-bonds, including carbamate–carbamate, carbamate–pyridine, carbamate–catechol, pyridine–catechol, and catechol–catechol interactions. The inset table illustrates the binding energy of an individual hydrogen bond. (c) Comparison of the lap shear strength and work of debonding of the polyurethane elastomer adhesives with other adhesives in previous literature. (d) A photograph showing a 4.5 kg bucket lifted by a strip made of the PUD20 elastomer. (e) A photograph exhibiting the PUD20 adhesive sandwiched by two stainless steel plates bearing a 60 kg adult. The adhesion area is only 300 mm2. | ||
Tensile behavior reflects the ductility and cohesive strength, which is important for the adhesion performance of an adhesive. Before studying the adhesive properties, tensile stress–strain curves of PUDx adhesives with different DMPA-DA values in the total chain extender were analyzed (Fig. 2a and Fig. S12, ESI†). Each PUDx elastomer performs excellent stretchability; however, their tensile strength is very different. A high percentage value of PDM (the H-bond moiety with high binding energy) in the total chain extender is found beneficial to the tensile strength, such as the PUD10 and PUD20 samples. This is likely that the quantities of H-bonds with high binding energy can hold the conformation of the soft segment's rearrangement and orientation, which creates the conditions for the possible phenomenon of strain-induced crystallization.38 Further with the decrease of PDM, the number of high-binding-energy hydrogen bindings decreases. The slip of soft segment gradually becomes easy to impede strain-induced crystallization, which leads to the obvious reduction in tensile strength. According to the typical stress–strain curve of PUD20 (Fig. 2b), the robust elastomer experiences three different stages during the unidirectional stretching process. At the initial stage, the elastomer is soft. With an increase of the tensile strain, the tensile stress increases slowly. At stage 2 where the strain exceeds about 400%, the slope of the tensile stress–strain curve becomes larger and larger. After reaching the third stage, the elastomer becomes much stiffer, and its modulus is almost constant. This mechano-responsive phenomenon is like the PUD20 elastomer can be self-reinforced during the tensile process, which makes it not only strong but also very ductile and tough (i.e., fracture energy of 174 kJ m−2, Fig. S13, ESI†). In Fig. 2c, at a large deformation, for example, 700% strain, the PUD20 sample is found to exhibit an obvious phenomenon of whitening by stretching. This whitening phenomenon can be reversible because once the stress is removed, the PUD20 elastomer will recover to the original transparent state. We speculate that this mechano-responsive self-reinforcing and whitening phenomenon is due to the rearrangement and then crystallization of the linear soft segment (PTMEG). To clarify this phenomenon, in Fig. 2d, optical microscopy is used to in situ observe the strain-induced crystallization during the stretching and releasing process. PUD20 is transparent in its original state; however, numerous nano- and micron-sized crystal chips (dark dots and regions) exist in the sample with large deformation of 700% strain. These crystal chips are found to disappear when the elastomer returns to the unstretched state, indicating the reversibility of such a strain-induced crystallization phenomenon. To further validate the crystallization of PTMEG, X-ray diffraction is applied to quantify the crystalline information of the PUD20 adhesive stretched to different strains. A new peak located at a 2θ value of ∼28° emerges and gradually grows, which is assigned to the characteristic crystalline plane in the monoclinic cell of the soft-segment chains. And the new peak disappears when the elastomer recovers to the unstretched state. This experimental finding is powerful evidence of PTMEG crystallization and the self-reinforcing ability of PUD20 in the stretching process.
 | ||
| Fig. 2 Tensile properties of the polyurethane elastomer adhesive. (a) Stress–strain curves of the PUDx adhesives with various percentage values of DMPA-DA in the total chain extender. (b) The typical stress–strain curve of the PUD20 adhesive is divided into three stages, showing the self-reinforcing characteristic at large deformations. (c) Optical photos showing the whitening and recoverable ability of the PUD20 adhesive when it is unidirectionally stretched. (d) Optical microscopy images showing an identical sample stretched to 700% and recovered to the original state. (e) X-ray diffraction results of the PUD20 adhesive stretched to various strains. | ||
After clarifying the characteristic of strain-induced crystallization, lap-shear adhesive properties are studied in Fig. 3. Fig. 3a illustrates the typical adhesive strength–displacement curves of the PUDx adhesives with various percentage values of DMPA-DA in the total chain extender. PUD20 exhibits the highest lap shear strength of 11.37 MPa, Fig. S14, ESI†) among the five different PUDx adhesives. Herein, the work of debonding is used to evaluate the adhesive toughness of the PUDx adhesives, which is the integral of the area of the typical lap shear strength–displacement curve. PUD20 is found to be a both strong and tough adhesive, whose work of debonding is as high as 10.32 kN m−1, outperforming most reported adhesives (Fig. 1c). The typical debonding process of the PUD20 adhesive is recorded by a commercial camera in Movie S1, ESI.† From the beginning to lap shear stretching until the bonding joint is completely broken, PUDx adhesive (i.e., PUD20) underwent a very large deformation process, especially in the thickness direction (Fig. S15, ESI†). In Fig. 3b, the lap shear force–strain (in the hypotenuse direction) curves of the different PUDx adhesives are recorded. It is found that PUDx (x is 10 to 40) is also a ductile adhesive, which can perform a very large debonding elongation in the hypotenuse direction. For example, the fracture strain of the PUD20 adhesive after the debonding process is 766%. This strain is high enough to allow the soft segment to perform the effect of strain-induced crystallization (Fig. 2b). As shown in Fig. 3c, the intrinsic high cohesive strength and large debonding deformation of the PUD20 adhesive can dissipate tremendous stress energy, with its energy release rate being calculated to be as high as 4.98 kJ m−2. In Table S3 (ESI†), the debonding type of the previously reported adhesives is analyzed, including gels, resins, and elastomers. Most strong and ductile adhesives undergo cohesive failure, sometimes accompanied by the phenomenon of interfacial failure. However, it is interesting that our PUD20 adhesive shows the different phenomenon of only interfacial failure yet still exhibiting superior adhesive strength and toughness. Although in comparison with most reported adhesives, the lap shear strength and work of debonding of the PUD20 adhesive are the most outstanding (Fig. 1c and Table S3, ESI†). This self-reinforcing PUD20 adhesive can be considered a new member of the family of strong and ductile adhesives. Its different debonding failure, which is found for the first time in this study, may be ascribed to its unique mechano-responsive merit and characteristic chemical structure, which needs more in-depth structural and performance analysis (Fig. 3d–h).
 | ||
| Fig. 3 Adhesive properties of the polyurethane elastomer. (a) Lap shear strength–displacement curves of the PUDx adhesives with various percentage values of DMPA-DA in the total chain extender. (b) Lap shear force–strain curves of the different PUDx adhesives. Noted that the strain refers to the hypotenuse direction. (c) The energy release rate of the different PUDx adhesives. (d) Synchronous and (e) asynchronous two-dimensional Fourier transform infrared images of the PUD20 adhesive. (f) Dielectric loss spectra of the PUD20 adhesive fitted via a combination of three H–N equations with DC conductivity at −90 °C. (g) Activation energies (Ea) of β- and γ-relaxation for the PUD20 and PU adhesives. (h) Schematic diagram illustrating the debonding process of the PUD20 adhesive. The blue, orange, and purple H-bond moieties refer to carbamate, pyridine, and catechol, respectively. | ||
According to Fig. 3d, e, and Fig. S16, ESI,† the absorption peaks at 3504, 3313, 1701, and 1598 cm−1 belong to ν(O–H), ν(N–H), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and ν(pyridine ring), respectively. Five positive cross-peaks are observed in the synchronous spectrum, including φ(1701, 3504), φ(1701, 3313), φ(1598, 3504), φ(1598, 3313), and φ(3504, 3504). While in the asynchronous spectrum, these peaks become negative cross-peaks or even vanished. This phenomenon indicates that the PUD20 adhesive is designed with dynamic H-bond moieties, and the binding energy of these H-bonds is hierarchical (molecular simulation results, Fig. 1b. The simulation details are provided in the ESI.†39,40 At the very beginning of the lap-shear process, H-bonds with the weak binding energy, such as carbamate–carbamate and catechol–catechol interaction, will disassociate and associate repeatedly in the lap-shear process, and the elastomer adhesive is allowed to undergo the corresponding deformation due to the favorable relaxation and slip of macromolecular chains (Fig. 3h). Since the H-bonds with the high binding energy, such as carbamate–pyridine and pyridine–catechol interaction, can withstand the aligned molecular configuration without any macroscopic material destruction, soft segment PTMEG will be sufficiently rearranged and begin to crystallize when the lap-shear strain approaches a certain value (Fig. 3h). Such a mechano-responsive characteristic is already clarified in Fig. 2, which can largely improve the intrinsic strength of the PUD20 elastomer, preventing itself from any cohesive failure. Therefore, the ultimate lap-shear strength of the PUD20 will mainly depend on the interfacial adhesive capability between the PUD20 and the double aluminum substrates. In Fig. S17, ESI,† the rheological master curve at a reference temperature of 25 °C following the principle of time–temperature superposition suggests that the characteristic relaxation time of the PUD20 is 886 s, demonstrating the fast chain dynamics of the PUD20 adhesive that will help the formation of H-bond interactions with the aluminum surface. More importantly, PUD20 is advantageous in a hanging adhesive moiety in its side chain, which is very favorable for the catechol moiety to interact with the aluminum substrate through secondary relaxations to enhance the interfacial adhesive capability. To verify this, broad-frequency dielectric spectrum measurements (BDS) are carried out for PUD20 and its control sample (PU, without DMPA-DA as the chain extender). According to Fig. 3f and Fig. S18, ESI,† every dielectric spectrum of PUD20 and PU can be fitted into two relaxation peaks via the Havriliak–Negami (H–N) function (see details in the ESI†), which are assigned to the motions of the hard segment (β relaxation) and the soft segment (γ relaxation), respectively. The Arrhenius function is employed to fit the active energies (Ea) for the different segmental motions at different temperatures. The results are plotted in Fig. 3g where the hard segment containing a hanging catechol moiety (Ea of 13 kJ mol−1) can relax more easily than that without such a side chain (Ea of 23 kJ mol−1). This much lower activation energy of the β relaxation in PUD20 signifies the insight that the hanging adhesive moiety (catechol) allows the adhesive to effectively associate with the target substrate through secondary side-chain relaxations, significantly enhancing the interfacial adhesive capability between the aluminum substrate and PUD20 adhesive. As a result, the PUD20 adhesive can keep itself intact due to the mechano-responsive characteristic while still withstanding a high lap-shear strength and dissipating tremendous stress energy due to the hanging catechol moiety and sacrificial H-bonds.
O), and ν(pyridine ring), respectively. Five positive cross-peaks are observed in the synchronous spectrum, including φ(1701, 3504), φ(1701, 3313), φ(1598, 3504), φ(1598, 3313), and φ(3504, 3504). While in the asynchronous spectrum, these peaks become negative cross-peaks or even vanished. This phenomenon indicates that the PUD20 adhesive is designed with dynamic H-bond moieties, and the binding energy of these H-bonds is hierarchical (molecular simulation results, Fig. 1b. The simulation details are provided in the ESI.†39,40 At the very beginning of the lap-shear process, H-bonds with the weak binding energy, such as carbamate–carbamate and catechol–catechol interaction, will disassociate and associate repeatedly in the lap-shear process, and the elastomer adhesive is allowed to undergo the corresponding deformation due to the favorable relaxation and slip of macromolecular chains (Fig. 3h). Since the H-bonds with the high binding energy, such as carbamate–pyridine and pyridine–catechol interaction, can withstand the aligned molecular configuration without any macroscopic material destruction, soft segment PTMEG will be sufficiently rearranged and begin to crystallize when the lap-shear strain approaches a certain value (Fig. 3h). Such a mechano-responsive characteristic is already clarified in Fig. 2, which can largely improve the intrinsic strength of the PUD20 elastomer, preventing itself from any cohesive failure. Therefore, the ultimate lap-shear strength of the PUD20 will mainly depend on the interfacial adhesive capability between the PUD20 and the double aluminum substrates. In Fig. S17, ESI,† the rheological master curve at a reference temperature of 25 °C following the principle of time–temperature superposition suggests that the characteristic relaxation time of the PUD20 is 886 s, demonstrating the fast chain dynamics of the PUD20 adhesive that will help the formation of H-bond interactions with the aluminum surface. More importantly, PUD20 is advantageous in a hanging adhesive moiety in its side chain, which is very favorable for the catechol moiety to interact with the aluminum substrate through secondary relaxations to enhance the interfacial adhesive capability. To verify this, broad-frequency dielectric spectrum measurements (BDS) are carried out for PUD20 and its control sample (PU, without DMPA-DA as the chain extender). According to Fig. 3f and Fig. S18, ESI,† every dielectric spectrum of PUD20 and PU can be fitted into two relaxation peaks via the Havriliak–Negami (H–N) function (see details in the ESI†), which are assigned to the motions of the hard segment (β relaxation) and the soft segment (γ relaxation), respectively. The Arrhenius function is employed to fit the active energies (Ea) for the different segmental motions at different temperatures. The results are plotted in Fig. 3g where the hard segment containing a hanging catechol moiety (Ea of 13 kJ mol−1) can relax more easily than that without such a side chain (Ea of 23 kJ mol−1). This much lower activation energy of the β relaxation in PUD20 signifies the insight that the hanging adhesive moiety (catechol) allows the adhesive to effectively associate with the target substrate through secondary side-chain relaxations, significantly enhancing the interfacial adhesive capability between the aluminum substrate and PUD20 adhesive. As a result, the PUD20 adhesive can keep itself intact due to the mechano-responsive characteristic while still withstanding a high lap-shear strength and dissipating tremendous stress energy due to the hanging catechol moiety and sacrificial H-bonds.
Traditional industrial adhesives are always permanent, which is disadvantageous for temporary adhesion or parts recovery. However, this strong and ductile PUD20 elastomer is an on-demand adhesive, that is toughly adhesive when applied while being easily detachable when someone needs to peel it off, since we expect that its adhesion through the formation of abundant H-bonds with the substrates can be switchable by regulating ambient temperature. In Fig. 4a and b, temperature-dependent Fourier transform infrared analysis is adopted to investigate the variation of H-bond interactions within the PUD20 adhesive. The hierarchical H-bond moieties are found very sensitive to ambient temperature, as the characteristic peaks including v(–OH), v(–NH), v(CO), and v(pyridine ring) all perform an obvious shift as a function of ambient temperature. At low temperatures near room temperature, these moieties will form H-bonds with each other, leading the elastomer cohesively strong and highly adhesive with the substrates. As an increase of ambient temperature, these H-bonds will disassociate and be free, making the PUD20 cohesively weak and detachable from the substrates. This analysis can be verified by the content variation of H-bonded moiety and free moiety as a function of temperature. Taking the CO moiety as an example, Fig. S19, ESI,† and Fig. 4c illustrate the typical content of H-bonded and free CO moiety and their respective variation as a function of temperature. The bonded counterpart will gradually disassociate upon a rise in the temperature, and more and more CO moieties will be free. Therefore, by controlling ambient temperature, the lap shear strength of the PUD20 adhesive can be on-demand regulated (Fig. 4d). For example, at low temperatures below 45 °C, the PUD20 is highly adhesive; once the temperature is above 60 °C, it will be easily detachable because of the breakage of the hydrogen bonds at the substrate–adhesive interface. Moreover, attributed to the favorable macromolecular relaxation, the adhesive properties of this PUD20 are manifested to be reversible. For instance, after four times of cycling bonding and detachment, its lap shear strength is almost constant (Fig. 4e). In Fig. 4f, various substrates are adopted for the lap shear adhesion test using the PUD20 adhesive. Its superior adhesive strength and toughness are demonstrated to be universal to these common substrates, including aluminum, stainless steel, copper, glass, epoxy, and rubber. It is noteworthy that for rigid and strong substrates, the PUD20 adhesive always undergo only the interfacial failure (Fig. S20, ESI†). However, for a stiff yet brittle epoxy or ductile yet weak nitrile rubber substrate, the PUD20 is found to be intact while the substrate is instead broken (Fig. S20, ESI†), which is attributed to the intrinsic weakness of the substrates. Besides, this mechano-responsive adhesive with only interfacial failure is regarded to be on-demand detachable with no residual adhesive on the target substrate, which will bring great convenience for the actual use and disassembly. In Fig. 4g and h, as a proof of concept, this PUD20 adhesive is applied to manifest its on-demand and reversible adhesion and debonding capability. A hairdryer with 2000 W is used to portably control the ambient temperature of the adhesive. At room temperature, this adhesive can firmly adhere to the aluminum substrates, even though this sandwiched sample is carrying a bucket (10 kg) that is more than 10000 times the own weight of the PUD20. After heating the hairdryer for 90 s, this PUD20 can be easily detached from these two aluminum substrates, without any residual adhesive on the target substrate. Since the H-bond interactions between the PUD20 and aluminum substrates are reversible and can be on-demanded controlled by ephemeral heating and then natural cooling, the strong and tough adhesion is restored again after the temperature regulation. The above application successfully demonstrated the reversible adhesion and debonding capability of the PUD20 adhesive. Along with its characteristic tough adhesion yet only interfacial-failure behavior, this new family member of the strong and ductile adhesive will bring inspiration for high-performance adhesives and their portable applications.
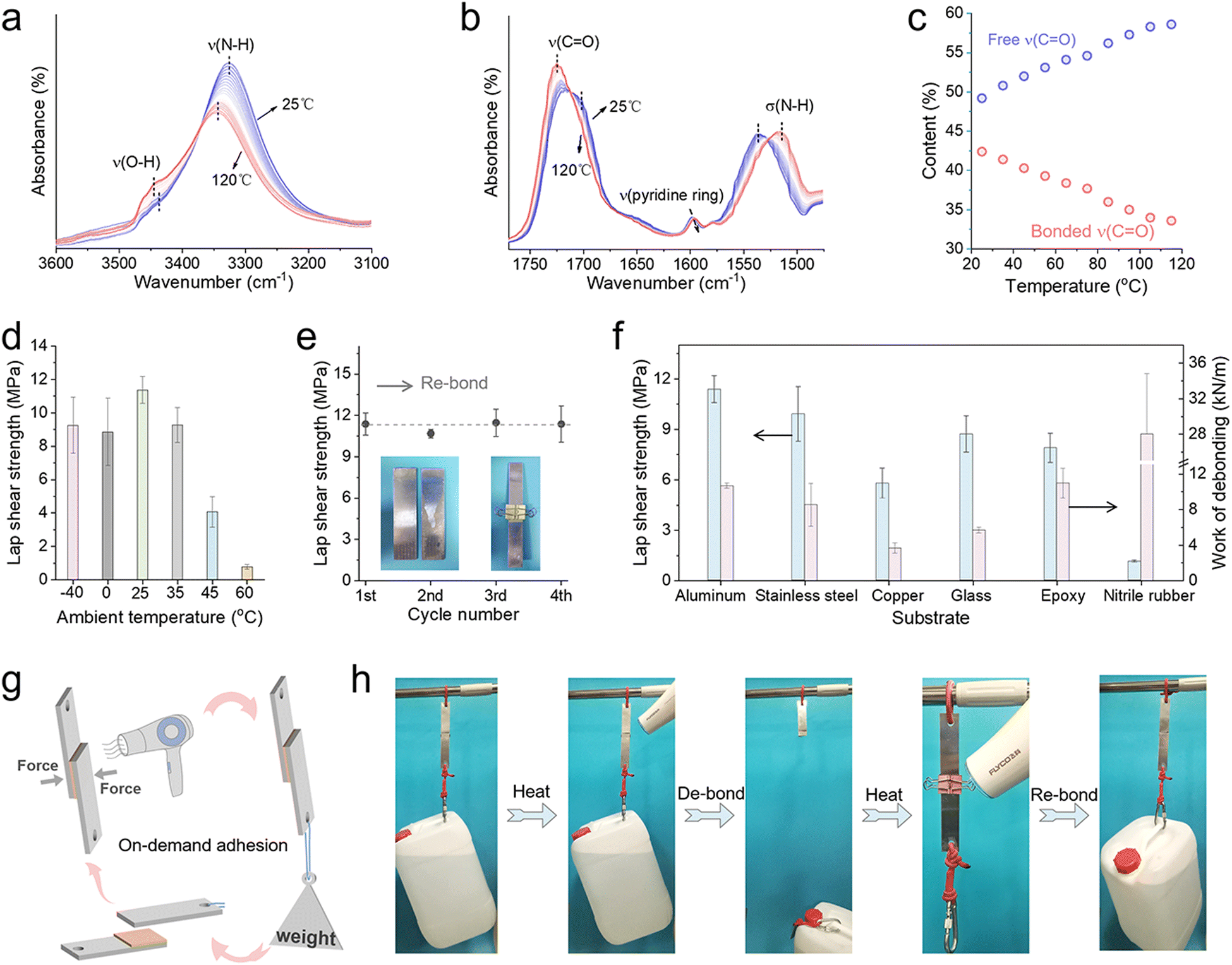 | ||
| Fig. 4 Reversible adhesion and debonding behavior of the polyurethane elastomer. Temperature-dependent Fourier transform infrared spectra of the PUD20 upon heating from 25–120 °C: (a) spectra from 3100–3600 cm−1; (b) spectra from 1450–1800 cm−1. (c) Content of H-bonded CO and free CO as a function of temperature from 25–115 °C. (d) Lap shear strength of the PUD20 adhesive at different ambient temperatures. (e) Lap shear strength of the PUD20 adhesive after cycling adhesion and debonding. (f) Lap shear strength of the PUD20 adhesive using different substrates. (g) Schematic image and the (h) corresponding photographs showing reversible adhesion and debonding of the PUD20 adhesive. | ||
3. Conclusions
In summary, this study reports a strong and ductile polyurethane adhesive that exhibits a distinctive debonding phenomenon of interfacial failure. Its lap shear strength and work of debonding are 11.37 Mpa and 10.32 kN m−1, respectively, which are superior to those of most of the reported tough adhesives exhibiting the phenomenon of cohesive failure. Attributed to its characteristic interfacial-failure behavior, this polyurethane adhesive can be conveniently detached from the substrates, with no adhesive remaining on the targets. Moreover, it can be reversibly adhered to and detached from the sandwiched substrates by on-demand control of ambient temperature. This kind of elastomer hot melt can be regarded as a new member of the family of strong and ductile adhesives, which will undoubtedly bring a lot of convenience to the construction and removal of the adhesives. Its molecular design and the mechano-responsive trait will open more opportunities for inspiring synthesizing modern and high-performance adhesives that are reliable in terms of operation and easy to be disassembled.4. Experimental section
4.1. Materials
Dopamine hydrochloride (DA·HCl) and trifluoroacetic acid (TFA) were purchased from Macklin. 2,2-Bis(hydroxymethyl)propionic acid (DMPA) and 2,2-dimethoxypropane (DMP) were purchased from Aladdin. N-Hydroxysuccinimide (NHS), p-toluenesulfonic acid (TsOH), triethylamine and ammonium hydroxide were purchased from Adamas. N-[3-(Dimethylamino)propyl]-N′-ethylcarbodiimide hydrochloride (EDC-HCl), sodium sulfate anhydrous, sodium bicarbonate and sodium chloride were purchased from Bide Pharmatech Ltd. Polytetramethylene ether glycol (PTMEG, Mn = 1000 g mol−1), dibutyltin dilaurate (DBTDL), isophorone diisocyanate (IPDI) and 2,6-pyridinedimethanol (PDM) were purchased from Adamas. All these chemical reagents were used without further purification. All solvents used in the experiment, such as acetone, methanol, ethanol, anhydrous tetrahydrofuran (THF), N,N-dimethylformamide (DMF) and dichloromethane (DCM) were purchased from Adamas.4.2. Synthesis of elastomer adhesive
The synthesis details of the chain extender (DMPA-DA) are provided in the ESI.† Before the synthesis of the elastomer adhesive, IPDI, PTMEG, PDM and DMPA-DA were first dried under vacuum at 70 °C overnight. Afterwards, PTMEG (4 g, 4 mmol) was added to a dried glass vessel equipped with a mechanical stirrer and a thermometer, and it was heated at 80 °C under nitrogen. Subsequently, IPDI (2.4 g, 10.8 mmol) and DBTDL (0.007 g) were successively added to the vessel and stirred at 80 °C for 3 h to obtain the NCO-terminated polyurethane prepolymer. Then, 15 mL of anhydrous THF was added to adjust the viscosity, and the PDM powder (0.76 g, 5.44 mmol) was directly added as the chain extender. After reacting for 3 h at 60 °C, 15 mL of an anhydrous THF solution of DMPA-DA (0.37 g, 1.36 mmol) was added and reacted for 4 h. The mixed solution was quickly transferred to a vacuum environment at 60 °C for 20 h. Finally, the bulk was dissolved in DCM and decanted into a rectangular mold to vaporize the solvent in a fume hood under ambient conditions for 48 h. The residual solvent was further removed in a vacuum oven at 60 °C for 24 h to obtain a polyurethane adhesive film (i.e., PUD20), where the value of 20 refers to the specific percentage of DMPA-DA in the total chain extender. Other adhesives, such as PUD10, PUD40, PUD60 and PUD100, were synthesized by identical processes except for the different molar ratios of PDM and DMPA-DA.4.3. Synthesis of the control sample (PU)
IPDI, PTMEG and PDM were dried under vacuum at 70 °C overnight. PTMEG (4 g, 4 mmol) was added to a dried glass vessel equipped with a mechanical stirrer and a thermometer, and it was heated at 80 °C under nitrogen. Subsequently, IPDI (2.4 g, 10.8 mmol) and DBTDL (0.007 g) were successively added to the vessel and stirred at 80 °C for 3 h to obtain the NCO-terminated polyurethane prepolymer. Then, 30 mL of anhydrous DMF was added to adjust the viscosity, and the PDM powder (0.95 g, 6.8 mmol) was directly added as the chain extender. After reacting for 8 h at 80 °C. Finally, the mixed solution was decanted into a rectangular mold and the solvent was vaporized at 80 °C for 48 h. The residual solvent was further removed in a vacuum oven at 80 °C for 24 h to obtain the control sample film (PU).4.4. Characterization
Details are available in the ESI.†Author contributions
K. Wu and C. Li co-designed the experiments. C. Li and W. Dong synthesized the extender agent. C. Li synthesized the elastomers and performed the experimental tests. C. Li and K. Wu analyzed and organized the data. L. Li, Z. Dou, Y. Li, L. Wei, and Q. Zhang helped with some experimental suggestions. K. Wu wrote the draft manuscript and revised the manuscript. Q. Zhang and K. Wu co-supervised the study.Conflicts of interest
The authors declare no competing financial or non-financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52103091), the National Key Research and Development Project of China (Grant No. 2022YFB3806900), and the State Key Laboratory of Polymer Materials Engineering (Grant No. sklpme2022-3-15).Notes and references
- E. A. Appel and O. A. Scherman, Nat. Mater., 2014, 13, 231–232 CrossRef CAS PubMed.
- L. M. Stapleton, A. N. Steele, H. Wang, H. Lopez Hernandez, A. C. Yu, M. J. Paulsen, A. A. A. Smith, G. A. Roth, A. D. Thakore, H. J. Lucian, K. P. Totherow, S. W. Baker, Y. Tada, J. M. Farry, A. Eskandari, C. E. Hironaka, K. J. Jaatinen, K. M. Williams, H. Bergamasco, C. Marschel, B. Chadwick, F. Grady, M. Ma, E. A. Appel and Y. J. Woo, Nat. Biomed. Eng., 2019, 3, 611–620 CrossRef CAS PubMed.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Chem. Rev., 2022, 122, 5604–5640 CrossRef CAS PubMed.
- M. M. Hasani-Sadrabadi, P. Sarrion, S. Pouraghaei, Y. Chau, S. Ansari, S. Li, T. Aghaloo and A. Moshaverinia, Sci. Transl. Med., 2020, 12, eaay6853 CrossRef CAS PubMed.
- F. Ershad, A. Thukral, J. Yue, P. Comeaux, Y. Lu, H. Shim, K. Sim, N.-I. Kim, Z. Rao, R. Guevara, L. Contreras, F. Pan, Y. Zhang, Y.-S. Guan, P. Yang, X. Wang, P. Wang, X. Wu and C. Yu, Nat. Commun., 2020, 11, 3823 CrossRef CAS PubMed.
- S. Baik, H. J. Lee, D. W. Kim, J. W. Kim, Y. Lee and C. Pang, Adv. Mater., 2019, 31, 1803309 CrossRef PubMed.
- Y. Zhang, Z. Chen, H. Zheng, R. Chen, C. Ma and G. Zhang, Adv. Sci., 2022, 9, 2200268 CrossRef CAS PubMed.
- R. Chen, Y. Zhang, Q. Xie, Z. Chen, C. Ma and G. Zhang, Adv. Funct. Mater., 2021, 31, 2011145 CrossRef CAS.
- Y. Liu, P. Wang, X. Su, L. Xu, Z. Tian, H. Wang, G. Ji and J. Huang, Adv. Mater., 2022, 34, 2108820 CrossRef CAS PubMed.
- Z. H. Zhao, P. C. Zhao, Y. Zhao, J. L. Zuo and C. H. Li, Adv. Funct. Mater., 2022, 32, 2201959 CrossRef CAS.
- Y. Yao, Z. Xu, B. Liu, M. Xiao, J. Yang and W. Liu, Adv. Funct. Mater., 2021, 31, 2006944 CrossRef CAS.
- S. Chen, Z. Li, Y. Wu, N. Mahmood, F. Lortie, J. Bernard, W. H. Binder and J. Zhu, Angew. Chem., Int. Ed., 2022, 134, e202203876 Search PubMed.
- J. Sun, L. Xiao, B. Li, K. Zhao, Z. Wang, Y. Zhou, C. Ma, J. Li, H. Zhang, A. Herrmann and K. Liu, Angew. Chem., Int. Ed., 2021, 60, 23687–23694 CrossRef CAS PubMed.
- W. Wang, Y. Li, H. Zhang, T. Chen, G. Sun, Y. Han and J. Li, Biomacromolecules, 2022, 23, 779–788 CrossRef CAS PubMed.
- P. Sun, Y. Li, B. Qin, J.-F. Xu and X. Zhang, ACS Mater. Lett., 2021, 3, 1003–1009 CrossRef CAS.
- G. Gao, F. Yang, F. Zhou, J. He, W. Lu, P. Xiao, H. Yan, C. Pan, T. Chen and Z. L. Wang, Adv. Mater., 2020, 32, 2004290 CrossRef CAS PubMed.
- J. Li, A. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. Seo, N. Vasilyev, J. Vlassak and Z. Suo, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- M. G. Mazzotta, A. A. Putnam, M. A. North and J. J. Wilker, J. Am. Chem. Soc., 2020, 142, 4762–4768 CrossRef CAS PubMed.
- L.-B. Mao, H.-L. Gao, H.-B. Yao, L. Liu, H. Cölfen, G. Liu, S.-M. Chen, S.-K. Li, Y.-X. Yan, Y.-Y. Liu and S.-H. Yu, Science, 2016, 354, 107–110 CrossRef CAS PubMed.
- J. Xu, T. Liu, Y. Zhang, Y. Zhang, K. Wu, C. Lei, Q. Fu and J. Fu, Matter, 2021, 4, 2474–2489 CrossRef CAS.
- U. G. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- H. Le Ferrand, F. Bouville, T. P. Niebel and A. R. Studart, Nat. Mater., 2015, 14, 1172–1179 CrossRef CAS PubMed.
- Z. Li, Y.-L. Zhu, W. Niu, X. Yang, Z. Jiang, Z.-Y. Lu, X. Liu and J. Sun, Adv. Mater., 2021, 33, 2101498 CrossRef CAS PubMed.
- Z. Yin, F. Hannard and F. Barthelat, Science, 2019, 364, 1260–1263 CrossRef CAS PubMed.
- Y. Oaki and H. Imai, Angew. Chem., Int. Ed., 2005, 44, 6571–6575 CrossRef CAS PubMed.
- X. Zhou, B. Guo, L. Zhang and G.-H. Hu, Chem. Soc. Rev., 2017, 46, 6301–6329 RSC.
- L. Eisoldt, A. Smith and T. Scheibel, Mater. Today, 2011, 14, 80–86 CrossRef CAS.
- A. Rising and J. Johansson, Nat. Chem. Biol., 2015, 11, 309–315 CrossRef CAS PubMed.
- F. Sun, L. Liu, T. Liu, X. Wang, Q. Qi, Z. Hang, K. Chen, J. Xu and J. Fu, Nat. Commun., 2023, 14, 130 CrossRef CAS PubMed.
- M. A. Rahman, C. Bowland, S. Ge, S. R. Acharya, S. Kim, V. R. Cooper, X. C. Chen, S. Irle, A. P. Sokolov and A. Savara, Sci. Adv., 2021, 7, eabk2451 CrossRef CAS PubMed.
- H. Chen, H. Lin, Z. Sun, H. Li, C. He and D. Mao, J. Mater. Chem. A, 2023, 11, 2443–2451 RSC.
- J. Zhang, Z. Chen, Y. Zhang, S. Dong, Y. Chen and S. Zhang, Adv. Mater., 2021, 33, 2100962 CrossRef CAS PubMed.
- Y. Li, W. Li, A. Sun, M. Jing, X. Liu, L. Wei, K. Wu and Q. Fu, Mater. Horiz., 2021, 8, 267–275 RSC.
- S. Li, J. Liu, Z. Wei, Q. Cui, X. Yang, Y. Yang and X. Zhang, Adv. Funct. Mater., 2023, 33, 2210441 CrossRef CAS.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Science, 2021, 372, 1078–1081 CrossRef CAS PubMed.
- C. B. Cooper, S. Nikzad, H. Yan, Y. Ochiai, J.-C. Lai, Z. Yu, G. Chen, J. Kang and Z. Bao, ACS Cent. Sci., 2021, 7, 1657–1667 CrossRef CAS PubMed.
- H. Xiong, T. Yue, Q. Wu, L. Zhang, Z. Xie, J. Liu, L. Zhang and J. Wu, Mater. Horiz., 2023, 10, 2128–2138 RSC.
- T. Guan, X. Wang, Y.-L. Zhu, L. Qian, Z. Lu, Y. Men, J. Li, Y. Wang and J. Sun, Macromolecules, 2022, 55, 5816–5825 CrossRef CAS.
- Y. Wang, S. Sun and P. Wu, Adv. Funct. Mater., 2021, 31, 2101494 CrossRef CAS.
- W. Zhang, B. Wu, S. Sun and P. Wu, Nat. Commun., 2021, 12, 4082 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00966a |
| This journal is © The Royal Society of Chemistry 2023 |