Stereoselective heterocycle synthesis through a reversible allylic alcohol transposition and nucleophilic addition sequence†
Youwei
Xie
and
Paul E.
Floreancig
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: florean@pitt.edu; Fax: +1 412-624-8611; Tel: +1 412-624-8727
First published on 14th September 2011
Abstract
The abilities of Re2O7 to promote non-stereoselective allylic alcohol transposition reactions and acetal ionization or enone activation have been coupled for a heterocycle synthesis in which thermodynamics dictate stereochemical outcomes. The stabilities of intermediate allylic cation and oxocarbenium ion intermediates dictate the efficiency of product equilibration. Long range stereoinduction can be observed in the synthesis of spiroketals and spirotricycles through this protocol.
Introduction
Rearrangement reactions are commonly employed in molecular syntheses to exploit strong thermodynamic driving forces that promote structural reorganization and/or to utilize well-defined transition states that provide efficient chirality transfer.1Rearrangements that lack a thermodynamic driving force can also be utilized to achieve these objectives when coupled to a subsequent step that is kinetically and thermodynamically favorable. This has been illustrated by the sequence of oxorhenium-mediated2 allylic alcohol transposition3,4 and trapping with electrophilic chlorosilanes5 or boronates.6Vanadium oxide-mediated transpositions have been driven by enzymatic acylation.7 Our interest in this strategy arose from our studies on the synthesis8 of leucascandrolide A, where we subjected allylic alcohol 1a to Re2O7 to effect the reversible conversion to 2 followed by the thermodynamically driven9 formation of macrolactol 3 (Scheme 1). Lactol 3 could also be accessed by exposing diastereomeric alcohol 1b to Re2O7, indicating that dynamic thermodynamic stereocontrol10 can be achieved under these reversible conditions due to the lack of stereochemical fidelity in the transposition process. The capacity to control stereochemistry through allylic alcohol transposition and reversible trapping was also recently described by Zakarian and co-workers11 in their elegant approach to benzylidene acetal formation.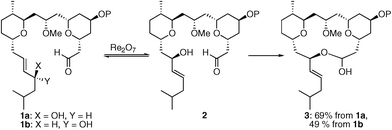 | ||
| Scheme 1 Stereoselective Re2O7-mediated allylic alcohol transposition and capture. | ||
Stereochemically defined secondary allylic alcohols can be prepared through a number of methods, including asymmetric additions of nucleophiles into aldehydes,12 kinetic resolution of racemic secondary alcohols,13 and stereoselective reduction of α,β-unsaturated ketones.14 These impressive methods, however, are labor and material intensive in comparison to using stereochemically undefined allylic alcohols as synthetic intermediates. The strategic benefits of employing diastereomeric mixtures as precursors to stereochemically pure products on improving the overall synthetic efficiencies led us to explore new facets of allylic alcohol isomerization. We report that the sequence of alcohol transposition and capture can be used in stereoselective heterocycle synthesis, and that the degree of stereocontrol is related to the thermodynamic differences between diastereomeric products and to the propensity of the individual steps toward reversal. In addition to syntheses of monocyclic products that are used to establish the scope of the method, we show that the process is also applicable to the preparation of bridged bicyclic and spirocyclic compounds. The latter cases demonstrate rare examples of 1,9- and 1,12-stereoinduction.
Results and discussion
We tested the applicability of the transposition/hemiacetal formation to form smaller rings (Scheme 2) by subjecting alcohol 4 to 5 mol% Re2O7 in CH2Cl2. Within 30 min at rt we observed complete starting material consumption and the formation of lactol 5, though the yield was only 20%. The major side products resulted from dehydrative coupling reactions of 5 arising from tetrahydrofuryl oxocarbenium ion formation11,15 and nucleophilic addition. This pathway was suppressed by conducting the reaction with acetal 6, whereby the Lewis acidity of Re2O7 acted to form a oxocarbenium ion 7, which trapped the transposed alcohol to form 8 in 83% yield. No stereocontrol was observed in this reaction, as expected based on thermodynamic considerations. We also exposed enone 9 to the reaction conditions to explore the viability of an allylic alcohol transposition and oxa-Michael reaction16 sequence and observed the formation of tetrahydropyran 10 as a single stereoisomer in nearly quantitative yield within 10 min at rt.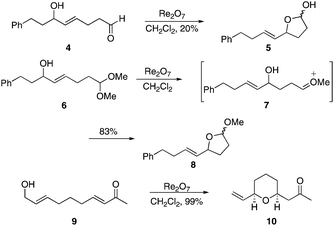 | ||
| Scheme 2 Initial cyclization studies. | ||
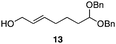
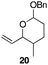
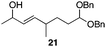
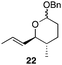
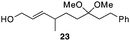
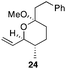
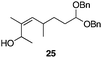
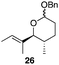
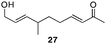
The scope of the process for monocyclization reactions is shown in Table 1. Entries 1–3 show that acetals containing primary allylic alcohols are suitable substrates and the formation of five-, six-, and seven-membered rings proceeds with nearly equivalent efficiency. As in Scheme 2, no stereoselectivity was observed in these reactions. Ketals react with even greater efficiency, as shown in entry 4 where 18 was formed as a single stereoisomer within 10 min. No stereocontrol is observed in acetal substrates that contain a stable stereocenter and a primary allylic alcohol (entry 5), but excellent stereocontrol arises from subjecting the corresponding secondary alcohols to the reaction conditions (entry 6). Ketal substrates that contain a stereocenter and a primary alcohol group show high stereocontrol (entry 7), with 24 being formed as nearly a single stereoisomer from 23 within 30 min. Trisubstituted alkene substrates are also viable participants in this reaction, as shown by the conversion of 25 to 26 (entry 8). Stereoselective transformations can be achieved with enone electrophiles, as seen in entries 9 and 10, though equilibration is much faster for secondary allylic alcohol 29 in comparison to primary alcohol 27.
| Entry | Substrateb | Product | Time | Yield (%)c |
|---|---|---|---|---|
| a Unless noted otherwise, Re2O7 (0.05 eq) was added to a solution of the substrate in CH2Cl2 (0.05–0.1 M) at rt and the mixture was stirred for the indicated time period. b Synthetic sequences for the substrates are provided in the Supporting Information†. c Combined yield of stereoisomers. d Reaction was initiated at 0 °C. e Ratio refers to the relative stereochemistry of the alkyl and vinyl groups. f Starting material was used as a mixture of diastereomers. g Yield was determined by 1H NMR with an internal standard. | ||||
| 1 |
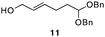
|
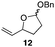
|
30 min | 84 |
| 2 |
|

|
30 min | 81 |
| 3 |
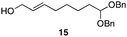
|
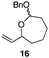
|
30 min | 81 |
| 4d |

|

|
10 min | 85 |
| 5 |
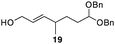
|
|
30 min | 99 dr = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1e :1e |
| 6f |
|
|
30 min | 86 dr > 10:1e |
| 7 |
|
|
30 min | 81 dr = 10:1e |
| 8f |
|
|
30 min | 91 |
| 9 |
|

|
48 h | 88g |
| 10f |
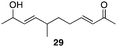
|
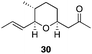
|
60 min | 90g |
Scheme 3 provides a consistent explanation based on fundamental principles for the results in Table 1. Rearrangement of the initially formed perrhenate ester (31) proceeds with little or no stereocontrol to form a mixture of esters 32 and 33. Cyclization follows oxocarbenium ion formation to yield 34 and 35, respectively, as the kinetic products. Ring formation is reversible, however, leading to thermodynamic equilibration in some cases. The equilibration most likely results from perrhenate ester ionization to form an allylic cation followed by ion pair collapse to yield a mixture of stereoisomers. Secondary allylic alcohols ionize more readily than primary allylic alcohols due to the cation-stabilizing effect of the additional alkyl group. Thus the superior stereocontrol in the cyclization of 21 relative to 19 (entries 6 and 5) can be ascribed to the greater potential for stereomutation in the short-lived acyclic intermediate. The heightened stability of the oxocarbenium ion from the ionization of ketals (X = alkyl) rather than acetals (X = H) promotes ring opening and enhances opportunities for stereochemical editing by conferring a longer lifetime to the acyclic intermediate. This difference accounts for the excellent level of stereocontrol that was observed in the cyclization of 23 (entry 7). Trisubstituted alkene substrates can yield products with a single alkene geometry when a sufficient energetic preference exists (entry 8). The cyclization of 25 is also noteworthy because the dehydrooxecane that would form from a direct cyclization of the starting alcohol into the intermediate oxocarbenium ion is not observed. The corresponding primary alcohol yields a mixture of tetrahydropyran and dehydrooxecane products (data not shown), again highlighting the benefits of utilizing secondary alcohols on isomerization kinetics. Thermodynamics dictate the stereochemical orientation of the anomeric centers, with isomers 36 and 37 having similar energies when X = H, and 36 being significantly less stable than 37 for X = alkyl. The same product mixtures are observed when secondary alcohol substrates are single stereoisomers or diastereomeric mixtures, confirming that stereocontrol arises from thermodynamic control rather than kinetic control. Ketone products equilibrate through retro-oxa-Michael reactions. Again, additional alkyl substitution facilitates ionization and leads to a more rapid equilibration.
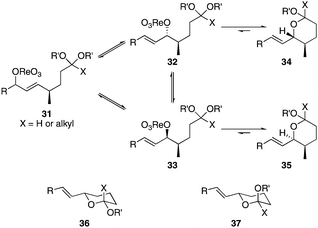 | ||
| Scheme 3 Pathways for stereochemical isomerization. | ||
Upon establishing the basic reactivity patterns of the process we turned our attention toward its application to the construction of more complex bicyclic structures. In these reactions the acetal group serves as an electrophile for consecutive nucleophilic additions. This is illustrated in Scheme 4 for the preparation of bridged bicyclic structures of the type that are found in a number of biologically active compounds such as the didemniserinolipids17 and the pinnatoxins.18 The reaction of diol acetal 38 with Re2O7 provided diastereomeric bicycles 39 and 40 as a separable 3:4 mixture after 21 h at rt. This reaction proceeded through an initial isomerization/cyclization to form 41, which underwent a second isomerization to yield 42 as a mixture of diastereomers. Ring closure provided the observed products. Resubjecting pure 40 to the reaction conditions yielded only a minimal amount of 39, indicating that the product ratio can be attributed to kinetic rather than thermodynamic factors. We prepared diol 43 as a mixture of stereoisomers in an effort to facilitate equilibration. Exposing 43 to the standard conditions resulted in the formation of bicycles 44 and 45 in 53% yield as a 7.3:1 mixture, indicating that the increased substitution promotes thermodynamic equilibration in accord with prior results. Resubjecting 45 to the reaction conditions provided a similar ratio of compounds. This ratio is consistent with the energetic difference between the isomers that was calculated by Spartan.19 We also examined the cyclization of ketone 46 based on our supposition that intramolecular addition into the oxocarbenium ion that formed from ionization of the initial lactol would be faster than the bimolecular addition that was observed in the reaction of 4. While 46 proceeded through the sequence with slightly lower efficiency than 38, the reaction yielded 39 and 40 in the same ratio.
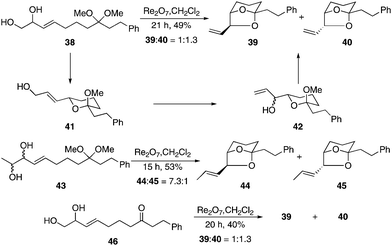 | ||
| Scheme 4 Double isomerization/cyclization route to bridged structures. | ||
This strategy can also be applied to the synthesis of spirocyclic structures,20 as shown in Scheme 5. Diol acetal 47 reacted with Re2O7 to provide a 1.1:1 mixture of spiroketals 48 and 49 within 30 min at rt. Allowing the reaction to proceed for 12 h improved the ratio of 48 to 49 to >20:1 in which the major product has the anomerically preferred stereochemical orientation at the ketal center and both vinyl groups have equatorial alignments. Ketones also serve as useful substrates for the process though the isomerization process is slower, with 50 providing 48 in a 61% yield after 48 h. Notably, equilibration in this reaction required the addition of MeOH. We postulate that the addition of MeOH facilitates the ring opening event to a greater degree than water or that water suppresses the Lewis acidity of Re2O7.3b As expected secondary diol 51 proceeded through the reaction and equilibrated more rapidly, providing 52 as a single stereoisomer within 1 h at 0 °C.
 | ||
| Scheme 5 Synthesis of spiroketals. | ||
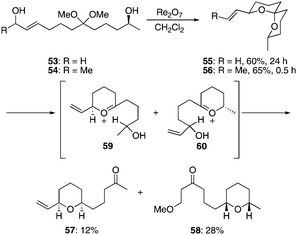
We prepared diol acetals 53 and 54 to study the influence of a non-equilibrating secondary alcohol on remote stereochemical induction. These compounds proceeded through the cyclization reactions (Scheme 6) efficiently to form spiroketals 55 and 56 in 60% and 65% yield, respectively. Consistent with prior studies, the reaction of secondary alcohol 54 converged to a single stereoisomer much more rapidly (30 min vs. 24 h) than the cyclization of primary alcohol 53. When enantiomerically pure 53 was used in the reaction minimal racemization (∼2%) was observed by HPLC detection. Thus this transformation demonstrates that a high level of relayed 1,9-stereoinduction can be obtained based on thermodynamic control. The isolation of tetrahydropyranyl ketones 57 and 58 as minor products from the cyclization of 53 is also noteworthy. These products arise from the ionization of the spiroketal to form oxocarbenium ions 59 and 60. Reduction of these ions by intramolecular hydride transfer leads to the tetrahydropyranyl ketones, with conjugate addition of MeOH to the intermediate enone ultimately providing 58. These transformations, while not resulting from predominant pathways, are nonetheless mechanistically intriguing because the stereocenter that directs the creation of the two stereocenters in the product is ultimately destroyed in the process.
 | ||
| Scheme 6 Remote stereoinduction and intramolecular redox isomerization. | ||
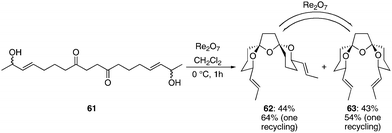
We explored the capacity for remote stereochemical induction across a spirotricyclic system by preparing diketone diol 61 through an oxidative enolate coupling reaction21 (see Supporting Information† for details). Exposing 61 to Re2O7 at 0 °C for 1 h provided spirotricycles 62 and 63 in 44% and 43% yield, respectively (see Scheme 7). Structural confirmation for these structures was provided by HPLC experiments where racemic mixture 62 showed 2 peaks of equal intensity when a chiral stationary phase was employed while meso isomer 63 showed only one peak. The formation of a diastereomeric mixture is a result of the lack of a strong thermodynamic preference for either compound that results from the numerous low energy conformations of the central tetrahydrofuran unit.22 Resubjecting either of the products to the reaction conditions quickly resulted in the formation of the thermodynamic mixture of diastereoisomers. Therefore one recycling of 63 provided 62 in an overall yield of 64% while one recycling of 62 provided 63 in 54% overall yield. The ratio of products that was formed in the equilibration reaction was slightly different from the ratio that was observed in the initial cyclization. We postulate that the liberation of water in the initial cyclization reaction could have a subtle influence on the rate of equilibration since water was excluded from the equilibration protocol. The important conclusion from this study is that either stereoisomer can be accessed in reasonable yield through a rapid sequence of reactions. The stereochemical arrangement around the tetrahydrofuran ring in 62 matches the orientation that is seen in azaspiracid-123 while the arrangement in 63 matches the pinnatoxins.17 Multiple studies have shown24 that the thermodynamic preferences in these spirotricycles can be manipulated by subtle structural changes, indicating that this protocol could be broadly applicable to the synthesis of this molecular class, particularly in consideration of the improved access to 1,4-diketones that has resulted from advances in oxidative enolate heterocoupling reactions.25 This sequence produced compounds in which stereocenters in the product arose from prochiral sites that had a 1,12-relationship in the starting material.
 | ||
| Scheme 7 Synthesis and equilibration of spirotricycles. | ||
Conclusions
We have demonstrated that the experimentally facile sequence of Re2O7-mediated allylic alcohol isomerization followed by nucleophilic addition into an oxocarbenium ion can be used to generate stereocenters with high levels of thermodynamic control. Stereocontrol is maximized when the energetic difference between diastereomeric products is sufficient to create a strong preference, when reversibility in the cyclization reaction is facilitated by increasing the stability of the oxocarbenium ion intermediate, and when stereochemical scrambling is promoted by increasing the substitution on the intermediate allyl cation. Applying these principles to more complex substrates led to the syntheses of bridged and spirocyclic ketals with good to excellent levels of stereocontrol. This method is particularly effective for setting remote stereocenters in spirocycle synthesis, where relayed 1,9- and 1,12-stereoinduction was demonstrated. Applying thermodynamic equilibration to establish remote stereocenters alleviates the need for labor- and material-intensive reagent-controlled asymmetric transformations, thereby enhancing the overall efficiency of complex molecule synthesis.Acknowledgements
This work was supported by generous funding from the National Institutes of Health (P50-GM06082).Notes and references
- For reviews of sigmatropic rearrangements, see: (a) E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133 RSC; (b) A. M. M. Castro, Chem. Rev., 2004, 104, 2939 CrossRef CAS; (c) Y. Chai, S.-P. Hong, H. A. Lindsay, C. McFarland and M. C. McIntosh, Tetrahedron, 2002, 58, 2905 CrossRef CAS; (d) H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 28, 43 RSC; (e) K. Tomooka, H. Yamamoto and T. Nakai, Liebigs Ann./Recl., 1997, 1275 CrossRef CAS; (f) K. Mikami and T. Nakai, Synthesis, 1991, 594 CrossRef CAS.
- For a review of rhenium oxides in synthesis, see: S. Bellemin-Lapponaz, ChemCatChem, 2009, 1, 357 CrossRef.
- (a) K. Narasaka, H. Kisama and Y. Hiyashi, Tetrahedron, 1992, 48, 2059 CrossRef CAS; (b) S. Bellemin-Laponnaz, H. Gisie, J. P. Le Ny and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1997, 36, 976 CrossRef CAS.
- For examples with other catalysts, see: (a) T. Hosogai, Y. Fujita, Y. Ninagawa and T. Nishida, Chem. Lett., 1982, 11, 357 CrossRef; (b) S. Matsubara, T. Okazoe, K. Oshima, K. Takai and H. Nozaki, Bull. Chem. Soc. Jpn., 1985, 58, 844 CrossRef CAS; (c) J. Belgacem, J. Kress and J. A. Osborn, J. Am. Chem. Soc., 1992, 114, 1501 CrossRef CAS; (d) F. R. Fronczek, R. L. Luck and G. Wang, Inorg. Chem. Commun., 2002, 5, 384 CrossRef CAS; (e) H. Zheng, M. Lejkowski and D. G. Hall, Chem. Sci., 2011, 2, 1305 RSC.
- (a) C. Morrill and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 2842 CrossRef CAS; (b) C. Morrill, G. L. Beutner and R. H. Grubbs, J. Org. Chem., 2006, 71, 7813 CrossRef CAS.
- E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2006, 128, 8142 CrossRef CAS.
- (a) S. Akai, K. Tanimoto, Y. Kanao, M. Egi, T. Yamamoto and Y. Kita, Angew. Chem., Int. Ed., 2006, 45, 2592 CrossRef CAS; (b) S. Akai, R. Hanada, N. Fujiwara, Y. Kita and M. Egi, Org. Lett., 2010, 12, 4900 CrossRef CAS.
- H. H. Jung, J. R. Seiders II and P. E. Floreancig, Angew. Chem., Int. Ed., 2007, 46, 8464 CrossRef CAS.
- Y. Wang, J. Janjic and S. A. Kozmin, J. Am. Chem. Soc., 2002, 124, 13670 CrossRef CAS.
- P. Beak, D. R. Anderson, M. D. Curtis, J. M. Laumer, D. J. Pippel and G. A. Weisenberger, Acc. Chem. Res., 2000, 33, 715 CrossRef CAS.
- A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962 CrossRef CAS.
- For representative examples, see: (a) H. Guo, C.-G. Dong, D.-S. Kim, D. Urabe, J. Wang, J. T. Kim, X. Liu, T. Sasaki and Y. Kishi, J. Am. Chem. Soc., 2009, 131, 15387 CrossRef CAS; (b) P. Wipf and S. Ribe, J. Org. Chem., 1998, 63, 6454 CrossRef CAS; (c) W. Oppolzer and R. N. Radinov, Helv. Chim. Acta, 1992, 75, 170 CrossRef CAS; (d) F. Langer, L. Schwink, A. Devasagayaraj, P.-Y. Chavant and P. Knochel, J. Org. Chem., 1996, 61, 8229 CrossRef CAS; (e) K. Soai, A. Ookawa, T. Kaba and K. Ogawa, J. Am. Chem. Soc., 1987, 109, 7111 CrossRef CAS.
- (a) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS; (b) D. C. Ebner, J. T. Bagdanoff, E. M. Ferreira, R. M. McFadden, D. D. Caspi, R. M. Trend and B. M. Stoltz, Chem.–Eur. J., 2009, 15, 12978 CrossRef CAS; (c) M. J. Schultz, S. S. Hamilton, D. R. Jensen and M. S. Sigman, J. Org. Chem., 2005, 70, 3343 CrossRef CAS; (d) J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492 CrossRef CAS; (e) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351 CrossRef CAS.
- E. J. Corey and C. J. Helal, Angew. Chem. Int. Ed., 1998, 37, 1987 Search PubMed.
- K. Tadpetch and S. D. Rychnovsky, Org. Lett., 2008, 10, 4839 CrossRef CAS.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218 RSC.
- (a) N. González, J. Rodríguez and C. Jiménez, J. Org. Chem., 1999, 64, 5705 CrossRef; (b) S. S. Mitchell, D. Rhodes, F. D. Bushman and D. J. Faulkner, Org. Lett., 2000, 2, 1605 CrossRef CAS.
- (a) D. Uemura, T. Chou, T. Haino, A. Nagatsu, S. Fukuzawa, S.-h. Zheng and H.-s. Chen, J. Am. Chem. Soc., 1995, 117, 1155 CrossRef CAS; (b) T. Chou, T. Haino, M. Kuramoto and D. Uemura, Tetrahedron Lett., 1996, 37, 4027 CrossRef CAS.
- Spartan (Wavefunction, Irvine, CA) calculations were performed by conducting a conformational search followed by semi-empirical energy calculations using the AM1 basis set.
- (a) B. R. Raju and A. K. Saikia, Molecules, 2008, 13, 1942 CrossRef CAS; (b) M. A. Brimble and D. P. Furkert, Curr. Org. Chem., 2003, 7, 1461 CrossRef CAS; (c) K. T. Mead and B. N. Brewer, Curr. Org. Chem., 2003, 7, 227 CrossRef CAS.
- Y. Ito, T. Konoike and T. Saegusa, J. Am. Chem. Soc., 1975, 97, 2912 CrossRef CAS.
- S. J. Angyal, Adv. Carbohydr. Chem. Biochem., 1984, 42, 15 CrossRef CAS.
- M. Satake, K. Ofuji, H. Naoki, K. J. James, A. Furey, T. McMahon, J. Silke and T. Yasumoto, J. Am. Chem. Soc., 1998, 120, 9967 CrossRef CAS.
- (a) T. Sugimoto, J. Ishihara and A. Murai, Tetrahedron Lett., 1997, 38, 7379 CrossRef CAS; (b) J. A. McCauley, K. Nagasawa, P. A. Lander, S. G. Mischke, M. A. Simones and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 7647 CrossRef CAS; (c) J. Ishihara, T. Sugimoto and A. Murai, Synlett, 1998, 603 CrossRef CAS; (d) S. Nakamura, J. Inagaki, T. Sugimoto, M. Kudo, M. Nakajima and S. Hashimoto, Org. Lett., 2001, 3, 4075 CrossRef CAS; (e) S. Sakamoto, H. Sakazaki, K. Hagiwara, K. Kamada, K. Ishii, T. Noda, M. Inoue and M. Hirama, Angew. Chem., Int. Ed., 2004, 43, 6505 CrossRef CAS; (f) C.-D. Lu and A. Zakarian, Org. Lett., 2007, 9, 3161 CrossRef CAS; (g) C. E. Stivala and A. Zakarian, J. Am. Chem. Soc., 2008, 130, 3774 CrossRef CAS; (h) R. Aroaz, D. Servent, J. Molgó, B. I. Iorga, C. Fruchart-Gaillard, E. Benoit, Z. Gu, C. Stivala and A. Zakarian, J. Am. Chem. Soc., 2011, 133, 10499 CrossRef.
- (a) B. M. Casey and R. A. Flowers, II, J. Am. Chem. Soc., 2011, 133, 11492 CrossRef CAS; (b) C. T. Avetta, Jr., L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern and R. J. Thomson, Org. Lett., 2008, 10, 5621 CrossRef; (c) M. P. DeMertino, K. Chen and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 11546 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: schemes for substrate synthesis, experimental procedures for all reactions, and compound characterization including stereochemical determination. See DOI: 10.1039/c1sc00570g |
| This journal is © The Royal Society of Chemistry 2011 |