
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versisterol, a new endophytic steroid with 3CL protease inhibitory activity from Avicennia marina (Forssk.) Vierh.†
Marwa Elsbaey *a,
Mahmoud A. A. Ibrahimb and
Mohamed-Elamir F. Hegazyc
*a,
Mahmoud A. A. Ibrahimb and
Mohamed-Elamir F. Hegazyc
aPharmacognosy Department, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: marwaelsebay1611@mans.edu.eg
bComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia 61519, Egypt
cChemistry of Medicinal Plants Department, National Research Centre, Giza 12622, Egypt
First published on 26th April 2022
Abstract
A new epoxy ergostane sterol, named versisterol, was isolated from Aspergillus versicolor, an endophytic fungus from Avicennia marina. The structure of the isolated compound was deduced by means of one- and two-dimensional NMR and high-resolution mass spectrometry. The absolute stereochemistry was elucidated by NOESY analysis, and experimental and calculated time-dependent density functional theory (TD-DFT) circular dichroism spectroscopy. Versisterol inhibited 3CL protease (3CLpro) with an IC50 value of 2.168 ± 0.09 μM. Binding affinities and molecular interactions of versisterol towards 3CLpro were scrutinized and compared to lopinavir with the help of the combination of docking computations and molecular dynamics (MD) simulation. In silico calculations demonstrated a comparable binding affinity of versisterol with a docking score of −9.4 kcal mol−1, and MM-GBSA binding energy over 200 ns MD simulation of −29.1 kcal mol−1, with respect to lopinavir (−9.8 and −32.2 kcal mol−1, respectively). These findings suggested that versisterol can be an auspicious prototype for developing new 3CLpro drug candidates against COVID-19.
1. Introduction
Owing to their diverse biosynthetic capacity, endophytic fungi represent an enormous pool for novel secondary metabolites. A literature review shows that the genus Aspergillus is the most dominant, representing an untapped source of diverse bioactive compounds.1 By way of example and not limitation, steroids,2 bisabolenes,3 indoloditerpens,4 meroterpenes,5 quinazolines,6 xanthones and anthraquinones,7 cyclopeptides and polyketides8 were previously reported from Aspergillus versicolor.Avicennia marina (Forssk.) Vierh. is a widespread mangrove in Egypt.9 Through our previous investigation on the endophytic diversity of A. marina, we have reported several secondary metabolites from A. versicolor,5,10 A. amstelodami11 and Cladosporium cladosporioides.12 In this manuscript, we report a new epoxy steroid from the rice culture of A. versicolor, an endophytic fungus isolated from A. marina fruits. In late 2019, the pandemic outbreak of coronavirus (COVID-19) appeared. A novel severe acute respiratory syndrome coronavirus (SARS-CoV-2) caused the pandemic.13 To date, there is no effective treatment; COVID-19 has not been only a major health problem, it is causing the worst economic crisis as well. The proteolytic processing of SARS-CoV-2 is mediated by 3-chymotrypsin-like protease (3CLpro), also named the main protease (Mpro). The 3CLpro has a main role in viral replication; therefore it is a prospective target for anticoronaviruses screening.13 In this work, the 3CLpro inhibitory activity of versisterol was investigated. Molecular interaction and binding affinity of versisterol with 3CLpro were foretold and compared to lopinavir (an antiviral protease inhibitor) with the assistance of molecular docking technique. The docked versisterol–3CLpro complex was thereafter subjected to molecular dynamics (MD) simulations throughout 200 ns. Besides, the corresponding binding affinity and steadiness were estimated.
2. Results and discussion
2.1. Identification of versisterol
Versisterol (Fig. 1) was purified as a rose-colored amorphous material. It was obtained as a minor metabolite from the fermented rice culture of A. versicolor. | ||
| Fig. 1 The structure of versisterol isolated from A. vsersicolor. | ||
The molecular formula of versisterol was determined as C28H40O3 based on HR-ESIMS peak at m/z 447.2925 [M + Na]+ (calcd 447.2875). The previous molecular formula suggested nine degrees of saturation. Interpretation of 1 and 2D NMR spectral data (Table 1, Fig. 2 and 3) established the presence of a steroidal nucleus with 6 methyls, 7 methylenes, 8 methines and 7 quaternary carbons. The data also showed the presence of four olefinic carbons at δC 159.3, 126.5, 136.0, and 134.4; in addition to two carbonyl carbons at δC 201.7, and 216.8.
| H/C | δC (m) | δH (m, J in Hz) |
|---|---|---|
| a Masked by solvent signal. | ||
| 1 | 40.0 (CH2) | 1β: 2.02 (m) |
| 1α: 2.07 (m) | ||
| 2 | 39.0 (CH2) | 2β: 2.64 (m) |
| 2α: 2.39 (m) | ||
| 3 | 201.7 (c) | — |
| 4 | 126.5 (CH) | 6.16 (br d) |
| 5 | 159.3 (c) | — |
| 6 | 41.4 (CH2) | 6β: 2.57 (d, 10.8) |
| 6α: 2.63 (d, 10.8) | ||
| 7 | 216.8 (c) | — |
| 8 | 66.9 (c) | — |
| 9 | 51.0 (CH) | 2.86 (dd, 14.1, 5.5) |
| 10 | 37.4 (C) | — |
| 11 | 25.9 (CH2) | 11β: 1.98 (m) |
| 11α: 2.12 (m) | ||
| 12 | 38.5 (CH2) | 12β: 1.91 (m) |
| 12α: 1.72 (m) | ||
| 13 | 55.7 (c) | — |
| 14 | 64.1 (c) | — |
| 15 | 35.2 (CH2) | 15β: 2.58 (m) |
| 15α: 2.38 (m) | ||
| 16 | 24.7 (CH2) | 16β: 1.71 (m) |
| 16α: 1.96 (m) | ||
| 17 | 49.7a (CH) | 1.70 (m) |
| 18 | 17.4 (CH3) | 0.99 (s) |
| 19 | 24.2 (CH3) | 1.29 (s) |
| 20 | 38.6 (CH) | 2.49 (m) |
| 21 | 24.1 (CH3) | 1.12 (d, 7.1) |
| 22 | 134.3 (CH) | 5.33 (m) |
| 23 | 136.0 (CH) | 5.33 (m) |
| 24 | 44.7 (CH) | 1.88 (m) |
| 25 | 34.4 (CH) | 1.48 (m) |
| 26 | 20.1 (CH3) | 0.84 (d, 6.8) |
| 27 | 20.5 (CH3) | 0.86 (d, 6.8) |
| 28 | 18.1 (CH3) | 0.94(d, 6.8) |
 | ||
| Fig. 2 Key COSY and HMBC correlations for versisterol. | ||
 | ||
| Fig. 3 Key NOESY correlations for versisterol. | ||
The presence of four methyls showing the doublet splitting pattern at δH 1.12 (CH3-21), 0.84 (CH3-26), 0.86 (CH3-27) and 0.94 (CH3-28), in addition to the olefinic carbons at δC 134.3 (C-22) and 136.0 (C-23) are a typical feature for the unsaturated alkyl side chain of ergosterol.14 This was established from the HMBC correlations of H3-21 to C-17, C-20, and C-22; H3-28 to C-23 and C-25; as well as H3-26 and H3-27 to C-24 and C-25. The methines at δH/C 2.86/51.0; 1.70/49.7; 2.49/38.6 were assigned to C-9, C-17, and C-20, respectively. This was established from the HMBC correlations of H3-19 to C-9; H3-18 and H3-21 to C-17; H3-21 and H-22 to C-20.
An α–β unsaturated carbonyl fragment was established from the shielded olefinic methine at δC 126.5 (C-4), deshielded olefinic quaternary carbon at δC 159.3 (C-5) and the carbonyl group at δC 201.7 (C-3). This was established from the HMBC correlations of H-4 to C-3 and C-5. In addition, another carbonyl group was assigned at position 7, resonating at δC 216.8 (C-7); this was established from the HMBC correlations of H-9 to C-7; and H-6 to C-7.
The two oxygenated quaternary carbons at δC 66.9 (C-8) and 64.1 (C-14) were assigned to an epoxy moiety at positions 8 and 14. This was confirmed from HMBC correlations of H-9, H3-18, H-12, and H-15 to C-14; and H-11/C-8.
The relative configuration was determined from the detailed analysis of the NOESY correlations (Fig. 3). The correlations of H3-19 with H-2β and 11β; H-11β with H3-18; H3-18 with H-15β; and H-15β with H-16β revealed that these protons are β-positioned. Meanwhile, the correlations of H-9 with H-1α, H-6α and H-12α; and H-17 with H-16α suggested that these protons are α-positioned. The correlations of H3-18 with H-20 as well as H3-21 with H-17 suggested (20R) configuration.15 Furthermore, regarding biogenesis, the 20S-epimer of ergosterol probably does not exist biologically. This theoretical restriction does not apply at C-24, and both C-24 epimers could naturally exist, although apparently not in fungi.16
In general, it is reported that fungi and algae produce sterols with the 24β configuration, whereas those produced in most vascular plants possess the 24α configuration.17 Furthermore, the chemical shifts of the side-chain carbons can permit the determination of the absolute configuration at C-24 in sterols. Referring to the C-24 epimers of 24-methylchlolesta-5,22-diene-3β-ol, the resonance of CH3-28 in versisterol (δC 18.10) was constituent with the 24β-epimer (δC 18.08), not with the 24α-epimer, in which CH3-28 is reported to specifically resonate at (δC 17.6 ± 0.1).17 Based on this evidence, the (24S) configuration was concluded.
The next step was to determine the configuration of the epoxy group. According to the literature, the oxirane ring is almost perpendicular to the molecular plane,15 and it was placed in the β-orientation based on the CD calculations. The absolute configuration of the compound was determined by the time-dependent density functional theory (TD-DFT) ECD calculations. To further confirm the β-orientation of the epoxy group, the chemical shift of C-8 and C-14 in versisterol were compared to the closely related (22E)-ergosta-22-ene-8,14-epoxy-3,7-dione (ehrenasterol).18 In versisterol, C-8 and C-14 resonated at δC 66.9 and 64.1, respectively; meanwhile, the α-oriented epoxy group in ehrenasterol resonated at δC 60.0 and 65.6, respectively. Furthermore, the deshielding of CH3-18 in versisterol (δC 17.4, δH 0.99) as compared to ehrenasterol (δC 15.3, δH 0.75) may suggest that CH3-18 and the epoxy group are oriented in the same side i.e. the β-orientation.19
The experimental ECD spectrum (Fig. 4) was in good agreement with the theoretical ECD of (22E,20R)-8β,14β-epoxy-ergosta-4,22-diene-3,7-dione.
 | ||
| Fig. 4 Experimental and TD-DFT-simulated electronic circular dichroism (ECD) in methanol for versisterol. | ||
Accordingly, the compound was elucidated as (22E,20R,24S)-8β,14β-epoxy-ergosta-4,22-diene-3,7-dione, and trivially named versisterol. The compound is a new derivative of ergosterol, which is the most abundant sterol in fungal cell membranes.20
2.2. 3CLpro inhibitory activity
To our current date, there is no effective treatment of COVID-19. Lopinavir and ritonavir are protease inhibitors that are commonly used for the treatment of HIV infection. The combination has been repurposed for COVID-19; however, it is still going under clinical trials.21Natural products have always been exploited in the pursuit of new drugs. The 3CLpro enzyme is a prospective target for anticoronaviruses drugs.13 Versisterol, a new ergosterol derivative isolated from the endophytic A. versicolor, in this study, was tested for its 3CLpro inhibition activity (Table 2). It showed an IC50 value of 2.168 ± 0.09 μM, which is about fifteen times of lopinavir (IC50 value of 0.148 ± 0.01 μM). It is worth noting that several steroids are reported to inhibit bovine chymotrypsin.22 In addition, ergosterol peroxide is reported to demonstrate antiviral and immunomodulatory activity against porcine delta coronavirus.23 The results, in addition to previous literature, suggest that versisterol is a promising skeleton for designing new drugs against COVID-19. These results prompted us to investigate the binding affinities and molecular interactions of versisterol towards 3CLpro and compare it to lopinavir employing molecular docking and molecular dynamics (MD) simulations.
| Compound | IC50 (μM) |
|---|---|
| a The data are expressed as mean ± standard deviation. | |
| Versisterol | 2.168 ± 0.09 |
| Lopinavir | 0.148 ± 0.01 |
2.3. Molecular docking
AutoDock4.2.6 software was used to anticipate the binding affinities and molecular features of the isolated versisterol and lopinavir complexed with 3CLpro. The portended binding affinities, 3D in addition to 2D visualizations of the docking poses of the investigated inhibitors within the binding pocket of 3CLpro are shown in Fig. 5. From the data in Fig. 5, versisterol elucidated an auspicious binding affinity towards 3CLpro with a docking score of −9.4 kcal mol−1. The prodigious potentiality of versisterol as a 3CLpro inhibitor is ascribed to its capacity to exhibit various hydrogen bonds, hydrophobic, in addition to van der Waals interactions with the most important amino acids within the binding pocket (Fig. 5). Precisely, the oxygen atom of the oxirane ring forms a hydrogen bond with the backbone NH of GLU166 with a bond length of 1.71 Å (Fig. 5). Besides, the CO group of cyclohexanone exhibits two hydrogen bonds with the hydroxy group of SER144 and the imidazole of HIS163 with bond lengths of 2.72 and 1.96 Å (Fig. 5). In comparison with versisterol, lopinavir demonstrated an almost identical binding affinity against 3CLpro with a docking score of −9.8 kcal mol−1, exhibiting four hydrogen bonds with LEU141, SER144, HIS163, as well as GLY143 with bond lengths in the range of 1.96 to 3.09 Å (Fig. 5).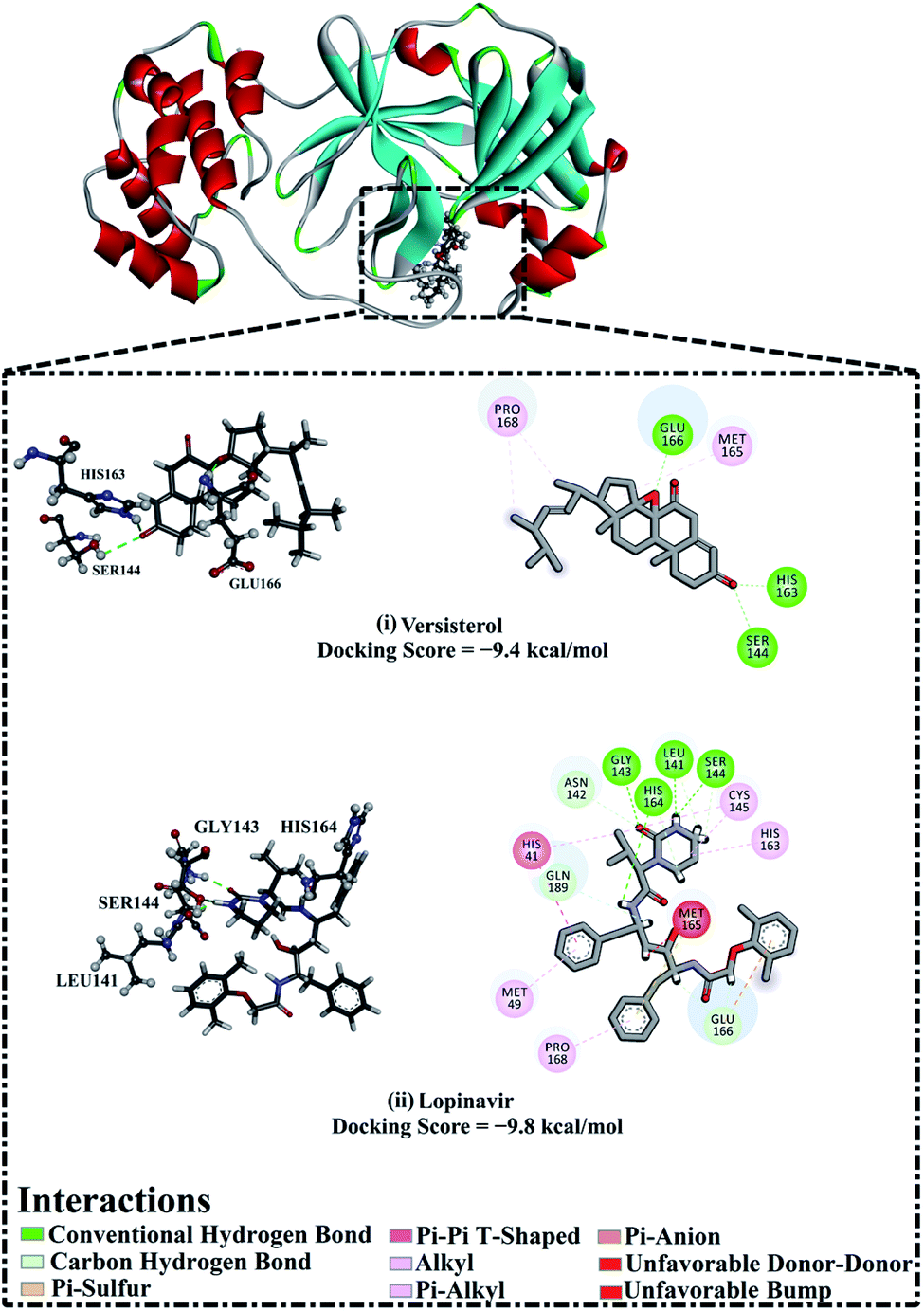 | ||
| Fig. 5 3D and 2D representations as well as the predicted docking scores of (i) versisterol and (ii) lopinavir with 3-chymotrypsin-like protease (3CLpro). | ||
2.4. Molecular dynamics simulations
Molecular dynamics (MD) simulations inspect the steadiness of the inhibitor–target complexes, structural specifics, orientational flexibilities, in addition to the thoroughness of inhibitor–target binding affinities.24,25 As a consequence, the investigated inhibitors complexed with 3CLpro were submitted to MD simulations pursued by binding free energy estimations using the molecular mechanics-generalized born surface area (MM-GBSA) approach. The computed MM-GBSA binding energies according to the gathered trajectories over 200 ns are represented in Fig. 6. As shown in Fig. 6, versisterol revealed a favorable binding affinity with an average ΔGbinding of −29.1 kcal mol−1 compared to lopinavir (calc. −32.2 kcal mol−1). The current results were in line with the experimental data, manifesting the comparable binding affinity of versisterol as a 3CLpro inhibitor. | ||
| Fig. 6 Decomposition of MM-GBSA binding energies for the inspected inhibitors complexed with 3-chymotrypsin-like protease (3CLpro) throughout 200 ns MD simulations. | ||
To explore the most significant interactions between the inhibitor and enzyme, MM-GBSA binding free energies of the scrutinized inhibitors complexed with 3CLpro were decomposed and depicted in Fig. 6. As shown in Fig. 6, it is evident that the binding affinities of versisterol and lopinavir were predominated via Evdw interactions with average values of −39.1 and −45.2 kcal mol−1, respectively. Eele interactions were convenient with average values of −5.4 and −21.1 kcal mol−1 for versisterol and lopinavir, respectively (Fig. 6). Together these findings supply quantitative data of the binding free energies of versisterol and lopinavir as anti-COVID-19 drug candidates.
2.5. Post-MD analyses
In order to otherwise establish the constancy and behavior of versisterol in complex with 3CLpro, structural and energetical analyses were accomplished throughout the simulation time of 200 ns and compared to those of lopinavir. Monitoring of the conformational immutability of the investigated complex was fulfilled via inspecting root-mean-square deviation (RMSD) and binding energy per-frame. | ||
| Fig. 7 Estimated MM-GBSA binding energy per-frame for versisterol (in black), and lopinavir (in light blue) with 3-chymotrypsin-like protease (3CLpro) throughout 200 ns MD simulations. | ||
 | ||
| Fig. 8 Root-mean-square deviation (RMSD) of the backbone atoms from the starting structure for versisterol (in black) and lopinavir (in light blue) with the 3-chymotrypsin-like protease (3CLpro) throughout 200 ns MD simulations. | ||
3. Experimental
3.1. General experimental procedures
Nuclear magnetic resonance (NMR) measurements were conducted on Varian INOVA-600 (600 MHz and 150 MHz for 1H and 13C, respectively). Chemical shifts (δ) are expressed in ppm and coupling constants (J) are in Hz. A Bruker microTOF mass spectrometer was used for high-resolution mass measurement. ECD spectra were recorded at room temperature on a Jasco J815 spectrophotometer in 1 cm cuvettes. A PerkinElmer Model 343 polarimeter was used for recording the optical rotation. Column chromatography was performed using silica gel G 60-230 (Merck, Germany). The thin-layer chromatographic screening was made with Merck precoated silica gel F254 plates.3.2. Fungal strain
The endophytic fungal strain was purified from the fruit of the mangrove Avicennia marina (Forssk.) Vierh. The mangrove was obtained from Kilo 17, Safaga, Red Sea, Egypt. It was identified as Aspergillus versicolor (GenBank accession no. LC431696). The detailed procedures for fungus isolation, purification and fermentation were described previously.53.3. Purification of versisterol
The fermentation media were extracted with EtOAc and defatted with n-hexane as previously described.5 The defatted ethyl acetate extract (9.7 g) was chromatographed through a silica gel column (28 × 4 cm, 164 g) using n-hexane/EtOAc as a solvent system. Fraction 3 (114.6 mg, eluted using 30% EtOAc in hexane gradient elution) was purified over a silica gel column (45 × 1.5, 120 g) using CH2Cl2/MeOH. Versisterol (1.6 mg) was eluted using 2% MeOH in CH2Cl2.3.4. ECD calculations
To predict the possible conformers for versisterol, conformational analysis was thoroughly performed within an energy window value set to 10 kcal mol−1 with the assistance of Omega2 software.26 Geometric optimization was first optimized for the resulted conformers at the B3LYP/6-31G*. Upon the optimized conformers, frequency calculations were then carried out to outline the nature of the local minima and evaluate the Gibbs free energies. The time-dependent density functional theory (TD-DFT) computations were then executed to calculate the first fifty excitation states by incorporating the polarizable continuum model (PCM) utilizing methanol as a solvent. The obtained electronic circular dichroism (ECD) spectra were Boltzmann-averaged, and SpecDis 1.71 was used to obtain ECD spectra.27,28 The Gaussian09 software was used to perform the quantum mechanical calculations.293.5. 3CLpro inhibitory activity
Inhibition of 3CLpro enzyme activity was measured using the Fluorogenic 3CLpro Assay Kit (BPS Bioscience #79955, San Diego CA, USA) following the guidelines of the manufacturer's instructions.30,313.6. In silico drug discovery
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000. Additionally, the number of genetic algorithm (GA) run was set to 250. Other docking parameter options were preserved at their default settings. The grid map utilizing a grid box was prepared with the assistance of the AutoGrid program. A grid box with dimensions 60 Å × 60 Å × 60 Å positioned at the coordinates (x, y, z) of −13.069, 9.740, and 68.490 was utilized to encompass the entire active site of 3CLpro. The grid spacing value was adjusted to 0.375 Å. The partial atomic charges of the investigated compounds were determined using the Gasteiger method.38 The foreseen docking poses for every investigated inhibitor were processed utilizing a tight clustering analysis through the root-mean-square deviation (RMSD) tolerance of 1.0 Å. Additionally, the lowest energy orientation from the largest cluster was selected as a representative binding mode.
000000. Additionally, the number of genetic algorithm (GA) run was set to 250. Other docking parameter options were preserved at their default settings. The grid map utilizing a grid box was prepared with the assistance of the AutoGrid program. A grid box with dimensions 60 Å × 60 Å × 60 Å positioned at the coordinates (x, y, z) of −13.069, 9.740, and 68.490 was utilized to encompass the entire active site of 3CLpro. The grid spacing value was adjusted to 0.375 Å. The partial atomic charges of the investigated compounds were determined using the Gasteiger method.38 The foreseen docking poses for every investigated inhibitor were processed utilizing a tight clustering analysis through the root-mean-square deviation (RMSD) tolerance of 1.0 Å. Additionally, the lowest energy orientation from the largest cluster was selected as a representative binding mode.The binding affinities of the inspected inhibitors complexed with 3CLpro were computed utilizing the molecular mechanical-generalized Born surface area (MM-GBSA) approach.49 For MM-GBSA computations, uncorrelated snapshots were recorded every 10 ps over the production stage. The MM-GBSA binding energy (ΔGbinding) can be conceptually summarized as:
| ΔGbinding = Gcomplex − (Gcompound + G3CLpro) |
4. Conclusion
A new epoxy steroid, versisterol, was purified from the endophytic fungus, A. versicolor. The absolute configuration was fully established by analyzing experimental and calculated TD-DFT ECD. Versisterol inhibited SARS-CoV-2 3CL protease (3CLpro) with IC50 value 2.168 ± 0.09 μM. Molecular docking and molecular dynamics (MD) simulations were utilized to predict the binding affinities and modes of versisterol against 3CLpro and compared to lopinavir (an antiviral protease inhibitor). Versisterol revealed good binding affinity against 3CLpro with a docking score of −9.4 kcal mol−1 and ΔGbinding = −29.1 kcal mol−1 over 200 ns MD simulations. On the other hand, lopinavir exhibited a docking score of −9.8 kcal mol−1 and ΔGbinding = −32.2 kcal mol−1. Energetical and structural analyses over the 200 ns MD simulations unveiled the constancy of versisterol and lopinavir with 3CLpro. This study provides some clues about versisterol as a candidate inhibitor for SARS-CoV-2 3CLpro.Author contributions
Marwa Elsbaey: conceptualization, investigation, methodology, writing – review & editing; Mahmoud A. A. Ibrahim: formal analysis, software, writing – review & editing; Mohamed-Elamir Hegazy: conceptualization and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computational work of this study was accomplished using the resources supported by the Science and Technology Development Fund, STDF, Egypt, grants no. 5480 & 7972 (granted to Mahmoud A. A. Ibrahim).References
- H. E. Ortega, D. Torres-Mendoza, Z. Caballero E and L. Cubilla-Rios, J. Fungi, 2021, 7, 570 CrossRef CAS PubMed.
- J. H. Ding, Z. G. Ding, J. Y. Zhao, M. G. Li, D. B. Hu, X. J. Jiang, D. S. Zhou, F. Wang and M. L. Wen, Nat. Prod. Res., 2019, 33, 1885 CrossRef CAS PubMed.
- X. D. Li, X. Li, X.-M. Li, X. L. Yin and B. G. Wang, Nat. Prod. Res., 2021, 35, 4265 CrossRef CAS PubMed.
- D. Zhang, W. Yi, H. Ge, Z. Zhang and B. Wu, Nat. Prod. Res., 2021, 35, 3114 CrossRef CAS PubMed.
- M. Elsbaey, C. Tanaka and T. Miyamoto, Phytochem. Lett., 2019, 32, 70 CrossRef CAS.
- Y. Zhuang, X. Teng, Y. Wang, P. Liu, G. Li and W. Zhu, Org. Lett., 2011, 13, 1130 CrossRef CAS PubMed.
- W. Ze-Hong, L. Dong, X. Ying, C. Jian-Liang and L. Wen-Han, Chin. J. Nat. Med., 2018, 16, 219 Search PubMed.
- Y. Zhuang, X. Teng, Y. Wang, P. Liu, H. Wang, J. Li, G. Li and W. Zhu, Tetrahedron, 2011, 67, 7085 CrossRef CAS.
- A. Gab-Alla, M. Fouda and W. Morsy, Egypt. J. Aquat. Biol. Fish., 2010, 14, 79 CrossRef.
- M. Elsbaey, C. Tanaka and T. Miyamoto, Nat. Prod. Res., 2022, 36, 760 CrossRef CAS PubMed.
- M. Elsbaey, A. Sallam, M. El-Metwally, M. Nagata, C. Tanaka, K. Shimizu and T. Miyamoto, Chem. Biodiversity, 2019, 16, e1900237 CrossRef PubMed.
- A. Sallam, M. El-Metwally, M. A. Sabry and M. Elsbaey, Nat. Prod. Res., 2021, 1 CrossRef PubMed.
- M. T. ul Qamar, S. M. Alqahtani, M. A. Alamri and L.-L. Chen, J. Pharm. Anal., 2020, 10, 313 CrossRef PubMed.
- R. Nowak, M. Drozd, E. Mendyk, M. Lemieszek, O. Krakowiak, W. Kisiel, W. Rzeski and K. Szewczyk, Molecules, 2016, 21, 946 CrossRef PubMed.
- J. H. Yu, S. J. Yu, K. L. Liu, C. Wang, C. Liu, J. y. Sun and H. Zhang, Steroids, 2021, 165, 108767 CrossRef CAS PubMed.
- J. H. Adler, M. Young and W. R. Nes, Lipids, 1977, 12, 364 CrossRef CAS PubMed.
- J. Sright, A. McInnes, S. Shimizu, D. Smith, J. Walter, D. Idler and W. Khalil, Can. J. Chem., 1978, 56, 1898 CrossRef.
- D. T. Youssef, J. M. Badr, L. A. Shaala, G. A. Mohamed and F. H. Bamanie, Phytochem. Lett., 2015, 12, 296 CrossRef CAS.
- S. H. Wu, R. Huang, C. P. Miao and Y. W. Chen, Chem. Biodiversity, 2013, 10, 1276 CrossRef CAS PubMed.
- M. L. Rodrigues, mBio, 2018, 9, e01755 CrossRef PubMed.
- A. B. Owa and O. T. Owa, J. Microbiol., Immunol. Infect., 2020, 53, 674 CrossRef CAS PubMed.
- M. Mayer, B. Neufeld and Z. Finci, Biochem. Pharmacol., 1982, 31, 2989 CrossRef CAS PubMed.
- C. Duan, X. Ge, J. Wang, Z. Wei, W. H. Feng and J. Wang, Int. Immunopharmacol., 2021, 93, 107317 CrossRef CAS PubMed.
- M. De Vivo, M. Masetti, G. Bottegoni and A. Cavalli, J. Med. Chem., 2016, 59, 4035 CrossRef CAS PubMed.
- J. E. Kerrigan, in Molecular dynamics simulations in drug design, ed. S. Kortagere, Humana Press, Totowa, NJ, 2013 Search PubMed.
- OMEGA 2.5.1.4, OpenEye Scientific Software, Santa Fe, NM, USA, 2013 Search PubMed.
- T. Bruhn, A. Schaumloffel, Y. Hemberger and G. Bringmann, SpecDis, University of Würzburg, Würzburg, Germany, 2012 Search PubMed.
- T. Bruhn, A. Schaumloffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E01, Gaussian Inc., Wallingford CT, USA, 2009 Search PubMed.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, ChemBioChem, 2020, 21, 730 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, C. Peng, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, R. Jiang, X. Yang, T. You, X. Liu, X. Yang, F. Bai, H. Liu, X. Liu, L. W. Guddat, W. Xu, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao and H. Yang, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33, 368 CrossRef PubMed.
- P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572 CrossRef CAS PubMed.
- SZYBKI 1.9.0.3, OpenEye Scientific Software, Santa Fe, NM, USA, 2013 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905 CrossRef CAS PubMed.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219 CrossRef CAS.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- M. A. A. Ibrahim, K. A. A. Abdeljawaad, A. H. M. Abdelrahman, O. R. Alzahrani, F. M. Alshabrmi, E. Khalaf, M. F. Moustafa, F. Alrumaihi, K. S. Allemailem, M. E. S. Soliman, P. W. Paré, M.-E. F. Hegazy and M. A. M. Atia, Antibiotics, 2021, 10, 934 CrossRef CAS PubMed.
- M. A. A. Ibrahim, A. H. M. Abdelrahman, M. A. M. Atia, T. A. Mohamed, M. F. Moustafa, A. R. Hakami, S. A. M. Khalifa, F. A. Alhumaydhi, F. Alrumaihi, S. H. Abidi, K. S. Allemailem, T. Efferth, M. E. Soliman, P. W. Paré, H. R. El-Seedi and M.-E. F. Hegazy, Mar. Drugs, 2021, 19, 391 CrossRef CAS PubMed.
- M. A. A. Ibrahim, A. H. M. Abdelrahman, T. A. Mohamed, M. A. M. Atia, M. A. M. Al-Hammady, K. A. A. Abdeljawaad, E. M. Elkady, M. F. Moustafa, F. Alrumaihi, K. S. Allemailem, H. R. El-Seedi, P. W. Pare, T. Efferth and M. F. Hegazy, Molecules, 2021, 26, 2082 CrossRef CAS PubMed.
- M. A. A. Ibrahim, E. A. A. Badr, A. H. M. Abdelrahman, N. M. Almansour, G. A. H. Mekhemer, A. M. Shawky, M. F. Moustafa and M. A. M. Atia, Mol. Inform., 2021, 40, 2060039 Search PubMed.
- M. A. A. Ibrahim, E. A. A. Badr, A. H. M. Abdelrahman, N. M. Almansour, A. M. Shawky, G. A. H. Mekhemer, F. Alrumaihi, M. F. Moustafa and M. A. M. Atia, Cell Biochem. Biophys., 2021, 79, 189 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269 CrossRef CAS.
- Dassault Systèmes BIOVIA, San Diego, CA, USA, 2019.
- I. Massova and P. Kollman, Perspect. Drug Discovery Des., 2000, 18, 113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00877g |
| This journal is © The Royal Society of Chemistry 2022 |