
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc(II)-mediated stereoselective construction of 1,2-cis 2-azido-2-deoxy glycosidic linkage: assembly of Acinetobacter baumannii K48 capsular pentasaccharide derivative†
Xiaoya
Zhao‡
a,
Han
Ding‡
 b,
Aoxin
Guo‡
b,
Xuemei
Zhong‡
a,
Siai
Zhou
a,
Guoqing
Wang
a,
Yuhua
Liu
c,
Akihiro
Ishiwata
*d,
Katsunori
Tanaka
de,
Hui
Cai
*a,
Xue-Wei
Liu
*b and
Feiqing
Ding
*a
b,
Aoxin
Guo‡
b,
Xuemei
Zhong‡
a,
Siai
Zhou
a,
Guoqing
Wang
a,
Yuhua
Liu
c,
Akihiro
Ishiwata
*d,
Katsunori
Tanaka
de,
Hui
Cai
*a,
Xue-Wei
Liu
*b and
Feiqing
Ding
*a
aSchool of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, China. E-mail: dingfq3@mail.sysu.edu.cn; caihui5@mail.sysu.edu.cn
bSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 637371, Singapore. E-mail: xuewei@ntu.edu.sg
cSchool of Physics and Electronic Engineering, Guangzhou University, Guangzhou 510006, China
dRIKEN Cluster for Pioneering Research, Wako, Saitama 3510198, Japan. E-mail: aishiwa@riken.jp
eDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, Tokyo, Japan
First published on 16th July 2024
Abstract
The capsular polysaccharide (CPS) is a major virulence factor of the pathogenic Acinetobacter baumannii and a promising target for vaccine development. However, the synthesis of the 1,2-cis-2-amino-2-deoxyglycoside core of CPS remains challenging to date. Here we develop a highly α-selective ZnI2-mediated 1,2-cis 2-azido-2-deoxy chemical glycosylation strategy using 2-azido-2-deoxy glucosyl donors equipped with various 4,6-O-tethered groups. Among them the tetraisopropyldisiloxane (TIPDS)-protected 2-azido-2-deoxy-D-glucosyl donor afforded predominantly α-glycoside (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β = >20:1) in maximum yield. This novel approach applies to a wide acceptor substrate scope, including various aliphatic alcohols, sugar alcohols, and natural products. We demonstrated the versatility and effectiveness of this strategy by the synthesis of A. baumannii K48 capsular pentasaccharide repeating fragments, employing the developed reaction as the key step for constructing the 1,2-cis 2-azido-2-deoxy glycosidic linkage. The reaction mechanism was explored with combined experimental variable-temperature NMR (VT-NMR) studies and mass spectroscopy (MS) analysis, and theoretical density functional theory calculations, which suggested the formation of covalent α-C1GlcN-iodide intermediate in equilibrium with separated oxocarbenium–counter ion pair, followed by an SN1-like α-nucleophilic attack most likely from separated ion pairs by the ZnI2-activated acceptor complex under the influence of the 2-azido gauche effect.
:β = >20:1) in maximum yield. This novel approach applies to a wide acceptor substrate scope, including various aliphatic alcohols, sugar alcohols, and natural products. We demonstrated the versatility and effectiveness of this strategy by the synthesis of A. baumannii K48 capsular pentasaccharide repeating fragments, employing the developed reaction as the key step for constructing the 1,2-cis 2-azido-2-deoxy glycosidic linkage. The reaction mechanism was explored with combined experimental variable-temperature NMR (VT-NMR) studies and mass spectroscopy (MS) analysis, and theoretical density functional theory calculations, which suggested the formation of covalent α-C1GlcN-iodide intermediate in equilibrium with separated oxocarbenium–counter ion pair, followed by an SN1-like α-nucleophilic attack most likely from separated ion pairs by the ZnI2-activated acceptor complex under the influence of the 2-azido gauche effect.
Introduction
Nosocomial infections caused by Acinetobacter baumannii, a Gram-negative opportunistic pathogenic bacterium, pose a major threat to public health. A. baumannii bacteria survive in the host for a long time and colonize the respiratory tract and circulatory system, causing pneumonia and other serious complications. The effectiveness of current antibiotic treatment for A. baumannii infections has been increasingly compromised by the emergence of drug resistance.1 The pathogenicity of A. baumannii is mediated by various virulence factors, including capsular polysaccharide (CPS), a complex long-chain glycopolymer anchored in bacterial cell walls by non-covalent interactions.2 Research indicates that CPS triggers immune responses producing specific antibodies against the pathogen and holds the potential to be developed as a vaccine precursor.3A. baumannii CPS features pathogen-associated molecular patterns (PAMPs) which interact with pattern recognition receptors (PRRs) on epithelial or immune cells, activating downstream signaling pathways leading to subsequent release of inflammatory cytokines. Currently, CPS molecules are procured from fermentation production as heterogeneous and low-purity materials, falling short of the requirements for vaccine development. The preparation of A. baumannii polysaccharide repeating unit antigens in pure form with well-defined structures, by either chemical or chemoenzymatic approaches, has attracted significant interest, such as Zhang's4 work for synthesizing O-antigens; Hashimoto,5 Kosma6 and Yin's7 works for lipopolysaccharide (LPS); and Gao,8 Seeberger,9 Ragains,10 Xiao's11 works for CPS.The K locus (KL) in A. baumannii gene clusters is responsible for CPS biosynthesis. Over 40 types of CPS K-unit structures have been determined in recent years, including the K48 capsule isolated from A. baumannii strain NIPH615. In 2015, Knirel's group12 elucidated the structure of the A. baumannii CPS K48 capsule type, which comprises two 1,2-cis amino glycosyl residues. The 1,2-cis 2-amino-2-deoxyglycoside structure occurs widely in various plant metabolites, anticoagulant drugs, and bacteria surface antigens among different serotypes, such as P. stuartii O44, A. baumannii CPS K47, K48 and K88 polysaccharides (Fig. 1A).13,14 While chemically constructing the 1,2-trans 2-amino-2-deoxyglycosidic bond is readily achieved by exploiting the neighboring group participation (NGP), construction of the 1,2-cis linkage remains difficult, and few direct syntheses of 1,2-cis 2-amino-2-deoxy glycosides have been reported. Indirect methods include the 2,3-cyclic protection strategies by Kerns15 and Manabe–Ito16 groups, which involve the 1,2-trans glycosylation with oxazolidinone-fused donors and the following anomerization of the glycosides. Nguyen et al.17 reported the strategy combining a C(2)-N-substituted benzylideneamino donor with a nickel triflate catalyst imparting 1,2-cis stereoselectivity. In 1978, Paulsen18 developed a 2-azido-2-deoxy pyranose donor without 2-NGP which undergoes the 1,2-cis glycosylation reaction, and the inert azido group was then converted to an amino group. Henceforth multiple new 2-azido-2-deoxy donors have been developed for the synthesis of 1,2-cis 2-amino-2-deoxy glycosides. Boons et al.19 developed an α-selective glycosylation adopting 2-azido-2-deoxyglucosyl trichloroacetimidate donors in the presence of thioether through the formation of β-anomeric sulfonium ion intermediates (Fig. 1B(a)). Gao et al.8 employed 2-azido-2-deoxy-1-thioglucoside donors armed with 6-O-TBS and 6-O-Bz groups under TolSCl/AgOTf conditions for 1,2-cis glycosylation through steric shielding and remote participation tactics (Fig. 1B(b) and (c)). Most of these strategies are restricted to relatively limited substrate scopes, albeit with moderate to excellent yields and selectivities.
 | ||
| Fig. 1 (A) Selected examples of natural polysaccharides containing 2-deoxy-2-amino-D-glucosidic bonds; (B) indirect approaches to the stereoselective synthesis of 1,2-cis 2-deoxy-2-azido glycosides; (C) our previous work for the construction of 1,2-cis glucosidic bond mediated by ZnI2; (D) presenting work about direct approach to 1,2-cis 2-deoxy-2-azido glycosides mediated by ZnI2. | ||
Our previous works have revealed that a mild Lewis acidic salt, namely ZnI2, effectively promotes cis glycosylation such as α-glucosylation,20 β-mannosylation,21 and β-rhamnosylation22 as well as 1,4/6-cis β-galactosylation in a selective manner (Fig. 1C).23 Built upon our established protocols and noticing the biological and medicinal relevance of 1,2-cis 2-amino-2-deoxy glucosyl skeleton, we envisioned that the zinc-mediated diastereoselective 1,2-cis glycosylation reaction could be extended to the stereoselective synthesis of α-2-deoxy-2-amino-glucoside structures (Fig. 1D). Moreover, Bols' work24 highlights the stereo-directing effects of “super-armed” silyl ether protecting groups on thioglycoside O-3, enhancing donor reactivities through silyl-assisted conformation shift of the pyranose ring from 4C1 to 1C4. Although there are sporadic reports of the cyclic disiloxane-assisted intramolecular aglycon delivery25 and arabinofuranosylation,26 stereoselective glycosylation with 2-amino-2-deoxy type glucosyl donors exploiting stereoelectronic effects of protecting groups remain underexplored by far. Drawing inspiration from previous works, we hypothesized that introducing a ring-conformation-restricting 4,6-O-cyclic protecting group and a sterically hindered O-3 protecting group such as silyl ethers can significantly improve α-stereoselectivity through synergistic stereoelectronic effects.
With these in mind, here we report a novel ZnI2-mediated chemical synthesis of 1,2-cis 2-azido-2-deoxyglycosides which employs a rationally designed 4,6-O-tethered-O-TIPDS-protected 2-azido-2-deoxy-D-glucosyl trichloroacetimidate donor, achieving exclusive α-stereoselectivity with a wide range of acceptor substrate scope. Contrary to the proposed Zn2+-mediated SN2-like directed nucleophilic attack involving the simultaneous coordination of Zn2+ with both benzyl ether on donor and hydroxy group on acceptor in our earlier works,20–23 results of our mechanistic studies combining experimental variable-temperature nuclear magnetic resonance (VT-NMR) characterization and theoretical density functional theory (DFT) calculations suggest that the new glycosylation reaction proceeds via a different mechanism, with the glycosyl oxocarbenium arising from activation of the donor preferentially adopting a conformation with the 3-silyl ether group blocking the β-face, leading to the following SN1-like nucleophilic attack by the acceptor from α-face exclusively. We showcased the applicability of our method with the synthesis of A. baumannii K48 capsular trisaccharide fragment, using this reaction as the key step. We further applied the method to the synthesis of a 2-amino-2-deoxy glucose-containing pentasaccharide repeating unit via a convergent [3 + 2] fragment coupling strategy.
Results and discussion
Donors optimization
To identify the most effective donor for the construction of 1,2-cis amino glucosyl linkage, we screened a variety of glycosyl donors with different protecting groups under standard conditions (Scheme 1). A 3,4,6-tri-O-benzyl 2-azido-2-deoxyglucosyl donor 2a was examined under optimized conditions (2.0 equiv. of ZnI2 in Et2O at 0.01 M of a concentration of the acceptor) for 1,2-cis glucosylation20 and a corresponding disaccharide 4a was obtained in a 40% yield (α:β = 1:2) although the 4,6-O-benzylidene-3-O-benzyl donor 2b resulted in 21% yield with a complete α-stereoselectivity. Another 4,6-O-benzylidene donor 2c with bulky TIPS-protected at C3 position gave α-product predominantly in a slightly increased yield of 35%. To enhance the obvious steric effect at C3–O protective group with 4,6-O-cyclic structure, the 4,6-O-TIPDS-protected donor 2d equipped with bulky TIPS group was then examined and gave 4d in 61% yield. As 3-O-benzoyl-4,6-O-TIPDS-protected donor 2e showed relatively low stereoselectivity (α:β = 3:1), the use of an bulky electron-donating group at C-3 is essential to the desired high 1,2-cis selectivity. Therefore, donor 2d was chosen for further optimization of the reaction conditions.
 | ||
| Scheme 1 Optimization of 2-azido-2-deoxy glucosyl donors. Donor 2 (2.0 equiv.), acceptor 3a (1.0 equiv.), promotor (1.0 equiv.), MS 4 Å (100 mg mL−1) were used unless otherwise specified. Combined yields of the anomeric mixture of corresponding glycosides were shown. Stereoselectivity was determined by the integration ratio obtained from 1H-NMR of crude mixture. | ||
Reaction conditions
The examination of the effects of different parameters on the stereoselective glycosylation of acceptor 3a with donor 2d was further carried out and the results are shown in Table 1. The results of screening of a series of representative Brønsted acids or Lewis acids (entries 1–9) pointed out ZnI2 to be the most effective promotor among them. Through the solvent screening, Et2O showed the highest 1,2-cis selectivity (entry 9, α:β = >20:1) and a satisfactory yield of 53% probably because of the optimum solubility of ZnI2 in Et2O and ether effect in directing the 1,2-cis glycosylation.20–23 Further optimizations of other various factors, such as temperature, substrate concentration, the equivalent of the promotor, and reaction time, were also conducted. Under the optimum conditions of 2.0 equiv. of ZnI2 at 0.01 M in Et2O for 72 h at 25 °C (entry 25), the 2-azido-2-deoxyglucosylation afforded the desired disaccharide 4d in 82% yield with high α-selectivity (α:β = >20:1). Notably, the yield of the desired product 4d decreased at lower temperature (entries 15–18), while the concentration of substrate and equivalent of promotor had no significant impact on stereoselectivity although the promoter loading was optimum at 2.0 equiv. for the yield of the product (entries 19–29).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Promotor | Equiv. | Time | [M] | Solvent | Temp. | Yieldb | α:βc |
| a Reaction conditions: donor 2d (2.0 equiv.), acceptor 3a (1.0 equiv.), MS 4 Å (100 mg mL−1). b Combined yield of the anomeric mixture of the corresponding glycoside. c Determined by the integration ratio obtained from 1H-NMR of crude mixture. | ||||||||
| 1 | TMSOTf | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 43% | 3:1 |
| 2 | TfOH | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 38% | 3:1 |
| 3 | Cu(OTf)2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 45% | 3:1 |
| 4 | ZnBr2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | Trace | — |
| 5 | Zn(OTf)2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 45% | 3:1 |
| 6 | ZnCl2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 32% | 10:1 |
| 7 | CuBr2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | Trace | — |
| 8 | B(C6F5)3 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 30% | 12:1 |
| 9 | ZnI2 | 1.0 | 72 h | 0.008 | Et2O | 25 °C | 53% | >20:1 |
| 10 | ZnI2 | 1.0 | 72 h | 0.008 | Toluene | 25 °C | 54% | 1:1 |
| 11 | ZnI2 | 1.0 | 72 h | 0.008 | DCM | 25 °C | 56% | 3:1 |
| 12 | ZnI2 | 1.0 | 72 h | 0.008 | MeCN | 25 °C | 21% | — |
| 13 | ZnI2 | 1.0 | 72 h | 0.008 | THF | 25 °C | Trace | — |
| 14 | ZnI2 | 1.0 | 72 h | 0.008 | 1,4-Dioxane | 25 °C | 64% | 9:1 |
| 15 | ZnI2 | 1.0 | 72 h | 0.008 | Et2O | −78 °C | Trace | — |
| 16 | ZnI2 | 1.0 | 72 h | 0.008 | Et2O | −40 °C | 19% | — |
| 17 | ZnI2 | 1.0 | 72 h | 0.008 | Et2O | −20 °C | 25% | — |
| 18 | ZnI2 | 1.0 | 72 h | 0.008 | Et2O | 0 °C | 50% | 8:1 |
| 19 | ZnI2 | 1.0 | 72 h | 0.01 | Et2O | 25 °C | 55% | >20:1 |
| 20 | ZnI2 | 1.0 | 72 h | 0.005 | Et2O | 25 °C | 50% | >20:1 |
| 21 | ZnI2 | 1.0 | 72 h | 0.003 | Et2O | 25 °C | 9% | >20:1 |
| 22 | ZnI2 | 1.0 | 72 h | 0.001 | Et2O | 25 °C | Trace | — |
| 23 | ZnI2 | 0.5 | 72 h | 0.01 | Et2O | 25 °C | 34% | >20:1 |
| 24 | ZnI2 | 1.0 | 72 h | 0.01 | Et2O | 25 °C | 61% | >20:1 |
| 25 | ZnI 2 | 2.0 | 72 h | 0.01 | Et 2 O | 25 °C | 82% |
>20:1 |
| 26 | ZnI2 | 3.0 | 72 h | 0.01 | Et2O | 25 °C | 45% | >20:1 |
| 27 | ZnI2 | 2.0 | 12 h | 0.01 | Et2O | 25 °C | 45% | >20:1 |
| 28 | ZnI2 | 2.0 | 24 h | 0.01 | Et2O | 25 °C | 55% | >20:1 |
| 29 | ZnI2 | 2.0 | 48 h | 0.01 | Et2O | 25 °C | 80% | >20:1 |

Substrate scope studies
With optimum conditions in hand, the substrate scope of optimized ZnI2-mediated 2-azido-2-deoxy glycosylation were explored (Scheme 2A). Firstly, with linear (3b and 3c), cyclic (3d and 3e) and branched (3f and 3g) aliphatic alcohols, the desired products 5b–g were obtained in excellent yields (81–99%) in all cases. Sterically hindered adamantanol (3h) and L-menthol (3k) were both successfully connected as corresponding 2-azido-2-deoxy glucoside with α-linkages in 80% and 93% yield, respectively (5h–k). ZnI2-promoted 1,2-cis 2-azido-2-deoxyglucosylation tolerated a variety of glycosyl acceptors including GlcO-2 (3l and 3m), GlcO-3 (3n), GlcNO-3 (3o), GlcO-4 (3p), GlcNO-4 (3q), GlcO-6 (3s and 3t) and GalO-6 (3r), resulting in good to excellent yields (5l–t). Next, the disaccharide acceptor GalO-3-β-(1→4)-Glc (3u) afforded corresponding α-trisaccharide 5u but only in 22% yield, probably due to the galactoside structure in the acceptor expected to coordinate with ZnI2 for deactivation.23 The amino acid such as protected L-serine derivatives 3i–j, and naturally occurring steroids such as cholesterol (3v) and diosgenin (3w) could be applied to afford the corresponding α-glycosides (5i–j and 5v–w), predominantly. Most of the acceptors tested resulted in complete α-stereoselectivities (α:β = >20:1) under the optimum conditions, except 1,2,3,4-tetra-O-acetyl-β-D-glucose 3r (GlcO-6) as less nucleophilic acceptor (5r, α:β = 2:1). In addition, to demonstrate the practicality of this methodology, the model glycosylation of 2d with 3a under the optimum conditions at a 0.1 mmol scale was performed and afforded GlcN-α-(1→6)-Glc disaccharide (4da) in 89% yield and complete stereoselectivity (α:β = >20:1). After that, the global deprotection for 4da was conducted by reduction of azido group, N-acetylation, desilylation and hydrogenolysis to afford methyl 2-acetamido-2-deoxy-α-D-glucopyranosyl-(1→6)-α-D-glucopyranoside (4db) in 55% yield over four steps (Scheme 2B). These results set a solid foundation for the synthesis of complex oligosaccharides using our methods.
 | ||
| Scheme 2 (A) Substrate scopes of ZnI2-directed 1,2-cis 2-azido-2-deoxy glycosylation.a (B) 0.1 mmol scale synthesis and global deprotection. aDonor 2d (2.0 equiv.), acceptor 3 (1.0 equiv.), promotor (2.0 equiv.), MS 4 Å (100 mg mL−1) were used unless otherwise specified. Combined yields of the anomeric mixture of corresponding glycosides were shown. Stereoselectivity was determined by the integration ratio obtained from 1H-NMR of crude mixture. | ||
Synthesis of A. baumannii CPS K48 pentasaccharide
Inspired by the effective construction of 1,2-cis 2-azido-2-deoxy glycosidic linkages, we applied this strategy to the unprecedented synthesis of A. baumannii K48 capsular pentasaccharide repeating fragment. Considering the complete and partial fragments' potential to be developed as vaccine precursors, the synthetic target was designed with a 5-aminopentyl spacer at the terminal α-glucose residue of core pentasaccharide, which could be attached to other biological molecules such as carrier protein for further immunological studies of these glycoconjugates. However, the synthesis of target molecules may be relatively tough due to the highly branching nature of the Gal unit, since the hydroxy groups at its C1, C3, and C4 positions are substituted in the meantime. Due to these predictable challenges, currently, there is no synthetic case reported about this CPS K-unit. Retrosynthetically, the desired pentasaccharide could be achieved by glycosylating disaccharide acceptor 19 with the trisaccharide donor 12 through a convergent [3 + 2] glycosylation (Scheme 3A). The 2-azido-2-deoxy α-glucosyl residue occurs in both fragments 12 and 19, and their key 2-azido-2-deoxy α-glucosidic linkages could be constructed by our developed facile and convenient ZnI2-mediated stereoselective glycosylation reaction to showcase the versatility and practicability of the methodology. | ||
| Scheme 3 (A) Retrosynthesis analysis of the A. baumannii CPS K48 polysaccharide repeating unit. (B) Stereoselective synthesis of building blocks and assembly of pentasaccharide. | ||
Our synthetic task commenced with the synthesis of trisaccharide building block 12 (Scheme 3B). The trimethylsilyl trifluoromethanesulfonate (TMSOTf)-mediated glycosylation between the perbenzoylated N-phenyl-trifluoroacetimidate (PTFAI) donor 6 and GalO-3 acceptor 7, affording the disaccharide with a moderate selectivity (α:β = 1:4) mainly via neighboring group participation (NGP) effect, and the desired β-linked product 8 was separated in 51% yield. The reductive ring-opening of benzylidene under BF3·Et2O–Et3SiH conditions gave C4–OH of Gal residue in disaccharide acceptor 9, which was glycosylated with 2d under standard ZnI2-promoted 1,2-cis 2-azido-2-deoxy glucosylation conditions to afford the branched trisaccharide 10 with satisfactory α-selectivity. The low yield (30%) was mainly attributed to both the significant steric hindrance of O-3 sugar substituent and the weak nucleophilicity of galactose C4–OH due to the electron-withdrawing effect of the axial-oriented hydroxyl group.27 Subsequent oxidative removal of the MP group of the trisaccharide 10 with the treatment of ceric ammonium nitrate (CAN) afforded the intermediate, which was then ready to be equipped with different leaving groups at C1 position for [3 + 2] glycosylation. The synthesis of GlcNO-3-α-(1→3)-Glc disaccharide building block 19 (Scheme 3B) commenced with the 4,6-O-naphthylidene thioglucoside which could be converted to trichloroacetimidate donor 15 and was glycosylated with 5-aminopentyl spacer 13 to afford 16 in 45% yield following our ZnI2-promoted α-glucosylation standard conditions as reported before (α:β > 20:1).20 After removal of C3–O-TIPS by fluoride-mediated desilylation to afford acceptor 17, the key ZnI2-promoted 1,2-cis 2-azido-2-deoxyglucosylation with 4,6-O-TIPDS-protected donor 2d afforded the desired disaccharide 18 in a complete α-stereoselectivity and 47% yield. Considering the difficulty of [3 + 2] segment ligation, the 4,6-O-TIPDS group of 10 was transformed to 4,6-O-benzylidene moiety 11 in two steps.
With A. baumannii CPS K48 α-GlcN3-linked branched trisaccharide fragment and disaccharide acceptor in hand, we further explored the optimum condition for the key [3 + 2] assembly of pentasaccharide derivative 20 (Table 2). At first, our commonly used trichloroacetimidate (TCA) donor was tried under the strong TMSOTf catalyst but failed mainly because of the instability of the imidate (Table 2, entry 1). Most of the TCA donor was hydrolyzed in the process of silica gel column chromatography. Although N-phenyltrifluoroacetimidate (PTFAI) donor could be prepared, its glycosylation turned out to be less effective, affording the desired pentasaccharide in only 21% yield (Table 2, entry 2). Hence, considering the instability of the imidate-type donor, we turned to using stable ester-type glycosyl donors to avoid unpleasant donor hydrolysis.28 Thus ortho-alkynylbenzoyl (ABz)29 (Table 2, entry 3) and ortho-(1-phenylvinyl)benzoyl (PVB)30 (Table 2, entry 4) groups were equipped to give the corresponding donors, but these two donors gave only trace amounts of the products. In view of the poor yields, the silyl-tethered trisaccharide 10 was converted to donor 12 in four steps (Scheme 3B). While the glycosyl ABz donor was barely effective (Table 2, entry 5), the glycosyl PVB donor 12 was able to be isolated (89%) and resulted in the formation of the corresponding glycoside 20 in 51% yield (Table 2, entry 6). Characterizations of both pentasaccharides with different protection patterns were supported by MALDI-TOF mass spectra, as confirmed by C138H161N7O32Si3Na at 2536.038 and C138H133N7O31Na at 2408.103, respectively. Both pentasaccharides were obtained with exclusive stereoselectivity under the effect of neighboring group participation (NGP). On the other hand, the coupling of PVB donor 12 with 5-(N-benzyl-N-benzyloxycarbonylamino)-1-pentanol spacer 13 afforded the desired trisaccharide unit 14 in 83% yield (α:β = 1:>20, Scheme 3B). These results of glycosylations of 12 suggested that disaccharide acceptor 19 attributed to the low [3 + 2] ligation efficiency because it is a weak and balky nucleophile compared to the spacer alcohol.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Leaving group (LG) | R | Promotor | Yieldb | α:βc |
| a Conditions: see ESI and scheme. b Combined yield of the anomeric mixture of the corresponding glycoside. c Determined by the integration ratio obtained from 1H-NMR of crude mixture. | |||||
| 1 | TCA | R1, R2 = TIPDS, R3 = TIPS | TMSOTf | — | — |
| 2 | PTFAI | R1, R2 = TIPDS, R3 = TIPS | TMSOTf | 21% | α:β 1:>20 |
| 3 | ABz | R1, R2 = TIPDS, R3 = TIPS | PPh3AuOTf | 9% | α:β 1:>20 |
| 4 | PVB | R1, R2 = TIPDS, R3 = TIPS | NIS, TMSOTf | 12% | α:β 1:>20 |
| 5 | ABz | R1 = R2 = R3 = Bn | PPh3AuOTf | — | — |
| 6 | PVB | R1 = R2 = R3 = Bn | NIS, TMSOTf | 51% | α:β 1:>20 |
TCA: C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)CCl3, PTFAI: C(NPh)CF3, ABz: NH)CCl3, PTFAI: C(NPh)CF3, ABz: |
 , , |
PVB: |

|
||

Mechanistic studies
Based on experimental results and controlled model experiments, the variable-temperature nuclear magnetic resonance experiments (VT-NMR)31 and density functional theory calculations were conducted for the proposed plausible mechanism of ZnI2-mediated 1,2-cis 2-azido-2-deoxy-glucosylation. As the model experiment, the reaction with both α- and β-isomers of 2-azido-2-deoxy-glucosyl trichloroacetimidate donor (2d-α and 2d-β) favored α-product selectively (Scheme 4A). In addition, the reactions of nucleophilic ethanol (3x) and less nucleophilic trifluoroethanol (3y) with donor 2d were performed under the standard conditions (Scheme 4B). The weak nucleophile trifluoroethanol cannot directly attack a covalent glycosyl intermediate in an SN2-like manner to an appreciable degree.32 Therefore, if our glycosylation proceeded with an SN2-like process, the stereoselectivity would be contaminated. Nevertheless, under the optimal conditions, the glycosylation of 3y invariably delivered the corresponding product 4y in excellent yield (98%) and complete α-selectivity. The result implies that the glycosylation reaction is less likely to proceed through an SN2 mechanism. Based on the literature and our experimental results, we proposed herein a mechanism involving the formation of a key intermediate of glycosyl iodide, which reversibly dissociates into ion pairs of glycosyl oxocarbenium with restricted conformation and close iodide anions. The oxocarbenium was then stereoselectively attacked by the acceptor nucleophile in an SN1-like manner.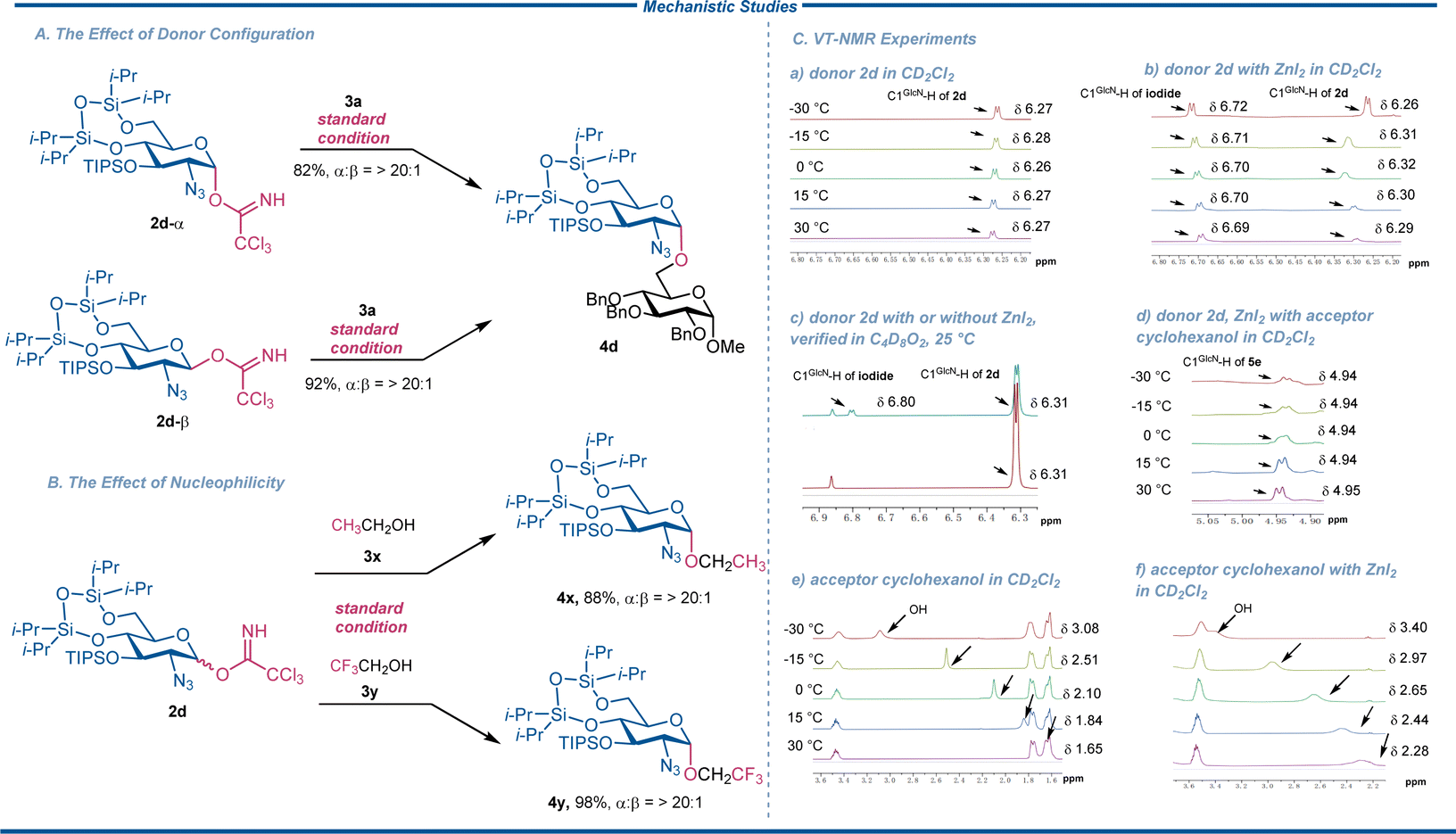 | ||
| Scheme 4 Mechanistic studies of 1,2-cis 2-azido-2-deoxy glycosylation. | ||
In the previous discussion for the ZnI2-mediated glycosylation with trichloroacetimidate donor,20–23 the initial generation of unstable glycosyl iodide intermediate has been considered. Although the formation of C1GlcN-iodide from 2d was expected, the intermediate was too unstable to isolate by silica gel chromatography separation. To confirm the existence of iodide, the VT-NMR studies were therefore performed at five temperature gradients from −30 °C to 30 °C (Scheme 4C). Considering the unavailability of Et2O-d10 and the melting point of dioxane-d8, dichloromethane-d2 was selected as the deuterated solvent for VT-NMR study. The equivalent of donor (38 mM), acceptor cyclohexanol (19 mM) and ZnI2 (38 mM) complied with the standard conditions for ZnI2-mediated 1,2-cis 2-azido-2-deoxy-glucosylation. The δ(C1GlcN–H) of donor 2d was monitored almost invariably around 6.27 ppm (Scheme 4C(a)) with or without zinc iodide addition. As expected, we observed δ(C1GlcN–H) of iodide at about 6.70 ppm (Scheme 4C(b)), supported by mass spectra of target C27H56N3O5Si3I at 713.40 and C27H56N3O5Si3INa at 736.80 (Fig. S6 and S7†). A further 1H-NMR experiment in dioxane-d8 instead of Et2O at 25 °C also showed δ(C1GlcN–H)2d at 6.31 ppm and δ(C1GlcN–H) of iodide at 6.80 ppm, as verified by VT-NMR experiment (Scheme 4C(c)). The coupling constants (3JH1–H2) of (C1GlcN–H) of iodide were 3.8 Hz in dichloromethane-d2 and 3.9 Hz in dioxane-d8, strongly confirming α-iodide formation according to the Karplus equation, while the expected peaks of β-iodide were not detectable in the solution. When the acceptor cyclohexanol was added to the mixture, the δ(C1GlcN–H) of product 5e could be observed at 4.94 ppm as α-glycoside (3JH1–H2 = 3.6 Hz, Scheme 4C(d)). Both control and VT-NMR experimental results clearly suggested that the reaction proceeded through α-iodide via initial SN1 reaction followed by subsequent SN1 reaction to afford the α-glycoside.
For further investigation of the function of ZnI2 in the reaction process, δ(OH) of acceptor cyclohexanol with or without ZnI2 addition were compared, and as a result, the δ(OH) shifted downfield from a range of 3.08–1.65 ppm to 3.40–2.28 ppm after addition of ZnI2 (Scheme 4C(e) and (f)). In contrast, the δ(NH) of donor 2d remained at 8.62–8.58 ppm after addition of ZnI2 (Scheme S1†), albeit a chemical shift of (C1GlcN–H)2d seemed to be slightly affected at the same time (Scheme 4C(b)) and the integrations of 1H peaks related to 2d decreased from −30 °C to 30 °C with a contrasting increase of those of α-iodide (Scheme S1†). The results indicated that ZnI2 preferentially coordinated with OH in the acceptor instead of NH in the trichloroacetimidate group, although α-iodide formation also proceeded by the addition of ZnI2 to 2d in the absence of acceptor alcohol.
Following the observations from 1H-NMR studies, we proposed a potential mechanism and explained the rationality by density functional theory (DFT) calculation (Schemes 5 and 6). Initially, the zinc cation activated the leaving group of the trichloroacetimidate donor; therefore, C1–O1 bond was weakened and the trichloroacetimidate ion departed from the glycosyl donor. The intermediate Zn-LG formed, accompanied by the dissociated iodide anion adding to the anomeric carbon through the oxocarbenium ion Int2 with 3H4 half-chair33 and producing α-glycosyl iodide intermediate Int3a, as we observed from VT-NMR. Calculations also confirmed that α-glycosyl iodide intermediate Int3a was more stable than the corresponding β-iodide intermediate Int3b (Int3avs.Int3b in Scheme 5). Meanwhile, the proton transferred from O–H bond of CH3OH to the intermediate Zn-LG and generated the nucleophilic reagent Zn-Nu in an exothermic fashion (Fig. S8†). Although the stable Int3a was observed for NMR analysis, Int2 should be used as the key intermediate to TS2. The transition states TS2gauche/TS2anti were proposed for the suggested second SN1-like displacement through glycosyl oxocarbenium ion Int2 by nucleophilic attack of the deprotonated alcohol as zinc methoxide complex to deliver the product. Computational results showed Zn-Nu α-nucleophilic attack of Zn-Nu took lower energy barrier than that towards oxocarbenium ion from β-face (+4.5 kcal mol−1vs. +17.7 kcal mol−1, from α vs. β, respectively) (Scheme 6). The conformation-directing nucleophilic attack of Zn-Nu in transition state TS2gauche might be attributed to the azido gauche effect, a preference that orients electronegative substituent to gauche form when adjacent azido group exists.34 Inspection of the structure of the TS2gauche pyranose ring also suggested that the bulky C3-OTIPS group effectively shields the β-side and prevents the acceptor from attacking from the β-face. It is also indicated that β-glycoside was +18.2 kcal mol−1 higher in energy than the α-selective product (P1avs.P1b, Scheme 6). According to the calculation results, the α-product was preferred in consistency with the experimental observation.
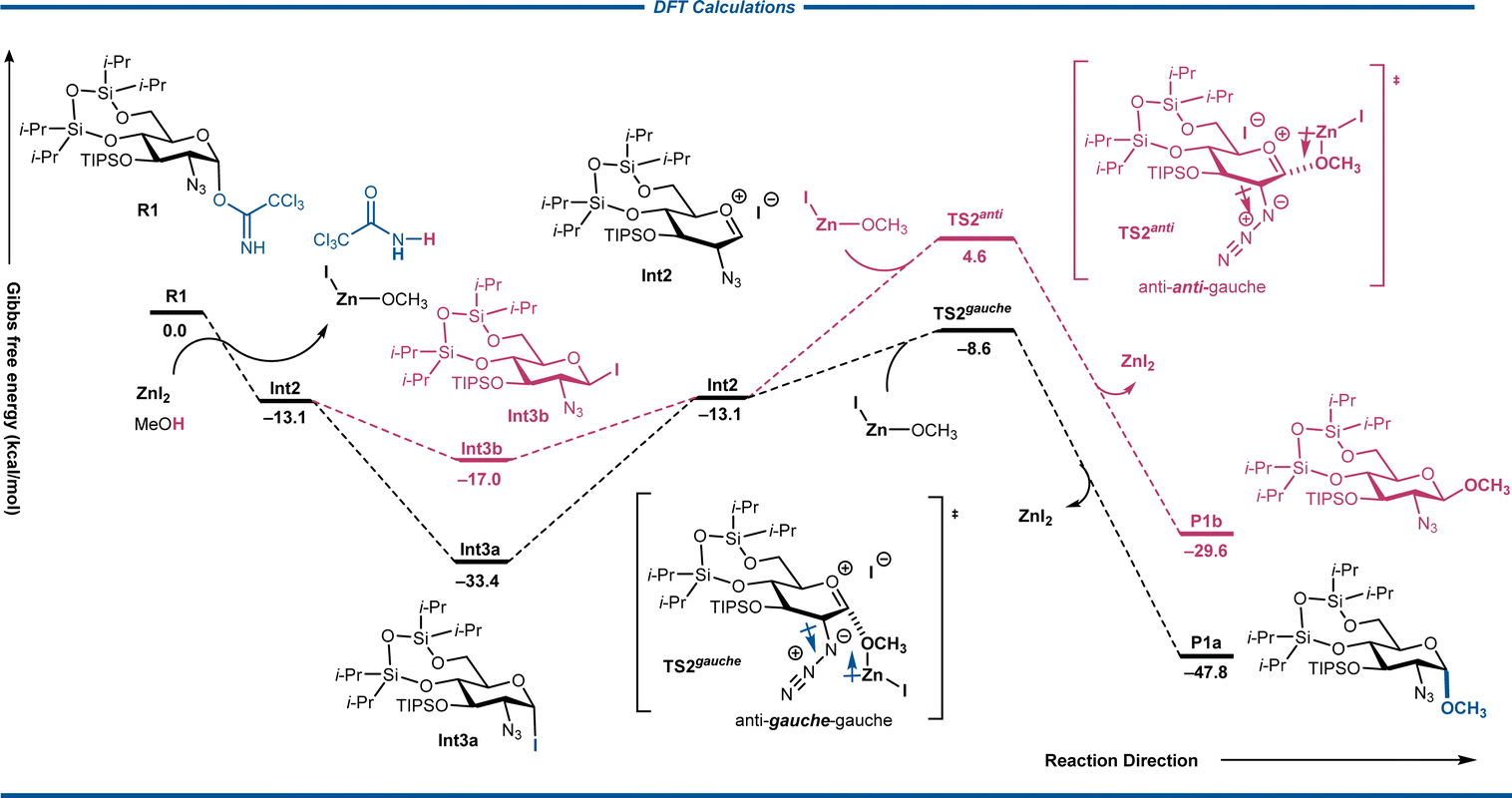 | ||
| Scheme 5 DFT calculations. | ||
 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In summary, we have successfully developed a ZnI2-mediated strategy for synthesizing 1,2-cis 2-azido glucosides with excellent stereoselectivities and wide acceptor substrate scope, employing the rationally designed 4,6-O-tethered-tetraisopropyldisiloxanylidene (TIPDS)-protected 2-deoxy-2-azido-D-glucosyl trichloroacetimidate donor 2d. We demonstrated the usefulness of the novel methodology by synthesizing protected antigenic branched trisaccharide 10 and disaccharide fragment 18 in the Acinetobacter baumannii K48 capsular polysaccharide and a pentasaccharide derivative 20 containing the antigenic trisaccharide structure through a convergent [3 + 2] glycosylation reaction. Mechanistic studies combining VT-NMR investigations and DFT calculations delineate a proposed mechanism involving the formation of a key intermediate α-glycosyl iodide Int2a upon donor activation with ZnI2, the reversible conversion of Int2a into glycosyl oxocarbenium in the solution, and the preferential α-face attack of the glycosyl oxocarbenium by the acceptor passing through transition state TS2gauche along an SN1-like pathway, under the influences of the stereo-directing azido gauche effect. Our lab is carrying out further experimental examinations focusing on the scale-up synthesis of A. baumannii K48 capsular pentasaccharide repeating fragment and investigations of the potential biological activity of deprotected moieties with macromolecule conjugates to the terminal residue, paving ways for future vaccine development.Data availability
Experimental procedures, characterisation data, and NMR spectra for new compounds can be found in the ESI.†Author contributions
Conceptualization: F. Q. D. and X. Y. Z.; methodology: X. Y. Z. and X. M. Z.; investigation: X. Y. Z., H. D., A. X. G., Y. H. L. and A. I.; resources: F. Q. D. and X. Y. Z.; original draft: F. Q. D., X. Y. Z. and A. I.; review and editing: all authors; funding acquisition: F. Q. D. and H. C.; project administration: F. Q. D. and H. C.; supervision: F. Q. D. and H. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Professor Guozhi Xiao from Kunming Institute of Botany, Chinese Academy of Sciences for generous support and helpful advice. This study was supported partly by the Shenzhen Science and Technology Program (No. GJHZ20220913142602004 to F. D.), the Guangdong Basic and Applied Basic Research Funding (No. 2024A1515010015 to F. D.).References
- C. M. Harding, S. W. Hennon and M. F. Feldman, Nat. Rev. Microbiol., 2018, 16, 91–102 CrossRef CAS
.
- D. Wong, T. B. Nielsen, R. A. Bonomo, P. Pantapalangkoor, B. Luna and B. Spellberg, Clin. Microbiol. Rev., 2017, 30, 409–447 CrossRef CAS PubMed
- C. Ma and S. McClean, Vaccines, 2021, 9, 570 CrossRef
- L. Duan, Q. Nie, Y. Hu, L. Wang, K. Guo, Z. Zhou, X. Song, Y. Tu, H. Liu, T. Hansen, J. Sun and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306971 CrossRef CAS
- R. Arihara, K. Kakita, K. Yamada, S. Nakamura and S. Hashimoto, J. Org. Chem., 2015, 80, 4278–4288 CrossRef CAS
- B. Pokorny and P. Kosma, Org. Lett., 2015, 17, 110–113 CrossRef CAS
- X. Zhou, L. Li, Z. Zhang, S. Duan, Y. Huang, Y. Luo, X. Mu, Z. Chen, Y. Qin, J. Hu, J. Yin and J. Yang, Angew. Chem., Int. Ed., 2022, 61, e202204420 CrossRef CAS
- Y. Zhang, H. Zhang, Y. Zhao, Z. Guo and J. Gao, Org. Lett., 2020, 22, 1520–1524 CrossRef CAS PubMed
- S. Zhang and P. H. Seeberger, Chem.–Eur. J., 2021, 27, 17444–17451 CrossRef CAS PubMed
- D. K. Njeri and J. R. Ragains, Org. Lett., 2022, 24, 3461–3465 CrossRef
- K. Shou, S. Liu, Y. Zhang and G. Xiao, Chin. J. Chem., 2024, 42, 1593–1598 CrossRef CAS
- A. S. Shashkov, J. J. Kenyon, N. P. Arbatsky, M. M. Shneider, A. V. Popova, K. A. Miroshnikov, N. V. Volozhantsev and Y. A. Knirel, Carbohydr. Res., 2015, 417, 81–88 CrossRef CAS PubMed
- N. A. Kocharova, O. G. Ovchinnikova, F. V. Toukach, A. Torzewska, A. S. Shashkov, Y. A. Knirel and A. Rozalski, Carbohydr. Res., 2005, 340, 1419–1423 CrossRef CAS
- A. S. Shashkov, J. J. Kenyon, N. P. Arbatsky, M. M. Shneider, A. V. Popova, K. A. Miroshnikov, R. M. Hall and Y. A. Knirel, Carbohydr. Res., 2016, 435, 173–179 CrossRef CAS PubMed
- K. Benakli, C. Zha and R. J. Kerns, J. Am. Chem. Soc., 2001, 123, 9461–9462 CrossRef CAS
- S. Manabe, H. Satoh, J. Hutter, H. P. Lüthi, T. Laino and Y. Ito, Chemistry, 2014, 20, 124–132 CrossRef CAS
-
(a) E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS
- H. Paulsen, Č. Kolář and W. Stenzel, Chem. Ber., 1978, 111, 2358–2369 CrossRef CAS
- J. Park, S. Kawatkar, J. H. Kim and G. J. Boons, Org. Lett., 2007, 9, 1959–1962 CrossRef CAS
- S. Zhou, X. Zhong, A. Guo, Q. Xiao, J. Ao, W. Zhu, H. Cai, A. Ishiwata, Y. Ito, X.-W. Liu and F. Ding, Org. Lett., 2021, 23, 6841–6845 CrossRef CAS PubMed
- X. Zhong, S. Zhou, J. Ao, A. Guo, Q. Xiao, Y. Huang, W. Zhu, H. Cai, A. Ishiwata, Y. Ito, X.-W. Liu and F. Ding, J. Org. Chem., 2021, 86, 16901–16915 CrossRef CAS PubMed
- J. Ao, X. Zhao, S. Zhou, Y. Guo, G. Wang, S. Fang, X. Yao, Y. Liu, A. Ishiwata, K. Tanaka, F. Ding and H. Cai, Org. Chem. Front., 2023, 10, 5610–5615 RSC
- S. Zhou, J. Ao, A. Guo, X. Zhao, N. Deng, G. Wang, Q. Yang, A. Ishiwata, X.-W. Liu, Q. Li, H. Cai and F. Ding, Org. Lett., 2022, 24, 8025–8030 CrossRef CAS
-
(a) C. M. Pedersen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2007, 129, 9222–9235 CrossRef CAS
- Y. Ito, Y. Ohnishi, T. Ogawa and Y. Nakahara, Synlett, 1998, 1102–1104 CrossRef CAS
- A. Ishiwata, H. Akao and Y. Ito, Org. Lett., 2006, 8, 5525–5528 CrossRef CAS
- P. K. Mondal, G. Liao, M. A. Mondal and Z. Guo, Org. Lett., 2015, 17, 1102–1105 CrossRef CAS
- H. Ding, J. Lyu, X. L. Zhang, X. Xiao and X. W. Liu, Nat. Commun., 2023, 14, 4010 CrossRef CAS PubMed
- B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed
- P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405 CrossRef CAS
- X. Liu, Y. Lin, W. Peng, Z. Zhang, L. Gao, Y. Zhou, Z. Song, Y. Wang, P. Xu, B. Yu, H. Sun, W. Xie and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202206128 CrossRef CAS PubMed
- X. Wang, P. Wang, D. Li and M. Li, Org. Lett., 2019, 21, 2402–2407 CrossRef CAS
- A. Franconetti, A. Ardá, J. L. Asensio, Y. Blériot, S. Thibaudeau and J. Jiménez-Barbero, Acc. Chem. Res., 2021, 54, 2552–2564 CrossRef CAS
-
(a) L. S. Sonntag, S. Schweizer, C. Ochsenfeld and H. Wennemers, J. Am. Chem. Soc., 2006, 128, 14697–14703 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental details. See DOI: https://doi.org/10.1039/d4sc03449j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |