
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu–Pt–Ni nanochains as dopamine catalysts: role of elements and their spatial distribution†
Hua
Fan
,
William
Le Boeuf
and
Vivek
Maheshwari
 *
*
Department of Chemistry, Waterloo Institute for Nanotechnology, 200 University Ave. West, Waterloo, N2L 3G1, ON, Canada. E-mail: vmaheshw@uwaterloo.ca
First published on 7th March 2023
Abstract
Multi-element materials can improve biosensing ability as each element can catalyze different steps in a reaction pathway. By combining Pt and Ni on self-assembled 1D gold nanochains and controlling their spatial distribution, a detailed understanding of each element’s role in dopamine oxidation is developed. In addition, the developed synthesis process provides a simple way to fabricate multi-element composites for electrocatalytic applications based on electrical double-layer formation on the surface of charged nanoparticles. The performance parameters of the catalyst, such as its sensitivity, limit of detection, and range of operation for dopamine sensing, are optimized by changing the relative ratios of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni and the morphology of the Pt and Ni domains, using the developed understanding. The morphology of the domains also affects the oxidation state of Ni, which is crucial to the performance of the electrocatalyst.
:Ni and the morphology of the Pt and Ni domains, using the developed understanding. The morphology of the domains also affects the oxidation state of Ni, which is crucial to the performance of the electrocatalyst.
Introduction
Detection of biologically relevant molecules using electrocatalysts is an intensely researched area due to its application in health monitoring and diagnostics. A variety of biomolecules such as glucose, dopamine, uric acid, ascorbic acid, lactic acid, and catechols, among others have been targeted for non-enzymatic sensing using electrocatalysts.1–7 The characteristics of an effective electrocatalytic material for bio-sensing share attributes with similar materials used in the areas of water splitting, methanol and ethanol oxidation, etc. The oxidation of biomolecules such as glucose and dopamine, similar to the oxidation of small organic molecules, results in multiple intermediates, and a multi-step process that requires adsorption and desorption of the intermediate molecules and their oxidation/reduction.1,3,8 Combining multiple elements in a composite catalyst has been an effective strategy for sensing of biomolecules. In particular, development of an electrocatalyst for dopamine sensing is a critical area of research. This is due to dopamine being a neurotransmitter which is linked to motor control and reinforced learning, with its abnormal concentration being related to neurological diseases like Parkinson, schizophrenia, and addiction.3,9,10 The oxidation pathway of dopamine at neutral pH consists of at least two distinct redox active steps (with intermediate steps).11 A number of catalysts have been reported for sensing dopamine. However, a fundamental understanding of the catalytic effect of each element (in the catalyst) on the sensing pathway is required to optimize its performance parameters such as sensitivity, range of operation and limit of detection (LoD), and linearity. In this work, we use a simple route for making a bi-elemental catalyst that allows for varying the relative ratio of the two elements and also their morphology. Using Pt- and Ni (metal, and metal oxide and hydroxide)-based catalyst for dopamine oxidation we show that while Pt is more active for the first step, Ni is favoured for the second step. Then by combining Pt and Ni in a single catalyst structure using the developed synthesis method, we show that the optimization of the performance is based on both the relative ratios of Pt:Ni, but also crucially on the morphology of the Pt and Ni domains. Based on the composition and the morphology of the catalyst, its sensitivity, LoD, and range of operation can be controlled to tailor its performance based on the detection requirement.
The catalyst is made by simple self-assembly of nanoparticles into micron-long chains, by the use of metal cations (Pt4+ and Ni2+). The cations are subsequently transformed into Pt and Ni (metallic) and Ni (2+ & 3+) domains while preserving the chain-like morphology. The method leads to the formation of a highly porous 3D structure of overlapping chains, resulting in high catalytic activity and stability due to effective mass transfer. The key aspect of the process is that the structural morphology of the chains can be modulated based on the kinetics of the self-assembly process. This is used for further optimization of the catalyst performance independent of its composition. Such structural modulations are key for the development of more effective catalysts.12–14
Dopamine undergoes a two-step oxidation process:11 the first redox step (a multi-step process) involves a two-electron oxidation process that transforms dopamine to dopamine-o-quinone (redox step I, RS1). The second step then transforms the quinone to aminochrome in a two-electron (multi-step) oxidation process (redox step II, RS2), Scheme 1a. By varying the catalyst composition and configuration, the sensitivity for dopamine detection can be varied from ∼133–1279 μA mM cm−2. The limit of detection can be varied from 1 nM to 34 nM, and the maximum detection can be as high as 371 μM. Maximum sensitivity is obtained from an optimized ratio of Ni:Pt (1:1) with predominantly domains of Pt0 and Ni0 and Ni3+ (and Pt–Ni alloy), while lower LoD and a wider range of operation are shown by the catalyst with a higher ratio of Ni compared to Pt and with a significant presence of Ni2+ and Ni3+. The effectiveness of the catalyst is dependent on the interplay between the rates for the two steps, RS1 and RS2. Being sequential in nature, a faster RS2 step (due to higher Ni content) leads to lower LoD and a larger range of operation as it promotes the irreversibility of the RS1 step.
 | ||
| Scheme 1 (a) The reaction pathway for dopamine oxidation has two key oxidation steps, R1 and R2. Each step involves a two-electron process. Between the two steps is the cyclization step, C1. (b) Based on the electrochemical results the Pt–Ni combined catalyst will have domains that have specific affinity to catalyze the first oxidation step (Pt) and the second step (Ni). | ||
Results and discussion
The schematic image in Fig. 1a shows the self-assembly process of Au nanoparticles (citrate capped) by using Pt4+ and/or Ni2+ cations. The 10–12 nm Au nanoparticles are stabilized in an aqueous medium due to the negatively charged citrate ions on their surface (zeta potential of ∼−40 mV). The self-assembly process is triggered by the addition of a limited amount of metal salt. The metal cations interact strongly with the negatively charged citrate groups on the Au surface and function as a linking bridge. This induces the electric dipole–dipole interactions causing the Au NPs to self-assemble into micron-long nanochains (zeta potential lowers to ∼−25 mV).15 | ||
| Fig. 1 (a) Schematic image of the preparation process of 1D Au–Ni–Pt nanochains; the TEM image of the gold nanochains (b) before and (c) after reduction. The inset images are related to HRTEM image and EELS elemental mapping (in 1 c), with Au shown in green and Pt in orange. The scale bar in (b) is 400 nm, in (c) 200 nm and the scale bar in the inset of (c) is 20 nm. (d) Typical UV-vis spectrum of self-assembled gold nanochains. | ||
Subsequently, they are chemically reduced by NaBH4 to obtain the continuous morphology of Au–Pt or Au–Ni nanochains with a micron length scale. The transmission electron microscopy (TEM) images of Fig. 1b and c show the typical morphology of these nanochains, before and after reduction. Before reduction, the nanochains show a discreet assembly of Au nanoparticles with a gap of 1–2 nm between adjacent particles (Fig. 1b). After reduction, the assembly is transformed to a continuous chain morphology due to the bridging of the gap between adjacent nanoparticles by metallic Pt and/or Ni (Fig. 1c). Further, the inset of Fig. 1c shows the elemental mapping of the Au–Pt chains by electron energy loss spectroscopy (EELS). The formation of a Pt shell around the Au nanoparticles and bridging of the gaps by thin Pt nanowires (∼3–4 nm in size) are clearly observed. The UV-vis absorption spectrum (Fig. 1d) illustrates the self-assembly process of the chains. The initial 10–12 nm Au nanoparticles show a plasmon resonance peak at 525 nm, which shifts to 620 nm, confirming the formation of ∼250 nm 1D branched chains. Based on our previous study, metal cations are adsorbed on the surface of Au NPs, and act as a linking bridge.16,17 After adding NaBH4, the metal cations are reduced into a metallic form (in the case of Pt) and to a mixed oxidation state (in the case of Ni). The ultra-thin nanowires morphology on the Au nanoparticle (Fig. 1c) template leads to high surface exposure of the active sites, which is crucial for electrocatalytic applications.
Dopamine oxidation was conducted on the Au–Pt and Au–Ni samples in neutral pH, to understand the effect of each element. The cyclic voltammetry plots ranging in scan rates from 20 mV s−1 to 200 mV s−1 are shown in Fig. 2 for Au–Pt and Au–Ni. For both materials, two sets of redox peaks are observed, P1(oxidation) and P2 (reduction) in the region ∼0.05–0.2 V and P3 (oxidation) and P4 (reduction) in the region ∼−0.25–−0.4 V. Peaks P1 and P2 are related to redox reaction step R1 and peaks P3 and P4 to redox reaction step R2. The reactions for these steps are listed below based on the established mechanism for dopamine oxidation (Scheme 1a).11 The reaction set R1 consists of two oxidation steps with potential inversion where the first step occurs at greater positive potential than the second step. There is an intermediate cyclization step between R1 and R2.11 The two materials show a critical difference in their dopamine oxidation behavior. For Au–Pt, relatively sharp oxidation and reduction peaks for step R1 are observed, and the peaks maintain their distinct sharp nature across the scan rate. For the peaks associated with step R2, however, with increasing scan rates they become more subdued. The cyclization of dopamine-o-quinone to leucoaminochrome is the non-redox step (C1) involved between steps R1 and R2.11 The suppression of the R2 peaks with increasing scan rates is in line with the cyclization step being rate limiting for the R2 redox step. This occurs as the C1 process has a timescale (rate constant of 0.2 s−1 to 1 min−1) similar to the voltage timescale based on the scan rate.11 In contrast, for the Au–Ni sample, the peaks associated with the R2 step become more prominent with increasing scan rates, signifying that the cyclization step is not rate limiting. Further, for the R1 step, the oxidation step occurs at a higher potential, and the peak is broader in comparison to the Au–Pt catalyst. The shift to higher potential indicates that the R1 oxidation step is less favored on the Ni surface compared to Pt. The broader peak structure can be related to the multiple Ni states (oxide, hydroxide, and oxyhydroxide), which can lead to variation in the oxidation process on the material. A faster cyclization step will lead to a less reversible R1 step. This is evident in the reduction peak (P2) for the R1 step with the Au–Ni catalyst, where this peak is subdued compared to the oxidation peak (P1). While for Au–Pt, it maintains a sharp and distinct profile. These key differences between Au–Pt and Au–Ni can be used to develop a hybrid catalyst that can improve the performance of dopamine sensing. The rate of the first redox step R1 (favored and rapid on Pt) and faster cyclization step (C1) leading to irreversibility in the R1 step (favored on Ni) will mutually affect the catalyst performance. A composite catalyst with close domains of Pt and Ni will therefore alter the catalyst performance and can lead to better sensitivity, range of operation, and LoD.
 | ||
| Fig. 2 The cyclic voltammetry plots of Au–Pt and Au–Ni catalysts with the scan rates of 20 mV s−1, 50 mV s−1, 100 mV s−1, 150 mV s−1, and 200 mV s−1 in PBS with 0.5 mM dopamine. | ||
To develop and optimize the catalytic performance, two sets of Pt- and Ni-based catalysts were synthesized. In the first composite catalyst (Au–Ni–Au–Pt), the chains of Au–Ni2+ and Au–Pt4+ were assembled separately in colloidal form. These chains were then mixed in the required elemental ratio of Ni:Pt and reduced to form the final Au–Ni–Au–Pt catalytic material. Fig. 3a and b show the CV scans for dopamine oxidation in neutral pH at 20 mV s−1 and 50 mV s−1 for three different Ni:Pt ratios (3:1; 1:1 and 1:3). The effect of increasing the relative Ni amount is observed in the reduced intensity and distinctness of the P1 and P2 peaks (in R1 step), where higher Ni amount (Ni:Pt ratio of 3:1) also shows a highly subdued P2 peak due to greater irreversibility of the R1 step (due to rapid cyclization). The difference is also observed in the redox peaks for the R2 step, where higher Ni amounts lead to more prominent peaks (P3 and P4). When the Ni amount is reduced to a ratio of 1:1 (Ni:Pt), the distinctness and intensity of the redox peaks in the R1 step are recovered, while the distinctness of the redox peaks is sustained for the R2 step to a greater degree. On reducing the Ni to a Ni:Pt ratio of 1:3, clear loss in the distinctness of the redox peaks in the R2 step is observed, while the intensity of the R1 step saturates.
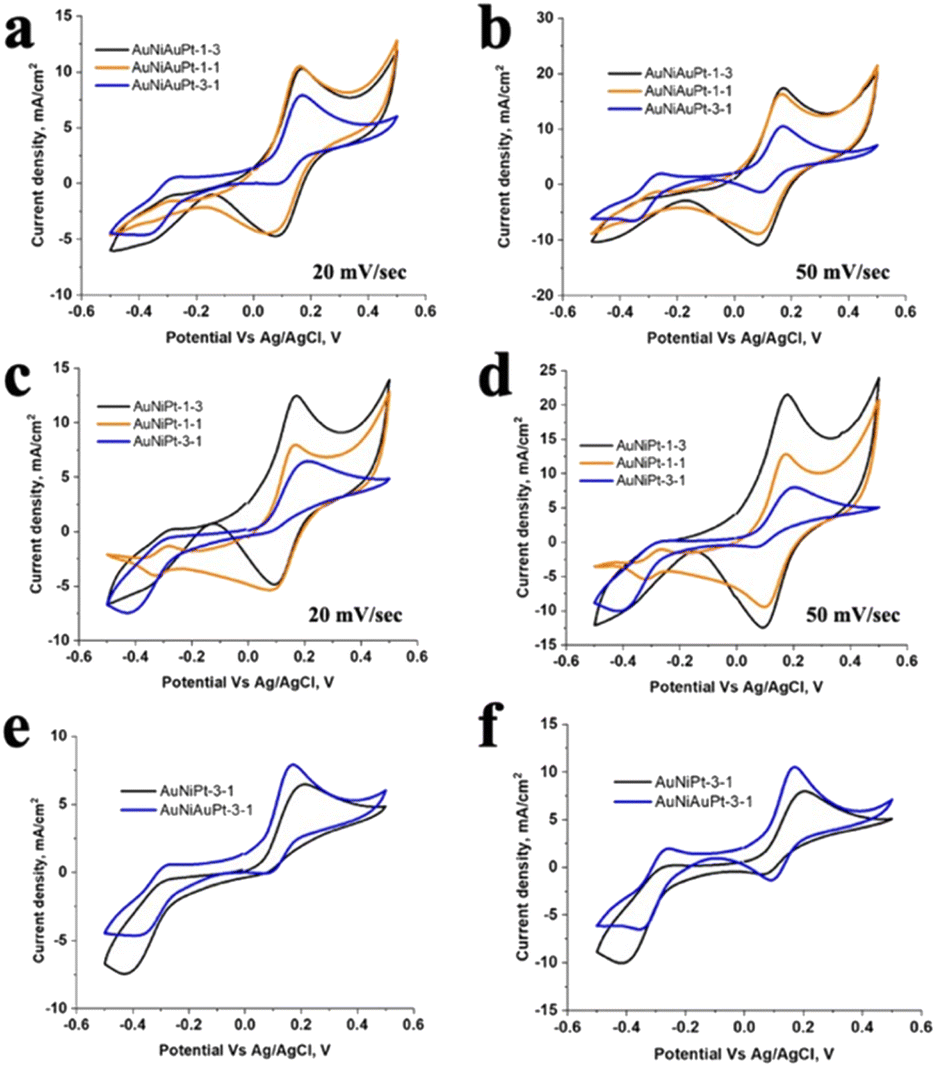 | ||
| Fig. 3 Cyclic voltammograms recorded from different Au–Ni–Pt and Au–Ni–Au–Pt catalysts in PBS with 0.5 mM dopamine at the scan rate of (a) and (c) 20 mV s−1, and (b) and (d) 50 mV s−1. Comparison of the two catalyst materials at a Ni:Pt ratio of 3:1 is shown in (e & f) at scan rates of 20 mV s−1 and 50 mV s−1. | ||
The second set of Pt- and Ni-based catalysts was synthesized by adding the Pt4+ and Ni2+ ions simultaneously to the Au nanoparticle solution for self-assembly. Following self-assembly, the solution is reduced to the final form of Au–Ni–Pt catalytic material. The elemental ratio between Pt and Ni is varied from 3:1, 1:1 to 1:3 (Pt:Ni). The CV scans at a rate of 20 mV s−1 and 50 mV s−1 at neutral pH for dopamine oxidation are shown in Fig. 3c and d. A similar trend is observed with increasing Ni ratio for the Au–Ni–Pt catalyst as was seen for the Au–Ni–Au–Pt material and discussed above. However, some key differences are observed in this material compared to Au–Ni–Au–Pt. A clear shift in the oxidation potential to higher values for the P1 peak is observed at a higher Ni:Pt ratio (3:1, higher relative amount of Ni), and the reduction peak P2 is highly subdued at higher Ni amounts. The P1 oxidation peak is also broader in the Au–Ni–Pt (3:1) sample than Au–Ni–Au–Pt (3:1). For better clarity, the CVs of the two materials Au–Ni–Pt and Au–Ni–Au–Pt (both Ni:Pt of 3:1) are presented in Fig. 3e and f for scan rates of 20 mV s−1 and 50 mV s−1. The dependence of the oxidation peak P1 and the reduction peak P2 on the scan rate, ν, was also plotted (Fig. S1†). P1 has a linear dependence on ν1/2, while P2 varies linearly with ν. The results indicate that oxidation (P1) is a diffusion-controlled process while reduction (P2) is a surface adsorption-controlled process. Based on these results the proposed reaction mechanism is shown in Scheme 1b. The first redox step R1 occurs on the Pt (or Pt–Ni alloy) domains due to its lower oxidation potential, which leads to the formation of dopamine-o-quinone. The cyclization process then leads to the transfer of leucoaminochrome to the Ni domains. This occurs as Ni domains are more favorable for step R2 where the end product aminochrome is formed in the final oxidation step.
To further understand the observed difference in the electrochemical performance of these materials, X-ray photoelectron spectroscopy was conducted (XPS). The results are presented in Fig. 4. The XPS spectrum was analyzed for Ni (2p), Pt (4f), Au (4f), and O (1s). For the Au (4f) spectrum (Fig. 4a), all the materials show identical peaks for 4f7/2 and 4f5/2 at binding energy (B. E.) of 84 and 87.7 eV. The B. E. and the separation of 3.7 eV between the two peaks confirm elemental Au in all the materials with no change in its state during the synthesis process. The Pt spectra (Fig. 4b) for Au–Pt show the two peaks for 4f7/2 and 4f5/2 at B. E. of 71.2 and 74.5 eV corresponding to metallic Pt (with 3.3 eV spin–orbit coupling).18 For Au–Ni–Pt and Au–Ni–Au–Pt, the two Pt peaks are shifted to slightly higher energy by 0.2 eV. This slight shift may indicate coupling between Pt and Ni domains.
 | ||
| Fig. 4 XPS spectra in the (a) Au 4f regions, (b) Pt 4f regions, (c) O 1s regions, and (d) Ni 2p regions for Au–Ni–Au–Pt, Au–Ni–Pt, Au–Ni, and Au–Pt samples. | ||
The O 1s spectra (Fig. 4c) show variation among the materials. For Au–Pt, a low B. E. peak is observed at 531.3 eV, corresponding to hydroxide species (M–OH).19 A high B. E. peak is observed at 532.6 eV, corresponding to adsorbed H2O. Among the Ni-based materials, a combination of peaks is also observed in the B. E. range of 534–531.3 eV. However, there is a difference among the three Ni-based materials, Au–Ni, Au–Ni–Pt, and Au–Ni–Au–Pt. These differences will also be coupled with the underlying O 1s spectra due to the Pt regions. In general, greater peak intensity is observed at lower B. E. for the Au–Ni–Au–Pt material, while for Au–Ni and Au–Ni–Pt, more prominent peaks are observed in the higher energy region. This points to oxide and defect sites in oxide being prominent with the Au–Ni–Au–Pt material. However Au–Ni and Au–Ni–Pt materials have a more prominent hydroxide form, with more uniformity in structure.
The Ni 2p3/2 and 2p1/2 spectra are presented in Fig. 4d. Metallic Ni peak is observed at B. E. of 852.9 eV in the Au–Ni–Pt and Au–Ni–Au–Pt sample, while no such peak is observed in the Au–Ni sample.18,20,21 The peak is also more prominent in the Au–Ni–Au–Pt sample than in Au–Ni–Pt. Further, all samples show peaks associated with oxidized Ni (Ni2+ and/or Ni3+). In Au–Ni, the peak is observed at 856.3 eV, which would indicate mostly Ni2+.19,20,22,23 In Au–Ni–Pt, the peak is shifted to higher energy at 856.7 eV. In Au–Ni–Au–Pt, instead of a single peak, at least two peaks are observed at 857 and 858.4 eV.20 The shift to higher energy is typically indicative of a higher oxidation state (Ni3+). In the case of Au–Ni–Au–Pt, the presence of two distinct peaks also indicates a high concentration of Ni3+ domains. Based on the XPS data, we can conclude that the Au–Pt sample is composed of metallic Pt domains with the presence of surface hydroxides (Pt-hydroxides). In the case of Au–Ni, oxidized domains of Ni are formed (Ni2+, NiO, and Ni(OH)2). For Au–Ni–Pt, metallic Pt is formed along with Ni being present in oxidized form (Ni2+ as the dominant phase, Ni3+ to a minor extent) and in Ni0 (to a minor extent as Ni–Pt alloy) state. The situation in Au–Ni–Au–Pt changes for Ni. While Pt is still in metallic form, the Ni0 phase is formed along with significant regions of both Ni2+ and Ni3+. The absence of Ni0 in the Au–Ni material indicates that the presence of Pt is required for the formation of a stable Ni0 phase, probably due to alloying.
From high-resolution transmission electron microscopy (HRTEM) images, we observe critical differences between the two materials, Au–Ni–Pt and Au–Ni–Au–Pt. In the case of Au–Ni–Pt, large amorphous regions corresponding to Ni2+ and Ni3+ are observed in between spherical particles (at their interface, highlighted in green), Fig. 5a. Small amorphous regions are also observed distributed at other locations along the chain-like structure, but most of them are covered with crystalline regions (highlighted in blue), resulting in limited exposure to the surface. For Au–Ni–Au–Pt, however, small-size amorphous regions (for Ni2+ and Ni3+) are observed with significant exposure to the surface, Fig. 5b. The interface regions between spherical nanoparticles in this material consist of crystalline phase (highlighted in blue) signifying it as being composed of Pt or Pt–Ni alloy. These morphological differences can also be observed in the EELS elemental maps of Fig. 5c and d, where large regions of Ni are observed at the interface of the nanoparticles for Au–Ni–Pt whereas small-size, dispersed Ni domains are observed for Au–Ni–Au–Pt. The X-ray diffraction analysis (XRD) is conducted on these samples to further confirm their structure. As seen in Fig. 5e, the Au nanoparticles show a diffraction peak at 38.2°, corresponding to the Au (111) plane.24 For Au–Ni, similarly, the only major peak observed is at 38.2°, and the spectrum is identical to that of Au nanoparticles. This matches well with the XPS results that the formation of Ni0 is not observed in the Au–Ni material, and only amorphous Ni2+ and Ni3+ phases are formed. In the case of Au–Pt, besides the Au peaks, we now observe new diffraction peaks at 39.8° (major) and 46.2° (minor), corresponding to Pt (111) and (200) planes.25 In the case of both Au–Ni–Au–Pt and Au–Ni–Pt (1:1 ratio of Ni:Pt), while the Au diffraction peak does not change, the Pt peak (major) shifts to a higher angle of 40.77° and 40.56°. The shift corresponds to the formation of the Pt–Ni alloy, which will result in a decrease in the lattice spacing due to the smaller size of Ni. The peak in Au–Ni–Au–Pt is also broader, and its slightly higher angle points to a greater degree of Pt–Ni alloy formation and smaller domain size, as is also observed in XPS and TEM data. Based on these characterization results the formation of the Pt–Ni on Au is proposed to occur through a modified Volmer–Weber mechanism. The initial layer of Pt–Ni (or just Pt or Ni) is formed on the Au chains as a homogenous layer (Frank–van der Merwe mode) based on core–shell type of structures visible in Fig. 1. This occurs due to the high concentration of the respective metal ions (Pt4+ and Ni2+) on the surface of Au nanoparticles due to strong interaction with the citrate capping. Following the formation of the first layer, the subsequent growth shifts as the Volmer–Weber mode leads to island-like growth.26 This occurs as the surface concentration of the metal cation is depleted and lattice mismatch occurs between Pt and Ni (Ni2+ and/or Ni3+). The use of Au nanoparticles as a template that has strong interaction with the metal cations, therefore, allows control over both the catalyst composition and structure (physical and chemical). Such a dual control is not possible with direct synthesis of Pt–Ni materials, where primarily only the composition can be controlled.
 | ||
| Fig. 5 The HRTEM image of (a) Au–Ni–Pt and (b) Au–Ni–Au–Pt (crystalline regions circled in blue and amorphous regions circled in green); EELS elemental mapping of (c) Au–Ni–Pt and (d) Au–Ni–Au–Pt (d) the XRD spectra of various composites. (e) The effect of interferents on the Au–Ni–Au–Pt (1:1) catalyst material. (f) The effect of interferents on other catalyst materials Au–Pt (no Ni) and Au–Ni–Au–Pt (7:1, high amount of Ni). | ||
The catalyst materials were also tested for the effect of interference molecules uric acid (UA), glucose, and ascorbic acid (AA). The Au–Ni–Au–Pt sample with a Ni:Pt ratio of 1:1 showed the greatest selectivity towards dopamine oxidation, with the negligible effect of interferents, as seen in Fig. 5f. However other material configurations such as Au–Ni–Au–Pt (7:1 ratio) or Au–Pt (Fig. 5g) showed significant effects from interferents. This further points to the interplay between the domain size of each phase and its composition, and its ability to interact with reactants and their intermediates. The Au–Ni–Au–Pt sample was also tested for stability and after being stored under ambient conditions for 4 months, the sensitivity is lowered by ∼7%, indicating good stability of the material (Fig. S4†).
The combined structural and electrochemical results provide a critical relationship between the catalyst structure and the oxidation of dopamine. Metallic domains (Pt and Pt–Ni alloy) are sensitive toward the R1 step. However the domains with higher oxidation states of Ni (Ni2+ and Ni3+) catalyze the R2 step more effectively. Based on this, a range of materials were prepared with varying ratios of Ni:Pt to optimize the sensitivity and the range of detection for dopamine oxidation. The results for dopamine oxidation for these materials are presented in Table 1. We observe that the Au–Ni catalyst performs worst in terms of sensitivity and range. As Pt is introduced, the sensitivity and range both improve due to an increase in the rate of step R1. The highest sensitivity for dopamine oxidation is shown by Au–Ni–Au–Pt with a Ni:Pt ratio of 1:1. The lowest LoD is shown by the material Au–Ni–Au–Pt with a Ni:Pt ratio of 7:1, along with the broadest operating range for dopamine detection. The highest sensitivity for the 1:1 ratio (Ni:Pt) shows that a balance between steps R1 and R2 is required for the oxidation of dopamine. A similar optimization is required for LoD and range of operation, but the ideal ratio of Ni:Pt changes. The difference lies in the interplay between the R1 and R2 steps. A faster R2 step leads to greater irreversibility in the R1 step and hence a broader range of operation and lower LoD. For high sensitivity, the R1 step needs to be dominating to ensure rapid and fast oxidation of dopamine in the first step, hence requiring a larger Pt phase in the catalyst structure. A comparison of the performance between this nanochain-based catalyst and some of the recently reported catalysts for dopamine oxidation is presented in Table S-5.† A better performance is observed from the nanochains compared to most other reported dopamine catalysts.27–32
| Sample | Sensitivity (μA mM−1 cm−2) | Range (μM) | R 2 | LoD (nM) | |
|---|---|---|---|---|---|
| AuNi-R | 133.6 | 1.68 | 36.5 | 0.9956 | 12.3 |
| AuPt-R | 330.3 | 11.6 | 371.8 | 0.9964 | 18.6 |
| Au–Ni–Pt (7:1) |
366.7 | 0.14 | 134.5 | 0.9999 | 2.1 |
| Au–Ni–Au–Pt (7:1) |
250.8 | 0.19 | 371.5 | 0.9976 | 1.05 |
| Au–Ni–Pt (3:1) |
368.9 | 0.439 | 134.5 | 0.9976 | 2.90 |
| Au–Ni–Au–Pt (3:1) |
430.1 | 0.19 | 85.75 | 0.9927 | 1.60 |
| Au–Ni–Pt (1:1) |
229.9 | 1.19 | 36.47 | 0.9929 | 10.9 |
| Au–Ni–Au–Pt (1:1) |
1279.3 | 0.140 | 36.47 | 0.9935 | 10.2 |
Experimental
Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :Ni ratios of 3:1, 1:1, and 1:3.
202.5/135/67.5 μL of 4.5 mg mL−1 PtCl4 and 35/70/105 μL of 4 mg mL−1 NiCl2 were mixed first and then added into 2 mL gold solution. When the color turned dark blue, the solution was reduced by 150 μL of 4 mg mL−1 NaBH4, which turned the color black and the colloidal chains could stay stable for over two months.
:Ni ratios of 3:1, 1:1, and 1:3.
1.5/1/0.5 mL of Au–Ni chains solution before reduction and 0.5/1/1.5 mL of Au–Pt chains solution before reduction were first mixed. Then 2 mL mixture was reduced by 150 μL of 4 mg mL−1 NaBH4, which turned the color black and the resulting solution was stable for over two months.
:Ni ratios of 3:1, 1:1, and 1:3.
202.5/135/67.5 μL of 4.5 mg mL−1 PtCl4 and 35/70/105 μL of 4 mg mL−1 NiCl2 were mixed first and then added into 2 mL gold solution. When the color turned dark blue, the solution was reduced by 150 μL of 4 mg mL−1 NaBH4, which turned the color black and the colloidal chains could stay stable for over two months.
:Ni ratios of 3:1, 1:1, and 1:3.
1.5/1/0.5 mL of Au–Ni chains solution before reduction and 0.5/1/1.5 mL of Au–Pt chains solution before reduction were first mixed. Then 2 mL mixture was reduced by 150 μL of 4 mg mL−1 NaBH4, which turned the color black and the resulting solution was stable for over two months.
Characterization section
Zeiss Ultraplus Field-emission Scanning Electron Microscopy (FESEM) was used to image the morphology of samples. Transmission electron microscopy (TEM) images were obtained with an LEO 912ab energy-filtered transmission electron microscope (EFTEM). The size distribution and zeta potential were measured using a Zeta Sizer Nano ZS90 from Malvern Instruments. UV-visible absorption spectra of samples were recorded using a PerkinElmer Lambda 750 spectrophotometer. X-ray photoelectron spectroscopy characterization was performed on a VGS ESCALab 250.Electrical measurements
Cyclic voltammetric (CV) and chronoamperometric experiments were performed with an Ivium CompactStat Electrochemical Analyzer electrochemical workstation. A conventional three-electrode system was adopted by using GCE as the working electrode, Ag/AgCl (1 M KCl) as the reference electrode and a Pt wire as the counter electrode. 15 μL gold chains solution was drop-cast on the GCE to do measurements. All experiments were performed at room temperature. All the dopamine detection experiments are performed using a stirrer with 500 rpm speed. CV scans were recorded in 0.01 M PBS from −0.5 to 0.5 V vs. the reference electrode at different scan rates. For chronoamperometry measurements, the amperometric response of the electrode toward successive additions of dopamine is recorded. A calibration curve is established by using the readout current versus dopamine concentration (the detection current is averaged over the last 15 s range). During the measurements, it was ensured that the baseline current (in the absence of dopamine) was stabilized before the addition of dopamine, and the current change with time is tracked at the working electrode as a function of increasing dopamine concentration. Interference tests measured electroactive compounds, such as 10 μM ascorbic acid (AA), 10 μM uric acid (UA), 10 μM acetaminophen (AP), and 250 μM glucose, which are commonly present in physiological samples and cause problems in the determination of dopamine (10 μM). The LoD was determined from the relationship, expressed as:| LoD = 3.3σ/S |
Conclusions
Multi-element materials have the ability to catalyze different steps in a reaction pathway, leading to better catalytic performance. For the detection of dopamine using a combination of Pt and Ni, we observe that the metallic phase (Pt and Pt–Ni alloy) and the metal oxide phase (Ni2+ and Ni3+) enhance the rate of different steps in the oxidation pathway. Both the elemental ratio (Ni:Pt) and also elemental state (Ni0, Ni2+, Ni3+) affect dopamine oxidation. The performance parameters of the catalyst can be tuned by controlling these structural effects in the catalyst. Based on the requirements, sensitivity, range of operation, and LoD, each shows optimized performance with different catalyst compositions and structures. We have also presented a simple synthesis method based on electrical double-layer formation on the surface of charged nanoparticles to control these structural parameters of the catalyst. Such structural optimization of catalysts with multi-element composition is required to enhance their performance in biosensing and electrochemical energy conversion reactions, directly impacting their application in medical diagnostics and treatment, H2 generation, CO conversion, fuel cells, and water splitting.
Author contributions
HF and VM planned the study. VM procured the funding and supervised the project. HF and WB conducted the experiments. HF and VM wrote the manuscript and analyzed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Waterloo, Canada Foundation for Innovation, Early researcher award from the Ministry of research and innovation and science, Ontario, and NSERC Canada.Notes and references
- M. Wei, Y. Qiao, H. Zhao, J. Liang, T. Li, Y. Luo, S. Lu, X. Shi, W. Lu and X. Sun, Chem. Commun., 2020, 56, 14553–14569 RSC.
- S. Y. Tee, C. P. Teng and E. Ye, Mater. Sci. Eng. C, 2017, 70, 1018–1030 CrossRef CAS PubMed.
- S. Lakard, I.-A. Pavel and B. Lakard, Biosensors, 2021, 11, 179 CrossRef CAS PubMed.
- R. Sha, N. Vishnu and S. Badhulika, Sens. Actuators B Chem., 2019, 279, 53–60 CrossRef CAS.
- X. Xiao, Z. Zhang, F. Nan, Y. Zhao, P. Wang, F. He and Y. Wang, J. Alloys Compd., 2021, 852, 157045 CrossRef CAS.
- A. S. Chang, N. N. Memon, S. Amin, F. Chang, U. Aftab, M. I. Abro, A. dad Chandio, A. A. Shah, M. H. Ibupoto, M. A. Ansari and Z. H. Ibupoto, Electroanalysis, 2019, 31, 1296–1303 CrossRef CAS.
- B. T. P. Quynh, J. Y. Byun and S. H. Kim, Sens. Actuators B Chem., 2015, 221, 191–200 CrossRef CAS.
- X. Liu and J. Liu, View, 2021, 2, 20200102 CrossRef.
- M. Sajid, N. Baig and K. Alhooshani, TrAC Trends Anal. Chem., 2019, 118, 368–385 CrossRef CAS.
- K. Jackowska and P. Krysinski, Anal. Bioanal. Chem., 2013, 405, 3753–3771 CrossRef CAS PubMed.
- R. P. Bacil, L. Chen, S. H. P. Serrano and R. G. Compton, Phys. Chem. Chem. Phys., 2020, 22, 607–614 RSC.
- Z. Liu, Y. Jiang and Z. Zhang, et al. , Nat. Synth., 2023, 2, 119–128 CrossRef.
- X. Wang, M. Chu and M. Wang, et al. , ACS Nano, 2022, 16, 16869–16879 CrossRef CAS PubMed.
- J. Feng, D. Xu, F. Yang, J. Chen, C. Wu and Y. Yin, Angew. Chem., Int. Ed., 2021, 60, 16958–16964 CrossRef CAS PubMed.
- H. Zhang and D. Wang, Angew. Chem., Int. Ed., 2008, 47, 3984–3987 CrossRef CAS PubMed.
- L. Pu, H. Fan and V. Maheshwari, Catal. Sci. Technol., 2020, 10, 2020–2028 RSC.
- H. Fan and V. Maheshwari, Adv. Funct. Mater., 2021, 31, 2106149 CrossRef CAS.
- A. Yadav, R. Pandey, T.-W. Liao, V. S. Zharinov, K.-J. Hu, J. Vernieres, R. E. Palmer, P. Lievens, D. Grandjean and Y. Shacham-Diamand, Nanoscale, 2020, 12, 6047–6056 RSC.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- K.-W. Park, J.-H. Choi, B.-K. Kwon, S.-A. Lee, Y.-E. Sung, H.-Y. Ha, S.-A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869–1877 CrossRef CAS.
- L. Ma, Q. Zhang, C. Wu, Y. Zhang and L. Zeng, Anal. Chim. Acta, 2019, 1055, 17–25 CrossRef CAS PubMed.
- A. P. Grosvenor, M. C. Biesinger, R. St, C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- S.-Q. Liu, H.-R. Wen, Y. Guo, Y.-W. Zhu, X.-Z. Fu, R. Sun and C.-P. Wong, Nano Energy, 2018, 44, 7–14 CrossRef CAS.
- T.-H. Lin, C.-W. Lin, H.-H. Liu, J.-T. Sheu and W.-H. Hung, Chem. Commun., 2011, 47, 2044–2046 RSC.
- W. Cheng, L. Xue, J. Wang, X. Chen, H. Sun, C. Huang, Q. Wu and W. Yao, ACS Appl. Nano Mater., 2021, 4, 8390–8398 CrossRef CAS.
- Q. Zhong, J. Feng, B. Jiang, Y. Fan, Q. Zhang, J. Chen and Y. Yin, J. Am. Chem. Soc., 2021, 48, 20513–20523 CrossRef PubMed.
- P. Zhao, C. Chen, M. Ni, L. Peng, C. Li, Y. Xie and J. Fei, Microchim. Acta, 2019, 186, 134 CrossRef PubMed.
- M. K. Alam, M. M. Rahman, M. M. Rahman, D. Kim, A. M. Asiri and F. Khan, J. Electroanal. Chem., 2019, 835, 329–337 CrossRef CAS.
- Z. Yang, X. Liu, X. Zheng and J. Zheng, Electroanal. Chem., 2018, 817, 48–54 CrossRef CAS.
- X. Li, Int. J. Electrochem. Sci., 2019, 14, 1082–1091 CrossRef CAS.
- S. Zhang, Y. Fu, Q. Sheng and J. Zheng, New J. Chem., 2017, 41, 13076–13084 RSC.
- Y. Liu, Y. Liu, H. Ou, D. Shi, L. Tian, Z. Chen, S. Bao, W. Xiao, X. Meng, R. Hu, J. Song, W. Chen, Z. Cheng and G. Zhao, J. Alloy Comp., 2022, 929, 167390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00932c |
| This journal is © The Royal Society of Chemistry 2023 |