DOI:
10.1039/D4RA07561G
(Paper)
RSC Adv., 2024,
14, 36902-36918
Synthesis, crystal structure, and antiviral evaluation of new imidazopyridine-schiff base derivatives: in vitro and in silico anti-HIV studies†
Received
22nd October 2024
, Accepted 13th November 2024
First published on 19th November 2024
Abstract
A series of Imidazo[1,2-a]pyridine-Schiff base derivatives were synthesized and characterized using 1H NMR, 13C NMR, Mass Spectrometry and FTIR techniques, and the structure of 4a was further confirmed through single-crystal X-ray diffraction analysis. Density Functional Theory (DFT) has been used to investigate the structural and electronic properties. The synthesized compounds were evaluated in vitro for their antiviral activity against human immunodeficiency virus type-1 (HIV-1) and human immunodeficiency virus type-2 (HIV-2) in MT-4 cells. Compound 4a displayed EC50 values of 82,02 and 47,72 μg ml−1 against HIV-1 and HIV-2, respectively. Molecular docking studies were conducted to gain insights into the interaction mechanism of the synthesized compounds with HIV-1 reverse transcriptase. ADME analysis suggested acceptable pharmacokinetic profiles, though solubility remains a limitation for these compounds, highlighting the need for further structural modifications to enhance bioavailability and therapeutic potential.
1. Introduction
Heterocyclic compounds featuring at least one carbon atom and one heteroatom, such as nitrogen or oxygen, are of enormous significance in diverse biological processes. 1 With their unique pharmacological properties and structural adaptability, they have emerged as pivotal components in developing innovative therapeutics.2 These heterocycles have risen to prominence as essential building blocks due to the unique characteristics and biological activities resulting from incorporating nitrogen atoms into their heterocyclic structures.3,4 Fused heterocyclic compounds with five or six members have garnered considerable attention in medicinal chemistry due to their versatile pharmacological and therapeutic implications.5 Within the realm of nitrogen-containing heterocycles, the fusion of imidazole and pyridine moieties, particularly in imidazo[1,2-a]pyridine derivatives, stands out as one of the most substantial nitrogen-bridged heterocyclic structures in organic compounds, especially in medicinal chemistry.6–9
The Imidazo[1,2-a]pyridine scaffold acts as a key pharmacophore motif for the identification and optimization of lead structures, owing to its manifold applications, therapeutic attributes, and facile binding with receptors and enzymes, recognized as privileged scaffold due to its well-established biological activities including antifungal,10,11 antibacterial,12–17 antiviral,18–20 anticancer,21–25 and anti-inflammatory activities,26,27 establishing the Imidazo[1,2-a]pyridine moiety as a focal point in medicinal chemistry.
On the other hand, Schiff bases stand out as versatile pharmacophores, occupying a prominent position within the realm of organic compounds and finding extensive utility in the design and development of a wide array of bioactive lead compounds.28–30 The imine group within these compounds plays a crucial role by forming hydrogen bonds with the active sites of receptors and enzyme constituents, lending these compounds a unique capability to intervene in cellular processes.31,32 These compounds showcase a remarkable spectrum of biological activities, spanning from antifungal,33,34 and antibacterial,35 to antimalarial,36 anticancer,37,38 antiviral,39 antitubercular,40 and antidepressant41 properties. Overall, the integration of imidazo[1,2-a]pyridine and Schiff base into a single pharmacophore could offer even more enhanced and interesting properties, paving the way for their use in a variety of fields.
Acquired Immunodeficiency Syndrome (AIDS) is a devastating infectious disease that poses a grave threat to human life caused by the human immunodeficiency virus (HIV), which attacks the immune system and weakens the body's ability to fight off infections and diseases.42 HIV exists in two main genotypes, HIV-1 and HIV-2, with HIV-1 being the primary pathogenic strain responsible for thousands of deaths each year.43 Although there is currently no cure for AIDS, combination antiretroviral therapy (cART) can control the virus and improve patient longevity and quality of life.44 Whereas HIV remains latent in the body and cannot be eradicated, necessitating lifelong treatment.45 Even so, a major problem is that the rapid emergence of drug-resistant variants limits the long-term efficacy of available anti-HIV drugs and remains one of the most important public health challenges worldwide.46 Hence, an urgent and desirable requirement for the development of innovative and potent anti-HIV entities has been a very active research field in recent years.
To our knowledge, the literature survey reveals there have been no anti-HIV studies conducted on Imidazo[1,2-a]pyridine-Schiff base derivatives. Therefore, as a part of our studies on nitrogen bridgehead heterocycles, we report our contribution to testing these moieties' anti-HIV activity against both HIV-1 and HIV-2 strains. The newly synthesized compounds were characterized using 1H NMR, 13C NMR, Mass Spectrometry, FTIR and single-crystal X-ray diffraction techniques. Additionally, electronic properties were estimated using the density functional theory at the B3LYP/6-311++G(d,p) level of theory to identify potential binding sites and predict chemical behavior. Moreover, the target compounds were screened for their antiviral activities in human T-lymphocyte (MT-4) cells infected with either HIV-1 (IIIB) or HIV-2 (ROD). Molecular docking was conducted to provide a comprehensive understanding of the mode of inhibition of the target compounds. Furthermore, drug-likeness and ADME examinations were performed and discussed.
2. Experimental methods
2.1. General information
Chemicals and solvents were reagent grade purchased from commercial suppliers and used as received with no further purification. The NMR spectra were gathered in Chloroform-D using the JNM-ECZ500R/S1 FT NMR SYSTEM (JEOL), operating at 500 MHz and 126 MHz for 1H and 13C, respectively. The following abbreviations were used to describe the multiplicity: s = singlet, d = doublet, t = triplet, m = multiplet, dd = doublet of doublets, ddd = doublet of doublets of doublets, td = triplet of doublets, tt = triplet of triplets and the coupling constants (J), were reported in the Hertz unit (Hz). Thermo Scientific TSQ 8000 Evo Triple Quadrupole GC-MS/MS spectrometry was employed to acquire the mass spectra. The FT-IR spectra were recorded across the range of 4000–400 cm−1 using JASCO FT/IR-4700 spectrophotometers. X-ray diffraction data were obtained from a shock-cooled single crystal at 150 K using a Bruker Venture Metaljet κ-geometry diffractometer. The diffractometer incorporated a Metal Jet with Helios MX Mirror Optics as a monochromator and a Bruker CMOS Photon III detector. Data collection involved the use of an Oxford Cryostream 700 low-temperature device with GaKα radiation (λ = 1.34139 Å). All collected data underwent integration through SAINT, and SADABS applied a multi-scan absorption correction. Structure determination employed intrinsic phasing methods with XT, followed by refinement through full-matrix least-squares against F2 using XL within the graphical user interface of OLEX2. Anisotropic displacement parameters were assigned to all non-hydrogen atoms, while hydrogen atom positions were determined from a difference Fourier map and refined isotropically.
2.2. Synthesis and crystal growth
The synthetic route leading to the target compounds 4a-d is depicted in Scheme 1. Initially, 5-chloropyridin-2-amine (1, 50 mmol) and phenacyl bromide (50 mmol) were subjected to stirring under solvent-free conditions at 80 °C for 1 h. The resulting precipitate was isolated via filtration and subsequently washed with acetone, affording 6-chloro-2-phenylimidazo[1,2-a]pyridine (2). The 3-amine derivative was synthesized via direct nitrosation, where a saturated solution of NaNO2 was added to compound 2 (50 mmol) in AcOH (50 ml) and stirred for 3 hours, yielding a green residue, followed by a reduction treatment involving the addition of a mixture of Zinc powder (2 eq) in EtOH (50 ml). The reaction mixture was stirred overnight at room temperature. The resulting solution was filtered, and the pH was adjusted to 9, leading to the precipitation of 6-chloro-2-phenylimidazo[1,2-a]pyridin-3-amine (3), which was subsequently recrystallized from absolute ethanol to obtain a sufficiently pure product. Subsequently, compound 3 (10 mmol) was condensed with Substituted aromatic aldehydes (10 mmol) in EtOH (50 ml), along with two drops of AcOH as a catalyst. The reaction mixture was stirred at room temperature for 24 hours, resulting in the formation of the corresponding compounds. To ensure high purity for subsequent analyses, recrystallization from absolute ethanol afforded analytically pure products.
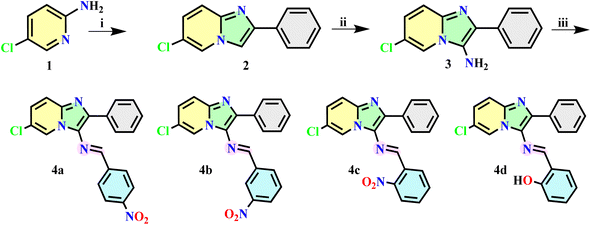 |
| | Scheme 1 The synthetic route of target compounds. Reagents and Conditions: (i) phenacyl bromide, solvent-free, 80 °C, 1 h; (ii) NaNO2, AcOH, rt, 3 h, then Zn Powder, AcOH, EtOH, overnight, rt; (iii) substituted aromatic aldehyde, AcOH, EtOH, rt, 24 h. | |
A single crystal appropriate for X-ray diffraction examination was generated via a mixed-solvent crystallization approach. A concentrated solution of 4a was produced in a methanol/dichloromethane mixture and subsequently filtered. After five days of evaporation at room temperature, suitable red crystals were obtained for further structural investigation.
2.3. Computational details
To establish a comprehensive link between the structure and its associated behaviors of the investigated compounds, the DFT method at the B3LYP/6-311++G(d,p) basis set was employed within the Gaussian 09 suite. Utilizing Gauss View 6 software, Frontier Molecular Orbitals (FMOs), and key reactivity descriptors, as well as Molecular Electrostatic Potential (MEP) Surface and Mulliken atomic charge were computed and visually represented to identify potential binding sites. Additionally, Electron Localization Function (ELF), Localized Orbital Locator (LOL) and Quantum Theory of Atoms in Molecule (QTAIM) analysis were conducted and visually represented using the Multiwfn code, VMD and Gnuplot tools.47
2.4. In vitro anti-HIV assay in MT-4 cells
In vitro anti-HIV assay evaluation of the antiviral activity of compounds 4a–e against HIV-1 (IIIB) and HIV-2 (ROD) strains in MT-4 cells was performed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method as described by Pannecouqueet al., 2008.48 In brief, stock solutions (10 times the final concentration) of the test compounds were added in 25 μl volumes to two series of triplicate wells to simultaneously assess their effects on mock and HIV-infected cells at the start of each experiment. Serial 5-fold dilutions of test compounds were made directly in flat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman instruments). Untreated HIV- and mock-infected cell samples were included as controls for each sample. HIV-1 (IIIB) or HIV-2 (ROD) stock (50 μl) at 100–300 CCID50 (50% cell culture infectious dose) or culture medium [10% heat-inactivated Fetal Calf Serum (FCS), 2 mM-glutamine, 0.1% sodium bicarbonate, and 20 μg ml−1 gentamicin] was added to either of the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effect of the test compound on uninfected cells in order to assess the cytotoxicity of the test compounds. Exponentially growing MT-4 cells were centrifuged for 5 min at 1000 rpm and the supernatant was discarded. The MT-4 cells were resuspended at 6 ×105 cells per mL, and 50 μL volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock- and HIV-infected cells was examined spectrophotometrically by the MTT method.
2.5. Molecular docking analysis
2.5.1. Protein preparation. The docking studies were conducted on HIV-1 Reverse Transcriptase (PDB ID: 1REV). The crystal structures of this key protein were obtained from the Protein Data Bank (PDB)49 and prepared for docking using AutoDock Tools.50 The preparation process involved the removal of all water molecules and co-crystallized ligands, the addition of polar hydrogens, and the assignment of Kollman. The grid box was centered at coordinates X = 3.678, Y = −40.856, Z = 23.497, with a box size of 23 × 23 × 23 Å. These parameters were selected to encompass the active sites of the protein, ensuring adequate coverage of the binding pockets during the docking process.
2.5.2. Ligand preparation. The synthesized compounds were not available in public databases such as PubChem due to their novel nature. Consequently, the three-dimensional structures of these compounds were obtained from CIF (Crystallographic Information File) format and optimized using the DFT method with the B3LYP/6-311++G(d,p) basis set, which was converted to PDB format using OpenBabel.51 The PDB files were then prepared for docking by optimizing the geometry, adding necessary hydrogens, and assigning Gasteiger charges and saved in PDBQT format. Along with the synthesized compounds, two control molecules, Nevirapine and Didanosine, were included in the study as control compounds. The SDF files for these reference ligands were retrieved from PubChem52 and then converted to PDB by OpenBabel and similarly prepared using the same protocol as for the synthesized compounds.
2.5.3. Docking procedure and molecular interactions analysis. Docking simulations were performed using AutoDock Vina.53 For each protein–ligand pair, the docking was conducted within the predefined grid box parameters to explore potential binding modes of the synthesized compounds and control molecules. The exhaustiveness of the search was set to 8 to ensure a comprehensive exploration of the binding conformations. The results were ranked based on binding affinity, with the lowest energy (kcal mol−1).The visualization of 2D interactions and 3D binding poses between the synthesized compounds and target proteins was conducted using BIOVIA Discovery Studio.54 Docking results from AutoDock Vina, comprising protein–ligand complexes for HIV-1 Reverse Transcriptase (PDB ID: 1REV), were imported into the software. Interaction analyses were focused on residues within a 5 Å radius of the ligand, capturing key interactions such as hydrogen bonds, hydrophobic interactions, and electrostatic contacts.
2.6. ADME profiling
The ADME properties of the Four synthesized compounds and two reference molecules, Nevirapine and Didanosine, were evaluated using the SwissADME web tool,55 with a specific focus on solubility. The molecular structures were prepared in PDB format and converted to SMILES format for input into SwissADME. The analysis predicted key pharmacokinetic parameters, including lipophilicity (XlogP3), water solubility (Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S), solubility class, gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeability, P-glycoprotein (Pgp) substrate status, cytochrome P450 inhibition (CYP2D6 and CYP3A4), and PAINS alerts. Particular attention was given to solubility parameters to assess the potential bioavailability and effectiveness of the compounds in biological environments.
S), solubility class, gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeability, P-glycoprotein (Pgp) substrate status, cytochrome P450 inhibition (CYP2D6 and CYP3A4), and PAINS alerts. Particular attention was given to solubility parameters to assess the potential bioavailability and effectiveness of the compounds in biological environments.
3. Results and discussion
3.1. Chemistry
In this study, novel Schiff base heterocycles derived from imidazo[1,2-a]pyridine have been synthesized using easy and conventional methods according to the literature procedure. Initially, 6-chloro-2-phenylimidazo[1,2-a]pyridine (2) was obtained by the condensation of commercially available 5-chloropyridin-2-amine (1) with Phenacyl bromide. Subsequently, the intermediate (2) was functionalized at the C-3 position by introducing an NO group and subsequently reduced to NH2 under the conditions of NaNO2/AcOH and Zn/AcOH/EtOH respectively, resulting in the formation of the 3-amino product, 6-chloro-2-phenylimidazo[1,2-a]pyridin-3-amine (3), which subjected to reaction with substituted aromatic aldehydes, leading to the synthesis of the target compounds 4a–d.
The structural assignments of all newly synthesized imines were validated through comprehensive analytical and spectral analyses. The 1H NMR spectra of compounds 4a–d exhibited the expected characteristic signals, notably a singlet observed at δ 8.73–9.23 ppm, attributable to the proton of the imine group (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). Additionally, in the 13C NMR spectra, a prominent peak emerged at δ 150.16–163.91 ppm, attributable to the carbon (NCH) of the imine moiety. The IR spectra of 4a–d provided additional confirmation, showing an absorption band around 1597–1703 cm−1, which is suggestive of the (CN) stretching vibration. Moreover, LC-MS analysis further confirmed the molecular weights of the compounds, thereby supporting the fact that the expected Schiff base heterocyclic derivatives have indeed been successfully formed (ESI file†).
CH). Additionally, in the 13C NMR spectra, a prominent peak emerged at δ 150.16–163.91 ppm, attributable to the carbon (NCH) of the imine moiety. The IR spectra of 4a–d provided additional confirmation, showing an absorption band around 1597–1703 cm−1, which is suggestive of the (CN) stretching vibration. Moreover, LC-MS analysis further confirmed the molecular weights of the compounds, thereby supporting the fact that the expected Schiff base heterocyclic derivatives have indeed been successfully formed (ESI file†).
3.1.1 (E)-6-Chloro-N-(4-nitrobenzylidene)-2-phenylimidazo[1,2-a]pyridin-3-amine (4a). Yield 94%. Mp; 218–220 °C, 1H NMR (500 MHz, Chloroform-D) δ 8.81 (s, 1H), 8.50 (dd, J = 2.1, 0.9 Hz, 1H), 8.27 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.8 Hz, 2H), 7.74 (dd, J = 8.3, 1.5 Hz, 2H), 7.54 (dd, J = 9.4, 0.9 Hz, 1H), 7.51–7.42 (m, 2H), 7.41 (tt, J = 7.3, 1.5 Hz, 1H), 7.25 (dd, J = 9.4, 2.1 Hz, 1H). 13C NMR (126 MHz, Chloroform-D) δ 152.56, 149.09, 142.09, 141.88, 136.95, 134.20, 129.21, 128.97, 128.81, 128.57, 128.29, 127.48, 124.20, 121.59, 121.49, 118.11. FT-IR: ν(C–H) = 3068, 3004 cm−1; ν(CN) = 1597 cm−1; ν(CC) = 1569 cm−1; ν(NO) = 1507, 1326 cm−1; ν(C–N) = 1230 cm−1; ν(C–C) = 1103 cm−1; ν(C–Cl) = 792 cm−1. LC-MS: m/z = 377.079 (M + 1).
3.1.2 (E)-6-Chloro-N-(3-nitrobenzylidene)-2-phenylimidazo[1,2-a]pyridin-3-amine (4b). Yield 86%. Mp; 211–213 °C, 1H NMR (500 MHz, Chloroform-D) δ 8.80 (s, 1H), 8.59 (t, J = 2.0 Hz, 1H), 8.50 (dd, J = 2.0, 0.9 Hz, 1H), 8.28 (ddd, J = 7.9, 2.0, 1.3 Hz, 1H), 8.11 (ddd, J = 7.9, 1.9, 1.3 Hz, 1H), 7.74 (dd, J = 8.6, 1.5 Hz, 2H), 7.62 (t, J = 7.9 Hz, 1H), 7.54 (dd, J = 9.5, 0.8 Hz, 1H), 7.46 (dd, J = 8.6, 7.2 Hz, 2H), 7.41 (tt, J = 7.2, 1.5 Hz, 1H), 7.24 (dd, J = 9.5, 2.0 Hz, 1H). 13C NMR (126 MHz, Chloroform-D) δ 153.29, 148.86, 141.84, 138.03, 136.37, 134.19, 133.64, 129.97, 129.23, 128.88, 128.50, 128.20, 127.24, 125.55, 122.84, 121.46, 121.43, 118.04. FT-IR: ν(C–H) = 3127, 3063 cm−1; ν(CN) = 1613 cm−1; ν(CC) = 1568 cm−1; ν(NO) = 1524, 1332 cm−1; ν(C–N) = 1228 cm−1; ν(C–C) = 1073 cm−1; ν(C–Cl) = 774 cm−1. LC-MS: m/z = 377.079 (M + 1).
3.1.3 (E)-6-Chloro-N-(2-nitrobenzylidene)-2-phenylimidazo[1,2-a]pyridin-3-amine (4c). Yield 89%. Mp; 220–222 °C, 1H NMR (500 MHz, Chloroform-D) δ 9.23 (s, 1H), 8.53 (dd, J = 2.0, 1.1 Hz, 1H), 8.34 (dd, J = 7.9, 1.5 Hz, 1H), 7.98 (dd, J = 8.2, 1.2 Hz, 1H), 7.77 (dd, J = 8.3, 1.4 Hz, 2H), 7.73 (ddd, J = 7.9, 7.3, 1.2 Hz, 1H), 7.60 (dd, J = 9.5, 1.1 Hz, 1H), 7.57 (ddd, J = 8.2, 7.3, 1.5 Hz, 1H), 7.49 (dd, J = 8.3, 7.3 Hz, 2H), 7.42 (tt, J = 7.3, 1.4 Hz, 1H), 7.27 (dd, J = 9.5, 2.0 Hz, 1H). 13C NMR (126 MHz, Chloroform-D) δ 150.16, 149.13, 141.78, 133.53, 133.33, 131.17, 131.02, 129.20, 129.09, 129.06, 128.72, 128.23, 127.53, 124.84, 121.63, 121.56, 118.01. FT-IR: ν(C–H) = 3079, 3024 cm−1; ν(CC) = 1562 cm−1; ν(NO) = 1513, 1330 cm−1; ν(C–N) = 1222 cm−1; ν(C–C) = 1092 cm−1; ν(C–Cl) = 806 cm−1. LC-MS: m/z = 377.079 (M + 1).
3.1.4 (E)-2-(((6-chloro-2-phenylimidazo[1,2-a]pyridin-3-yl)imino)methyl)phenol (4d). Yield 83%. Mp; 130–132 °C, 1H NMR (500 MHz, Chloroform-D) δ 12.33 (s, 1H), 8.73 (s, 1H), 8.20 (dd, J = 2.0, 0.8 Hz, 1H), 7.75 (dd, J = 8.4, 1.4 Hz, 2H), 7.65 (dd, J = 9.5, 0.8 Hz, 1H), 7.43 (dd, J = 8.5, 7.4 Hz, 2H), 7.40–7.37 (m, 1H), 7.36 (tt, J = 7.4, 1.4 Hz, 1H), 7.22 (dd, J = 9.5, 2.0 Hz, 1H), 7.09 (dd, J = 7.6, 1.7 Hz, 1H), 7.05 (dd, J = 8.4, 1.2 Hz, 1H), 6.90 (td, J = 7.6, 1.2 Hz, 1H). 13C NMR (126 MHz, Chloroform-D) δ 176.12, 163.91, 160.86, 141.10, 134.80, 134.10, 133.21, 132.69, 129.25, 128.68, 128.22, 127.00, 121.85, 120.24, 119.84, 119.03, 118.12, 117.43. FT-IR: ν(O–H) = 3393 cm−1; ν(C–H) = 3078, 3024 cm−1; ν(CN) = 1703 cm−1; ν(CC) = 1623 cm−1; ν(C–O) = 1444 cm−1; ν(C–N) = 1267 cm−1; ν(C–Cl) = 751 cm−1. LC-MS: m/z = 349.092 (M + 1).
3.2. Single crystal XRD studies
Given the importance of molecular structure and conformation in dictating biological activities, particularly in interactions with enzyme or receptor active sites, compound 4a was subjected to single crystal X-ray diffraction analysis. This approach aims to provide a comprehensive and definitive validation of the structural assignment of these compounds.
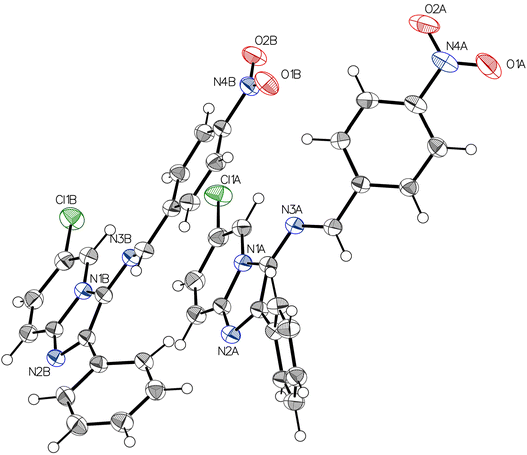
The grown crystal of 4a was subjected to single crystal X-ray diffraction studies, solved and refined. Table 1 shows a detailed report of the structure refinement. The compound 4a crystallizes in the monoclinic system with the space group P21/c with eight molecules in a unit cell. The unit cell dimensions observed are a = 10.7755(6) A°, b = 26.7963(15) A°, c = 12.0718(6) A°, α = 90°, β = 102.178(3)°, γ = 90°. Fig. 1 depicts the molecular structure of 4a using 50% probability thermal displacement ellipsoids, with hydrogen atoms depicted as spheres of arbitrary size for clarity. The figure provides a separate view of the two molecules of the asymmetric unit, along with the atomic numbering scheme. Fig. 2 displays a view of the two molecules of the asymmetric unit within the unit cell. The adjacent molecules are interconnected through hydrogen bonding interactions of C–H⋯N and C–H⋯O types (Table 2), resulting in the formation of a dimer, as depicted in Fig. 3. The imidazopyridine ring of the molecules in the dimer forms dihedral angles of 34.29° and 27.87° with the phenyl plane (C8A–C13A) and (C8B–C13B), respectively. In molecule B, the phenyl group (C15B–C20B) is nearly planar with the imidazopyridine ring (1.70°), while in molecule A, it forms a dihedral angle of 73.40°. The configuration of the CN bond in the imine is described as “anti” to reduce overall steric hindrance. The crystal lattice reveals that the molecules are stacked in the form of parallel packs along the c-axis direction (Fig. 3). In Table 3, selected bond distances and bond angles of compound 4a have been presented.
Table 1 Crystallographic Data and Refinement Parameters for 4a
| Parameter |
Values |
| Empirical formula |
C20H13ClN4O2 |
| Formula weight |
376.79 |
| Temperature [K] |
150 |
| Crystal system |
Monoclinic |
| Space group (number) |
P21/c (14) |
| a [Å] |
10.7755(6) |
| b [Å] |
26.7963(15) |
| c [Å] |
12.0718(6) |
| α [°] |
90 |
| β [°] |
102.178(3) |
| γ [°] |
90 |
| Volume [Å3] |
3407.2(3) |
| Z |
8 |
| ρcalc [g cm−3] |
1.469 |
| μ [mm−1] |
1.441 |
| F(000) |
1552 |
| Crystal size [mm3] |
0.04 × 0.13 × 0.13 |
| Crystal colour |
Clear light red |
| Crystal shape |
Plate |
| Radiation |
GaKα (λ = 1.34139 Å) |
| 2θ range [°] |
7.12 to 158.31 (0.68 Å) |
| Index ranges |
−15 ≤ h ≤ 15 |
| −39 ≤ k ≤ 39 |
| −17 ≤ l ≤ 15 |
| Reflections collected |
114388 |
| Independent reflections |
11104 |
| Rint = 0.0493 |
| Rsigma = 0.0248 |
| Completeness to θ = 53.594° |
100.0% |
| Data/restraints/parameters |
11104/0/591 |
| Goodness-of-fit on F2 |
1.064 |
| Final R indexes [I ≥ 2σ(I)] |
R1 = 0.0407 wR2 = 0.1019 |
| Final R indexes [all data] |
R1 = 0.0508 wR2 = 0.1088 |
| Largest peak/hole [e Å−3] |
0.28/−0.39 |
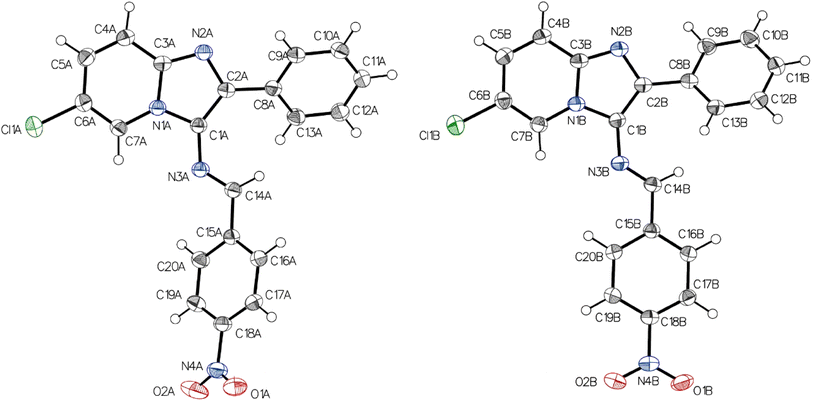 |
| | Fig. 1 The molecular structure of 4a, showing the atom-numbering scheme. Ellipsoids are drawn at the 50% probability level, and hydrogen atoms are shown as spheres of arbitrary size. | |
 |
| | Fig. 2 ORTEP representation of the dimer of two molecular units of 4a. Ellipsoids are drawn at the 50% probability level, and hydrogen atoms are shown as spheres of arbitrary size. | |
Table 2 Hydrogen bonds for 4a
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
DHA |
| Symmetry transformations used to generate equivalent atoms: 1 − X, 1 − Y, −Z. +X, 0.5 − Y, −0.5 + Z. |
| C4A–H4A⋯N2Ba |
0.970(16) |
2.445(16) |
3.4088(14) |
172.7(14) |
| C14A–H14A⋯O1Bb |
0.982(16) |
2.604(16) |
3.5192(15) |
155.0(12) |
| C4B–H4B⋯N2Aa |
0.971(16) |
2.669(17) |
3.6324(14) |
171.6(14) |
 |
| | Fig. 3 Molecular packing of the title compound, Projection along the c-axis. | |
Table 3 Some bond distances (Å) and bond angles (°) of 4a
| Bond lengths (Å) |
| Cl1A–C6A |
1.7349(12) |
C16A–C17A |
1.3930(15) |
O2B–N4B |
1.2276(15) |
| O1A–N4A |
1.2238(18) |
C17A–H17A |
0.962(18) |
N2B–C2B |
1.3681(14) |
| N1A–C1A |
1.3912(14) |
C17A–C18A |
1.3867(17) |
N2B–C3B |
1.3339(15) |
| N1A–C3A |
1.3890(13) |
C18A–C19A |
1.3836(19) |
C2B–C8B |
1.4704(16) |
| N1A–C7A |
1.3760(14) |
C19A–H19A |
0.946(17) |
N3B–C14B |
1.2812(15) |
| C1A–C2A |
1.3997(14) |
C19A–C20A |
1.3846(16) |
C3B–C4B |
1.4113(16) |
| C1A–N3A |
1.3796(13) |
C20A–H20A |
0.964(16) |
N4B–C18B |
1.4707(13) |
| O2A–N4A |
1.2295(17) |
Cl1B–C6B |
1.7353(12) |
C4B–H4B |
0.971(16) |
| N2A–C2A |
1.3712(14) |
O1B–N4B |
1.2253(15) |
C4B–C5B |
1.3690(17) |
| N2A–C3A |
1.3328(15) |
N1B–C1B |
1.3948(14) |
C5B–H5B |
0.942(18) |
| C2A–C8A |
1.4698(16) |
N1B–C3B |
1.3863(13) |
C5B–C6B |
1.4209(16) |
| N3A–C14A |
1.2821(15) |
N1B–C7B |
1.3702(15) |
C6B–C7B |
1.3568(17) |
| C3A–C4A |
1.4134(16) |
C1B–C2B |
1.4068(14) |
C7B–H7B |
0.989(15) |
| N4A–C18A |
1.4690(14) |
C1B–N3B |
1.3738(13) |
C8B–C9B |
1.4001(15) |
| Bond angles (°) |
| C3A–N1A–C1A |
107.18(9) |
N1A–C7A–H7A |
116.8(9) |
C3B–N2B–C2B |
106.46(9) |
| C7A–N1A–C1A |
129.44(9) |
C6A–C7A–N1A |
117.27(10) |
C1B–C2B–C8B |
130.84(10) |
| C7A–N1A–C3A |
123.06(10) |
C6A–C7A–H7A |
126.0(9) |
N2B–C2B–C1B |
110.55(10) |
| N1A–C1A–C2A |
104.81(9) |
C9A–C8A–C2A |
118.83(10) |
N2B–C2B–C8B |
118.48(9) |
| N3A–C1A–N1A |
115.79(9) |
C13A–C8A–C2A |
122.41(10) |
C14B–N3B–C1B |
124.36(10) |
| N3A–C1A–C2A |
139.33(11) |
C13A–C8A–C9A |
118.65(11) |
N1B–C3B–C4B |
118.82(10) |
| C3A–N2A–C2A |
106.02(9) |
C8A–C9A–H9A |
117.9(9) |
N2B–C3B–N1B |
110.95(9) |
| C1A–C2A–C8A |
130.26(10) |
C3B–N1B–C1B |
107.34(9) |
N2B–C3B–C4B |
130.24(10) |
| N2A–C2A–C1A |
110.86(10) |
C7B–N1B–C1B |
129.51(9) |
O1B–N4B–O2B |
123.62(10) |
| N2A–C2A–C8A |
118.85(9) |
C7B–N1B–C3B |
123.14(10) |
O1B–N4B–C18B |
118.19(10) |
| C14A–N3A–C1A |
122.62(10) |
N1B–C1B–C2B |
104.64(9) |
O2B–N4B–C18B |
118.19(10) |
| N1A–C3A–C4A |
118.76(10) |
N3B–C1B–N1B |
115.35(9) |
C3B–C4B–H4B |
119.7(10) |
| N2A–C3A–N1A |
111.09(9) |
N3B–C1B–C2B |
139.92(11) |
C5B–C4B–C3B |
119.01(10) |
| N2A–C3A–C4A |
130.12(10) |
C3B–N1B–C1B |
107.34(9) |
C5B–C4B–H4B |
121.3(10) |
3.3. Density functional theory (DFT) calculations
3.3.1. Geometry and frontier molecular orbitals (FMOs) investigations. The utilization of DFT in quantum chemical computations offers significant advantages in discerning optimal molecular structures and predicting diverse properties, encompassing total energy and electronic structure to establish a connection between the molecular structure under investigation and their associated behaviors. Molecular structure optimization of 4a-d was conducted to identify the most stable ground state geometry for each molecule. Subsequently, vibrational frequency calculations were executed to validate the absence of negative or imaginary frequencies, thereby affirming the optimized geometry as representing an energy minimum configuration.56 Consequently, the stable configurations were successfully determined and are illustrated in Fig. 4.
 |
| | Fig. 4 The optimized molecular structures of 4a-d. | |
Frontier Molecular Orbitals (FMOs) are a key concept in theoretical chemistry, particularly in the field of organic chemistry.57 They play a crucial role in understanding and predicting the reactivity of organic molecules. Central to FMO analysis are the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO), often referred to as the “frontier” orbitals due to their decisive roles in chemical interactions. The HOMO signifies the electron-donating capacity of a molecule, while the LUMO represents its electron-accepting capacity. The energy difference between these orbitals, known as the HOMO–LUMO gap (ΔEgap), serves as a critical parameter governing the molecule's reactivity and stability. A small ΔEgap suggests heightened reactivity, whereas a wider gap implies diminished reactivity and increased stability. This concept holds particular significance in biological systems, where ΔEgap determines a molecule's propensity to engage in chemical interactions, such as binding to proteins or enzymes. Consequently, a profound understanding of FMOs and the HOMO–LUMO gap is indispensable for forecasting the behavior of compounds in biological settings and assessing their potential efficacy as active agents. The graphical illustrations of FMOs for the optimized configurations are presented in Fig. 5. The green and red areas indicate MOs with opposing phases, the positive phase is illustrated in red, while the negative phase is displayed in green. It is evident that the HOMOs are distributed across the entire molecule. Conversely, the LUMOs are typically positioned on the substituted phenyl nuclei.58–60
 |
| | Fig. 5 Correlation diagram between HOMO and LUMO of 4a–d. | |
In the investigation of molecular reactivity and electronic structure, FMOs analysis was used to derive the global reactivity descriptors. Table 4 presents a comparative overview of these descriptors, providing valuable insights into the electronic properties and potential reactivity of the studied compounds. Notably, compounds 4a exhibit smaller ΔEgap values, suggesting heightened reactivity and potential for chemical interactions, whereas compounds 4d exhibit larger gaps, generally correlating with higher stability and reduced reactivity. Compound 4a showcases the lowest hardness (1.40 eV) and highest softness (0.72 eV), suggesting a relatively more reactive and responsive to changes in electron density. Furthermore, the electronegativity (χ) and electrophilicity index (ω) values shed light on the molecules' interaction capabilities. Specifically, 4a demonstrates the highest χ (4.68061 eV) and ω (7.83946 eV) values, suggesting its enhanced ability to engage in electron transfer processes and interact with receptors, which could be beneficial for biological applications. Overall, the variations in these descriptors among the compounds underscore their distinct reactivity and stability profiles.59–66
Table 4 Global reactivity descriptors of 4a–d
| Quantum parameters |
4a |
4b |
4c |
4d |
| EHOMO (eV) |
−6.07792 |
−5.99220 |
−5.97234 |
−5.52879 |
| ELUMO (eV) |
−3.28331 |
−3.07324 |
−3.02481 |
−2.21581 |
| ΔEgap (eV) |
2.79460 |
2.91895 |
2.94753 |
3.31297 |
| η (eV) |
1.39730 |
1.45947 |
1.47376 |
1.65648 |
| σ (eV−1) |
0.71566 |
0.68517 |
0.67853 |
0.60368 |
| χ (eV) |
4.68061 |
4.53272 |
4.49857 |
3.87230 |
| ω (eV) |
7.83946 |
7.03867 |
6.86581 |
4.52606 |
3.3.2. Molecular electrostatic potential (MEP) analysis. Molecular electrostatic potential (MEP) analysis is a fundamental tool in computational chemistry used to visualize and understand the distribution of electron density within a molecule. MEP offers a valuable understanding of the electrostatic interactions that occur during chemical reactions, binding processes, and molecular recognition events.67 By mapping the MEP onto the molecular surface and utilizing a color spectrum to differentiate various electrostatic potential energy levels, regions of positive and negative electrostatic potential can be identified. Positive regions indicate areas characterized by electron deficiency or electrophilic nature, depicted in blue, while negative regions represent areas with electron-rich or nucleophilic character shown in red. These electrostatic features play a crucial role in molecular recognition, such as ligand binding to receptors or enzymes, and can provide valuable information for drug design and molecular docking studies.68The MEP maps generated at the optimized geometries are depicted in Fig. 6. In 4a–d, the areas surrounding the nitrogen atom N-6 of the imidazole moiety, in conjunction with the NO2 groups, exhibit pronounced electron-rich characteristics, indicating a strong affinity for electrophiles. Consequently, these regions are expected to serve as sites for nucleophilic attack. Conversely, regions of electron deficiency are observed around the hydrogen atoms, notably around the hydrogen of the OH group in 7d. These regions signify electrophilic sites capable of attracting nucleophilic entities. MEP analysis is particularly useful for predicting sites of electrophilic or nucleophilic attack, identifying potential binding sites for substrates or inhibitors, and understanding the reactivity and selectivity of chemical reactions.
 |
| | Fig. 6 MEP surface of 4a–d. | |
3.3.3. Mulliken charge analysis. Mulliken charge analysis is a computational technique used in quantum chemistry to assess the distribution of electron density within a molecule. It involves calculating the partial charges associated with each atom in a molecule based on its electronic structure.69 Mulliken charges provide valuable insights into the electronegativity and reactivity of individual atoms within a molecular system. These charges can help identify regions of electron accumulation or depletion, aiding in the understanding of chemical bonding, molecular interactions, and reactivity patterns.70The Mulliken atomic charge analysis, as presented in Fig. 7 and Table 5, offers insight into the electron distribution within these compounds. The computed atomic charges, expressed in atomic units (a.u.), reveal the varying degrees of electron density surrounding each atom. Carbon atoms typically exhibit a wide range of charges, reflecting their diverse bonding environments and electronic interactions. The presence of negative charges on C-5 suggests an accumulation of electron density, which may stem from the electron-donating tendencies of neighboring atoms. Notably, carbon atoms directly bonded to electronegative species like nitrogen or oxygen often display more positive charges, indicative of electron withdrawal.71 Conversely, nitrogen atoms generally show slightly positive charges, suggesting a partial donation of electron density primarily attributed to the involvement in resonance structures. Hydrogen atoms attached to these molecules tend to have positive charges, consistent with their typical behavior as electron donors. Interestingly, the presence of halogen atoms such as chlorine alters the charge distribution, leading to more positively charged neighboring carbon atoms.72
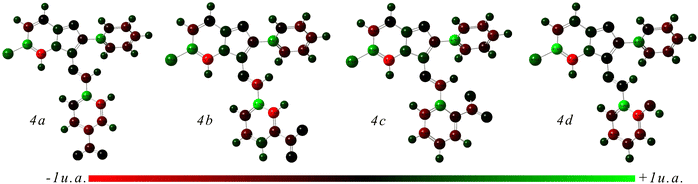 |
| | Fig. 7 Optimized structures with Mulliken atomic charges designated by a color change on atoms. | |
Table 5 The Mulliken atomic charges of 4a–d
| 4a |
4b |
4c |
4d |
| Atom |
Charge(a.u.) |
Atom |
Charge(a.u.) |
Atom |
Charge(a.u.) |
Atom |
Charge(a.u.) |
| C-1 |
0.203994 |
C-1 |
0.185665 |
C-1 |
0.226520 |
C-1 |
0.146311 |
| C-2 |
−0.332893 |
C-2 |
−0.299092 |
C-2 |
−0.400394 |
C-2 |
−0.329245 |
| C-3 |
0.624179 |
C-3 |
0.545881 |
C-3 |
0.692295 |
C-3 |
0.851660 |
| C-4 |
−1.374555 |
C-4 |
−1.312930 |
C-4 |
−1.436671 |
C-4 |
−1.772958 |
| N-5 |
0.153961 |
N-5 |
0.159470 |
N-5 |
0.172304 |
N-5 |
0.161004 |
| N-6 |
0.038980 |
N-6 |
0.032879 |
N-6 |
0.040087 |
N-6 |
0.030745 |
| C-7 |
−0.122225 |
C-7 |
−0.121464 |
C-7 |
−0.189184 |
C-7 |
−0.033622 |
| C-8 |
0.109309 |
C-8 |
0.110638 |
C-8 |
0.061244 |
C-8 |
0.057807 |
| C-9 |
0.671143 |
C-9 |
0.726014 |
C-9 |
0.752757 |
C-9 |
0.685316 |
| C-10 |
−0.270178 |
C-10 |
−0.307890 |
C-10 |
−0.200300 |
C-10 |
−0.316664 |
| C-11 |
−0.272456 |
C-11 |
−0.290127 |
C-11 |
−0.197174 |
C-11 |
−0.304427 |
| C-12 |
−0.317998 |
C-12 |
−0.309185 |
C-12 |
−0.302677 |
C-12 |
−0.286754 |
| C-13 |
−0.325595 |
C-13 |
−0.334433 |
C-13 |
−0.361800 |
C-13 |
−0.389736 |
| C-14 |
−0.121458 |
C-14 |
−0.115391 |
C-14 |
−0.313337 |
C-14 |
−0.039869 |
| N-15 |
0.035789 |
N-15 |
0.024729 |
N-15 |
−0.007140 |
N-15 |
0.018148 |
| C-16 |
−0.285280 |
C-16 |
−0.624930 |
C-16 |
−0.244269 |
C-16 |
−0.063480 |
| C-17 |
0.660660 |
C-17 |
1.185587 |
C-17 |
0.603634 |
C-17 |
0.812139 |
| C-18 |
−0.080022 |
C-18 |
−0.197777 |
C-18 |
−0.453849 |
C-18 |
−0.233247 |
| C-19 |
−0.252564 |
C-19 |
−0.323868 |
C-19 |
−0.486856 |
C-19 |
−0.466159 |
| C-20 |
−0.389093 |
C-20 |
0.169558 |
C-20 |
−0.201515 |
C-20 |
−0.206736 |
| C-21 |
−0.374148 |
C-21 |
−0.186389 |
C-21 |
−0.394111 |
C-21 |
−0.420447 |
| C-22 |
−0.641921 |
C-22 |
−1.420155 |
C-22 |
−0.053781 |
C-22 |
−0.793863 |
| H-23 |
0.197587 |
H-23 |
0.197378 |
N-23 |
−0.347589 |
H-23 |
0.188161 |
| H-24 |
0.176843 |
H-24 |
0.177603 |
O-24 |
0.030812 |
H-24 |
0.186352 |
| H-25 |
0.179643 |
H-25 |
0.179711 |
O-25 |
0.048861 |
H-25 |
0.176253 |
| H-26 |
0.163104 |
H-26 |
0.163272 |
H-26 |
0.196746 |
H-26 |
0.156113 |
| H-27 |
0.177306 |
H-27 |
0.175969 |
H-27 |
0.193655 |
H-27 |
0.168074 |
| H-28 |
0.206470 |
H-28 |
0.205354 |
H-28 |
0.199368 |
H-28 |
0.202671 |
| H-29 |
0.136942 |
H-29 |
0.165680 |
H-29 |
0.164000 |
H-29 |
0.241514 |
| H-30 |
0.156492 |
H-30 |
0.132973 |
H-30 |
0.171107 |
H-30 |
0.144382 |
| H-31 |
0.256758 |
H-31 |
0.203500 |
H-31 |
0.197519 |
H-31 |
0.181830 |
| H-32 |
0.253485 |
H-32 |
0.193186 |
H-32 |
0.114975 |
H-32 |
0.166625 |
| H-33 |
0.196087 |
H-33 |
0.251664 |
H-33 |
0.152131 |
H-33 |
0.128124 |
| N-34 |
−0.243941 |
N-34 |
−0.235905 |
H-34 |
0.185726 |
H-34 |
0.179658 |
| O-35 |
−0.004043 |
O-35 |
0.019690 |
H-35 |
0.182535 |
O-35 |
−0.225554 |
| O-36 |
0.004856 |
O-36 |
0.010521 |
H-36 |
0.241816 |
H-36 |
0.266016 |
| H-37 |
0.180713 |
H-37 |
0.251873 |
H-37 |
0.227196 |
C-37 |
0.121869 |
| Cl-38 |
0.438589 |
C-38 |
−0.013044 |
Cl-38 |
0.430822 |
H-38 |
0.191083 |
| C-39 |
−0.011488 |
C-39 |
0.195209 |
C-39 |
0.104876 |
Cl-39 |
0.420906 |
| H-40 |
0.196967 |
Cl-40 |
0.428578 |
H-40 |
0.199660 |
|
|
3.3.4. ELF and LOL analysis. The analysis conducted using the Electron Localization Function (ELF) and the Localized Orbital Locator (LOL) offers valuable insights into the electron density and bonding characteristics within molecules.73 In this investigation, color-filled maps representing ELF and LOL were generated through the utilization of the Multiwfn 3.8 program. The ELF color-filled map displayed in Fig. 8 features a color scale bar spanning from 0.000 to 1.000. Regions exhibiting ELF values below 0.500, as depicted by the blue color surface, signify the presence of delocalized electrons surrounding carbon and oxygen atoms. Conversely, regions of pronounced electron localization, particularly in the vicinity of hydrogen atoms, particularly associated with the Imidazopyridine ring, are highlighted in red, indicating elevated ELF values exceeding 0.500.74
 |
| | Fig. 8 ELF and LOL maps of 4a–d. | |
In contrast, the LOL color-filled map showcased in Fig. 8 reveals smaller LOL values, represented by blue and white regions, primarily observed in valence regions such as those surrounding carbon, nitrogen, and chlorine atoms, suggesting the potential existence of electron depletion zones. Conversely, larger LOL values, signifying heightened electron localization, are evident in the inner surface regions, notably around hydrogen atoms, depicted in red, emphasizing the impact of electron localization in these regions. Overall, the ELF and LOL analyses provide comprehensive information about electron distribution and bonding characteristics within the molecule, aiding in a deeper understanding of its structural properties.75,76
3.3.5. QTAIM analysis. The Quantum Theory of Atoms in Molecules (QTAIM) analysis is a potent computational chemistry tool designed to explore non-covalent interactions (NCI) in molecular systems. Built upon electron density principles, the RDG analysis method utilizes this data to craft detailed 3D isosurfaces and scatter plots, unveiling the spatial distribution and intensity of weak interactions, employing color-coded graphs to distinguish interaction types, intricately depict robust interactions in blue (typically hydrogen bonding) and weaker interactions in green (resembling van der Waals forces), while red highlights regions affected by steric hindrance effects.68,77 In this investigation, the utilization of QTAIM molecular graph analysis facilitated the exploration and visualization of NCI. The examined compounds displayed a range of intra-molecular interactions, notably van der Waals interactions indicated by (H…C) connections, characterized by sign (λ2)ρ values below −0.01 a.u. Moreover, (O….H) connections were identified in 4c (ortho-NO2) and 4d (ortho-OH), with 4b showcasing a distinct (Cl….H) connection. These interactions, identified via critical points (CPs) and intra-molecular hydrogen bond interactions highlighted by green bonds between specific atom pairs like N⋯H and O⋯H, significantly influence the stability and physical properties of the compounds. The presence of red coloration at the cores of phenyl, imidazole, and pyridine rings implies strong steric repulsion, evident from the peak presence at positive sign (λ2)ρ values exceeding 0.02 a.u. (Fig. 9).63,78
 |
| | Fig. 9 NCI and RDG plot of 4a–d. | |
3.4. In vitro anti-HIV assays
Compounds 4a–d were assessed for their ability to inhibit HIV-1 (strain IIIB) and HIV-2 (strain ROD), with monitoring of the virus-induced cytopathic effects in MT-4 cells using the MTT method. The results are presented in Table 6, where Didanosine and Nevirapine were used as positive drugs. The cytotoxicity of the compounds in MT-4 cells was also evaluated alongside their antiviral activity.
Table 6 In vitro anti-HIV-1a and HIV-2b activity of 4a-d
| Compound |
Virus strain |
av. EC50c (μg ml−1) |
av. CC50d (μg ml−1) |
SIe |
| Anti-HIV-1 activity measured against strain IIIB. Anti-HIV-2 activity measured against strain ROD. Compound dose required to achieve 50% protection of MT-4 cells from HIV-1 and HIV-2-induced cytopathogenic effects. Compound dose that reduces the viability of mock-infected MT-4 cells by 50%. Selectivity index (SI) (CC50/EC50). SI value: X1 stand for 1 or <1. Values were averaged from at least three independent experiments. |
| 4a |
IIIB |
82.02 |
>125.00 |
>2 |
| ROD |
47.72 |
>125.00 |
>3 |
| 4b |
IIIB |
>82.11 |
82.11 |
<1 |
| ROD |
>82.11 |
82.11 |
<1 |
| 4c |
IIIB |
>125.00 |
>125.00 |
X1 |
| ROD |
>125.00 |
>125.00 |
X1 |
| 4d |
IIIB |
>17.91 |
17.91 |
<1 |
| ROD |
>17.91 |
17.91 |
<1 |
| Didanosine |
IIIB |
2.89 |
>50 |
>17 |
| ROD |
3.39 |
>50 |
>15 |
| Nevirapine |
IIIB |
0.08 |
>4 |
>48 |
| ROD |
>4.00 |
>4 |
X1 |
All the compounds lacked significant activity and demonstrated limited in vitro efficacy against HIV-1 and HIV-2 compared to the standard anti-HIV drugs. Compound 4a displayed an EC50 values of 82.02 and 47.72 μg ml−1 against HIV-1 and HIV-2. Respectively, while 4d was inactive at concentrations below 17.91 μg ml−1.
Due to the ease of synthetic accessibility and structural modifications, these findings provide a preliminary basis for further exploration, with the potential to refine these compounds for future anti-HIV applications. However, despite the limited in vitro activity, molecular docking and ADME studies were conducted to gain insights from both in vitro and in silico analyses.
3.5. Molecular docking studies
In this study, molecular docking simulations were conducted to assess the binding affinities of newly synthesized compounds against the HIV-1 Reverse Transcriptase (PDB ID: 1REV). The choice of HIV-1 Reverse Transcriptase as the target receptor was driven by its critical role in the HIV replication cycle, where it facilitates the conversion of viral RNA into DNA for viral integration into the host genome.79 This enzyme is a well-established target in antiviral drug development, making it a suitable model for evaluating the potential inhibitory effects of new compounds.80 The docking results revealed promising binding affinities for all compounds (Table 7), with docking scores of −9.8 kcal mol−1 for 4a, −11.7 kcal mol−1 for 4b, −11.2 kcal mol−1 for 4c, and −12.7 kcal mol−1 for 4d. When compared to the control molecules, Nevirapine81 and Didanosine,82 which displayed docking scores of −9.5 kcal mol−1 and −8.0 kcal mol−1 respectively, it is evident that the synthesized compounds exhibit superior binding potential, particularly compound 4d, which showed the highest binding affinity. The high docking score of 4d is particularly noteworthy because it correlates with the relatively better in vitro activity observed for this compound. Although the in vitro results for 4d were not as strong as those for the reference drugs, 4d showed a somewhat effective interaction with HIV-1 Reverse Transcriptase, with an EC50 value of >17.91 μg ml−1. This suggests that the strong in silico binding affinity may indeed translate into some level of biological activity. The observed moderate in vitro efficacy of 4d, despite its high docking score, highlights the complexity of drug interactions within a biological system. While the docking simulations predict strong binding based on static protein models,83,84 they do not account for the dynamic and multifaceted nature of protein–ligand interactions in vivo.85 Factors such as protein conformational flexibility,86 compound solubility,87 and stability in the biological environment86,88 can significantly influence the actual inhibitory potential of a compound.
Table 7 Docking scores of 4a–d against 1REV receptor with control drugs
| Compound |
Docking score (kcal mol−1) |
| 4a |
−9.8 |
| 4b |
−11.7 |
| 4c |
−11.2 |
| 4d |
−12.7 |
| Nevirapine |
−9.5 |
| Didanosine |
−8 |
3.6. Molecular interactions analysis
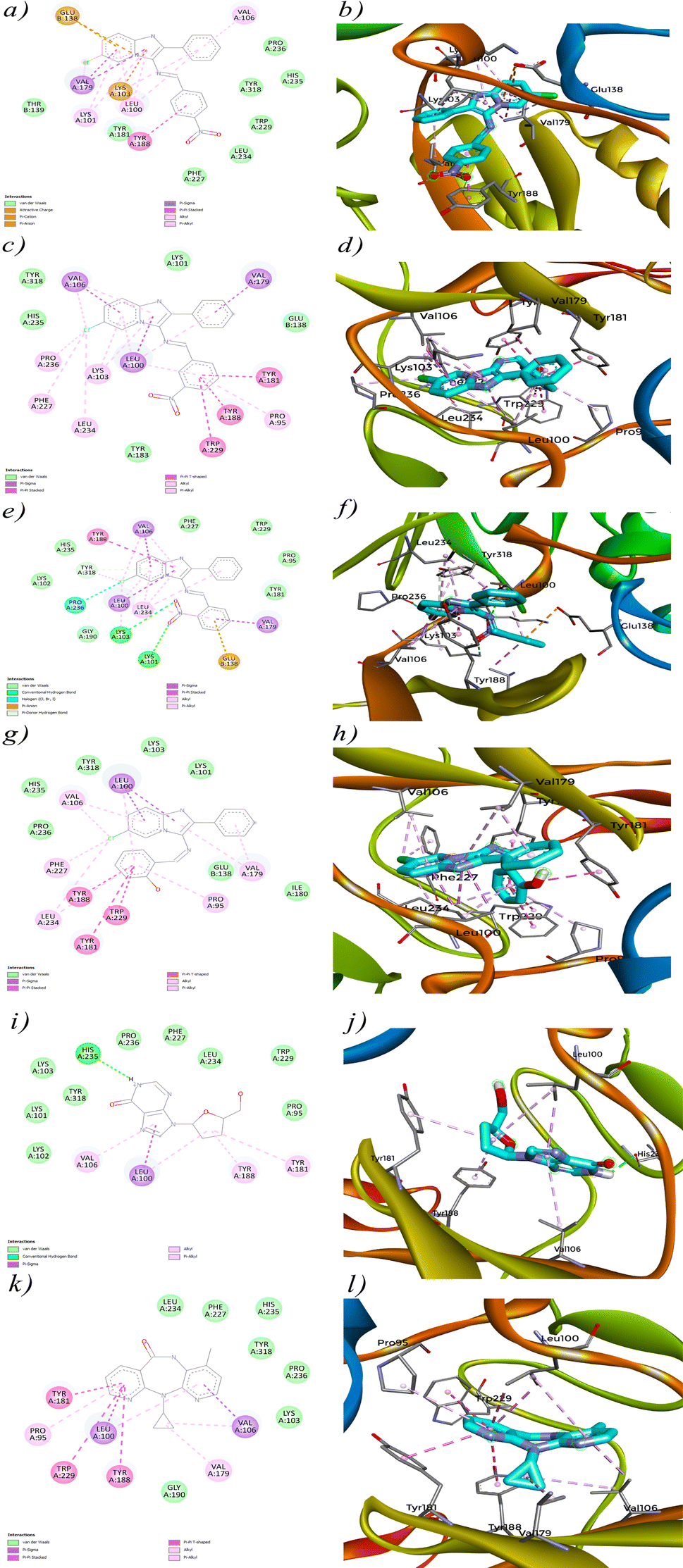
The interaction analysis of 4a–d against HIV-1 Reverse Transcriptase (PDB ID: 1REV) reveals crucial insights into how these molecules engage with the enzyme's active site. Each compound demonstrates a unique interaction profile, which is likely to influence its binding affinity and potential inhibitory activity. Starting with 4a (Fig. 10a), the interaction diagram highlights significant Pi-alkyl interactions with residues such as VAL A:106, LEU A:100, and LYS A:101. These interactions contribute to the stabilization of the compound within the enzyme's hydrophobic pockets,89,90 suggesting effective binding to the active site. Additionally, 4a exhibits a Pi-cation interaction with LYS A:103 and a Pi-anion interaction with GLU B:138, which further stabilize its binding. These interactions align with its docking score of −9.8 kcal mol−1. However, despite these promising interactions, the in vitro activity of 4a was not as strong, indicating potential issues such as poor solubility or inadequate membrane permeability that might prevent the compound from achieving effective concentrations in a biological environment.
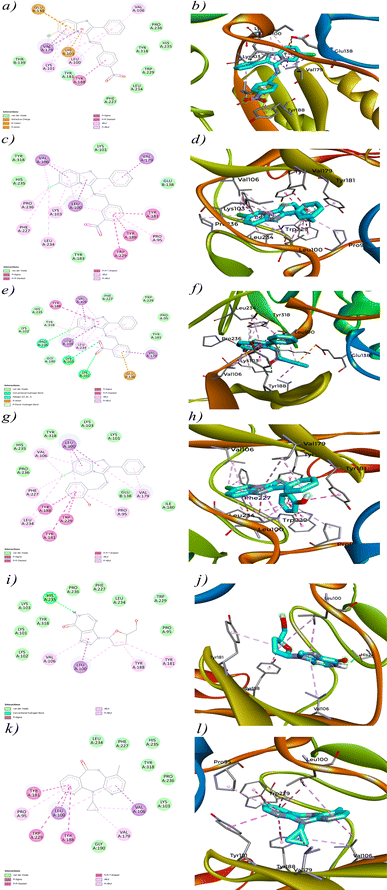 |
| | Fig. 10 2D and 3D view of interaction between compounds (4a–d), control drugs and protein (PDB ID: 1REV). | |
4b (Fig. 10c) shows an even stronger interaction profile, characterized by multiple Pi–Pi stacked and Pi–Pi T shaped interactions. Notably, residues such as TYR A:181, TRP A:229, and TYR A:181 are involved in these interactions, which are crucial for stabilizing the ligand within the active site.91 The Pi–Pi stacked interactions, in particular, are significant for stabilizing aromatic rings within the enzyme's active site.92 The presence of additional Pi-sigma interactions with LEU A:100, VAL A:106 and VAL A:179 enhances the binding strength of 4b, correlating with its high docking score of −11.7 kcal mol−1. However, similar to 4a, the lack of significant in vitro efficacy may be due to factors like protein flexibility in a living system or the compound's pharmacokinetic properties that were not accounted for in the docking simulations. 4c (Fig. 10e) presents a combination of Pi–Pi stacked, Pi-alkyl and conventional hydrogen bond interactions. The compound engages in strong Pi–Pi stacked and Pi-aninon interactions with TYR A:188 and GLU B:138, helping anchor it within the active site. Hydrogen bonding with LYS A:101 and LYS A:103 suggests that 4c also engages in traditional polar interactions, contributing to its binding affinity. Despite these interactions, the docking score of −11.2 kcal mol−1 for 4c is slightly lower than that of 4b, which might indicate a less optimal binding orientation or fewer critical contacts within the active site. The moderate in vitro performance of 4c could stem from similar factors that affected 4a and 4b, such as solubility issues or the dynamic nature of the protein–ligand interactions in a biological context. 4d (Fig. 10g) stands out with the strongest interaction profile among the synthesized compounds, consistent with its highest docking score of −12.7 kcal mol−1. The compound forms extensive Pi–Pi stacked, T shaped and Pi-alkyl interactions with multiple key residues, including TYR A:188, TRP A:229, TYR A:181, PRO A:95 and VAL A:106. The significant Pi-sigma interaction with LEU A:100 likely enhances the electrostatic stability of 4d within the binding site. The variety and density of these interactions suggest that 4d is highly effective at engaging the active site of HIV-1 Reverse Transcriptase, contributing to its superior binding affinity. This extensive interaction profile is similar to that observed for the reference molecule Nevirapine, which also relies on strong hydrophobic and electrostatic interactions within the active site. The promising in vitro activity of 4d, albeit not as strong as Nevirapine, reinforces its potential as a candidate for further development, particularly because its interaction profile closely resembles that of the control molecule. The comparison between the synthesized compounds and the reference molecules, Didanosine (Fig. 10i) and Nevirapine (Fig. 10k), highlights the superior interaction profiles of the synthesized compounds, especially 4d. Nevirapine primarily relies on Pi-alkyl, Pi-sigma and Pi–Pi stacked/T shaped interactions with key residues like PRO A:95, TYR A:181, TYR A:188, LEU A:100 and VAL A:106, while Didanosine, despite its lower docking score, forms conventional hydrogen bonds and Pi-sigma interactions that are less extensive than those observed for the synthesized compounds. However, the lack of significant in vitro efficacy for most of these compounds, despite strong docking scores, suggests a non-correlation between the in silico predictions and actual biological activity. One possible hypothesis for this discrepancy is that docking simulations, which typically involve static models of the protein, may not fully capture the dynamic nature of proteins in vivo. Additionally, factors such as poor solubility, inadequate membrane permeability, or compound instability in biological conditions could have prevented these compounds from achieving effective concentrations at the target site, leading to reduce in vitro efficacy.
3.7. In silico prediction of ADME properties
The ADME analysis of 4a–d alongside the control molecules provides critical insights into their drug-likeness (Table 8) and pharmacokinetic profiles (Table 9).55 These properties are essential for understanding how these compounds might behave in a biological system, particularly with regard to their solubility, absorption, distribution, and potential metabolic interactions.93 Starting with solubility, which is a key factor influencing the bioavailability of a drug,87,94 the synthesized compounds show varying levels of solubility. Compounds 4a, 4b, and 4c all fall into the “poorly soluble” category, with LogS values of −6.85. Poor solubility can significantly limit the effectiveness of a drug, as it may hinder the compound's ability to dissolve in the biological fluids and reach sufficient concentrations at the target site. This limitation is likely a contributing factor to the less promising in vitro results observed for these compounds, despite their high docking scores. In contrast, compound 4d stands out with a LogS value of −5.78, placing it in the “moderately soluble” category. This enhanced solubility compared to the other synthesized compounds suggests that 4d may have a better chance of achieving effective concentrations in vivo, which could partially explain its relatively better in vitro activity. The moderately soluble nature of 4d, coupled with its strong docking interactions, makes it a promising candidate for further development. Additionally, 4d shows an advantage in permeability, being BBB permeant, which could potentially make it more effective in crossing biological barriers to reach the target site. When comparing the synthesized compounds to the control molecules, Nevirapine and Didanosine demonstrate superior solubility with LogS values of −2.92 and 0.02, respectively, classifying them as “soluble” and “highly soluble.” These solubility profiles are indicative of their well-established efficacy in clinical use, as higher solubility typically correlates with better absorption and bioavailability.95 Nevirapine's ability to be BBB permeant96 further enhances its pharmacokinetic profile, ensuring it can reach its target effectively. Didanosine, although not BBB permeant, benefits from its excellent solubility, which facilitates its distribution and action within the body. Beyond solubility, the ADME analysis also highlights other important factors such as lipophilicity (XlogP3),97 gastrointestinal absorption,98 and potential interactions with metabolic enzymes.99 All the synthesized compounds, including 4d, have high lipophilicity (XlogP3 > 5 for 4a, 4b, and 4c; 5.33 for 4d), which is generally favorable for membrane permeability but can sometimes lead to issues such as poor solubility or increased potential for off–target interactions. The control molecules, Nevirapine and Didanosine, exhibit lower lipophilicity, which, combined with their superior solubility, likely contributes to their well-balanced pharmacokinetic profiles. In terms of gastrointestinal absorption, all compounds, including the synthesized ones, are predicted to have high GI absorption, which is advantageous for oral administration. Finally, the absence of alerts in the PAINS filter across all compounds suggests that they are unlikely to interfere with or generate false-positive results in biological assays,100 further supporting their suitability for drug development. In summary, while the synthesized compounds show limitations due to poor solubility, 4d emerges as a more promising candidate due to its moderate solubility, high permeability, and strong docking interactions. The superior solubility and pharmacokinetic profiles of the control molecules, Nevirapine and Didanosine, underscore the importance of optimizing solubility and other ADME properties in the development of new antiviral agents. Further optimization of 4d's properties, particularly in improving its solubility and ensuring effective in vivo performance, could enhance its potential as a therapeutic agent.
Table 8 Lipinski's rule of five 4a–d with control drugs
| Compound |
Lipinski's rule |
H-bond acceptors |
H-bond donors |
Molecular weight |
MLOGP |
| 4a |
YES |
4 |
0 |
376.8 |
4.12 |
| 4b |
YES |
4 |
0 |
376.8 |
4.12 |
| 4c |
YES |
4 |
0 |
376.8 |
4.12 |
| 4d |
YES |
3 |
1 |
347.8 |
3.39 |
| Nevirapine |
YES |
3 |
1 |
266.3 |
2.14 |
| Didanosine |
YES |
5 |
2 |
236.23 |
−0.2 |
Table 9 ADME properties of 4a–d along with control drugs
| Compound |
XlogP3 |
LogS |
Solubility class |
GIA |
BBB |
P-gp |
CYP2D6 inhibitor |
CYP3A4 inhibitor |
PAINS alert |
| 4a |
5.51 |
−6.85 |
Poorly soluble |
High |
No |
No |
No |
No |
0 Alert |
| 4b |
5.51 |
−6.85 |
Poorly soluble |
High |
No |
No |
No |
No |
0 Alert |
| 4c |
5.51 |
−6.85 |
Poorly soluble |
High |
No |
No |
No |
No |
0 Alert |
| 4d |
5.33 |
−5.78 |
Moderately soluble |
High |
Yes |
No |
Yes |
No |
0 Alert |
| Nevirapine |
1.96 |
−2.92 |
Soluble |
High |
Yes |
Yes |
No |
No |
0 Alert |
| Didanosine |
−1.47 |
0.02 |
Highly soluble |
High |
No |
No |
No |
No |
0 Alert |
3.8. Aqueous solubility
Aqueous solubility is a critical factor that influences a drug's absorption, distribution, metabolism, and excretion properties in vivo. It is well-established that poor water solubility can result in unfavorable pharmacokinetic profiles, such as low oral bioavailability and a short half-life, ultimately limiting a drug's therapeutic efficacy.101,102 Therefore, assessing the solubility of drug candidates has become essential in optimizing their pharmacological potential.
The aqueous solubility of the synthesized imidazopyridine-Schiff base derivatives was evaluated at pH 7.0 to understand its impact on their observed anti-HIV activity. The solubility data presented in Table 10 indicate a noticeable correlation with antiviral efficacy. Compound 4d, which exhibited the highest solubility (125 μg ml−1), demonstrated relatively better antiviral activity, with EC50 values of 17.91 μg ml−1. This suggests that enhanced solubility may facilitate better interaction with viral targets, leading to improved activity. Compounds 4a and 4b, with moderate solubility (25 μg ml−1), showed limited antiviral effects, for instance, compound 4a achieved an EC50 of 82.02 μg ml−1 against HIV-1 and 47.72 μg ml−1 against HIV-2. In contrast, compound 4c, which had very low solubility (<1 μg ml−1), showed no anti-HIV activity, with EC50 values exceeding 125 μg ml−1 for both viral strains. These observations suggest that solubility plays a crucial role in the bioavailability and antiviral performance of the compounds. Therefore, optimizing aqueous solubility could be a key factor in enhancing the antiviral potential of future derivatives.
Table 10 Aqueous solubility of 4a–d at pH = 7.0
| Compound |
4a |
4b |
4c |
4d |
| Solubility (μg mL−1) |
25 |
25 |
<1 |
125 |
4. Conclusion
In this study, new imidazo[1,2-a]pyridine-Schiff base derivatives were synthesized and characterized using various spectroscopic techniques, with the structure of compound 4a further confirmed by single-crystal X-ray diffraction analysis. Additionally, DFT calculations were employed to explore the electronic and structural properties of the compounds, providing valuable insights into their potential reactivity and interaction with biological targets. The in vitro antiviral evaluation against HIV-1 and HIV-2 revealed limited efficacy compared to standard anti-HIV drugs. Despite this, molecular docking analyses demonstrated notable binding affinities for HIV-1 reverse transcriptase, surpassing those of the reference drugs. However, the promising docking results did not translate into significant in vitro activity, likely due to poor solubility and suboptimal pharmacokinetic properties, as suggested by ADME analysis and aqueous solubility. Consequently, further structural optimization, particularly focusing on improving solubility and bioavailability, is essential to enhance the therapeutic potential of these compounds.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compound 4a has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition Number 2391520 and can be obtained from https://www.ccdc.cam.ac.uk/structures/Search?ccdc=2391520.
Author contributions
Mohamed Azzouzi: validation, writing – original draft. Abderrahim Ait Ouchaoui: investigation, methodology. Omar Azougagh: software, formal analysis. Salah Eddine El Hadad: software, methodology. Mohamed Abou-salama: resources, formal analysis. Adyl Oussaid: data curation, methodology. Christophe Pannecouque: methodology, investigation, validation, reviewing and editing. Taoufik Rohand: validation, supervision.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank their respective institutions for providing the necessary facility to complete this work.
References
- P. N. Kalaria, S. C. Karad and D. K. Raval, Eur. J. Med. Chem., 2018, 158, 917–936 CrossRef CAS.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- B. R. Smith, C. M. Eastman and J. T. Njardarson, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed.
- W.-Y. Fang, L. Ravindar, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam, N. S. Alharbi and H.-L. Qin, Eur. J. Med. Chem., 2019, 173, 117–153 CrossRef CAS.
- B. Narasimhan, D. Sharma and P. Kumar, Med. Chem. Res., 2011, 20, 1119–1140 CrossRef CAS.
- A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC.
- K. Pericherla, P. Kaswan, K. Pandey and A. Kumar, Synthesis, 2015, 47, 887–912 CrossRef CAS.
- R. Goel, V. Luxami and K. Paul, RSC Adv., 2015, 5, 81608–81637 RSC.
- C. Enguehard-Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888–899 CrossRef CAS PubMed.
- M. H. Fisher and A. Lusi, J. Med. Chem., 1972, 15, 982–985 CrossRef CAS PubMed.
- Y. Rival, G. GRASSY and G. MICHEL, Chem. Pharm. Bull., 1992, 40, 1170–1176 CrossRef CAS PubMed.
- A. Nordqvist, M. T. Nilsson, O. Lagerlund, D. Muthas, J. Gising, S. Yahiaoui, L. R. Odell, B. R. Srinivasa, M. Larhed, S. L. Mowbray and A. Karlén, MedChemComm, 2012, 3, 620–626 RSC.
- T. H. Al-Tel and R. A. Al-Qawasmeh, Eur. J. Med. Chem., 2010, 45, 5848–5855 CrossRef CAS.
- G. C. Moraski, L. D. Markley, P. A. Hipskind, H. Boshoff, S. Cho, S. G. Franzblau and M. J. Miller, ACS Med. Chem. Lett., 2011, 2, 466–470 CrossRef CAS PubMed.
- S. Kumar, N. Sharma, I. K. Maurya, A. K. K. Bhasin, N. Wangoo, P. Brandão, V. Félix, K. K. Bhasin and R. K. Sharma, Eur. J. Med. Chem., 2016, 123, 916–924 CrossRef CAS.
- N. P. Mishra, S. Mohapatra, C. R. Sahoo, B. P. Raiguru, S. Nayak, S. Jena and R. N. Padhy, J. Mol. Struct., 2021, 1246, 131183 CrossRef CAS.
- C. Wei, J. Huang, Y. Luo, S. Wang, S. Wu, Z. Xing and J. Chen, Pestic. Biochem. Physiol., 2021, 175, 104857 CrossRef CAS PubMed.
- G. Puerstinger, J. Paeshuyse, E. De Clercq and J. Neyts, Bioorg. Med. Chem. Lett., 2007, 17, 390–393 CrossRef CAS.
- C. Li, R. Song, S. He, S. Wu, S. Wu, Z. Wu, D. Hu and B. Song, J. Agric. Food Chem., 2022, 70, 7375–7386 CrossRef CAS PubMed.
- J.-M. Chezal, J. Paeshuyse, V. Gaumet, D. Canitrot, A. Maisonial, C. Lartigue, A. Gueiffier, E. Moreau, J.-C. Teulade, O. Chavignon and J. Neyts, Eur. J. Med. Chem., 2010, 45, 2044–2047 CrossRef CAS PubMed.
- N. Dahan-Farkas, C. Langley, A. L. Rousseau, D. B. Yadav, H. Davids and C. B. de Koning, Eur. J. Med. Chem., 2011, 46, 4573–4583 CrossRef CAS PubMed.
- N. Masurier, E. Debiton, A. Jacquemet, A. Bussière, J.-M. Chezal, A. Ollivier, D. Tétégan, M. Andaloussi, M.-J. Galmier, J. Lacroix, D. Canitrot, J.-C. Teulade, R. C. Gaudreault, O. Chavignon and E. Moreau, Eur. J. Med. Chem., 2012, 52, 137–150 CrossRef CAS.
- J. Dam, Z. Ismail, T. Kurebwa, N. Gangat, L. Harmse, H. M. Marques, A. Lemmerer, M. L. Bode and C. B. de Koning, Eur. J. Med. Chem., 2017, 126, 353–368 CrossRef CAS PubMed.
- S. Endoori, K. C. Gulipalli, S. Bodige, P. Ravula and N. Seelam, J. Heterocycl. Chem., 2021, 58, 1311–1320 CrossRef CAS.
- M. Reddy Gangireddy, M. Mantipally, R. Gundla, V. Nayak Badavath, K. Paidikondala and A. Yamala, ChemistrySelect, 2019, 4, 13622–13629 CrossRef CAS.
- M. Hieke, C. B. Rödl, J. M. Wisniewska, E. la Buscató, H. Stark, M. Schubert-Zsilavecz, D. Steinhilber, B. Hofmann and E. Proschak, Bioorg. Med. Chem. Lett., 2012, 22, 1969–1975 CrossRef CAS PubMed.
- J. I. Serrano-Contreras, M. E. Meléndez-Camargo, Y. K. Márquez-Flores, M. P. Soria-Serrano and M. E. Campos-Aldrete, Toxicol. Res., 2022, 11, 730–742 CrossRef.
- A. L. Berhanu, Gaurav, I. Mohiuddin, A. K. Malik, J. S. Aulakh, V. Kumar and K.-H. Kim, TrAC, Trends Anal. Chem., 2019, 116, 74–91 CrossRef CAS.
- M. N. Uddin, S. S. Ahmed and S. M. R. Alam, J. Coord. Chem., 2020, 73, 3109–3149 CrossRef CAS.
- C. Verma and M. A. Quraishi, Coord. Chem. Rev., 2021, 446, 214105 CrossRef CAS.
- B. D. Nath, M. M. Islam, M. R. Karim, S. Rahman, M. A. A. Shaikh, P. E. Georghiou and M. Menelaou, ChemistrySelect, 2022, 7, 202104290 CrossRef.
- A. Kajal, S. Bala, S. Kamboj, N. Sharma and V. Saini, J. Catal., 2013, 2013, 893512 Search PubMed.
- A. Hamad, Y. Chen, M. A. Khan, S. Jamshidi, N. Saeed, M. Clifford, C. Hind, J. M. Sutton and K. M. Rahman, MicrobiologyOpen, 2021, 10, 1218 CrossRef PubMed.
- L. Wei, J. Zhang, W. Tan, G. Wang, Q. Li, F. Dong and Z. Guo, Int. J. Biol. Macromol., 2021, 179, 292–298 CrossRef CAS PubMed.
- C. M. da Silva, D. L. da Silva, L. V. Modolo, R. B. Alves, M. A. de Resende, C. V. B. Martins and Â. de Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- M. S. Tople, N. B. Patel, P. P. Patel, A. C. Purohit, I. Ahmad and H. Patel, J. Mol. Struct., 2023, 1271, 134016 CrossRef CAS.
- N. Uddin, F. Rashid, S. Ali, S. A. Tirmizi, I. Ahmad, S. Zaib, M. Zubair, P. L. Diaconescu, M. N. Tahir, J. Iqbal and A. Haider, J. Biomol. Struct. Dyn., 2020, 38, 3246–3259 CrossRef CAS.
- S. Shekhar, A. M. Khan, S. Sharma, B. Sharma and A. Sarkar, Emergent Mater., 2022, 5, 279–293 CrossRef CAS.
- S. Kaushik, S. K. Paliwal, M. R. Iyer and V. M. Patil, Med. Chem. Res., 2023, 32, 1063–1076 CrossRef CAS PubMed.
- R. Cordeiro and M. Kachroo, Bioorg. Med. Chem. Lett., 2020, 30, 127655 CrossRef CAS PubMed.
- N. Yuldasheva, N. Acikyildiz, M. Akyuz, L. Yabo-Dambagi, T. Aydin, A. Cakir and C. Kazaz, J. Mol. Struct., 2022, 1270, 133883 CrossRef CAS.
- Z. Wang, Z. Yu, D. Kang, J. Zhang, Y. Tian, D. Daelemans, E. De Clercq, C. Pannecouque, P. Zhan and X. Liu, Bioorg. Med. Chem., 2019, 27, 447–456 CrossRef CAS.
- S. Xu, L. Sun, A. Dick, W. A. Zalloum, T. Huang, M. E. Meuser, X. Zhang, Y. Tao, S. Cherukupalli, D. Ding, X. Ding, S. Gao, X. Jiang, D. Kang, E. De Clercq, C. Pannecouque, S. Cocklin, X. Liu and P. Zhan, Eur. J. Med. Chem., 2022, 227, 113903 CrossRef CAS PubMed.
- D. M. Margolis and D. J. Hazuda, Curr. Opin. HIV AIDS, 2013, 8, 230–235 CrossRef CAS PubMed.
- D. Kang, Z. Huo, G. Wu, J. Xu, P. Zhan and X. Liu, Expert Opin. Ther. Pat., 2017, 27, 383–391 CrossRef CAS.
- L. Sun, T. Huang, A. Dick, M. E. Meuser, W. A. Zalloum, C.-H. Chen, X. Ding, P. Gao, S. Cocklin, K.-H. Lee, P. Zhan and X. Liu, Eur. J. Med. Chem., 2020, 190, 112085 CrossRef CAS.
- M. Azzouzi, O. Azougagh, A. A. Ouchaoui, S. e. El hadad, S. Mazières, S. E. Barkany, M. Abboud and A. Oussaid, ACS Omega, 2024, 9, 837–857 CrossRef CAS.
- C. Pannecouque, D. Daelemans and E. De Clercq, Nat. Protoc., 2008, 3, 427–434 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids
Res., 2015, 44, 1202–1213 CrossRef.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS.
- U. Baroroh, M. Biotek, Z. S. Muscifa, W. Destiarani, F. G. Rohmatullah and M. Yusuf, Indonesian Journal of Computational Biology, 2023, 2, 22–30 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- G. Vijayakumari, N. Iyandurai, M. Raja, V. Vetrivelan, A. Thamarai, S. Javed and S. Muthu, Chem. Phys. Impact, 2023, 6, 100190 CrossRef.
- J. Yu, N. Q. Su and W. Yang, JACS Au, 2022, 2, 1383–1394 CrossRef CAS PubMed.
- H. M. Abd El-Lateef, M. M. Khalaf, A. A. Amer, M. Kandeel, A. A. Abdelhamid and A. Abdou, ACS Omega, 2023, 8, 25877–25891 CrossRef CAS.
- J. S. Al-Otaibi, Z. Ullah, Y. S. Mary, Y. S. Mary, S. Soman, M. Thirunavukkarasu and H. W. Kwon, Vietnam J. Chem., 2022, 60, 636–652 CrossRef CAS.
- S. M. Al-Muntaser, A. A. Al-Karmalawy, A. M. El-Naggar, A. K. Ali, N. E. A. Abd El-Sattar and E. M. Abbass, RSC Adv., 2023, 13, 12184–12203 RSC.
- F. Rizwana B, S. Muthu, J. C. Prasana, C. S. Abraham and M. Raja, Chem. Data Collect., 2018, 17–18, 236–250 CrossRef.
- J. D. D. Tarika, X. D. D. Dexlin, S. Madhankumar, D. D. Jayanthi and T. J. Beaula, Spectrochim. Acta, Part A, 2021, 259, 119907 CrossRef CAS.
- V. Puthanveedu and K. Muraleedharan, Comput. Theor. Chem., 2023, 1225, 114126 CrossRef CAS.
- G. Jeyanthi and C. Gnana Sambandam, J. Mol. Struct., 2021, 1243, 130879 CrossRef CAS.
- S. Janani, H. Rajagopal, S. Sakthivel, S. Kadaikunnan, G. Abbas and S. Muthu, J. Mol. Struct., 2023, 1289, 135832 CrossRef CAS.
- Y. Kubota, S. Tanaka, K. Funabiki and M. Matsui, Org. Lett., 2012, 14, 4682–4685 CrossRef CAS PubMed.
- D. S. El Sayed and E.-s. M. Abdelrehim, Spectrochim. Acta, Part A, 2021, 261, 120006 CrossRef CAS PubMed.
- O. Azougagh, S. Essayeh, N. Achalhi, A. El Idrissi, H. Amhamdi, M. Loutou, Y. El Ouardi, A. Salhi, M. Abou-Salama and S. El Barkany, Carbohydr. Polym., 2022, 276, 118737 CrossRef CAS.
- A. Prabakaran, V. Vijayakumar, N. Radhakrishnan, R. Chidambaram and S. Muthu, Polycyclic Aromat. Compd., 2022, 42, 925–941 CrossRef CAS.
- I. Azad, Y. Akhter, T. Khan, M. I. Azad, S. Chandra, P. Singh, D. Kumar and M. Nasibullah, J. Mol. Struct., 2020, 1203, 127285 CrossRef CAS.
- A. Jeelani, S. Muthu, B. R. Raajaraman and S. Sevvanthi, Spectrosc. Lett., 2020, 53, 778–792 CrossRef CAS.
- J. Geethapriya, A. Shanthidevi, M. Arivazhagan, N. Elangovan and R. Thomas, J. Indian Chem. Soc., 2022, 99, 100418 CrossRef CAS.
- S. Sundaram, V. N. Vijayakumar and V. Balasubramanian, Comput. Theor. Chem., 2022, 1217, 113920 CrossRef CAS.
- S. Sevvanthi, S. Muthu, S. Aayisha, P. Ramesh and M. Raja, Chem. Data Collect., 2020, 30, 100574 CrossRef CAS.
- K. Arulaabaranam, G. Mani and S. Muthu, Chem. Data Collect., 2020, 29, 100525 CrossRef CAS.
- S. Janani, H. Rajagopal, S. Muthu, S. Javed and A. Irfan, J. Indian Chem. Soc., 2022, 99, 100438 CrossRef CAS.
- M. Azzouzi, Z. E. Ouafi, O. Azougagh, W. Daoudi, H. Ghazal, S. E. Barkany, R. Abderrazak, S. Mazières, A. E. Aatiaoui and A. Oussaid, J. Mol. Struct., 2023, 1285, 135525 CrossRef CAS.
- M. Thirunavukkarasu, P. Prabakaran, A. Saral, N. S. Alharbi, S. Kadaikunnan, A. S. Kazachenko and S. Muthu, J. Mol. Liq., 2023, 380, 121714 CrossRef CAS.
- W. S. Hu and S. H. Hughes, Cold Spring Harbor Perspect. Med., 2012, 2, a006882 Search PubMed.
- E. De Clercq, J. Clin. Virol., 2004, 30, 115–133 CrossRef CAS.
- A. Milinkovic and E. Martínez, Expert Rev. Anti-Infect. Ther., 2004, 2, 367–373 CrossRef CAS PubMed.
- M. J. M. Hitchcock, Clin. Infect. Dis., 1993, 16, 16–21 CrossRef PubMed.
- L. G. Ferreira, R. N. Dos Santos, G. Oliva and A. D. Andricopulo, Molecules, 2015, 20, 13384–13421 CrossRef CAS PubMed.
- L. Pinzi and G. Rastelli, Int. J. Mol. Sci., 2019, 20, 4331 CrossRef CAS.
- J. Wang, A. Bhattarai, H. N. Do and Y. Miao, QRB Discovery, 2022, 3, 13 CrossRef.
- K. Henzler-Wildman and D. Kern, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- J. A. McCammon, Curr. Opin. Struct. Biol., 1998, 8, 245–249 CrossRef CAS.
- R. Patil, S. Das, A. Stanley, L. Yadav, A. Sudhakar and A. K. Varma, PLoS One, 2010, 5, 12029 CrossRef PubMed.
- R. Vaidyanathan, S. Murugan Sreedevi, K. Ravichandran, S. M. Vinod, Y. Hari Krishnan, L. K. Babu, P. S. Parthiban, L. Basker, T. Perumal, V. Rajaraman, G. Arumugam, K. Rajendran and V. Mahalingam, JCIS Open, 2023, 12, 100096 CrossRef.
- J. Ribas, E. Cubero, F. J. Luque and M. Orozco, J. Org. Chem., 2002, 67, 7057–7065 CrossRef CAS PubMed.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS PubMed.
- F. Yamashita and M. Hashida, Drug Metab. Pharmacokinet., 2004, 19, 327–338 CrossRef CAS.
- T. J. Hou, K. Xia, W. Zhang and X. J. Xu, J. Chem. Inf. Comput. Sci., 2004, 44, 266–275 CrossRef CAS.
- A. K. Shah and S. A. Agnihotri, J. Controlled Release, 2011, 156, 281–296 CrossRef CAS PubMed.
- Y. Yuan, F. Zheng and C.-G. Zhan, AAPS J., 2018, 20, 54 CrossRef.
- R. Datta, D. Das and S. Das, Chemom. Intell. Lab. Syst., 2021, 213, 104309 CrossRef CAS.
- N. Basant, S. Gupta and K. P. Singh, Comput. Biol. Chem., 2016, 61, 178–196 CrossRef CAS PubMed.
- T. Lynch and A. Price, Am. Fam. Physician, 2007, 76, 391–396 Search PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS.
- J.-S. Wang, K.-X. Zhao, K. Zhang, C. Pannecouque, E. De Clercq, S. Wang and F.-E. Chen, Bioorg. Chem., 2024, 147, 107340 CrossRef CAS.
- H. Chen, C. Khemtong, X. Yang, X. Chang and J. Gao, Drug Discovery Today, 2011, 16, 354–360 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Abderrahim Ait Ouchaouibc,
Omar Azougagh
*a,
Abderrahim Ait Ouchaouibc,
Omar Azougagh