
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiscale simulations reveal the driving forces of p53C phase separation accelerated by oncogenic mutations†
Yawei
Yu
,
Qian
Liu
,
Jiyuan
Zeng
,
Yuan
Tan
 ,
Yiming
Tang
and
Guanghong
Wei
*
,
Yiming
Tang
and
Guanghong
Wei
*
Department of Physics, State Key Laboratory of Surface Physics, and Key Laboratory for Computational Physical Sciences (Ministry of Education), Fudan University, Shanghai 200438, People's Republic of China. E-mail: ghwei@fudan.edu.cn
First published on 15th July 2024
Abstract
Liquid–Liquid phase separation (LLPS) of p53 to form liquid condensates has been implicated in cellular functions and dysfunctions. The p53 condensates may serve as amyloid fibril precursors to initiate p53 aggregation, which is associated with oncogenic gain-of-function and various human cancers. M237I and R249S mutations located in p53 core domain (p53C) have been detected respectively in glioblastomas and hepatocellular carcinoma. Interestingly, these p53C mutants can also undergo LLPS and liquid-to-solid phase transition, which are faster than wild type p53C. However, the underlying molecular basis governing the accelerated LLPS and liquid-to-solid transition of p53C remain poorly understood. Herein, we explore the M237I/R249S mutation-induced structural alterations and phase separation behavior of p53C by employing multiscale molecular dynamics simulations. All-atom simulations revealed conformational disruptions in the zinc-binding domain of the M237I mutant and in both loop3 and zinc-binding domain of the R249S mutant. The two mutations enhance hydrophobic exposure of those regions and attenuate intramolecular interactions, which may hasten the LLPS and aggregation of p53C. Martini 3 coarse-grained simulations demonstrated spontaneous phase separation of p53C and accelerated effects of M237I/R249S mutations on the phase separation of p53C. Importantly, we find that the regions with enhanced intermolecular interactions observed in coarse-grained simulations coincide with the disrupted regions with weakened intramolecular interactions observed in all-atom simulations, indicating that M237I/R249S mutation-induced local structural disruptions expedite the LLPS of p53C. This study unveils the molecular mechanisms underlying the two cancer-associated mutation-accelerated LLPS and aggregation of p53C, providing avenues for anticancer therapy by targeting the phase separation process.
Introduction
Biomolecular condensates, often recognized as membraneless organelles due to their ability to sequester related cellular macromolecules, play a crucial role in diverse biological functions, including stress response, cellular signaling, metabolism, and RNA storage.1–4 Numerous studies indicate that these biomolecular condensates form through liquid–liquid phase separation (LLPS).5 An increasing number of investigations demonstrate that intrinsically disordered proteins (IDPs) have a high propensity to undergo LLPS owing to their weak, dynamic, and multivalent interactions.6–8 The liquid condensates formed by some IDPs (such as TDP-43, FUS, and tau) can mature into amyloid fibrils under appropriate conditions, which has been related to neurodegenerative diseases.9–14 Recently, it has been reported that the oncogenic p53 protein can also undergo LLPS and form liquid condensates,15–19 and these condensates can transit into fibrillary aggregates through liquid-to-solid transition.20,21 Some factors, such as mutation, denaturant, and hydrostatic pressure can cause p53 protein to undergo abnormal LLPS and irreversible aggregation,22–24 compromising its normal functions.25 Similar to the fibrillary aggregates formed by the aforementioned IDPs associated with neurodegenerative diseases, amyloid-like aggregates of p53 are observed in various cancers, including ovarian, lung, breast, and skin cancers.26–28 Targeting the p53 LLPS and aggregation pathways has emerged as a potential therapeutic strategy against cancers.29 For example, it is found that polyanions (such as heparin)19 and DNA/RNA can modulate p53 LLPS and aggregation.30–32 Heparin apparently stabilizes the liquid condensates of p53 protein,19 a high stoichiometric RNA: p53 ratio inhibits aggregation of p53 protein,31 and cognate DNA stabilizes p53 and prevents its misfolding and aggregation.30The p53 is a multidomain protein consisting of 393 amino acids. It comprises three functional domains: the N-terminal transactivation domain (p53NTD, residues 1–93), the core domain (p53C, residues 94–290), and the intrinsically disordered C-terminal domain (p53CTD, residues 291–393). As the “guardian of the genome”,33–35 the p53 protein plays a pivotal role in regulating various biological functions, including cell cycle arrest, autophagy, DNA repair, and apoptosis under stress conditions.36–38 Significantly, p53 serves as a major barrier against cancer initiation and progression.37,39 However, mutations can cause abnormal LLPS and subsequent aggregation of p53 protein, resulting in the impairment of p53 antitumor activity.40–45 For example, p53CTD can undergo LLPS readily and tends to form stable liquid condensates, whereas oncogenic mutations in the p53 tetramerization domain (located inside p53CTD) impair the formation of liquid condensates, reducing the activation of p53 target genes and promoting cancer progression.18 In addition, both full-length p53 and its thermostable mutants could form liquid condensates, with the mutant condensates showing an increased fibrillary propensity, which impairs the anticancer ability of p53 protein.46,47 More recently it has been reported that cancer-related M237I and R249S mutants of p53C can undergo LLPS, with the process evolving a solid-like phase transition faster than that in the case of p53C WT.19 However, the molecular basis regarding the faster LLPS and liquid-to-solid transition induced by the two mutations remain mostly unknown.
Inspired by these experimental studies, we aim to elucidate the impacts of M237I and R249S mutations on p53C conformations and LLPS behavior, and the underlying molecular mechanisms using multiscale molecular dynamics (MD) simulations by combining all-atom (AA) and coarse-grained (CG) models. MD simulation methods have been widely used in studying the conformational properties, aggregation propensity and LLPS behavior of proteins.48–52 For instance, Ingólfsson et al.49 explored the intramolecular and intermolecular interaction dynamics of TDP-43, and revealed the dominant physical interactions that drive the formation of biomolecular condensates. Utilizing the hybrid resolution protein model, Zhang et al.48 revealed the coupling between the secondary structure and phase separation propensity of the TDP-43 conserved region. We recently unraveled the mechanisms underlying R248Q mutation-enhanced aggregation of p53C, and explored the small molecule inhibitor resveratrol (RSV) to prevent the conversion of p53C into aggregation-prone conformations.53 In this study, we conducted AA monomer simulations and CG phase coexistence simulations on p53C WT and its two mutants (M237I and R249S). Our AA simulations reveal that M237I mutation induces the disruption of the zinc-binding domain and the increase of loop2 and loop3 flexibility, and thus results in the overall structural destabilization of p53C and its adoption of aggregation-prone molten globule states, potentially speeding the LLPS and liquid-to-solid transition of p53C. Furthermore, the R249S mutation attenuates intramolecular interactions and enhances the hydrophobic exposure of loop3 and zinc-binding domain, which may accelerate the LLPS and aggregation of p53C. Moreover, our Martini 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 CG simulations demonstrate M237I/R249S mutation-facilitated LLPS behavior of p53C. Importantly, we find that the regions with enhanced intermolecular interactions observed in CG simulations align with the destabilized regions observed in AA simulations, indicating the crucial role of the disrupted regions in accelerating the LLPS of p53C. This multiscale MD simulation study bridges the gap between M237I/R249S mutation-induced conformational disruption and phase separation behavior of p53C, and provides for the first time a microscopic explanation for the mutation-accelerated LLPS of p53C, which paves a way for anticancer therapy by targeting LLPS and the aggregation process of p53C.
54 CG simulations demonstrate M237I/R249S mutation-facilitated LLPS behavior of p53C. Importantly, we find that the regions with enhanced intermolecular interactions observed in CG simulations align with the destabilized regions observed in AA simulations, indicating the crucial role of the disrupted regions in accelerating the LLPS of p53C. This multiscale MD simulation study bridges the gap between M237I/R249S mutation-induced conformational disruption and phase separation behavior of p53C, and provides for the first time a microscopic explanation for the mutation-accelerated LLPS of p53C, which paves a way for anticancer therapy by targeting LLPS and the aggregation process of p53C.
Materials and methods
All-atom MD simulations of p53C monomer conformations
Three systems were simulated, namely wild type (WT), M237I mutant (M237I), and R249S mutant (R249S) of p53C. The initial coordinate of the WT monomer (Fig. 1a) was extracted from the X-ray diffraction structure (PDB ID: 2AC0),55 which consists of the p53C (residues: 94–290) tetramer binding with two 12-bp DNA. In our simulations, we selected chain A as the initial structure of the p53C monomer in the WT system (Fig. 1a). The M237I and R249S mutants were generated using Pymol56 by mutating the residues at the two corresponding sites. The N- and C-termini of each p53C were capped with acetyl (–CH3CO) and amino (–NH2) groups, respectively, to maintain comparable systems with both terminal residues uncharged in full-length p53. The p53C monomer comprises three loops (L1, L2, and L3), two α-helixes (H1 and H2), one β-sandwich, and one Zn ion. In Fig. 1a, L1 (red cartoon), L2 (yellow cartoon), L3 (green cartoon), and Zn ion (red ball) in p53C are labeled. | ||
| Fig. 1 Initial structures and AA-to-CG mapping of p53C in AA and CG simulations. (a) The structure of the p53C monomer, highlighting loops L1, L2, and L3, helixes H1 and H2, and Zn ion. Residues M237 and R249 are depicted in stick representation. (b) Mapping from the AA structure to CG structure. (c) The initial state of CG simulation for p53C LLPS, which was obtained after a series of equilibrium steps, with each CG molecule colored differently. | ||
The AA simulations were conducted using the Gromacs-2018.3 software package.57 Amber14SB58 force fields were employed to characterize the p53C protein. Initially, each p53C monomer was placed at the center of a 7.0 × 7.0 × 7.0 nm3 simulation box and solvated with TIP3P59 water molecules. Each system was neutralized by adding Na+ and Cl− ions to achieve a physiological salt concentration of 0.15 M. The total number of atoms in each AA system of the p53C monomer was approximately 33690. The lengths of all chemical bonds of the p53C monomer were constrained using the LINCS algorithms,60 while the bond lengths of water molecules were constrained by SETTLE methods.61 Electrostatic interactions were treated with the Particle Mesh Ewald (PME) method, using a real space cutoff of 1.2 nm.62 The cutoff for van der Waals interactions was set at 1.2 nm with a Verlet buffer.63 All systems underwent initial energy minimization using the steepest descent method, and convergence was achieved when the maximum force in the system was smaller than 100 kJ mol−1 nm−1. Subsequently, the energy-minimized systems were equilibrated in the NVT ensemble and further equilibrated in the NPT ensemble. The simulation systems were coupled to a temperature bath of 310 K using a velocity rescale method64 with a coupling constant of 0.1 ps. The pressure of each system was maintained at 1 bar using the Parrinello–Rahman method65 with a coupling time constant of 1.0 ps. The time step of integration was set to 2 fs, and the neighbor list was updated every 10 steps. Ultimately, all AA simulations were conducted in the NPT ensemble without constraints. Periodic boundary conditions were used in all AA simulations. Three independent 2.2 μs MD simulations were performed for each system (WT, M237I, and R249S), with an accumulated simulation time of 19.8 μs.
Martini 3 coarse-grained MD simulations of p53C spontaneous phase separation
Three systems, namely WT, M237I, and R249S, were constructed using the Martini 354 CG model. The martinize2 program within Vermouth66 was employed to map the AA structures into CG structures and generate Martini 354 topologies (Fig. 1b). Unless specified, the initial structures for AA-to-CG mapping were selected from the AA simulations (the conformations at t = 2.0 μs). Secondary structure assignment was performed on the basis of the AA structure using the DSSP (Definition of Secondary Structure of Proteins)67 program. To preserve the tertiary structure of the AA p53C monomer in the CG model, the elastic network68 was utilized during AA-to-CG mapping. Within the elastic network model, elastic bonds were added for any two CG beads within a distance of 0.9 nm, using a force constant of 500 kJ mol−1 nm−2.
After constructing the CG p53C model, the simulation system with 32 p53C molecules was set up in an elongated box with a 2 × 2 × 8 configuration (i.e., there are 2, 2, and 8 p53C molecules in x, y, z directions separated by a minimum distance of 2 nm). The LLPS of several proteins have been investigated by a number of CG-MD phase coexistence simulation studies, and the protein concentration ranges from 1.7 to 10.6 mM.69–72 In accordance with those previous computational studies, we used 32 molecules to carry out the phase coexistence simulations of p53C with a protein concentration of 8.9 mM. Each system underwent initial energy minimization under vacuum conditions, followed by solvation, charge neutralization, and the addition of 0.15 M NaCl using the Gromacs-2018.357 software package. The total number of beads in each CG system was approximately 65500. Subsequently, a series of optimization steps were conducted in each system, including energy minimization under solvation conditions, 5 ns equilibrium steps in the NVT ensemble, and two equilibrium steps using 2 fs and 20 fs time steps in the NPT ensemble totaling 35 ns (Fig. 1c). In the equilibrium steps, the velocity rescale method64 and Berendsen method73 were used respectively for maintaining the system at a temperature of 310 K and a pressure of 1 bar. In the production run, the velocity rescale method64 and Parrinello–Rahman method65 were used respectively for temperature coupling and pressure coupling. Two individual 10 μs CG simulations were performed for each system (WT, M237I, and R249S), with an accumulated simulation time of 60 μs. A summary of the AA and CG simulation setup details of the six systems is provided in Table 1.
| MD simulations | Systems | # of MD runs | # of atoms | Box size (nm3) | Simulation time |
|---|---|---|---|---|---|
| All-atom | WT | 3 | ∼33690 |
7 × 7 × 7 | 2200 ns × 3 |
| M237I | 3 | ∼33690 |
7 × 7 × 7 | 2200 ns × 3 | |
| R249S | 3 | ∼33690 |
7 × 7 × 7 | 2200 ns × 3 | |
| Coarse-grained | WT | 2 | ∼65500 |
12 × 12 × 45 | 10000 ns × 2 |
| M237I | 2 | ∼65500 |
12 × 12 × 45 | 10000 ns × 2 |
|
| R249S | 2 | ∼65500 |
12 × 12 × 45 | 10000 ns × 2 |
Data analysis
The analysis of our simulation data was carried out using in-house developed codes and tools implemented in the Gromacs package.57 Statistical analyses were performed using data generated in the last 700 ns trajectory (i.e., 1500–2200 ns) of AA simulations and the last 4000 ns trajectory (i.e., 6000–10000 ns) of CG simulations. In AA simulations, several parameters, including backbone root mean square deviation (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), and contact number, were used to characterize the conformational properties of p53C. An atomic contact was considered when the distance between two carbon atoms is within 0.54 nm, and the distance between any other two atoms is within 0.46 nm. In CG simulations, a bead contact was considered when the distance between two beads is within 0.7 nm.74–77 A hydrogen bond (HB) was deemed to be formed when the distance between the HB donor (D) and acceptor (A) is ≤ 0.35 nm, and the D–H⋯A angle is ≥150°. A cation–π interaction was considered to exist when the distance between the centroid of an aromatic ring and the cation group NH3+ of Arg/Lys sidechain is ≤0.60 nm.78
To estimate the extent of hydration between p53C and water molecules, we calculated the water number around p53C residues with a cutoff of 0.35 nm. The radial distribution function (RDF) describes the density distribution of a specific atom that varies as a function of distance from a reference atom. In our study, we analyzed the density distribution of the oxygen atom of water molecule using RDF, and the atom of p53C was used as the reference atom. Unless specified, all the results presented below are the average of three independent MD runs for AA simulations and the average of two independent MD runs for CG simulations. Structure visualization was performed using the Pymol software.56
Results and discussion
M237I mutation reduces the structural stability of p53C by mainly disrupting the zinc-binding domain
We initially investigated the impact of M237I mutation on the structure of p53C. The time evolution of the backbone RMSD value of M237I mutant (∼0.3 nm) is similar to that of WT (Fig. S1a–c†). The RMSD value matrix shows that WT and M237I mutant sample multiple conformations with the largest RMSD value of 0.3 nm (Fig. S1d and e†). However, the RMSD matrix of M237I with respect to WT shows overall much larger RMSD values (with the largest RMSD value of 0.51 nm) (Fig. S1f†), which indicates the structural alterations of p53C caused by M237I mutation. We further examined the total contact number, SASA, and Rg of p53C. It is found that the contact number of M237I mutant is smaller than that of WT (Fig. 2a), and the SASA and Rg values of M237I mutant are larger than those of WT (Fig. 2b and c), which indicates the partial structural features of aggregation-prone molten globule states. These findings suggest that M237 mutation weakens the molecular interactions and disrupts the structural stability of p53C.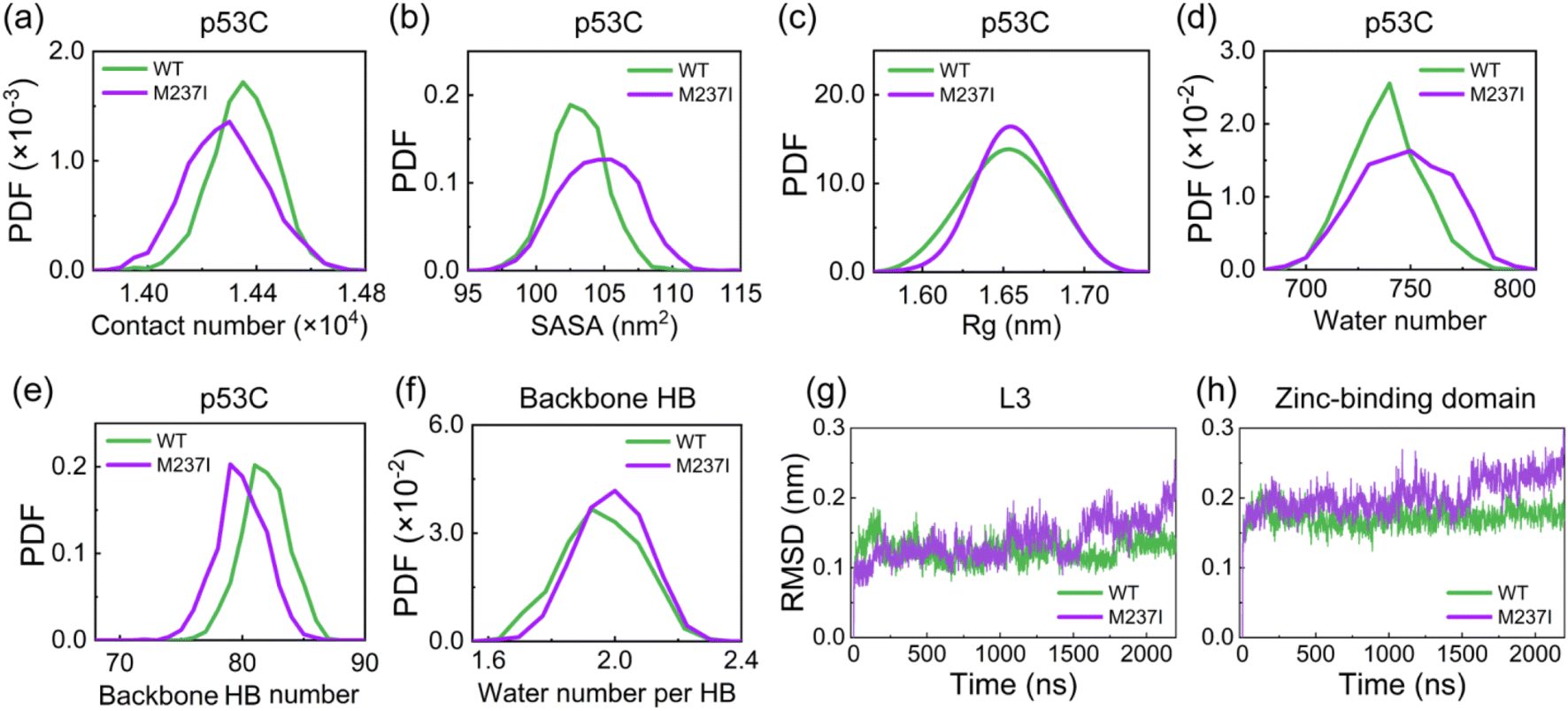 | ||
| Fig. 2 Impact of M237I mutation on the structural stability of p53C. (a–c) PDF of the (a) total contact number, (b) SASA value, and (c) Rg value of p53C. (d) PDF of the water number around p53C residues with a cutoff of 0.35 nm. (e and f) PDF of (e) backbone HB number and (f) average water number around backbone HB of p53C. (g and h) Time evolution of the backbone RMSD values of (g) L3 and (h) zinc-binding domain. These data suggest that M237I mutation reduces the structural stability of p53C, particularly disrupting the zinc-binding domain. | ||
Considering the significance of water molecules in biological macromolecular systems,79–81 we explored the interactions between p53C and water molecules. Our data indicate that M237I mutation increases the water number around p53C (Fig. 2d), manifesting enhanced water exposure of p53C induced by M237I mutation. It is reported that backbone HBs and their hydrophobic protection play a crucial role in proteins' resistance against water molecule intrusion.82,83 Therefore, we calculated the backbone HB number and the average water number around backbone HB. The probability density function (PDF) of backbone HB number in M237I mutant exhibits a peak around values of ∼83, while that of WT has a peak at reduced values (∼77) (Fig. 2e). The decrease in backbone HB number might result from the damage of hydrophobic interaction around HB. We can see that the average water number around backbone HB in M237I mutant is larger than that in WT (Fig. 2f). Consequently, the backbone of M237I mutant displays reduced resistance to water attacks, which is detrimental to the structural stability of p53C. To identify regions with the most significant structural destruction in p53C, we calculated the time evolution of RMSD for different regions of p53C, including β-sandwich, L1, L2, L3, and zinc-binding domain (i.e., the coordinated region of Zn ion, including residues C176, P177, H178, and H179 in loop L2, C238, N239, S240, S241, and C242 in loop L3). The RMSD values of L1, L2, and β-sandwich in M237I mutant are similar to those in WT (Fig. S2a–c†), while the RMSD values of L3 and zinc-binding domain continuously increase during the last 500 ns trajectory in M237I mutant, and become larger than WT (Fig. 2g and h). These results demonstrate that M237I mutation induces structural destabilization of p53C, primarily in the zinc-binding domain.
How does M237I mutation disrupt the zinc-binding domain and destabilize p53C?
To examine how M237I mutation causes structural disruptions of the zinc-binding domain and of p53C, we analyzed the differences in the L3–L3 contact number map between M237I and WT (Fig. 3a). It can be seen that most residue pairs in M237I mutant exhibit a decreased contact number, primarily involving main chain–main chain (MC–MC) contacts (Fig. S3a†), although an increase of contact number is observed for a few residue pairs, such as I237–C238 (Fig. 3a, green rectangle). This increase of I237–C238 interaction competes for the interaction between residues C238 and M246, resulting in a decrease of the contact number of C238–M246 (Fig. 3a, purple rectangle). Additionally, the N-terminal and C-terminal residues of loop L3 show reduced contact numbers, especially residue pairs S240–M246, C242–M246 (Fig. S3b†), and S241–R248 (Fig. 3a, red rectangle). These reduced pairwise contacts induce an increase in the center of mass of Cα distance between N-terminal residues (I237–M243) and C-terminal residues (G244–P250) (Fig. S3c†), leading to the structural disruption of L3. Representative snapshots of L3 in WT (Fig. 3b) and M237I (Fig. 3c) illustrate the corresponding structural changes. Furthermore, we assessed the differences of L3–L2 contact number map between M237I and WT (Fig. 3d). It should be noted that the contact number of most residue pairs in M237I mutant is decreased (Fig. 3d, red rectangle). These reduced contacts mostly correspond to HB interactions in the following residue pairs of I237–R175 (MC–SC HB) (Fig. S3d†), R249–H168 (SC–SC HB) (Fig. S3e†), and R249–E171 (SC–SC HB) (Fig. S3f†). The disrupted L3–L2 HB interactions can be seen from the representative snapshots in Fig. 3e and f. Recent studies showed that the native interactions of L3–L2 are crucial for the coordinated stability of Zn ion in p53C (Fig. 3g).84–86 We found that in our simulations, M237I mutation causes the structural disruption of the zinc-binding domain. This result is supported by the increase of SASA and Rg values of the zinc-binding domain (Fig. 3h and i) and of the contact number between the zinc-binding domain and water molecules in M237I mutant (Fig. 3j), as well as the increase of water molecules around Zn ion seen in the RDF of Zn ion and the water oxygen atom (Fig. S4†). We further analyzed the contact number of the zinc-binding domain and contact number between L2 and L3 (Fig. S5†). It is shown that the contact number starts to decrease around t = 750 ns in M237I mutant, while fluctuating around a constant value in WT p53C (Fig. S5†). These results indicate that the disruption of the zinc-binding domain occurs concurrently with the decrease of L2–L3 interactions. Taken together, our results demonstrate that M237I mutation disrupts both the zinc-binding domain and the L2–L3 interactions, and thus results in the overall structural destabilization of p53C and its adoption of aggregation-prone molten globule states, which may potentially accelerate the LLPS and aggregation ability of p53C. | ||
| Fig. 3 Effects of M237I mutation on the interactions in L2, L3, and zinc-binding domains and the resulting conformational alterations. (a) Differences in L3–L3 contact number map between M237I and WT (i.e., M237I minus WT, L3 residues: M237I–P250). (b and c) Representative snapshots of loop L3 showing reduced interactions in (c) M237I compared to (b) WT (reduced interactions: C238–M246, S240–M246, C242–M246, and S241–R248). (d) Differences in L3–L2 contact number map between M237I and WT (i.e., M237I minus WT, L2 residues: Y163–C176, L3 residues: M237I–P250). Three residue pairs display reduced contact numbers (marked by red rectangles, i.e., M237–R175, R249–H168, and R249–E171). (e and f) Representative snapshots of L3 and L2 showing disrupted interactions in (f) M237I compared to (e) WT (disrupted interactions: M237–R175, R249–H168, R249–E171). (g) A representative snapshot of the zinc-binding domain showing the Zn ion coordination in p53C. Zn ion is shown with a red ball and the coordinated residues are indicated (zinc-binding domain residues: C176, P177, H178, and H179 in L2, C238, N239, S240, S241, and C242 in L3). (h and i) PDF of (h) SASA and (i) Rg values of the zinc-binding domain. (j) PDF of the contact number between the zinc-binding domain and water molecules. These results indicate that M237I mutation induces the disruption of the zinc-binding domain, leading to the global structural destabilization of p53C. | ||
R249S mutation destabilizes p53C by primarily destructing both loop L3 and zinc-binding domain
To assess the impact of R249S mutation on the structure of p53C, we analyzed the time evolution of backbone RMSD value for both WT and R249S. In R249S mutant, the time evolution of the RMSD value (∼0.3 nm) is similar to that in WT (Fig. S6a†), whereas there is an increase in the fluctuation of RMSD value (Fig. S6b and c†), indicating structural perturbation caused by R249S mutation. The RMSD value matrix illustrates that WT and R249S sample diverse conformations with the largest RMSD value of 0.35 nm (Fig. S6d and e†). In contrast, the RMSD matrix of R249S with respect to WT shows overall much larger RMSD values (with the largest RMSD value of 0.51 nm) (Fig. S6f†), again reflecting structural disturbance of p53C by R249S mutation. To delve deeper, we examined the total contact number, SASA, and Rg of WT and R249S p53C. It is found that R249S mutant shows a decrease in total contact number (∼1.42 × 104) compared to WT (∼1.44 × 104) (Fig. 4a). Moreover, the SASA and Rg values of R249S mutant are larger than those of WT (Fig. 4b and c), and the water molecule number around p53C also becomes larger (Fig. 4d). These data indicate that R249S mutation reduces intramolecular interactions and enhances hydrophobic exposure of p53C. Further analysis indicates that the structural perturbation of p53C is attributed to the disruption of backbone HBs and of its hydrophobic protection. More specifically, in R249S mutant, the backbone HB number decreases (Fig. 4e), and the average water number around backbone HBs increases (Fig. 4f). Backbone HB is crucial for preserving protein native structure.87,88 However, R249S mutation disrupts the hydrophobic protection of backbone HB, thus reducing the structural stability of p53C. | ||
| Fig. 4 The influences of R249S mutation on p53C structure. (a–c) PDF of (a) total contact number, (b) SASA value, and (c) Rg value of p53C. (d) PDF of the water number around p53C residues with a cutoff of 0.35 nm. (e and f) PDF of (e) backbone HB number and (f) average water number around backbone HB of p53C. Time evolution of backbone RMSD value of (g) L3 and (h) zinc-binding domain. These data show that R249S mutation reduces the structural stability of p53C by predominantly disrupting loop L3 and the zinc-binding domain. | ||
To identify the destabilized regions in R249S mutant, we calculated the time evolution of RMSD values for key p53C regions (i.e., L1, L2, L3, β-sandwich, and zinc-binding domain). Loop L1 has a rather large RMSD values in both WT and R249S mutant, manifesting its significant conformational fluctuations (Fig. S7a†) and the adoption of diverse conformations, including collapsed and extended states (Fig. S7b†). Our finding is in good agreement with previous studies showing that L1 possesses both recessed and extended states.89,90 In addition, the RMSD values of L2 and β-sandwich regions remain mostly unchanged in both systems (Fig. S7c and d†). However, the RMSD values of L3 and zinc-binding domain show a notable increase in R249S mutant (Fig. 4g and h), suggesting that the structural damage of p53C caused by R249S mutation is primarily localized in L3 and zinc-binding domain.
How does R249S mutation disrupt both loop L3 and zinc-binding domain and destabilize p53C?
To explore how R249S mutation causes structural damage of loop L3 and the zinc-binding domain, we compared the L3–L3 contact number map of R249S with WT and displayed the difference in Fig. 5a. After mutation, the total contact number (including MC–MC, SC–SC, and SC–MC) for most residue pairs decreases (particularly MC–MC contacts, Fig. S8a†). Significant decrease of contacts (Fig. 5a, green rectangle) is observed between the mutation site R/S249 and residues G245, M246, and N247 (Fig. S8b and c,† SC–MC HB interaction). These diminished contacts around the mutation site disrupt the intact interactions of L3 (Fig. 5a, red rectangle). The main disrupted residue pairs in loop L3 are C242–G245/M246/N247(with a decrease of MC–MC HB number) (Fig. S8d†) and S241–R248 (with an increase of Cα–Cα distance) (Fig. S8e†). Representative snapshots are provided for WT (Fig. 5b) and R249S (Fig. 5c) to show these structural disruptions. To further examine the R249S-induced conformational changes of L3, we compared the L3–L2 contact number map between R249S and WT (Fig. 5d). Strikingly, the mostly disrupted interactions are all involved the residue at the mutation site R/S249. The corresponding interactions are SC–SC hydrogen-bonding interaction between R/S249 and E171 (Fig. S8f†), SC–SC hydrogen-bonding interaction between R/S249 and H168 (Fig. S8g†), and cation–π interaction between R/S249 and Y163 (Fig. S8h†). The disruption of those interactions is illustrated in representative snapshots for WT (Fig. 5e) and R249S (Fig. 5f). In short, R249S mutation damages the native interaction networks for loop L3, leading to its structural destabilization. As depicted in Fig. 5g, L3 unfolds and becomes more exposed to water in R249S mutant, with a gradual increase of the average Cα–Cα distance between its N-terminal (M237–M243) and C-terminal (G244–P250) residues (Fig. 5h). Consequently, the disrupted conformation of L3 leads to an increase of contacts between L3 backbone atoms and water molecules (contact number changing from 48 to 62, Fig. 5i). The RDF curve between the midpoint of the Cα–Cα distance of C242 and M246 in the central region of L3 and water molecules indicates a noticeable increase in the number of water molecules in the 0.5 nm shell around L3 (Fig. 5j). In R249S mutant, the L3 region is more invaded by water molecules, compromising structural stability and intensifying the hydrophobic exposure of loop L3. | ||
| Fig. 5 Analysis of R249S mutation-induced disruption of native interactions and enhancement of hydration on L3 and zinc-binding domain. (a) Differences in L3 contact number map between R249S and WT (i.e., R249S minus WT, L3 residues: M237I–P250). (b and c) Representative snapshots of loop L3 showing reduced interactions in (c) R249S and (b) WT. (d) Differences in L3–L2 contact number map between R249S and WT (i.e. R249S minus WT, L2 residues: Y163–C176, L3 residues: M237I–P250). (e and f) Representative snapshots of L3 and L2 showing the disrupted interactions in (f) R249S and (e) WT. (g) Representative snapshot showing the conformational difference of L3 in WT and R249S mutant (10 representative snapshots were superposed with each other). (h) Time evolution of the distance between the center of mass of Cα atoms in N-terminal (residues M237–M243) and that of Cα atoms in C-terminal (residues G244–P250) of L3. (i) PDF of the contact number between the backbone atoms of L3 residues and water molecules. (j) RDF between the midpoint of Cα–Cα distance of residues C242 and M246 in the central region of L3 and the oxygen atom of water molecules. These results demonstrate that R249S mutation induces more hydrophobic exposure and hydration of L3 and zinc-binding domain. | ||
In addition, we observed the damage of the zinc-binding domain in R249S mutant, evident in the increased hydrophobic contact number between the residues of the zinc-binding domain (residues C176, P177, H178, H179, C238, N239, S240, S241, C242) and water molecules (Fig. S9a†) and the heightened RDF peak of water molecules around Zn ion (Fig. S9b†). R249S mutation induces the SASA and Rg values of the zinc-binding domain to become larger (Fig. S9c and d†), indicating an enhanced hydrophobic exposure of the zinc-binding domain in R249S mutant. Mounting evidence suggests that the hydrophobic exposure and hydration of protein play important roles in protein LLPS and aggregation.30,91,92 Our simulations demonstrate attenuated intramolecular interactions and increased hydrophobic exposure in both loop L3 and zinc-binding domain, which may hasten the LLPS and aggregation of p53C.
M237I and R249S mutations increase intermolecular interactions and facilitate the LLPS of p53C
To explore how M237I/R249S mutation affects the phase separation behavior of p53C, we investigated the spontaneous LLPS process of p53C WT and the two mutants by conducting Martini 3 coarse-grained simulations. Throughout two individual 10 μs simulations, MD runs of the WT system concluded with a loosely organized configuration of all 32 molecules (Fig. 6a), indicating the slow LLPS process of p53C. In contrast, two dense protein clusters are observed in the M237I system at 10 μs (Fig. 6b). Notably, one dense phase and one dilute phase are observed in the R249S system at 10 μs, indicating that R249S mutant undergoes LLPS faster than both M237I and WT systems (Fig. 6c). Strong protein intermolecular interactions usually facilitate its LLPS and aggregation.93 To examine whether M237I/R249S mutation impacts intermolecular interactions of p53C, we calculated the PDF of the protein–protein contact number in the three systems. It is found that the peak value of p53C–p53C contact number is approximately 8100 in the WT system (Fig. 7a), while it increases to 8400 and 9600 in M237I and R249S mutants, respectively (Fig. 7b and c). In addition, the time evolution of the average intermolecular contact number over the two MD runs (Fig. 7d–f, results for each of the two MD trajectories are given in Fig. S10†) shows that the contact number increases rapidly within the first 2 μs, and reaches 6000 in WT p53C and 8000 in the two mutants. Afterwards, the contact number increases gradually to 7500 in WT p53C, 8000 in M237I mutant, and 8200 in R249S mutant at t = 6 μs. During the last 4 μs, the contact number in WT, M237I and R249S p53C respectively fluctuates at a constant value of 8100, 8400, and 9600. These data demonstrate that at the same simulation time point, both M237I and R249S mutants have a larger intermolecular contact number than WT p53C, implying their faster LLPS compared to WT p53C at 37 °C. Qualitatively similar results are observed at a lower temperature of 25 °C (Fig. S11†). Altogether, these results indicate that during the early stage of p53C phase separation, both M237I and R249S mutants phase separate faster than WT p53C. | ||
| Fig. 6 Dynamics of p53C phase separation in CG simulations. (a–c) Representative snapshots illustrating the LLPS process in WT (a), M237I (b), and R249S (c) systems (representative timestamps at 0, 4, 8, and 10 μs). Our data show that both M237I and R249S mutations accelerate the LLPS of p53C. | ||
 | ||
| Fig. 7 Effect of M237I/R249S mutation on the LLPS behavior of p53C. (a–c) PDF of intermolecular contact number of p53C in WT (a), M237I (b), and R249S (c) systems. (d–f) Time evolution of average intermolecular contact number over two individual trajectories for WT (d), M237I (e), and R249S (f) p53C. (g–i) Intermolecular contact number map of WT (g), M237I (h), and R249S (i) systems, with the enhanced contacts labeled by dashed rectangles. The orange and purple rectangles highlight respectively the L1 region and Turn S3/S4 region (the turn structure between β-strand 3 and β-strand 4). In M237I mutant (h), the green rectangle highlights the zinc-binding domain and part of L3 regions and the red rectangle highlights the L3 region. In R249S mutant (i), the green rectangle highlights the zinc-binding domain plus L2 and the red rectangle highlights the zinc-binding domain plus the L3 region. The regions with enhanced intermolecular interactions are consistent with the disrupted regions with weakened intramolecular interactions observed in AA simulations. Our data show that M237I and R249S mutations hasten the LLPS of p53C. | ||
To elucidate the driving force of M237I/R249S mutation-accelerated LLPS of p53C, we calculated the intermolecular contact number maps of p53C (Fig. 7g–i). The differences of intermolecular contacts between M237I/R249S and WT (Fig. S12a and b†) show that four dominant regions in the map of M237I/R249S mutant have an increased contact number compared to the WT system (Fig. 7g–i). Those regions involve L1, Turn S3/S4, zinc-binding domain, L2, and L3 (labeled by a dashed rectangle). Among them, the zinc-binding domain in M237I mutant (Fig. 7h, labeled by green and red rectangles), corresponds to the most disrupted regions (i.e., zinc-binding domain) of p53C monomer observed in M237I AA simulations (Fig. 3). Intriguingly, in R249S mutant, the regions (L3 and zinc-binding domain, Fig. 7i, labeled by green and red rectangles) with enhanced intermolecular interactions observed in CG simulations coincide with the most disrupted regions with weakened intramolecular interactions observed in AA simulations. An increasing number of experimental and computational studies reveal that the LLPS of biomolecules is driven by intermolecular electrostatic, cation–π, hydrophobic, π–π, and hydrogen bonding interactions.2,4,94,95 Our results from multiscale simulations reaffirm the importance of hydrophobic, hydrogen bonding, and cation–π interactions among the dominant regions of M237I/R249S in triggering the LLPS of p53C. Specifically, our AA simulations revealed that M237I/R249S mutation induces the disruptions of hydrophobic, hydrogen bonding, and cation–π interactions in L2, L3 regions and Zn-binding domain of p53C. We found that these mutation-induced disruptions of intramolecular interactions led to an increase of intermolecular interactions in our CG simulations. More specifically, in M237I mutant, L1, Turn S3/S4, zinc-binding domain and loop L3 (Fig. 7h) show a notable increase of intermolecular interactions involving charged residues (K101, R110, K120, D148, E221, E224, D228, and R248), hydrophobic residues (V122, V147, P152, P153, P222, P223, V225 and M243), and aromatic residues (Y103, Y107, F113, H115, W146, Y220 and H233). In R249S mutant, L1, Turn S3/S4, L2 and L3 regions display increased multivalent intermolecular interactions (Fig. 7i) that are similar to those in M237I mutant. And increased intermolecular interactions are also seen among other residues, namely charged residues K139, R175, and D184, hydrophobic residues V97, L114, M169, L188, and M237, and aromatic residues H168, H179, and H193. It is conceivable that the enhancement of these multivalent intermolecular interactions would accelerate the phase separation process of p53C. These results reveal that M237I/R249S mutation-induced local structural disruptions enhance the intermolecular interactions and in turn accelerate the LLPS of p53C. In addition, we found that R249S mutant displays a faster LLPS process than M237I mutant. The expedited LLPS of M237I/R249S mutant may facilitate its fast liquid-to-solid phase transition and fibrillization. Several studies have reported that the aggregation-prone segment 251ILTIITL257 (i.e., S9) plays a crucial role in the aggregation of p53C.45,96–98 We further analyzed the impact of M237I and R249S mutations on the solvent exposure of this aggregation-prone segment in our simulations. As shown in Fig. S13a,† in AA simulations, both M237I and R249S mutations increase the solvent exposure of segment S9, potentially increasing the aggregation propensity of the two p53C mutants. In CG simulations, the two mutations led to an increase of hydrophobic contact number between segment S9 and p53C (Fig. S13b†). Moreover, S9 exhibits a reduced hydrophobic SASA in both M237I and R249S mutants (Fig. S13c†), albeit with a different extent of reduction. This decrease is likely due to the increased intermolecular interactions between segment S9 and p53C (Fig. S13b†), which masks the hydrophobic exposure of segment S9. These results suggest that segment S9 involves the liquid-to-solid transition and fibril formation accelerated by M237/R249S mutations. Therefore, in addition to the zinc-binding domain and loop3, the aggregation-prone segment S9 also plays a role in the M237I/R249S mutation-accelerated liquid-to-solid transition and aggregation of p53C. In summary, our results provide an interpretation for the M237I/R249S mutation-accelerated LLPS, liquid-to-solid phase transition and fibril formation reported in recent experimental studies.19
Conclusions
In summary, we investigated the underlying molecular basis governing the accelerated p53C LLPS by M237I and R249S mutations through multiscale MD simulations. In AA simulations, we explored the effects of the two mutations on the structural properties and conformational dynamics of p53C. Our simulations revealed that M237I mutation disrupts the backbone hydrogen bonds of p53C, and thus induces structural destabilization of p53C, especially in the zinc-binding domain. M237I mutation also increases the flexibility of L2 and L3 regions and the hydrophobic exposure of p53C. Furthermore, the R249S mutation disrupts the intramolecular interactions and backbone hydrogen bonds of p53C, and in turn causes the structural destabilization of p53C. R249S mutation induces the hydrophobic exposure primarily in loop L3 and zinc-binding domain. These physical property alterations suggest that p53C M237I/R249S mutant tends to adopt aggregation-prone molten globule states, potentially speeding the LLPS and further liquid-to-solid transition of p53C (Fig. 8). Compared to M237I mutation, the destabilized extent of p53C caused by R249S mutation is more pronounced. In R249S mutant, the reduced intramolecular interactions and increased hydrophobic exposure are larger than those in M237I mutant. Consequently, p53C R249S mutant may undergo faster LLPS and liquid-to-solid transition than M237I mutant. Martini 3 CG simulations show that the two mutants possess stronger intermolecular interactions than WT p53C, with the R249S mutant showing greater enhancement than M237I mutant. Increased intermolecular interactions facilitate p53C LLPS, which is evidenced by the denser phase of p53C observed in both mutants compared to the WT system (Fig. 8). Importantly, we find that the regions with enhanced intermolecular interactions observed in CG simulations coincide with the disrupted regions with weakened intramolecular interactions observed in AA simulations, indicating the crucial role of the disrupted regions in accelerating the LLPS of p53C. In conclusion, our multiscale MD study bridges the gap between M237I/R249S mutation-induced conformational disruption and phase separation of p53C, and offers an in-depth understanding of the molecular mechanisms underlying M237I/R249S mutation-accelerated LLPS of p53C, which fosters the development of anticancer therapy by targeting the LLPS and aggregation process. | ||
| Fig. 8 Schematic representation of p53C LLPS and aggregation induced by M237I and R249S mutations. Upper left panel: M237I mutation induces the disruption of the zinc-binding domain, promotes the formation of aggregation-prone molten globule states in p53C. Lower left panel: R249S mutation induces the disruption of both L3 and zinc-binding domain, resulting in the overall structural destabilization of p53C. Right panel: these molecular-level structural disruptions accelerate the LLPS and subsequent fibrillization of p53C in M237I and R249S mutants. | ||
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. Y. and G. W. conceived the project. Y. Y. performed the simulations. Y. Y., Q. L., J. Z., Y. T., Y. T., and G. W. analyzed the simulation data. Y. Y. drafted the manuscript, and G. W. reviewed and edited the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported financially by the National Key Research and Development Program of China (Grant No. 2023YFF1204402), the National Natural Science Foundation of China (Grant No. 12374208 and 12074079), and the Natural Science Foundation of Shanghai (Grant No. 22ZR1406800). All simulations are performed using the GPU Cluster at Fudan University.References
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- A. S. Holehouse and R. V. Pappu, Biochemistry, 2018, 57, 2415–2423 CrossRef CAS PubMed.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed.
- A. A. Hyman, C. A. Weber and F. Jüelicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- T. Mittag and R. Parker, J. Mol. Biol., 2018, 430, 4636–4649 CrossRef CAS.
- S. Clark, J. B. Myers, A. King, R. Fiala, J. Novacek, G. Pearce, J. Heierhorst, S. L. Reichow and E. J. Barbar, Elife, 2018, 7, e36258 CrossRef.
- P. Mohanty, J. Shenoy, A. Rizuan, J. F. Mercado-Ortiz, N. L. Fawzi and J. Mittal, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305625120 CrossRef CAS.
- D. L. J. Lafontaine, J. A. Riback, R. Bascetin and C. P. Brangwynne, Nat. Rev. Mol. Cell Biol., 2021, 22, 165–182 CrossRef CAS.
- S. Alberti and D. Dormann, Annu. Rev. Genet., 2019, 53, 171–194 CrossRef CAS.
- E. Gomes and J. Shorter, J. Biol. Chem., 2019, 294, 7115–7127 CrossRef CAS.
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin and L. Van den Bosch, et al. , Trends Cell Biol., 2018, 28, 420–435 CrossRef CAS PubMed.
- X. Liu, Z. Lao, X. Li, X. Dong and G. Wei, Phys. Chem. Chem. Phys., 2022, 24, 16263–16273 RSC.
- Z. Lao, X. Dong, X. Liu, F. Li, Y. Chen, Y. Tang and G. Wei, J. Chem. Inf. Model., 2022, 62, 3227–3238 CrossRef CAS PubMed.
- I. Ghodke, M. Remisova, A. Furst, S. Kilic, B. Reina-San-Martin, A. R. Poetsch, M. Altmeyer and E. Soutoglou, Mol. Cell, 2021, 81, 2596–2610 CrossRef CAS PubMed.
- K. Kamagata, A. Hando, M. Ariefai, N. Iwaki, S. Kanbayashi, R. Koike and K. Ikeda, Sci. Rep., 2023, 13, 5648 CrossRef CAS PubMed.
- S. Usluer, E. Spreitzer, B. Bourgeois and T. Madl, Int. J. Mol. Sci., 2021, 22, 11431 CrossRef CAS.
- C. Chen, G. H. Fu, Q. Q. Guo, S. Xue and S. Z. Luo, Int. J. Biol. Macromol., 2022, 222, 207–216 CrossRef CAS.
- E. C. Petronilho, M. M. Pedrote, M. A. Marques, Y. M. Passos, M. F. Mota, B. Jakobus, G. D. de Sousa, F. P. da Costa, A. L. Felix and G. D. S. Ferretti, et al. , Chem. Sci., 2021, 12, 7334–7349 RSC.
- J. L. Silva, T. C. Vieira, Y. Cordeiro and G. A. P. de Oliveira, Curr. Opin. Struc. Biol., 2022, 73, 102346 CrossRef CAS.
- R. Wilcken, G. Z. Wang, F. M. Boeckler and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13584–13589 CrossRef CAS.
- D. Ishimaru, L. R. Andrade, L. S. P. Teixeira, P. A. Quesado, L. M. Maiolino, P. M. Lopez, Y. Cordeiro, L. T. Costa, W. M. Heckl and G. Weissmüller, et al. , Biochemistry, 2003, 42, 9022–9027 CrossRef CAS PubMed.
- D. Ishimaru, L. F. Maia, L. M. Maiolino, P. A. Quesado, P. C. M. Lopez, F. C. L. Almeida, A. P. Valente and J. L. Silva, J. Mol. Biol., 2003, 333, 443–451 CrossRef CAS.
- I. Lima, A. Navalkar, S. K. Maji, J. L. Silva, G. A. P. de Oliveira and E. A. Cino, Biochem. J., 2020, 477, 111–120 CrossRef CAS.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef.
- A. P. D. A. Bom, L. P. Rangel, D. C. F. Costa, G. A. P. de Oliveira, D. Sanches, C. A. Braga, L. M. Gava, C. H. I. Ramos, A. O. T. Cepeda and A. C. Stumbo, et al. , J. Biol. Chem., 2012, 287, 28152–28162 CrossRef.
- Y. Yang-Hartwich, M. G. Soteras, Z. P. Lin, J. Holmberg, N. Sumi, V. Craveiro, M. Liang, E. Romanoff, J. Bingham and F. Garofalo, et al. , Oncogene, 2015, 34, 3605–3616 CrossRef CAS PubMed.
- J. L. Silva, C. V. D. Gallo, D. C. F. Costa and L. P. Rangel, Trends Biochem. Sci., 2014, 39, 260–267 CrossRef CAS PubMed.
- J. L. Silva, E. A. Cino, I. N. Soares, V. F. Ferreira and G. A. P. de Oliveira, Acc. Chem. Res., 2018, 51, 181–190 CrossRef CAS PubMed.
- D. Ishimaru, A. P. D. A. Bom, L. M. T. R. Lima, P. A. Quesado, M. F. C. Oyama, C. V. D. Gallo, Y. Cordeiro and J. L. Silva, Biochemistry, 2009, 48, 6126–6135 CrossRef CAS PubMed.
- P. S. Kovachev, D. Banerjee, L. P. Rangel, J. Eriksson, M. M. Pedrote, M. M. D. C. Martins-Dinis, K. Edwards, Y. Cordeiro, J. L. Silva and S. Sanyal, J. Biol. Chem., 2017, 292, 9345–9357 CrossRef CAS PubMed.
- Q. Y. Chen, Y. P. Wu, Z. J. Dai, Z. Q. Zhang and X. R. Yang, Int. J. Biol. Macromol., 2023, 230, 123221 CrossRef CAS PubMed.
- D. P. Lane, Nature, 1992, 358, 15–16 CrossRef CAS PubMed.
- A. J. Levine and M. Oren, Nat. Rev. Cancer, 2009, 9, 749–758 CrossRef CAS.
- A. J. Levine, R. Tomasini, F. D. McKeon, T. W. Mak and G. Melino, Nat. Rev. Mol. Cell Biol., 2011, 12, 259 CrossRef CAS PubMed.
- J. Lei, R. Qi, Y. Tang, W. Wang, G. Wei, R. Nussinov and B. Ma, FASEB J., 2019, 33, 4225–4235 CrossRef CAS.
- H. L. Wang, M. Guo, H. D. Wei and Y. H. Chen, Signal Transduct. Ther., 2023, 8, 92 CrossRef CAS.
- H. F. Chen and R. Luo, J. Am. Chem. Soc., 2007, 129, 2930–2937 CrossRef CAS PubMed.
- O. Hassin and M. Oren, Nat. Rev. Drug Discov., 2023, 22, 127–144 CrossRef CAS PubMed.
- P. A. J. Muller and K. H. Vousden, Nat. Cell Biol., 2013, 15, 2–8 CrossRef CAS PubMed.
- D. W. Meek, Nat. Rev. Cancer, 2009, 9, 714–723 CrossRef CAS.
- M. S. Safari, Z. Wang, K. Tailor, A. B. Kolomeisky, J. C. Conrad and P. G. Vekilov, Iscience, 2019, 12, 342–355 CrossRef CAS.
- M. A. Marques, G. A. P. de Oliveira and J. L. Silva, Essays Biochem., 2022, 66, 1023–1033 CrossRef CAS PubMed.
- X. J. Jin, M. Zhou, S. X. Chen, D. Q. Li, X. L. Cao and B. D. Liu, Cell. Mol. Life Sci., 2022, 79, 380 CrossRef CAS PubMed.
- D. S. Yang, A. Saeedi, A. Davtyan, M. Fathi, M. B. Sherman, M. S. Safari, A. Klindziuk, M. C. Barton, N. Varadarajan and A. B. Kolomeisky, et al. , Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015618118 CrossRef CAS PubMed.
- K. Kamagata, S. Kanbayashi, M. Honda, Y. Itoh, H. Takahashi, T. Kameda, F. Nagatsugi and S. Takahashi, Sci. Rep., 2020, 10, 580 CrossRef CAS.
- Z. J. Dai, G. L. Li, Q. Y. Chen and X. R. Yang, Biochim. Biophys. Acta, Gene Regul. Mech., 2022, 1865, 194827 CrossRef CAS.
- Y. Zhang, S. Li, X. Gong and J. Chen, J. Am. Chem. Soc., 2024, 146, 342–357 CrossRef CAS PubMed.
- H. I. Ingólfsson, A. Rizuan, X. K. Liu, P. Mohanty, P. C. T. Souza, S. J. Marrink, M. T. Bowers, J. Mittal and J. Berry, Biophys. J., 2023, 122, 4370–4381 CrossRef PubMed.
- A. C. Murthy, G. L. Dignon, Y. Kan, G. H. Zerze, S. H. Parekh, J. Mittal and N. L. Fawzi, Nat. Struct. Mol. Biol., 2019, 26, 637–648 CrossRef CAS.
- V. H. Ryan, G. L. Dignon, G. H. Zerze, C. V. Chabata, R. Silva, A. E. Conicella, J. Amaya, K. A. Burke, J. Mittal and N. L. Fawzi, Mol. Cell, 2018, 69, 465–479 CrossRef CAS PubMed.
- A. E. Conicella, G. H. Zerze, J. Mittal and N. L. Fawzi, Structure, 2016, 24, 1537–1549 CrossRef CAS.
- Q. Liu, L. Li, Y. Yu and G. Wei, J. Phys. Chem. B, 2023, 127, 7708–7720 CrossRef CAS PubMed.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks and T. A. Wassenaar, et al. , Nat. Methods, 2021, 18, 382 CrossRef CAS.
- M. Kitayner, H. Rozenberg, N. Kessler, D. Rabinovich, L. Shaulov, T. E. Haran and Z. Shakked, Mol. Cell, 2006, 22, 741–753 CrossRef CAS PubMed.
- W. L. Delano. The PyMOL Molecular Graphics System Version 2.4.1, Schrodinger LLC, 2021 Search PubMed.
- M. J. Abrahama, T. Murtolad, R. Schulz, S. Pall, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. v. Gunsteren and J. Hermans, Intermol. Forces, 1981, 331–342 CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- D. Y. Tom Darden and L. Pedersen, J. Chem. Phys., 1993, 132, 154104 Search PubMed.
- S. Páll and B. Hess, Comput. phys. commun., 2013, 184, 2641–2650 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Chem. Phys., 1981, 123, 234505 Search PubMed.
- P. C. Kroon, F. Grünewald, J. Barnoud, M. van Tilburg, P. C. Souza, T. A. Wassenaar and S.-J. Marrink, arXiv, 2023, preprint, arXiv:2212.01191 [q-bio.QM], DOI:10.48550/arXiv.2212.01191.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- X. Periole, M. Cavalli, S. J. Marrink and M. A. Ceruso, J. Chem. Theory Comput., 2009, 5, 2531–2543 CrossRef CAS.
- B. S. Schuster, G. L. Dignon, W. S. Tang, F. M. Kelley, A. K. Ranganath, C. N. Jahnke, A. G. Simpkins, R. M. Regy, D. A. Hammer and M. C. Good, et al. , Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11421–11431 CrossRef CAS.
- R. M. Regy, G. L. Dignon, W. Zheng, Y. C. Kim and J. Mittal, Nucleic Acids Res., 2020, 48, 12593–12603 CrossRef CAS PubMed.
- U. Kapoor, Y. C. Kim and J. Mittal, J. Chem. Theory Comput., 2023, 20, 1717–1731 CrossRef.
- H. I. Ingolfsson, A. Rizuan, X. Liu, P. Mohanty, P. C. T. Souza, S. J. Marrink, M. T. Bowers, J. Mittal and J. Berry, Biophys. J., 2023, 122, 4370–4381 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Vangunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- X. W. Dong, S. Bera, Q. Qiao, Y. M. Tang, Z. H. Lao, Y. Luo, E. Gazit and G. H. Wei, J. Phys. Chem. Lett., 2021, 12, 2576–2586 CrossRef CAS PubMed.
- C. D. Zhan, Z. H. Lao, Y. M. Tang, Q. Qiao and G. H. Wei, Chem. Commun., 2021, 57, 4267–4270 RSC.
- X. H. Li, X. W. Dong, G. H. Wei, M. Margittai, R. Nussinov and B. Y. Ma, Chem. Commun., 2018, 54, 5700–5703 RSC.
- R. X. Qi, Y. Luo, G. H. Wei, R. Nussinov and B. Y. Ma, J. Phys. Chem. Lett., 2015, 6, 3276–3282 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- A. De Simone, G. G. Dodson, C. S. Verma, A. Zagari and F. Fraternali, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7535–7540 CrossRef CAS PubMed.
- S. Fischer and C. S. Verma, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9613–9615 CrossRef CAS.
- D. Hamelberg and J. A. McCammon, J. Am. Chem. Soc., 2004, 126, 7683–7689 CrossRef CAS PubMed.
- E. A. Cino, I. N. Soares, M. M. Pedrote, G. A. P. de Oliveira and J. L. Silva, Sci. Rep., 2016, 6, 32535 CrossRef CAS PubMed.
- A. Fernandez and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 113–118 CrossRef CAS PubMed.
- M. M. Pedrote, M. F. Motta, G. D. S. Ferretti, D. R. Norberto, T. C. L. S. Spohr, F. R. S. Lima, E. Gratton, J. L. Silva and G. A. P. de Oliveira, Iscience, 2020, 23, 100820 CrossRef CAS.
- J. X. Duan and L. Nilsson, Biochemistry, 2006, 45, 7483–7492 CrossRef CAS PubMed.
- J. S. Butler and S. N. Loh, Biochemistry, 2003, 42, 2396–2403 CrossRef CAS PubMed.
- A. Fernandez, T. R. Sosnick and A. Colubri, J. Mol. Biol., 2002, 321, 659–675 CrossRef CAS.
- A. Fernandez and R. Scott, Biophys. J., 2003, 85, 1914–1928 CrossRef CAS.
- R. Vainer, S. Cohen, A. Shahar, R. Zarivach and E. Arbely, J. Mol. Biol., 2016, 428, 3013–3025 CrossRef CAS PubMed.
- T. J. Petty, S. Emamzadah, L. Costantino, I. Petkova, E. S. Stavridi, J. G. Saven, E. Vauthey and T. D. Halazonetis, EMBO J., 2011, 30, 2167–2176 CrossRef CAS PubMed.
- J. L. Silva, T. C. R. G. Vieira, M. P. B. Gomes, P. A. Bom, L. M. T. R. Lima, M. S. Freitas, D. Ishimaru, Y. Cordeiro and D. Foguel, Acc. Chem. Res., 2010, 43, 271–279 CrossRef CAS.
- L. M. Lima and J. L. Silva, J. Biol. Chem., 2004, 279, 47968–47974 CrossRef CAS.
- M. Poudyal, K. Patel, L. Gadhe, A. S. Sawner, P. Kadu, D. Datta, S. Mukherjee, S. Ray, A. Navalkar and S. Maiti, et al. , Nat. Commun., 2023, 14 CAS.
- G. L. Dignon, R. B. Best and J. Mittal, Annu. Rev. Phys. Chem., 2020, 71, 53–75 CrossRef CAS PubMed.
- Y. Hong, S. Najafi, T. Casey, J. E. Shea, S. I. Han and D. S. Hwang, Nat. Commun., 2022, 13, 7326 CrossRef PubMed.
- J. Xu, J. Reumers, J. R. Couceiro, F. De Smet, R. Gallardo, S. Rudyak, A. Cornelis, J. Rozenski, A. Zwolinska and J. C. Marine, et al. , Nat. Chem. Biol., 2011, 7, 285–295 CrossRef CAS PubMed.
- S. Ghosh, D. Ghosh, S. Ranganathan, A. Anoop, P. S. Kumar, N. N. Jha, R. Padinhateeri and S. K. Maji, Biochemistry, 2014, 53, 5995–6010 CrossRef CAS PubMed.
- G. Z. Wang and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2634–E2643 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03645j |
| This journal is © The Royal Society of Chemistry 2024 |