Controlled network structure and its correlations with physical properties of polycarboxyl octaphenylsilsesquioxanes-based inorganic–organic polymer nanocomposites†
Zhanbin
Wang
,
Zhonggang
Wang
*,
Hao
Yu
,
Linni
Zhao
and
Jing
Qu
Department of Polymer Science and Materials, School of Chemical Engineering. Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: zgwang@dlut.edu.cn
First published on 8th February 2012
Abstract
Polycarboxyl octaphenylsilsesquioxanes (polycarboxyl-OPS) containing different numbers of reactive carboxyl groups per OPS molecule were synthesized, which were then utilized as nanofillers and macromolecular cross-linkers to cure diglycidyl ether of bisphenol-A (DGEBA) to yield a series of novel inorganic/organic polymer nanocomposites. The network structures and cross-linking densities were modulated by varying the contents of polycarboxyl-OPS in the system. The interface between OPS and DGEBA matrix was remarkably enhanced by the strong covalent bonds as evidenced by the complete disappearance of the carboxyl absorption at 1699 cm−1, the epoxy group at 915 cm−1 and the newly emerged strong peak of ester bond at 1725 cm−1 in the FTIR spectra. All the OPS/DGEBA cured samples are transparent, and the wide angle X-ray diffractions (WAXD) exhibited similar amorphous patterns to the pure DGEBA resin, whereas the peak at ∼8° corresponding to OPS aggregations were absent, indicating a homogeneous dispersion of OPS within the DGEBA matrix. The correlations between network structures, viscoelastic behaviors, cross-linking densities, inter-segmental distances, glass transition temperatures, flexural properties and thermal stability of OPS/DGEBA products were systematically investigated by means of dynamic mechanical analysis (DMA), DSC, TGA, scanning electron microscopy (SEM) and WAXD methods.
Introduction
The preparation of nanocomposites by uniformly dispersing inorganic fillers within a polymeric matrix at nanometre scale has been proved to be an efficient tool to create a novel material with the combination of advantages of several components that can not be achieved by the organic polymer alone. The physical properties of the nanocomposites are strongly influenced by the type, size, shape and physicochemical nature of the fillers. Over the last decade, the most intensely studied fillers for organic–inorganic hybrid nanocomposites include carbon naonotubes,1 layered silicates,2,3 graphene4 and polyhedral oligomeric silsesquioxanes (POSS).5–12Among them, POSS receive great attention because of their unique symmetric cage structure, precisely controlled monodispersed nanometre size (ca. 1–2.5 nm in diameter), and the strong molecular tailoring and designing capability. The high silicon content and rigid structure of POSS make the POSS-modified polymers typically have excellent flame-retardancy, thermal oxidation stability, electrical insulating property, low thermal expansion coefficient, and good resistance to atomic oxygen erosion in near-earth orbit environments.13–21The interfacial compatibility between the inorganic filler and polymer matrix is of essential importance for organic–inorganic nanocomposites. The organic groups surrounding the inorganic silica cores are beneficial to the enhanced affinity of POSS for polymer. Moreover, through the control of reaction condition, POSS can be modified to yield functional POSS precursors such as acrylate-POSS, methacrylate-POSS, vinyl-POSS, styryl-POSS, norbornyl-POSS, epoxy-POSS or amino-POSS,22–29 which are utilized as macromonomers to copolymerize with other monomers to form linear, branching or cross-linking organic–inorganic copolymers. Thus, POSS are attached to organic polymer matrix through strong covalent bonds, and the organic–inorganic interfacial interactions are greatly improved.30–32
In addition, the previous studies show that the influencing degree of POSS on the physical properties of a polymer composite can also be altered by a number of other factors, including the type and number of functional groups of the POSS macromonomer, the copolymerization monomer, the content of POSS in the copolymer, the polymerization method and the reaction conditions. For monofunctional POSS, like vinyl-POSS, its copolymerization with methylacrylate or styrene lead to linear polymethylacrylates or polystyrenes with bulky POSS groups dangling on the polymer backbone,16–18,21,22 while in the case of a polyfunctional POSS compound, the reactive groups can act as claws to anchor the polymer segments onto the rigid POSS core. In this way, the POSS fillers are more tightly tethered with the polymer matrix through multiple covalent bonds so as to achieve more significant enhancement in physical properties for the nanocomposites.33,34
Epoxy resins are one of the most important class of thermosetting polymers, and have been widely used in many fields because of their low cost and excellent comprehensive properties, such as strong adhesion ability with various substrates, high mechanical strength and modulus, easy processing, good dimensional stability and chemical resistance.35–39 However, the densely cross-linked structure makes the cured products often suffer from brittleness, poor crack resistance and low fracture toughness, which should be improved before they are considered for some high-tech applications, e.g. advanced composite materials in aeronautics and astronautics industries.
Herein, the work was undertaken to synthesize polycarboxyl octaphenylsilsesquioxane (polycarboxyl-OPS) containing different numbers of carboxyl group per OPS molecule, which were then mixed with an DGEBA epoxy resin with various polycarboxyl-OPS contents. Upon heating, polycarboxyl-OPS act as macromolecular curing agents, and the carboxyls react with the epoxy groups in DGEBA to create covalently cross-linked OPS/epoxy networks. The influences of the copolymerization reaction, carboxyl content and OPS/DGEBA composition on the network structure, viscoelastic behavior, thermal properties and fracture toughness for the nanocomposites were investigated by means of Fourier transform infrared (FTIR), dynamic mechanical analysis (DMA), differential scanning calorimetry (DSC), thermal gravimetric analysis (TGA), scanning electron microscopy, wide angle X-ray diffraction (WAXD) and mechanical measurements.
Experimental
Materials
Phenyltrichlorosilane (C6H5SiCl3) was purchased from J&K-Chemical Company and used without further purification. Octaphenylsilsesquioxane (OPS) was synthesized in our laboratory via hydrolysis and condensation of phenyltrichlorosilane according to the procedure in the literature40 with 87% yield. DGEBA was supplied by Dalian Qihua Chemical Company with an epoxy equivalent weight of 183 g mol−1. 4-Methylhexahydrophthalic anhydride (MHHPA) and curing accelerator 2-ethyl-4-methylimidazole (EMI) were obtained from Shanghai Darui Fine Chemical Co. and used as received. N,N-Dimethylformamide (DMF) was dried and distilled under reduced pressure prior to use. Tetrahydrofuran (THF) was purified by refluxing over sodium with the indicator of benzophenone complex.The polybromo-OPS with relatively low bromine content (polybromo-OPS-LB) was prepared via a similar procedure to the above except that the amount of Br2 charged in the system was 8.3 mL (0.16 mol) instead of 11.9 mL (0.23 mol). Yield: 90%. FTIR (cm−1): 3069, 1578, 1108, 1069, 1010, 817. 1H-NMR (DMSO-d6, ppm): 6.9–8.0 ppm.
The synthesis procedure of polycarboxyl-OPS with low carboxyl content is similar to the above except for the replacement of polybromo-OPS-HB with polybromo-OPS-LB, and the charged amount of BuLi was reduced from 60 mL to 42 mL. FT-IR (cm−1): 3700–2800, 3064, 1697, 1596, 1189, 1107; 1H-NMR (D2O/NaOH, ppm): 6.9–8.0; 13C-NMR (D2O/NaOH, ppm): 174–185, 124–147.
As a comparison, using MHHPA as the curing agent and EMI as the catalyst, the cured sample of pure DGEBA was also prepared. In this case, a stoichiometric amount of MHHPA and 0.5 wt% of EMI were mixed with DGEBA, and the obtained mixture was poured into the mould and then degassed under vacuum for 30 min. Other curing conditions are the same as that of the polycarboxyl-OPS/DGEBA systems.
Instruments
The FTIR spectra were recorded on a Nicolet 20DXB FTIR spectrometer in the 400–4000 cm−1 region. All the samples were mixed with the KBr powder and then pressed into the flakes.The 1H-NMR and 13C-NMR were performed on a Varian INOVA400 spectrometer at room temperature. 1H-NMR spectra of polybromo-OPS were measured in deuterated dimethyl sulfoxide (DMSO-d6). The NOE-suppressed quantitative 13C-NMR spectra of polycarboxyl-OPS were obtained with 0.5 s acquisition times at a 2500 Hz sweep width in D2O/NaOH.
The dynamic mechanical tests were conducted on a TA Q800 series thermal analysis system at a frequency of 1 Hz, the samples were heated from 40 to 200 °C at a heating rate of 3 °C min−1 under a nitrogen atmosphere. The samples were cut to dimensions of 18 × 7.5 × 1 mm3 prior to tests.
The DSC measurements were carried out on a NETZSCH DSC204 with indium metal as a standard. All samples (about 10 mg in weight) were heated from 40 to 250 °C at a heating rate of 10 °C min−1 in nitrogen atmosphere. The glass transition temperatures (Tg) were read at the middle of the change in the heat capacity.
The thermal stability were investigated by NETZSCH TG209C under nitrogen atmosphere in the 40–650 °C region and conducted with a heating rate of 20 °C min−1.
The three point bending test was run on the Reger3050 electronic universal testing machine at room temperature. According to the national standard (GB/T9341-2000), the samples were cut to dimensions of 100 × 8 × 3 mm3 prior to test. The span was 64 mm and the loading speed was 2 mm min−1.
Scanning electron microscopy (SEM) images of the fractured surfaces of samples were recorded on a JEOL JSM-5600LV scanning electron microscope at a voltage of 10 kV. The surfaces were sputter coated with gold before taking the micrographs.
Wide-angle X-ray diffraction (WAXD) measurements were performed at room temperature on a Rigku D/max-2400 X-ray diffractometer using Cu-Kα radiation at a wavelength of 1.54 Å (40 kV, 15 mA). The scanning rate was 2°·min−1. over a range of 2θ = 2.5 ∼ 50°.
Results and discussion
Synthesis and characterization of polycarboxyl-OPS with different carboxyl contents
The syntheses of polycarboxyl-OPS are presented in Scheme 1. Firstly, the phenyls of OPS were brominated in good yields (85–90%), using iron powder as catalyst. Subsequently, through the successive procedures of lithiation by BuLi and carboxylation by CO2, the polybromo-OPS were successfully converted to polycarboxyl-OPS. The number of bromine-substituents in the precursor polybromo-OPS was controlled by the bromine amount charged in the reaction system, which then determined the content of carboxyl group in polycarboxyl-OPS synthesized. | ||
| Scheme 1 Synthesis route of polycarboxyl-OPS. | ||
The FTIR spectra of brominated products are shown in Fig. 1S (ESI†). The absorptions at 3069, 3027 and 1578 cm−1 are corresponded to the benzene rings. The peaks at 1110 and 819 cm−1 are characteristics of the unsymmetrical stretching vibrations of Si–O–Si in POSS cage. The absorption of aromatic C–Br vibration at 1067 cm−1 confirms that OPS have been successfully brominated.
The real bromine number per polybromo-OPS molecule is quantitatively determined by the 1H-NMR spectra using tetrahydrofuran (THF) as a standard, and the weight ratios of THF to polybromo-OPS in the two samples are kept the same. As shown in Fig. 1, the chemical shifts of –CH2– and –O–CH2– of THF are at 1.73 and 3.59 ppm, respectively, whereas those for the aromatic protons in brominated phenyls are at 7.2–8.1 ppm. Relative to THF, the peak area of the protons on the benzene ring for polybromo-OPS-HB is apparently lower than that of polybromo-OPS-LB, indicating that more protons on the phenyls of polybromo-OPS-HB are replaced by bromine. According to the peak areas, the numbers of bromine substituents for the two brominated OPS products are calculated, and the values are 6.4 and 9.0, respectively.
 | ||
| Fig. 1 1H NMR spectra of polybromo-OPS with THF as internal standard of (a) polybromo-OPS-HB, (b) polybromo-OPS-LB. | ||
It should be noted that, for the polybromo-OPS, the phenyls can be mono-substituted, di-substituted, and even small amount of trisubstituted, and the bromine replacements can be at ortho-, meta- or para-position. It has been demonstrated in the previous reports42,43 that, for polybromo-aromatic compounds, once bromine has been lithiated by BuLi, the electropositive effect of lithium-cation makes the further lithiation reaction of other bromines on the same phenyl can not take place. Accordingly, after the carboxylation reaction of polybromo-OPS, there is only one carboxyl per phenyl group and the positions of carboxyl substituents on the phenyls of polycarboxyl-OPS may be different from one another. In order to determine the replacement position of carboxyl group in phenyl ring, the Si–C bonds of polycarboxyl-OPS were cleaved by KF/H2O2, and the organic fragments were collected and analyzed by LC-MS and the data are presented in Table 1S and Table 2S (ESI†).
As shown in Fig. 2S (ESI†), the FTIR spectra of polycarboxyl-OPS show a wide peak between 2800–3700 cm−1 due to the vibration of OH in the carboxyl group. The peak at 1699 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption of hydrogen-bonded carboxyl group. The characteristic band of the Si–O–Si vibration appears at 1107 cm−1.
O absorption of hydrogen-bonded carboxyl group. The characteristic band of the Si–O–Si vibration appears at 1107 cm−1.
NOE-suppressed quantitative 13C-NMR spectra of the two polycarboxyl-OPS are illustrated in Fig. 2. The spectra look very complicated because of the various substituted phenyls as revealed by the LC-MS results. The characteristic signals of carboxyl CO are found at 170–185 ppm, whereas the aromatic carbon atoms are at 125–150 ppm. From the integrated areas, the average number of carboxyl groups in each OPS molecule can be calculated. As a result, the carboxyl values of two polycarboxyl-OPS compounds are 5.5 and 7.0, which are consistent with the results obtained from the LC-MS data in Table 1S and Table 2S.† Correspondingly, for the sake of brevity, the two polycarboxyl-OPS are named hereafter as COOH5.5OPS and COOH7.0OPS, respectively.
 | ||
| Fig. 2 NOE-suppressed quantitative 13C NMR spectra of polycarboxyl-OPS of (a) COOH5.5OPS, (b) COOH7.0OPS. | ||
Crosslinking reactions between polycarboxyl-ops and dgeba epoxy resin
COOH5.5OPS and COOH7.0OPS are utilized as nanofillers to improve the physical properties of DGEBA epoxy resin. The presence of multiple reactive carboxyl groups makes them be not simply blended, but copolymerized with DGEBA to generate an integrative cross-linking network as depicted in Scheme 2, where 4-methylhexahydrophthalic anhydride (MHHPA) is used as co-curing agent, and the amounts of COOH7.0OPS in the copolymers are 0, 3, 6, 12 wt%, respectively. As a comparison, the sample of COOH5.5OPS/DGEBA/MHHPA with COOH5.5OPS of 6 wt% was also cured in order to investigate the influence of carboxyl content of polycarboxyl-OPS on the properties of copolymerization networks. | ||
| Scheme 2 Synthesis routes to the OPS/DGEBA copolymerized nanocomposites. | ||
The typical dynamic DSC curves of the COOH7.0OPS/DGEBA/MHHPA sample with COOH7.0OPS of 6 wt% in the absence and presence of the catalyst EMI are illustrated in Fig. 3. For the reaction system without catalyst, the exothermic peaks appeared at 165.5 °C with the reaction heat of 499.7 J g−1. After adding small amount of catalyst EMI, the exothermic peak temperature decreased obviously from 165.5 °C to 145.7 °C, while the reaction heat remarkably increased from 499.7 J g−1 to 4191 J g−1, indicating that the curing reaction could be effectively catalyzed by EMI.
 | ||
| Fig. 3 Dynamic DSC curves for epoxy resin with 10 wt% of COOH7.0OPS for (a) without EMI, (b) with 0.5 wt% of EMI. | ||
Furthermore, the copolymerization reactions of COOH7.0OPS/DGEBA/MHHPA samples with different COOH7.0OPS contents were examined by FTIR spectroscopy (Fig. 4). The spectra of pure COOH7.0OPS and DGEBA are also presented to make comparisons. After curing, the characteristic bands of epoxy groups at 915 cm−1 in DGEBA and carboxyl groups at 1699 cm−1 in COOH7.0OPS disappear completely. Instead, a strong peak at 1725 cm−1 corresponding to ester group appears, demonstrating that, under the present curing condition, the carboxyl groups of COOH7.0OPS indeed participate into the curing reaction with epoxy groups of DGEBA to generate covalently cross-linked OPS/epoxy networks. In addition, it is observed that the absorption intensities at 1110 cm−1 corresponding to the Si–O–Si groups of silsesquioxanes cage skeleton grow progressively with COOH7.0OPS content in the system.
 | ||
| Fig. 4 FTIR spectra of (a) uncured pure DGEBA, (b) cured epoxy with 3 wt% of COOH7.0OPS, (c) cured epoxy with 6 wt% of COOH7.0OPS, (d) cured epoxy with 12 wt% of COOH7.0OPS, (e) pure COOH7.0OPS. | ||
Viscoelastic behaviors and the correlation with network structures
In order to understand the viscoelastic behavior and microstructural information of OPS/DGEBA cross-linked networks, the dynamic mechanical properties of the cured products were measured, and the plots of storage modulus and tanδ as a function of temperature are illustrated in Fig. 5. According to the polymer elasticity theory, the cross-linking densities (ρ) of cross-linked networks are related to the storage modulus in the rubber region, and can be calculated according to the equation: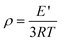 | (1) |
 | ||
| Fig. 5 Dynamic mechanical analysis curves of epoxy with different content of polycarboxyl-OPS. | ||
The storage modulus in both glassy and rubbery regions, the calculated cross-linking densities, glass transition temperatures as well as the height and width of the tanδ peaks are summarized in Table 1. Among the five samples, the pure epoxy resin has the highest storage modulus in glassy region because of its high cross-linking density and dense packing of epoxy chains, which result in its brittleness. Compared to the pure epoxy, the incorporation of 3 wt% of COOH7.0OPS gives rise to a considerable reduction of cross-linking density from 1.20 × 10−3 to 0.51 × 10−3 mol cm−3, i.e., there are much fewer net nodes in unit volume of COOH7.0OPS/DGEBA cured product. The reason is attributed to the rigidity and bulkiness of OPS molecule, which can open up the epoxy segments, and the resultant high free volume or loose network is advantageous for the segmental mobility ability, reflecting in the decreased storage modulus and glass transition temperature. Similar phenomena that the incorporation of POSS result in the reduction of glass transition temperature of POSS/polymer composites have also been reported previously, such as POSS-PEG-PMMA,32 poly(vinyl cinnamate)-POSS,46 epoxy-POSS,47 poly(hydroxystyrene-co-vinylpyrrolidone)-POSS hybrids.48
| Storage Modulus (MPa) | |||||||
|---|---|---|---|---|---|---|---|
| a Storage modulus at 50 °C. b Storage modulus at Tg-DMA + 50 °C. c ρ refers to the cross-linking density of the network. d Width of tanδ peak at its half height. | |||||||
| Sample | T g-DSC (°C) | T g-DMA (°C) | Glass regiona | Rubber regionb | ρ c (10−3 mol cm−3) | Height of tanδ | Width of tanδd (°C) |
| Pure epoxy | 125.2 | 126.1 | 2743 | 16.5 | 1.20 | 0.96 | 22.3 |
| 3 wt% COOH7.0OPS | 121.4 | 116.5 | 2634 | 7.05 | 0.51 | 1.12 | 23.6 |
| 6 wt% COOH7.0OPS | 114.6 | 111.3 | 2603 | 13.2 | 0.96 | 0.90 | 24.3 |
| 12 wt% COOH7.0OPS | 97.1 | 85.8 | 2487 | 20.2 | 1.47 | 0.56 | 28.4 |
| 6 wt% COOH5.5OPS | 119.3 | 121.5 | 2350 | 21.5 | 1.56 | 0.65 | 24.4 |
As expected, with the further increase of COOH7.0OPS content in the system, it is observed that the cross-linking densities of the cured samples grow again since the presence of multiple reactive carboxyl groups in COOH7.0OPS. However, the high cross-linking density doses not always imply the dense packing of polymer segments for a polymer network. For the OPS/epoxy network, the increased cross-linking density means that the polymer segments between cross-linking joints of network becomes short, which are more easier to be propped up by the bulky and rigid OPS moieties, and consequently, the enlarged inter-segmental spacing is favorable for the motion of epoxy segments, leading to the decrease of modulus. Moreover, the varying trend of storage modulus listed in Table 1 agrees with the glass transition temperature.
On the other hand, the effect of carboxyl number per OPS molecule on the properties of polycarboxyl-OPS/epoxy nanocomposites is examined. Similar to COOH7.0OPS, COOH5.5OPS can also be copolymerized with DGEBA to yield covalently cross-linked networks, but their viscoelastic behaviors and network structures are considerably different. The incorporation of COOH5.5OPS results in the apparently higher cross-linking density and glass transition temperature of the cured product than the case of COOH7.0OPS. For example, as shown in Table 1, after introducing 6 wt% polycarboxyl-OPS in the system, the epoxy sample modified by COOH5.5OPS has the cross-linking density and glass transition temperature of 1.56 × 10−3 mol cm−3 and 119 °C, whereas those for the product modified by COOH7.0OPS are only 0.96 × 10−3 mol cm−3 and 114 °C, respectively. The reason may be that, at the same OPS content, the number of epoxy segments covalently bonded on the OPS molecule for the COOH5.5OPS/DGEBA is fewer than that in the COOH7.0OPS/DGEBA system. As a consequence, compared to COOH7.0OPS, the influencing degree of COOH5.5OPS on the packing of DGEBA epoxy segments is reduced, and the spacing between epoxy segments can not be enlarged significantly by the OPS moieties like that in COOH7.0OPS/DGEBA network. However, contrary to the cross-linking density, it is interesting to note that the storage modulus of COOH5.5OPS/DGEBA network in the glass region greatly decreases, which is the lowest among the five samples, implying that, for COOH7.0OPS/DGEBA sample, the epoxy segments are too tightly tethered on the rigid OPS molecule, whereas the OPS nanoparticles attaching with epoxy network via slightly fewer chemical bonds, e.g., the COOH5.5OPS/DGEBA sample, can relax the external force more effectively.
The tanδ is the ratio of viscous components to elastic components, so the height and width of the tanδ peak can reveal some useful structural information of the epoxy network. As shown in Fig. 5, compared to the pure epoxy, after adding 3 wt% of COOH7.0OPS in the system, the tanδ peak becomes wider and higher since the segments are mutually pulled by the bulky OPS cross-linking points and the packing of epoxy segments is disrupted to some extent, leading to the glass transitions (or α relaxation) covering over a wider temperature region. With the further increment of COOH7.0OPS content, the widths of tanδ peak continue to increase, but the heights start to decrease, suggesting that the elasticity component play a more important role in the event of there being high percentage of OPS in the network owing to the hard nature of OPS molecule.
The packing of polymer segments in the OPS/epoxy networks with different contents of polycarboxyl-OPS are investigated by the wide-angle X-ray diffractogram method. As illustrated in Fig. 6, all the cured networks are amorphous in nature due to the highly cross-linked structures. Moreover, no diffraction peak at 2θ angle of ∼8° corresponding to the aggregation of OPS moeties20,49,50 can be detected. Together with the fact that all the samples are transparent, it proved that the OPS are covalently bonded and homogeneously distributed within the epoxy network. In addition, the cured COOH5.5OPS/DGEBA sample exhibits a very similar diffraction halo to pure epoxy, meaning that the incorporation of COOH5.5OPS does not significantly affect the aggregation structure of epoxy resin network. However, in the event of polycarboxyl-OPS with more carboxyl group per OPS molecule being used, e.g., COOH7.0OPS, the things are different. Relative to the pure epoxy resin, it can be seen that the halos of the cured COOH7.0OPS/DGEBA samples shift toward smaller diffraction angles, and the shifting degree is more pronounced for the sample with the higher content of COOH7.0OPS in the system. For example, the diffraction of maximum peak of the sample containing 12 wt% COOH7.0OPS occurs at the apparently smaller angle of 13.7° compared to 17.1° of pure epoxy sample. Moreover, the pattern becomes broadened. Besides the main diffraction peak at 13.7°, another small wide one at around 28.8° appears, indicating that the packing of epoxy segments are greatly disrupted by the higher content of bulky COOH7.0OPS molecule.
 | ||
| Fig. 6 Wide angle X-ray diffractograms of epoxy resins with (a) 0 wt% of OPS, (b) epoxy with 6 wt% of COOH5.5OPS, (c) epoxy with 3 wt% of COOH7.0OPS, (d) epoxy with 6 wt% of COOH7.0OPS, (c) epoxy with 12 wt% of COOH7.0OPS. | ||
The position of the diffraction halo has been successfully used for the characterization of spacing of chains or segments of amorphous polymers such as polyimides,51 poly(amide–imide)s,52 polyarylates,53 and poly(aryletherketone)s.54 From the angle of the peak maxima (θ), the intersegmental distance (d-spacing) can be calculated according to the Bragg's equation: λ = 2dsinθ, where λ is the X-ray radiation wavelength, and d the intersegmental distance. For this series of OPS/DGEBA cured samples, the results show that the d-spacing values rank in the order: COOH7.0OPS/DGEBA (12 wt%) (6.46 Å) ≫ COOH7.0OPS/DGEBA (6 wt%) (5.68 Å) > COOH7.0OPS/DGEBA (3 wt%) (5.33 Å) > COOH5.5OPS/DGEBA (6 wt%) (5.24 Å) ∼ pure epoxy (5.24 Å). The above WAXD findings are consistent well with the DMA results that the bulky COOH7.0OPS molecule can effectively prop up the epoxy segments and the openness degree increases with the content of COOH7.0OPS in the system.
Mechanical and thermal properties of OPS-modified epoxy networks
The flexural stress-strain curves of the pure epoxy resin and its OPS-modified products are presented in Fig. 7. As was seen, the stress of pure epoxy increased with strain to the maximum value without obvious yielding, implying the brittle fracture. After incorporation of polycarboxyl-OPS, the flexural moduli start to decrease gradually with the OPS content, which trend agrees well with that of storage moduli as measured by DMA method. Correspondingly, relative to the pure epoxy, the OPS-modified samples exhibit apparently improved toughness, as reflected by the increased strain at break. | ||
| Fig. 7 Flexural stress–strain curves of the epoxy resins with different contents of polycarboxyl-OPS. | ||
Relative to COOH7.0OPS, there are fewer reactive carboxyl groups per COOH5.5OPS molecule. After its copolymerization with epoxy resin, the OPS molecules are not attached with the epoxy segments as tightly as that in COOH7.0OPS/DGEBA samples. As a consequence, it is advantageous for the homogeneously dispersed OPS nanoparticles to buffer or release the stress imparted by the external force so that the cured COOH5.5OPS/DGEBA exhibits the larges stain value at break among all the samples.
The scanning electron microscopy is used here to microscopically observe the morphology of fractural surface of samples. As displayed in Fig. 8, for the pure epoxy resin, the fracture surface was rather smooth, without any plastic deformation, indicating that the crack propagate was uninterrupted and there was no energy dissipation mechanism occurring. As a contrast, after incorporation of polycarboxyl-OPS, the fracture surfaces of the samples became rough, indicative of the occurrence of plastic deformation to some extent. The OPS molecule is an organic–inorganic nanoparticle with multiple reactive carboxyl groups surrounding the hard inorganic silica core, while the robust OPS molecule has higher ability to withstand the impact and tearing of external or internal force. Because of the chemical bonding between OPS and the epoxy matrix, the formation of the ductile cross-linked network between OPS and thermosetting epoxy resin results in the cracks in the epoxy segment being inhibited through the energy transfer from epoxy segment to OPS particles, and this effect is more significant for the COOH5.5OPS/DGEBA sample.
 | ||
| Fig. 8 Scanning electron micrographs of the fracture surfaces of the epoxy resins with (a) 0 wt% of COOH7.0OPS, (b) 3 wt% of COOH7.0OPS, (c) 6 wt% of COOH7.0OPS, (d) 12 wt% of COOH7.0OPS, and (e) 6 wt% of COOH5.5OPS. | ||
The thermal degradation curves of the modified samples are presented in Fig. 9. The five samples exhibit the similar maximum weight-loss temperatures at about 415 °C, but the residual weights at above 600 °C for the OPS-modified samples are significantly higher than that of the pure DGEBA epoxy resin. For example, the residual weight of pure epoxy is only 3.5 wt%, whereas that of the sample containing 12 wt% of COOH7.0OPS is as high as 27 wt%, which are caused by not only the inorganic nature of silica core but also the covalent bonding of multiple epoxy chains on the OPS molecule. At the same polycarboxyl-OPS content, compared to the COOH5.5OPS/DGEBA sample, the cured COOH7.0OPS/DGEBA exhibits apparently higher residual weight since there are more epoxy segments chemically boned on the OPS core.
 | ||
| Fig. 9 TGA curves of cured pure epoxy and polycarboxyl-OPS/DGEBA copolymer networks. | ||
Conclusions
In this paper, octaphenylsilsesquioxane (OPS) with different numbers of carboxyl groups per OPS molecule (COOH5.5OPS and COOH7.0OPS) were synthesized, which were then utilized as reactive macromolecular precursors to cure with DGEBA to generate novel covalently-cross-linked OPS/epoxy nanocomposites. The chemical structures of carboxyl number of polycarboxyl-OPS were confirmed by FTIR, 1H NMR 13C NMR and LC-MS analysis methods. The FTIR results demonstrated that the carboxyls of polycarboxyl-OPS had indeed reacted with epoxy groups of DGEBA to form ester bonds. All the synthesized nanocomposites with polycarboxyl-OPS content from 0 to 12 wt% are transparent. In the WAXD diffractograms, no peak corresponding to the OPS aggregation at diffraction angle of ∼8° can be detected, indicating that OPS are covalently bonded onto the epoxy segments, and homogeneously dispersed within the epoxy networks. Moreover, it is the first time to use the WAXD method to characterize the intersegmental distance in POSS/epoxy network, and the results were well consistent with the variation trend of viscoelastic behavior, glass transition temperature and fractural toughness. For the COOH7.0OPS/DGEBA system, with the increase of OPS incorporated in the epoxy network, the diffraction halos shift apparently toward small angle, reflecting that the rigid and bulky multifunctional OPS molecule can significantly open up the epoxy segments between two cross-linking nodes, leading to an increased intersegmental distance, which accounts for the experimental facts that the OPS-modified epoxy resins exhibit increased segmental motion ability, decreased mechanical modulus and improved toughness.Acknowledgements
We thank the National Science Foundation of China (nos. 51073030 and 20874007) and the Program for New Century Excellent Talents in University of China (No. NCET-06-0280) for financial support of this research.References
- A. Goeldel, A. Marmur, G. R. Kasaliwal, P. Potschek and G. Heinrich, Macromolecules, 2011, 44, 6094–6102 CrossRef.
- S. S. Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef CAS.
- E. P. Giannelis, Adv. Mater., 1996, 8, 29–35 CrossRef CAS.
- C. Y. Wan, M. Frydrych and B. Q. Chen, Soft Matter, 2011, 7, 6159–6166 RSC.
- F. Zhao and Y. D. Huang, J. Mater. Chem., 2011, 21, 3695–3703 RSC.
- V. K. Daga, E. R. Anderson, S. P. Gido and J. J. Watkins, Macromolecules, 2011, 44, 6793–6799 CrossRef CAS.
- F. K. Wang, X. H. Lu and C. B. He, J. Mater. Chem., 2011, 21, 2775–2782 RSC.
- B. Alvarado-Tenorio, A. Romo-Uribe and P. T. Mather, Macromolecules, 2011, 44, 5682–5692 CrossRef CAS.
- J. Wu, T. S. Haddad and P.T. Mather, Macromolecules, 2009, 42, 1142–1152 CrossRef CAS.
- J. Wang, A. Sutti, X. G. Wang and T. Lin, Soft Matter, 2011, 7, 4364–4369 RSC.
- Z. B. Wang, S. W. Leng, Z. G. Wang, G. Y. Li and H. Yu, Macromolecules, 2011, 44, 566–574 CrossRef CAS.
- S. Kraus-Ophir, I. Jerman, B. Orel and D. Mandler, Soft Matter, 2011, 7, 8862–8869 RSC.
- S. A. Madbouly and J. U. Otaigbe, Prog. Polym. Sci., 2009, 34, 1283–1332 CrossRef CAS.
- S. Jin, T. Hirai, B. Ahn, Y. Rho, K. W. Kim, M. Kakimoto, P. Gopalan, T. Hayakawa and M. Ree, J. Phys. Chem. B, 2010, 114, 8033–8042 CrossRef CAS.
- A. Tuteja, W. Choi, M. L. Ma, J. M. Mabry, S. A. Mazzella, G. C. Rutledge, G. H. McKinley and R. E. Cohen, Science, 2007, 318, 1618–1622 CrossRef CAS.
- W. A. Zhang and A. H. E. Muller, Macromolecules, 2010, 43, 3148–3152 CrossRef CAS.
- N. C. Escude and E. F. X. Chen, Chem. Mater., 2009, 21, 5743–5753 CrossRef CAS.
- A. L. Goffin, E. Duquesne, J. M. Raquez, H. E. Miltner, X. X. Ke, M. Alexandre, G. Van Tendeloo, B. Van Mele and P. Dubois, J. Mater. Chem., 2010, 20, 9415–9422 RSC.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS.
- Q. Chen, R. W. Xu, J. Zhang and D. S. Yu, Macromol. Rapid Commun., 2005, 26, 1878–1882 CrossRef CAS.
- S. Xiao, M. Nguyen, X. Gong, Y. Cao, H. Wu, D. Moses and A. J. Heeger, Adv. Funct. Mater., 2003, 13, 25–29 CrossRef CAS.
- E. G. Shockey, A. G. Bolf, P. F. Jones, J. J. Schwab, K. P. Chaffee, T. S. Haddad and J. D. Lichtenhan, Appl. Organomet. Chem., 1999, 13, 311–327 CrossRef CAS.
- C. M. Leu, Y. T. Chang and K. H. Wei, Macromolecules, 2003, 36, 9122–9127 CrossRef CAS.
- H. Y. Xu, B. H. Yang, J. F. Wang, S. Y. Guang and C. Li, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5308–5317 CrossRef CAS.
- A. Sellinger and R. M. Laine, Macromolecules, 1996, 29, 2327–2330 CrossRef CAS.
- B. Decker, C. Hartmann-Thompson, P. I. Carver, S. Keinath and P. R. Santurri, Chem. Mater., 2010, 22, 942–948 CrossRef CAS.
- A. Tsuchida, C. Bolln, F. G. Sernetz, H. Frey and R. Mulhaupt, Macromolecules, 1997, 30, 2818–2824 CrossRef CAS.
- H. E. Miltner, N. Watzeels, A. L. Goffin, E. Duquesne, S. Benali, P. Dubois, H. Rahier and B. Van Mele, J. Mater. Chem., 2010, 20, 9531–9542 RSC.
- Q. H. Zhang, H. He, K. Xi, X. Huang, X. H. Yu and X. D. Jia, Macromolecules, 2011, 44, 550–557 CrossRef CAS.
- P. H. Lin and R. Khare, Macromolecules, 2009, 42, 4319–4327 CrossRef CAS.
- Y. L. Liu and H. C. Lee, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4632–4623 CrossRef CAS.
- O. Monticelli, A. Fina, A. Ullah and P. Waghmare, Macromolecules, 2009, 42, 6614–6623 CrossRef CAS.
- E. Markovic, S. Clarke, J. Matisons and G. Simon, Macromolecules, 2008, 41, 1685–1692 CrossRef CAS.
- C. Zhang and R. M. Laine, J. Am. Chem. Soc., 2000, 122, 6979–6988 CrossRef CAS.
- S. J. Garcia, A. Serra and J. Suay, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2316–2367 CrossRef CAS.
- T. M. Lee and C. C. M. Ma, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 757–768 CrossRef CAS.
- Q. P. Guo, P. Figueiredo, R. Thomann and W. Gronski, Polymer, 2001, 42, 10101–10110 CrossRef CAS.
- P. Jain, V. Choudhary and I. K. Varma, Eur. Polym. J., 2003, 39, 181–189 CrossRef CAS.
- B. Francis, G. V. Poel and F. Posada, Polymer, 2003, 44, 3687–3699 CrossRef CAS.
- J. F. Brown, L. H. Vogt and P. I. Prescott, J. Am. Chem. Soc., 1964, 86, 1120–1125 CrossRef CAS.
- C. M. Brick, R. Tamaki, S. G. Kim, M. Z. Asuncion, M. Roll, T. Nemoto, Y. Ouchi, Y. Chujo and R. M. Laine, Macromolecules, 2005, 38, 4655–4660 CrossRef CAS.
- J. H. Fournier, X. Wang and J. Wuest, Can. J. Chem., 2003, 81, 376–379 CrossRef CAS.
- B. F. Zhang and Z. G. Wang, Chem. Commun., 2009, 33, 5027–5029 RSC.
- S. Park and F. L. Jin, Polym. Degrad. Stab., 2004, 86, 515–520 CrossRef CAS.
- B. Francis, S. Thomas, J. Jose, R. Ramaswamy and V. L. Rao, Polymer, 2005, 46, 12372–12385 CrossRef CAS.
- Y. Ni and S. Zheng, Chem. Mater., 2004, 16, 5141–5148 CrossRef CAS.
- H. Z. Liu, S. X. Zheng and K. M. Nie, Macromolecules, 2005, 38, 5088–5097 CrossRef CAS.
- H. Y. Xu, S. W. Kuo, J. S. Lee and F. C. Chang, Polymer, 2002, 43, 5117–5124 CrossRef CAS.
- J. Choi, A. F.Yee and R. M. Laine, Macromolecules, 2003, 36, 5666–5682 CrossRef CAS.
- J. Choi, R. Tamaki, S. G. Kim and R. M. Laine, Chem. Mater., 2003, 15, 3365–3375 CrossRef CAS.
- X. R. Wang and V. J. Foltz, Polymer, 2006, 47, 5090 CrossRef CAS.
- Z. K. Xu, M. Bohning, J. D. Schultze, G. T. Li and J. Springer, Polymer, 1997, 38, 1573 CrossRef CAS.
- I. Kresse, A. Usenko, J. Springer and V. Privalko, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 2183 CrossRef CAS.
- B. F. Zhang, Z. G. Wang, Z. B. Wang, J. F. Li, S. W. Leng and C. J. Shen, Polymer, 2009, 50, 2025 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra00021k |
| This journal is © The Royal Society of Chemistry 2012 |