DOI:
10.1039/D5SC02937F
(Edge Article)
Chem. Sci., 2025,
16, 10714-10721
iso-TEtraQuinoline (i-TEQ): an inherently chiral N4 macrocyclic quinoline tetramer†
Received
22nd April 2025
, Accepted 15th May 2025
First published on 20th May 2025
Abstract
Chiral macrocycles are attracting growing interest due to their broad applicability as ligands in asymmetric catalysis and as host molecules for chiral recognition. Robustness and high thermodynamic stability can be effectively achieved by strategically linking aromatic panels to construct an axially chiral macrocyclic framework. Cyclic concatenation of four quinoline units affords a fully sp2-hybridized, non-planar macrocycle featuring four inwardly oriented, coordinatively active pyridyl nitrogen atoms. The previously reported tetramer TEtraQuinoline (TEQ) exhibits head-to-tail connectivity of its quinoline units, affording an achiral architecture possessing S4 symmetry. Herein, we report the design and synthesis of iso-TEtraQuinoline (i-TEQ), an inherently chiral analogue featuring head-to-head connectivity at the 2,2′- and 8,8′-positions, which gives rise to a D2-symmetric architecture. Detailed comparative investigations of the connectivity isomers i-TEQ and TEQ revealed an array of distinct characteristics, including the overall architecture, intrinsic macrocyclic strain, spatial orientation of nitrogen lone pairs, thermodynamic stability, racemisation behaviour, metal complex stability, and circularly polarised luminescence.
Introduction
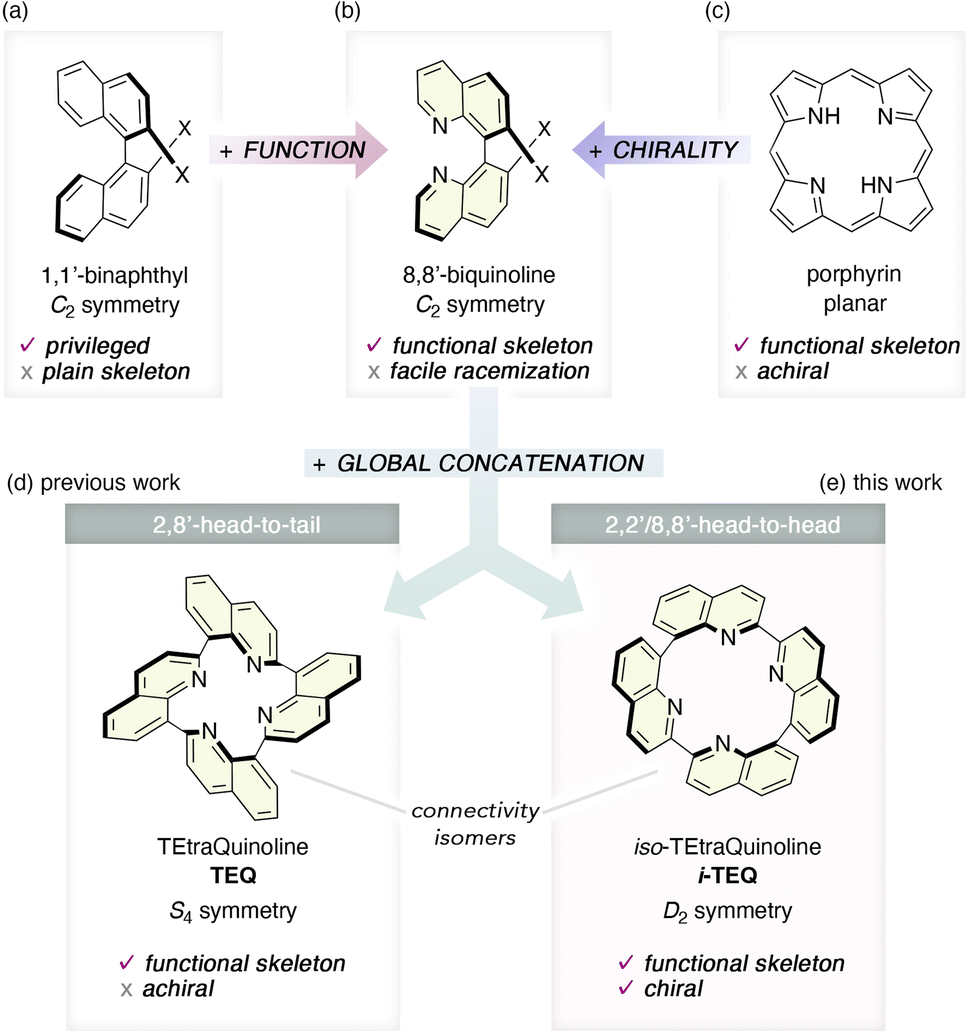
The chemical space occupied by axially chiral entities has remarkably expanded over the past several decades owing to their diverse applications as chiral ligands,1 chiral sensing materials,2 and biologically active natural or artificial products.3 Due to their high thermodynamic stability and structural robustness, axially chiral molecules composed of sp2-hybridized aromatic units have attracted considerable attention for the construction of well-defined chiral environments in catalysis and sensing applications. The 1,1′-binaphthyl unit represents a privileged scaffold for this purpose and is extensively utilized in catalyst design by incorporating chalcogen and pnictogen atoms at 2,2′-positions to promote enantioselective metal- and organocatalysis, and molecular recognition4 (Fig. 1a). In contrast, the 8,8′-biquinoline framework, which is structurally analogous to the 1,1′-binaphthyl but features an N(sp2) atom in place of one C(sp2)–H unit, is utilized only sparingly due to its relatively low racemisation barrier5 (Fig. 1b). Incorporating the quinoline unit is particularly advantageous, as it not only imparts axially chirality but also introduces a functional motif capable of coordinating metal cations, thereby offering dual functionality in a single framework. Inspired by porphyrin—a representative functional framework ubiquitous in nature that coordinates metal cations through four sp2-hybridized nitrogen atoms—we previously designed a tetrameric quinoline construct, TEtraQuinoline (TEQ), as a non-planar analogue of porphyrin.6 While its cyclic concatenation effectively suppresses the flipping of the linked quinoline units, the head-to-tail assembly through the 2- and 8-positions imparts an overall achiral, S4-symmetric rigid architecture to the TEQ framework. Given the diverse chemistry exhibited by TEQ in metal coordination, photophysical behaviour, and catalytic applications,7 the development of its chiral variant is highly desirable. Herein, we report the re-design of a quinoline-based, axially chiral molecular framework that functions as a tetradentate nitrogen ligand: iso-TEtraQuinoline (i-TEQ).8 By switching the ring connectivity from head-to-tail to head-to-head at the 2,2′- and 8,8′-positions, i-TEQ adopts a D2-symmetric, inherently chiral architecture.9 Detailed comparative structural analyses uncovered similarities and differences between these connectivity isomers, highlighting the impact of the concatenation pattern on the chemical and photophysical properties.
 |
| | Fig. 1 Hierarchical structural modulation with respect to axial chirality and intrinsic functionality. (a–c) Structure and properties of 1,1′-binaphthyl, 8,8′-biquinoline, and porphyrin. (d) Previously studied TEtraQuinoline (TEQ) and its architectural features. (e) The newly developed macrocyclic quinoline framework, iso-TEtraQuinoline (i-TEQ), embodies both intrinsic functionality and inherent chirality. | |
Results and discussion
Synthesis of iso-TEtraQuinolines (i-TEQs)
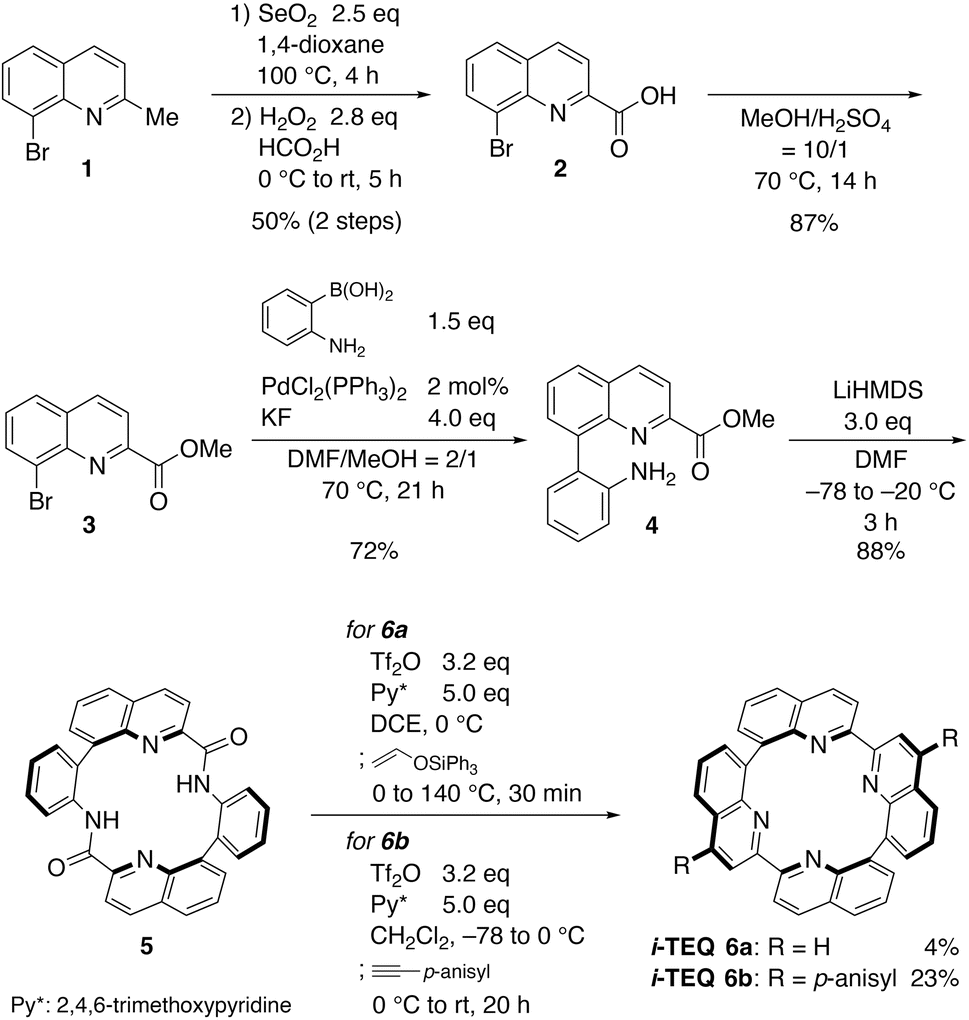
Inspired by the TEQ framework, we designed a synthetic plan utilizing cyclic diamide 5 with two quinoline units as a key precursor to i-TEQvia a two-fold quinoline forming reaction (Scheme 1). The synthesis of i-TEQ was initiated by a two-step benzylic oxidation of 8-bromo-2-methylquinoline 1 mediated by SeO2 and H2O2/HCO2H to afford carboxylic acid 2. After converting 2 into methyl ester 3 with H2SO4 in MeOH, Suzuki–Miyaura cross-coupling with (2-amino)phenylboronic acid afforded aminoester 4.10 Following brief screening,11 simple cryogenic and highly diluted conditions with LiHMDS engaged 4 in smooth amidative dimerization, furnishing the requisite cyclic diamide 5 in 88% yield. According to Movassaghi's protocol12 and our previous modification for quinoline formation from macrocyclic amides,6a,h,i electrophilic activation of the amide functionality using Tf2O and 2,4,6-trimethoxypyridine and subsequent formal cycloaddition afforded the i-TEQ architecture. Quinoline formation with (triphenylsiloxy)ethylene furnished non-substituted D2-symmetric i-TEQ6a, but the yield was only 4%. More reactive p-anisylacetylene allowed for a more efficient ring reinforcement reaction to give C2-symmetric bis-p-anisylated i-TEQ6b in 23% yield. The p-anisyl groups of i-TEQ6b can be smoothly demethylated, enabling further structural elaboration.13
 |
| | Scheme 1 Synthesis of i-TEQ6a, 6b. | |
A detailed comparative structural analysis was performed on the thus-obtained i-TEQ6b—a connectivity isomer of p-anisylated TEQ7b sharing the same atomic composition (Fig. 2). To minimize structural deviations caused by crystal packing effects, DFT-optimized structures of both isomers were compared using the B3LYP-D3/6-31G(d,p) level of theory with the IEFPCM(CHCl3) solvation model (Fig. 2a).14 Comparison of the top views of these structures revealed a distinct difference in the inner hollow topology: i-TEQ6b features a diamond-shaped 16-membered macrocycle, while TEQ7b exhibits a square-like counterpart. In contrast, the spatial arrangement of the four nitrogen atoms is similar, with the distances between diagonal nitrogen atoms being nearly identical, ranging from 4.35 to 4.47 Å. Additionally, the τ′4 values15 are close to zero, suggesting that i-TEQ6b, like TEQ7b, is capable of accommodating metal cations that prefer a square planar coordination mode (Fig. 2a and b). The diamond-shaped cavity of i-TEQ6b arises from the head-to-head (2,2′ and 8,8′) linkage of the quinoline units, leading to an overall laterally distorted structure characterized by reduced dihedral angles and an increased vertical dimension (i-TEQ6b: 6.56 Å vs.TEQ7b: 6.01 Å). Whereas the D2-symmetric i-TEQ skeleton is more stable than the S4-symmetric TEQ skeleton (ΔG = 2.2 kcal mol−1), enthalpic evaluation via a homodesmotic reaction16 and StrainViz analysis17 indicated that i-TEQ adopts a more strained architecture (difference in total strain: 9.0 kcal mol−1), with the local structures of the intrinsic quinoline units being more distorted (Fig. 2b–d). This seemingly contradictory outcome is attributed to electronic repulsion between the lone pairs on the inwardly oriented nitrogen atoms; those of i-TEQ are oriented in a mutually more skewed fashion to prevent undesired overlapping. This electronic factor likely outweighs the skeletal distortion, rendering i-TEQ more thermodynamically stable than TEQ, as supported by TG-DTA analysis; i-TEQ6b remains stable above 300 °C, whereas TEQ7b begins to decompose above 250 °C.18
 |
| | Fig. 2 Structural comparison of connectivity isomers i-TEQ6b and TEQ7b sharing a tetrameric quinoline macrocyclic architecture and two peripheral diagonal p-anisyl groups. (a) Optimized structures are calculated at the B3LYP-D3/6-31G(d,p)/IEFPCM(CHCl3) level of theory. Colour codes; carbon: grey, nitrogen: blue, oxygen: red, hydrogen atoms are omitted for clarity. aVertical size of the main framework is calculated by the distance between the midpoint of the two quinoline C5 carbons on the upper side and the midpoint of the two quinoline C5 carbons on the lower side. (b) Analysis of local structures. bLSP: least squares plane. More detailed analysis is summarized in ESI.† (c) StrainViz analysis and NBO of nitrogen lone pairs. StrainViz calculation was carried out at the B3LYP/6-31G(d) level of theory. (d) Comparison of Gibbs free energy for the formation of non-substituted i-TEQ6a and TEQ7a. | |
Physicochemical properties of i-TEQs
The structural rigidity of the i-TEQ framework, a cyclic assembly of four quinoline panels with a hitherto unknown pattern of connectivity, is great of interest. Racemic samples of D2-symmetric non-substituted i-TEQ6a and C2-symmetric p-anisylated i-TEQ6b were optically resolved via preparative HPLC on a chiral stationary phase (Fig. 3a). 6a and 6b were successfully resolved with base-peak separation under identical conditions (DAICEL CHIRALPAK IB-N5, nhexane/CH2Cl2 mixed solvent eluent). The initially eluted enantiomers of both 6a and 6b displayed a positive Cotton effect in a range of 330–350 nm, corresponding to HOMO–LUMO excitation (Fig. 3b).19 DFT simulations of electronic CD spectra suggested the absolute configuration shown in Fig. 3a.20 Intriguingly, optical rotation of the first eluents of 6a and 6b were opposite (6a: dextrorotatory, 6b: levorotatory), presumably because the closely located diagonal p-anisyl groups of 6b affected optical rotation at the wavelength of sodium D-lines.
 |
| | Fig. 3 (a) Partial HPLC chromatogram for optical resolution of rac-i-TEQ6a and 6b separated by DAICEL CHIRALPAK IB-N5 column with nhexane/CH2Cl2 eluent. (b) Circular dichroism (CD) spectra of optically pure samples of non-substituted (+)- and (−)-i-TEQ6a, and anisylated (+)- and (−)-i-TEQ6b in CH2Cl2 (25 μM). | |
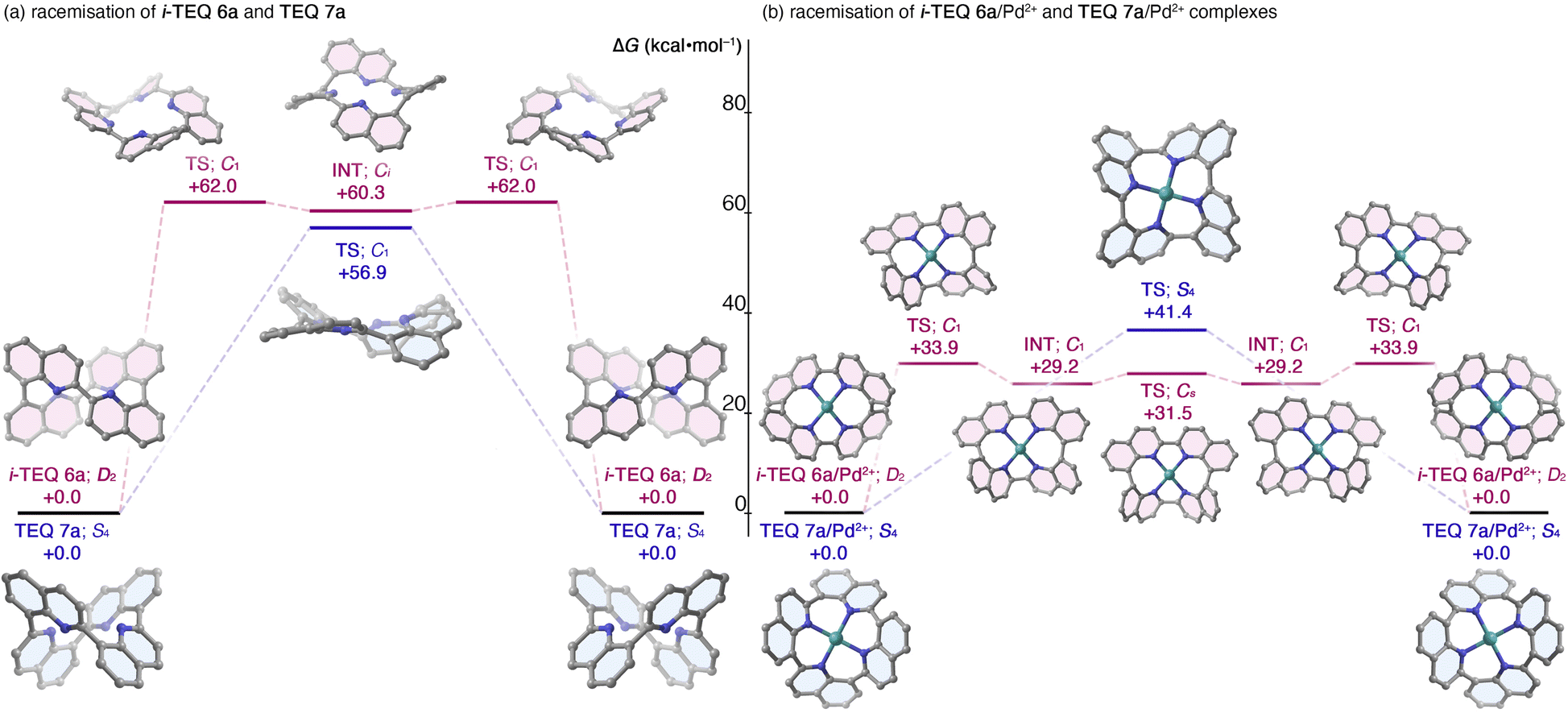
The stereochemical integrity of (−)-i-TEQ6b was traced at a 1 mg mL−1 concentration in diphenyl ether under various conditions (Table 1). The initial racemisation attempt without any additive confirmed that the stereochemistry of the i-TEQ architecture is quite stable; no decomposition or erosion of enantiopurity was observed even at 220 °C for 24 h (Entry 1). DFT calculations on the non-substituted i-TEQ6a successfully reproduced the experimentally observed high racemisation barrier, yielding an inversion barrier of 58.0 kcal mol−1 for the transition state at the B3LYP-D3/6-311+G(2d,p)/SMD(diethyl ether) level of theory (Fig. 4a). Given that structurally similar acyclic azaBINOL is readily racemised (28.4 kcal mol−1),5a the unusual retention of axial chirality of i-TEQ is likely due to its unique concatenated structure. Notably, the inversion barrier of i-TEQ6a is even higher than that of the configurationally stable TEQ7a (56.9 kcal mol−1 at the B3LYP-D3/6-31G(d,p) level of theory), and the inversion process proceeds via a stepwise mechanism involving a Ci-symmetric intermediate, in contrast to the single-step flipping observed for TEQ7a (Fig. 4a). We reasoned that mitigating the repulsive orbital interactions of the four inwardly oriented nitrogen atoms enhanced the racemisation. While racemisation of (−)-i-TEQ6b barely proceeded in the presence of 1 equivalent of trifluoroacetic acid (TFA) at 140 °C, a higher temperature (160 °C) induced slow the racemisation (t1/2 = ca. 37 days) (Table 1, Entries 2,3). Quinoline ring flipping was significantly accelerated by the addition of [Pd(MeCN)4](BF4)2 with t1/2 of 2.2 h at 140 °C (20% ee after 2.5 h), which corresponds to an empirically determined flipping barrier of 32.1 kcal mol−1 (Entries 4,5). Intriguingly, p-anisylated (−)-TEQ7b, featuring head-to-tail concatenation, was significantly more reluctant to racemise and remained at 88% ee even after 24 h of stirring under identical conditions (with Pd salt at 140 °C).21 This distinct response to the Pd2+ cation was reproduced by DFT calculations (Fig. 4b); the pathway from the i-TEQ6a/Pd2+ complex to its antipode involves a series of local minima connected by partially flipped C1- and Cs-symmetric transition states, which significantly lower the inversion barrier. In contrast, the TEQ7a/Pd2+ complex undergoes a high-energy S4-symmetric transition state to achieve stereoinversion. Other azophilic metal cations featuring a square planar coordination mode, e.g. Co2+ or Cu2+ rendered racemisation, whereas acceleration was significantly smaller than that of Pd2+ and no racemization occurred at a temperature not exceeding 100 °C (Table 1, Entries 6–9). A plot of enantiomeric excess and elapsed time clearly demonstrates the substantial rigidity and characteristic response to Pd2+ in the racemisation of (−)-i-TEQ6b (Fig. 5). DFT calculations located energetically more favourable transition states by protonation or metal coordination.22
Table 1 Racemisation study of i-TEQ6b in the absence or presence of additives
|

|
| Entry |
Additivea |
Temp. (°C) |
Time (h) |
eeb (%) |
Rate constant (× 10–7 s–1) |
t
1/2 (h) |
ΔG‡exp (kcal·mol–1) |
ΔG‡calcc (kcal·mol–1) |
|
1 Equivalent of additives was added.
Determined by chiral stationary phase HPLC analysis.
ΔG‡calc was calculated on non-substituted i-TEQ6a at the B3LYP-D3/6-311+G(2d,p)-SDD(Co,Cu,Pd)/SMD(diethyl ether) level of theory. Compared with Fig. 4, a more extended basis set was employed and an appropriate solvation model was applied.
No racemisation occurred.
Calculated on the monoprotonated model.
Calculated on the diprotonated model.
|
| 1 |
None |
220 |
24 |
>99 |
—d |
— |
— |
58.0 |
| 2 |
TFA |
140 |
24 |
>99 |
—d |
— |
— |
50.8e, 46.3f |
| 3 |
160 |
56 |
91 |
2.18 |
883 |
38.9 |
| 4 |
[Pd(MeCN)4](BF4)2 |
100 |
30 |
63 |
20.2 |
95.3 |
31.8 |
33.3 |
| 5 |
140 |
2.5 |
20 |
888 |
2.2 |
32.1 |
| 6 |
CoCl2 |
100 |
24 |
>99 |
—d |
— |
— |
30.6 |
| 7 |
140 |
56 |
88 |
2.99 |
644 |
36.8 |
| 8 |
CuCl2 |
100 |
24 |
>99 |
—d |
— |
— |
34.4 |
| 9 |
140 |
30 |
72 |
14.8 |
130 |
35.5 |
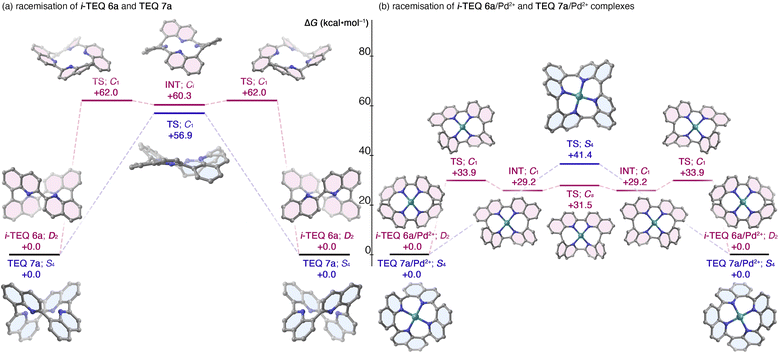 |
| | Fig. 4 Stereoinversion of non-substituted quinoline tetramers in the absence and presence of Pd2+ cation. Calculated at the B3LYP-D3/6-31G(d,p)-SDD(Pd) level of theory. (a) i-TEQ6a and TEQ7a, and (b) i-TEQ6a/Pd2+ complex and TEQ7a/Pd2+ complexes. | |
 |
| | Fig. 5 Racemisation profile of enantiopure (−)-i-TEQ6b in the presence of additives (1 eq) at 140 °C in diphenyl ether (1 mg mL−1). A plot of enantiomeric excess (%) versus time is shown. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. | |
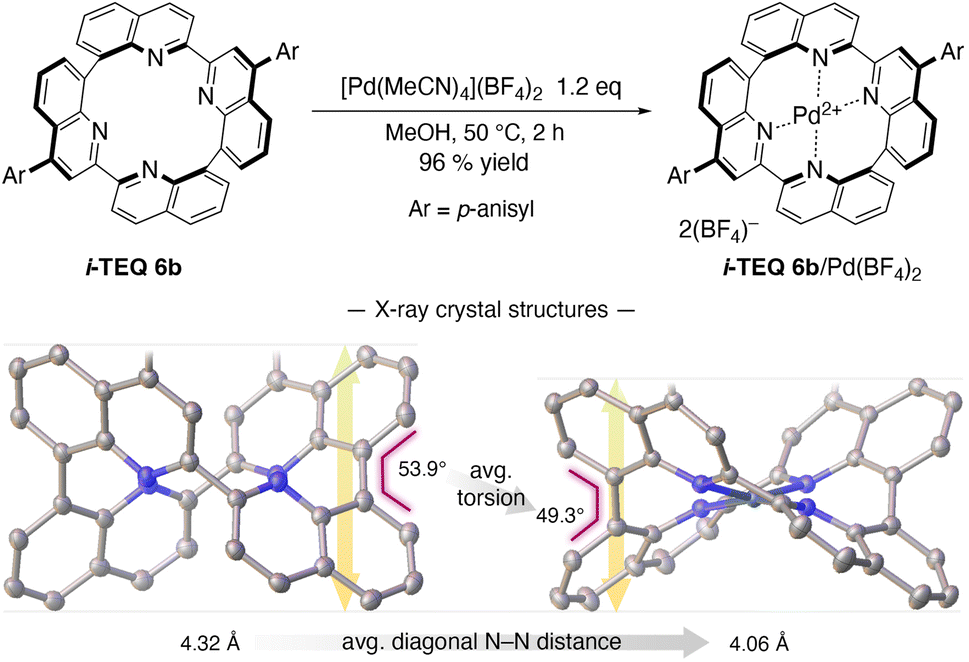
The accelerated racemisation of (−)-i-TEQ6b in the presence of Pd2+ demonstrates that i-TEQ is capable of accommodating Pd2+ cations despite the highly twisted configuration of the lone pair orbitals of the four quinoline nitrogen atoms (Fig. 2c). Indeed, treatment of i-TEQ6b with [Pd(MeCN)4](BF4)2 in MeOH at 50 °C furnished the i-TEQ6b/Pd(BF4)2 complex in excellent yield (Scheme 2). Comparative X-ray diffraction analysis of the crystal structures of i-TEQ6b and i-TEQ6b/Pd(BF4)2 revealed that both the averaged torsion angle at 8,8′-biquinoline units and the averaged distance of the diagonal nitrogen atoms decreased upon complexation from 53.9° to 49.3° and from 4.32 to 4.06 Å, respectively, leading to a vertically collapsed architecture.
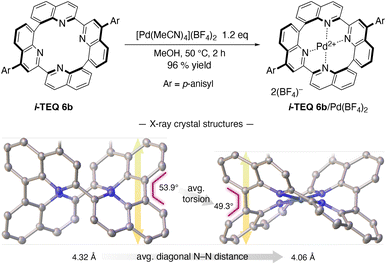 |
| | Scheme 2 Complexation of i-TEQ6b with [Pd(MeCN4)](BF4)2, and crystal structures of the 6b and 6b/Pd(BF4)2 complex. ORTEP drawings are shown at 50% probability ellipsoids. Colour codes; carbon: grey, nitrogen: blue, oxygen: red, palladium: dark blue. Hydrogen atoms, p-anisyl substituents, and counter anions were omitted for clarity. | |
To probe the thermodynamic stability of the Pd2+ complexes of i-TEQ6b and TEQ7b, structural isomers with an identical elemental composition, a ligand metathesis experiment was conducted (Scheme 3). The i-TEQ6b/Pd(BF4)2 complex and metal-free TEQ7b were mixed in CDCl3 and heated under reflux. 1H NMR revealed that the signals derived from both the i-TEQ6b/Pd(BF4)2 complex and the TEQ7b completely disappeared (Scheme 3, (i,ii) vs. (iii)). Instead, newly observed peaks corresponded to the free forms of i-TEQ6b and the TEQ7b/Pd(BF4)2 complex (Scheme 3, (iii) vs. (iv, v)). In contrast, under identical conditions, a mixture of the free form of i-TEQ6b with the TEQ7b/Pd(BF4)2 complex displayed virtually no change in 1H NMR analysis, confirming the unidirectional Pd2+ cation swapping from i-TEQ6b to TEQ7b (Scheme 3 (iv,v) vs. (vi)). The empirically observed higher stability of the Pd2+ complex of TEQ7b over that of i-TEQ6b was reproduced by DFT calculations at the B3LYP-D3/6-31G(d,p)-SDD(Pd)/IEFPCM(CHCl3) level of theory; [i-TEQ6b + TEQ7b/Pd2+] is 27.7 kcal mol−1 more stable than [i-TEQ6b/Pd2+ + TEQ7b].
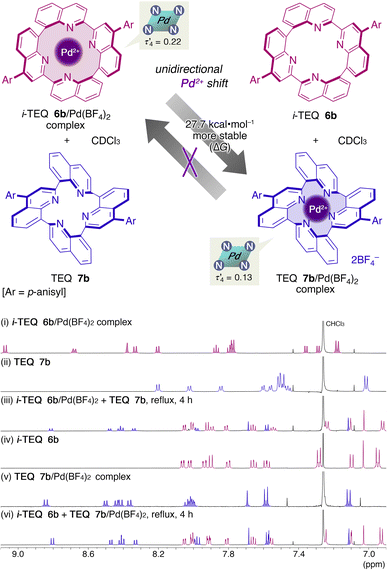 |
| | Scheme 3 Unidirectional Pd2+ cation swapping between i-TEQ6b and TEQ7b. Partial 1H NMR spectra in CDCl3 (i) i-TEQ6b/Pd(BF4)2; (ii) TEQ7b; (iii) i-TEQ6b/Pd(BF4)2 + TEQ7b at reflux temperature after 4 h; (iv) i-TEQ6b; (v) TEQ7b/Pd(BF4)2; (vi) TEQ7b/Pd(BF4)2 + i-TEQ6b at reflux temperature after 4 h. | |
The photophysical properties of i-TEQ6b were systematically investigated. Although i-TEQ6b is inherently barely emissive, the addition of TFA induced a pronounced fluorescence enhancement in an equivalence-dependent manner (Fig. 6). In CH2Cl2, the fluorescence intensity of i-TEQ6b increased progressively with the incremental addition of TFA, reaching a plateau at 4 equivalents (460 nm, ΦF = 0.41). This acid-responsive fluorescence behaviour, combined with the inherent chirality of 6b, prompted us to explore its potential in circularly polarized luminescence (CPL). Enantiomerically pure samples of (+)-6b and (−)-6b exhibited mirror-image CPL spectra upon the addition of 4 equivalents of TFA, with an absolute luminescence dissymmetry factor (|glum|) of 4.95 × 10−3.23 Notably, this value is an order of magnitude greater than that of ring-connectivity isomer TEQ7b,6a highlighting the strong influence of the concatenation pattern on the resultant physicochemical properties.24
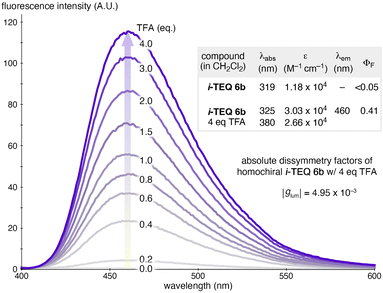 |
| | Fig. 6 Photophysical properties of i-TEQ6b. Fluorescence spectra of i-TEQ6b in CH2Cl2 (20 μM) were recorded upon incremental addition of TFA in CH2Cl2 (excitation at 385.5 nm). | |
Derivatization from cyclic diamide
Cyclic diamide 5, a key intermediate in the synthesis of i-TEQs, serves as a flexible N4 macrocyclic scaffold (Scheme 4). Partial reduction of the amide moiety of 5 using the Zr-based Schwartz reagent25 afforded diimine 8 in 54% yield. Reduction to secondary amines was achieved via Ir-catalysed silane conditions,26 affording tetradentate ligand 9. While electrophilic activation of the amide with Tf2O followed by formal cycloaddition with alkynes furnishes the quinoline ring en route to i-TEQs (vide supra), a two-fold intramolecular cyclization by the neighbouring quinoline proceeded in the absence of an external nucleophile to afford a warped, tetraaza dicationic π-material 10. A brief base screening identified electron-deficient 2-chloropyridine as optimal, providing 10 in 79% yield. 10 was readily reduced to furnish a more stable, neutral fused polycyclic material 11,27 whose unique three-dimensional structure was unambiguously determined by X-ray crystallographic analysis.
 |
| | Scheme 4 Derivatization from cyclic diamide 5. | |
Conclusions
Here, we describe the design and synthesis of a D2-symmetric iso-TEtraQuinoline (i-TEQ) as an inherently, three-dimensional analogue of porphyrin. i-TEQ is a head-to-head quinoline tetramer and a connectivity isomer of previously reported S4-symmetric, achiral TEQ, in which four quinoline units are concatenated in a head-to-tail fashion. Comprehensive and systematic comparable analyses of i-TEQ and TEQ revealed the unique features of these closely related molecular entities in terms of their overall architecture, stereochemical integrity, intrinsic ring strain, metal complexation abilities, and photophysical behaviour. Given its built-in chirality, i-TEQ represents a unique molecular scaffold that will serve as a cornerstone in the development of chiral porphyrinoids, thereby expanding the chemical space of porphyrin-based materials.
Data availability
Experimental and characterization data, including crystallographic data [5 (CCDC 2392427), 6b (CCDC 2392429), and 6d (CCDC 2392433), 6b/Pd(BF4)2 (CCDC 2392430), 11 (CCDC 2392428)], photophysical measurements, and NMR spectra, as well as computational investigations. The data supporting this article have been included as part of the ESI.†
Author contributions
N. K. and W. X. conceived and directed the project. R. Y. and W. X. synthesised, analysed, and characterised all compounds. T. K. performed theoretical calculations with partial support from N. K. Y. N. carried out CPL measurement and related data analysis. The draft manuscript was written by W. X. and N. K., and all the authors contributed to finalize manuscript though proofreading. All authors approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by KAKENHI grant JP22K19037 (Grant-in-Aid for Exploratory Research; to N. K.), JP23H01952 (Grant-in-Aid for Scientific Research (B); to N. K.), and MEXT KAKENHI grant 23H03809 (Grant-in-Aid for Transformative Research Areas (B); to N. K.). N. K. thanks the Mitsubishi Foundation and Mukai Science and Technology Foundation for financial support. W. X. thanks TOBE MAKI foundation for financial support. T. K. thanks Izumi Science and Technology Foundation for financial support. Dr Tomoyuki Kimura at the Institute of Microbial Chemistry is gratefully acknowledged for X-ray crystallographic analysis. Computational calculations were performed using the resources of the Research Center for the Computational Science at Okazaki, Japan (Project No. 23-IMS-C081, 24-IMS-C077).
Notes and references
-
(a) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed;
(b) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed;
(c) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 Search PubMed.
-
(a) L. Pu, Chem. Rev., 1998, 98, 2405–2494 CrossRef CAS PubMed;
(b) L. Pu, Acc. Chem. Res., 2012, 45, 150–163 CrossRef CAS PubMed.
-
(a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed;
(b) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005–7022 Search PubMed;
(c) M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904–2919 CrossRef CAS PubMed;
(d) B. A. Lanman, A. T. Parsons and S. G. Zech, Acc. Chem. Res., 2022, 55, 2892–2903 Search PubMed;
(e) Z. Wang, L. Meng, X. Liu, L. Zhang, Z. Yu and G. Wu, Eur. J. Med. Chem., 2022, 243, 114700 CrossRef CAS PubMed.
- For selected reviews on asymmetric catalysis with 1,1’-bi-2-naphthol (BINOL);
(a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 Search PubMed;
(b) L. Eberhardt, D. Armspach, J. Harrowfield and D. Matta, Chem. Soc. Rev., 2008, 37, 839–864 RSC;
(c) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077–2118 Search PubMed;
(d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 Search PubMed;
(e) L. Pu, Chem. Rev., 2024, 124, 6643–6689 Search PubMed . For lanthanum–lithium–BINOL (LLB) complexes;
(f) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187–2210 CrossRef CAS PubMed;
(g) N. Kumagai, M. Kanai and H. Sasai, ACS Catal., 2016, 6, 4699–4709 CrossRef CAS . For the other phosphines, amides, and others;
(h) Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3466 Search PubMed;
(i) M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 Search PubMed;
(j) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 Search PubMed;
(k) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed ; For 1,1’-binaphthyl-based macrocycles;
(l) R. Ning, H. Zhou, S.-X. Nie, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 10894–10898 Search PubMed;
(m) G. Sun, X. Zhang, Z. Zheng, Z.-Y. Zhang, M. Dong, J. L. Sessler and C. Li, J. Am. Chem. Soc., 2024, 146, 26233–26242 CrossRef CAS PubMed.
-
(a) P. R. Blakemore, C. Kilner and S. D. Milicevic, J. Org. Chem., 2006, 71, 8212–8218 CrossRef CAS PubMed;
(b) J. Xiao and T.-P. Loh, Org. Lett., 2009, 11, 2876–2879 CrossRef CAS PubMed;
(c) S.-M. Sephton, C. Wang, L. N. Zakharov and P. R. Blakemore, Eur. J. Org Chem., 2012, 3249–3260 CrossRef CAS;
(d) J. V. Chocholoušová, J. Vacek, A. Andronova, J. Míšek, O. Songis, M. Šámal, I. G. Stará, M. Meyer, M. Bourdillon, L. Pospíšil and I. Starý, Chem.–Eur. J., 2014, 20, 877–893 CrossRef PubMed.
-
(a) W. Xu, Y. Nagata and N. Kumagai, J. Am. Chem. Soc., 2023, 145, 2609–2618 CrossRef CAS PubMed . Other examples of quinoline-based macrocycles;
(b) P. S. Shirude, E. R. Gillies, S. Ladame, F. Godde, K. Shin-ya, I. Huc and S. Balasubramanian, J. Am. Chem. Soc., 2007, 129, 11890–11891 CrossRef CAS PubMed;
(c) T. Shimasaki, R. Kuroda, M. Akao, T. Akimoto, T. Ishikawa, T. Iwanaga, N. Teramoto and M. Shibata, Chem. Lett., 2019, 48, 133–136 CrossRef CAS;
(d) S. Adachi, M. Shibasaki and N. Kumagai, Nat. Commun., 2019, 10, 3820 CrossRef PubMed;
(e) D. Yang, J. L. Greenfield, T. K. Ronson, L. K. S. von Krbek, L. Yu and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 19856–19861 CrossRef CAS PubMed;
(f) T. Kobayashi and N. Kumagai, Angew. Chem., Int. Ed., 2023, 62, e202307896 CrossRef CAS PubMed;
(g) T. Kobayashi, T. Sakurai and N. Kumagai, Bull. Chem. Soc. Jpn., 2023, 96, 1139–1143 CrossRef CAS;
(h) K. Kihara, T. Kobayashi, W. Xu and N. Kumagai, Chem.–Eur. J., 2024, 30, e202304176 CrossRef CAS PubMed;
(i) T. Karimata, W. Xu and N. Kumagai, Chem.–Eur. J., 2025, 31, e202404335 CrossRef CAS PubMed.
- M. Nishiwaki, W. Xu and N. Kumagai, Asian J. Org. Chem., 2023, 12, e202300261 CrossRef CAS.
- For the naphthalene analogue;
(a) Y. Nojima, M. Hasegawa, N. Hara, Y. Imai and Y. Mazakia, Chem. Commun., 2019, 55, 2749–2752 RSC;
(b) Y. Nojima, M. Hasegawa, N. Hara, Y. Imai and Y. Mazaki, Chem.–Eur. J., 2021, 27, 5923–5929 CrossRef CAS PubMed.
- For other inherently chiral macrocyclic compounds, see:
(a) S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed;
(b) H. Han, X.-G. Wang, S. Tong, J. Zhu and M.-X. Wang, ACS Catal., 2025, 15, 6018–6024 CrossRef CAS;
(c) S. J. Nemat, H. Jędrzejewska, A. Prescimone, A. Szumna and K. Tiefenbacher, Org. Lett., 2020, 22, 5506–5510 CrossRef CAS PubMed;
(d) G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed;
(e) F. Sannicolò, P. R. Mussini, T. Benincori, R. Cirilli, S. Abbate, S. Arnaboldi, S. Casolo, E. Castiglioni, G. Longhi, R. Martinazzo, M. Panigati, M. Pappini, E. Q. Procopio and S. Rizzo, Chem.–Eur. J., 2014, 20, 15298–15302 CrossRef PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- See ESI Section 2.6† for details..
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254–14255 CrossRef CAS PubMed.
- See ESI Section 2.9 and 2.10† for details..
- X-ray crystallographic analysis of i-TEQ6b and TEQ7b revealed that crystal structures and calculated structures of the main frameworks are almost identical..
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- See ESI Section 7.7† for details..
- C. E. Colwell, T. W. Price, T. Stauch and R. Jasti, Chem. Sci., 2020, 11, 3923–3930 RSC.
- See ESI Fig. S28 and S29.†.
- See ESI Table S4† for details..
- See ESI Fig. S17 and S18.†.
- See ESI Section 5.3† for details..
- See ESI Section 6† for details..
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed . See ESI Section 7.5† for experimental details..
- See ESI Section 7.6† for calculational details..
- J. Schwartz and J. A. Labinger, Angew. Chem., Int. Ed., 1976, 15, 333–340 CrossRef.
- C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304–11307 CrossRef CAS PubMed.
- For fused polycyclic material see following reviews or latest articles;
(a) H. Ito, K. Ozaki and K. Itami, Angew. Chem., Int. Ed., 2017, 56, 11144–11164 CrossRef CAS PubMed;
(b) S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed;
(c) G. G. Miera, S. Matsubara, H. Kono, K. Murakami and K. Itami, Chem. Sci., 2022, 13, 1848–1868 RSC;
(d) S. M. Elbert, O. T. A. Paine, T. Kirschbaum, M. P. Schuldt, L. Weber, F. Rominger and M. Mastalerz, J. Am. Chem. Soc., 2024, 146, 27324–27334 CrossRef CAS PubMed . For aza-substituted warped nano-sheets;
(e) S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed;
(f) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed;
(g) S. Higashibayashi, P. Pandit, R. Haruki, S. Adachi and R. Kumai, Angew. Chem., Int. Ed., 2016, 55, 10830–10834 CrossRef CAS PubMed;
(h) K. Oki, M. Takase, S. Mori, A. Shiotari, Y. Sugimoto, K. Ohara, T. Okujima and H. Uno, J. Am. Chem. Soc., 2018, 140, 10430–10434 Search PubMed;
(i) T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2020, 26, 14560–14564 CrossRef CAS PubMed;
(j) P. An, R. Li, B. Ma, R.-Y. He, Y.-K. Zhang, M.-J. Xiao and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24478–24483 Search PubMed;
(k) J. Wagner, P. Z. Crocomo, M. A. Kochman, A. Kubas, P. Data and M. Lindner, Angew. Chem., Int. Ed., 2022, 61, e202202232 CrossRef CAS PubMed;
(l) M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Chem. Sci., 2023, 14, 2353–2360 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Toi
Kobayashi
a,
Toi
Kobayashi