
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn alternating copolymer of phenothiazine and ethylenedioxythiophene for perovskite solar cells: effects of flexible and rigid substituent alternation†
Bing
Zhang‡
 ,
Yaohang
Cai‡
,
Lifei
He‡
,
Niansheng
Xu
*,
Yi
Yuan
,
Jing
Zhang
,
Yuyan
Zhang
* and
Peng
Wang
*
,
Yaohang
Cai‡
,
Lifei
He‡
,
Niansheng
Xu
*,
Yi
Yuan
,
Jing
Zhang
,
Yuyan
Zhang
* and
Peng
Wang
*
State Key Laboratory of Silicon and Advanced Semiconductor Materials, Department of Chemistry, Zhejiang University, Hangzhou 310058, China. E-mail: nshxu@zju.du.cn; yuyanzhang@zju.edu.cn; pw2015@zju.edu.cn
First published on 24th September 2024
Abstract
Developing p-type polymeric semiconductors with exceptional electrical performance, heat tolerance, and cost-effectiveness is pivotal for advancing the practical application of n-i-p perovskite solar cells. Here, we employed direct arylation polycondensation to synthesize an alternating copolymer of phenothiazine and 3,4-ethylenedioxythiophene, featuring a high glass transition temperature (175 °C). In addition to the alternation of conjugated units within the main chain, the copolymer features alternating flexible (2-octyldodecyl) and rigid (trimethylphenyl) substituents at the nitrogen positions of the phenothiazine moiety. Compared to reference polymers with solely flexible or rigid substituents, the alternating use of these moieties resulted in the polymeric semiconductor composite film with smoother morphology and enhanced hole mobility. By employing this polymer with a distinct distribution of substituents and an innovative main chain structure as a hole transport material, we fabricated perovskite solar cells achieving an average efficiency of 25.1%. These cells also exhibited excellent stabilities under conditions of 85 °C thermal storage and 45 °C operation.
1. Introduction
For high-efficiency n-i-p perovskite solar cells (PSCs) (Fig. 1A), the electron transport layer is typically fabricated from a low-cost tin dioxide colloidal solution, while the hole transport layer (HTL) commonly utilizes the molecular semiconductor spiro-OMeTAD (Fig. 1B).1–3 spiro-OMeTAD possesses a relatively shallow highest occupied molecular orbital (HOMO) energy level, facilitating facile doping via fluoroalkyl sulfonyl imide salt-assisted air oxidation to achieve the moderate conductivity necessary for efficient devices. Maintaining the optimal hole density in the device, typically around 1018 cm−3, is crucial to minimize rapid charge recombination at the perovskite/HTL interface.4 However, the HTL composed of spiro-OMeTAD and fluoroalkyl sulfonyl imide salt typically exhibits a glass transition temperature below 85 °C. Extended exposure to high temperatures can degrade the HTL morphology, significantly reducing device efficiency.5–7 Additionally, the conduction band offset between tin dioxide and formamidinium lead triiodide (FAPbI3)-based perovskite is only a few tens of milli-electron volts. This small offset means the interfacial charge separation rate is not significantly faster than the radiative and non-radiative recombination rates in FAPbI3.8 Therefore, selecting an HTL with a sufficiently shallow HOMO energy level is crucial for achieving efficient long-range charge separation in the device. Moreover, the p-type organic semiconductor used in n-i-p PSCs must exhibit excellent film-forming property and sufficient hole conductivity, maintain its morphology under prolonged thermal stress, and effectively prevent the ingress of external species because lead halide perovskites are sensitive to moisture and tend to undergo slow decomposition via outgassing reactions when heated, and ions probably migrate under high electric fields.9 To date, many polymeric semiconductors have been employed in HTLs of n-i-p PSCs, but materials that combine high performance and low cost remain scarce.10–27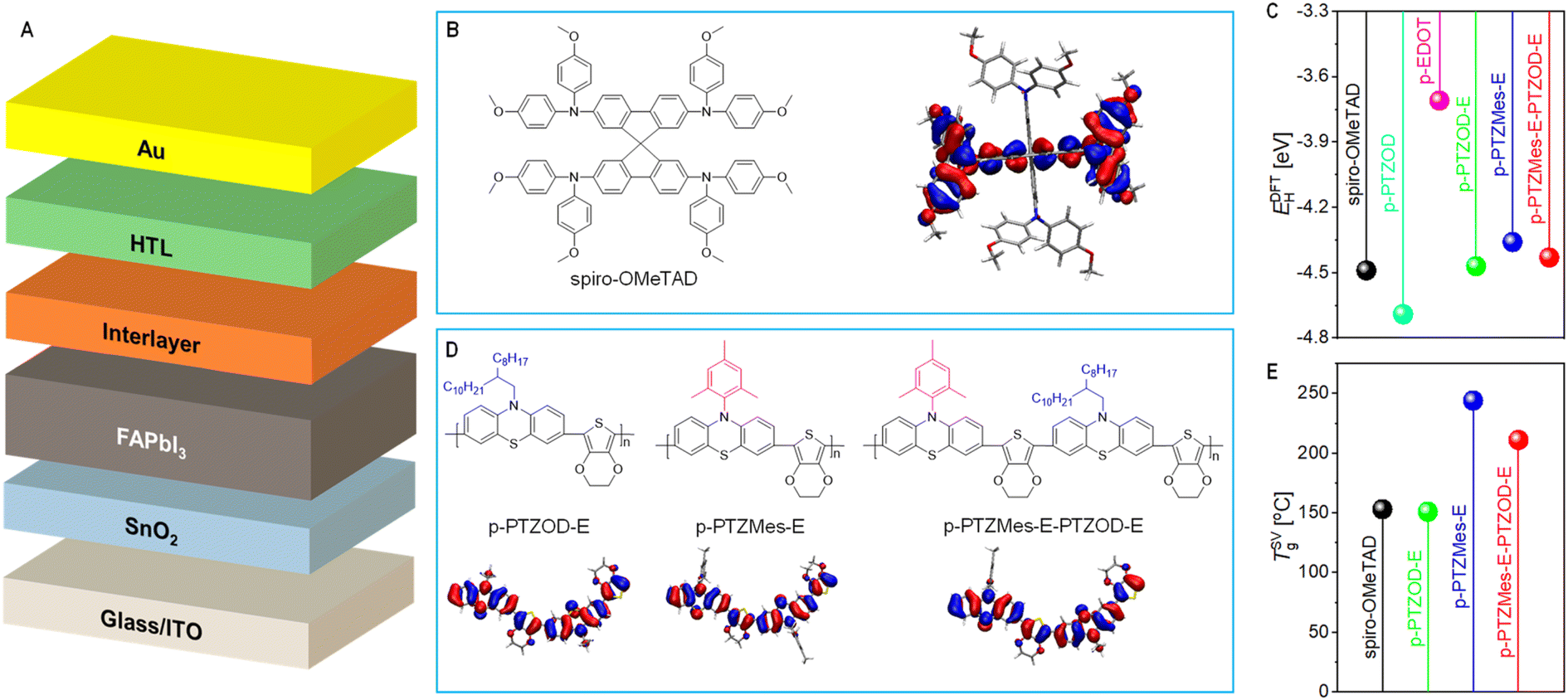 | ||
| Fig. 1 (A) Schematic representation of a multilayer n-i-p perovskite solar cell. ITO denotes indium tin oxide, SnO2 serves as the electron transport layer, FAPbI3 is the light-absorbing layer, HTL stands for the hole transport layer, and Au represents the gold electrode. (B) Chemical structure of spiro-OMeTAD and contour plot of its highest occupied molecular orbital (HOMO). Isovalue: 0.02. (C) HOMO energy levels (EDFTH) obtained from density functional theory calculations. (D) Chemical structures of three phenothiazine–ethylenedioxythiophene alternating copolymers and contour plots of their HOMOs. Isovalue: 0.02. To improve computational efficiency, 2-octyldodecyl is replaced with ethyl. (E) Glass transition temperatures (TSVg) derived from molecular dynamics simulations. | ||
Phenothiazine (PTZ) and ethylenedioxythiophene (EDOT) are commercially available and inexpensive fine chemicals. In single crystals, PTZ molecules adopt a non-planar butterfly conformation with a dihedral angle of 158.5° between the two benzene rings.28 The dioxane fragment in EDOT dimers assumes a distorted chair conformation, with the sp3 carbon atom and the thiophene ring being non-coplanar.29 In both molecules, the lone pair electrons on nitrogen, sulfur, and oxygen atoms can engage in multi-electron p–π conjugation with π-orbital electrons. The nitrogen atom in PTZ is prone to alkylation reaction, while the para position to nitrogen on the benzene ring is susceptible to electrophilic substitution reaction. Meanwhile, the C–H bonds in the thiophene ring of EDOT can undergo highly selective activation via palladium catalysis.30–39
In our previous study, it was found that the alternating copolymer constructed from PTZ with EDOT and aza[5]helicene showed a too deep HOMO energy level, leading to a very low efficiency of 18.4% in n-i-p PSCs.40 Density functional theory calculations (Fig. 1C) reveal that the HOMO energy level (EDFTH) of the homopolymer of 2-octyldodecyl-substituted PTZ (p-PTZOD, structure shown in Fig. S1†) is −4.69 eV, while that of the homopolymer of EDOT (p-EDOT, Fig. S1†) is −3.71 eV. The alternating copolymer of 2-octyldodecyl-substituted PTZ and EDOT (p-PTZOD-E, Fig. 1D) exhibits an EDFTH of −4.47 eV, notably closest to that of spiro-OMeTAD (−4.49 eV) among these polymers. Molecular dynamics simulations (Fig. 1E) demonstrate that the theoretical glass transition temperature (TSVg) of p-PTZOD-E is just comparable to that of spiro-OMeTAD. Encouragingly, when 2-octyldodecyl is replaced by trimethylphenyl, the corresponding polymer (p-PTZMes-E, Fig. 1D) exhibits a TSVg as high as 244 °C. This change in substituents also significantly alters the HOMO energy level, with p-PTZMes-E having an EDFTH of −4.36 eV, which is 110 meV higher than that of p-PTZOD-E (Fig. 1C). As anticipated, when every other 2-octyldodecyl group in p-PTZOD-E is replaced with a trimethylphenyl group, the resulting polymer p-PTZMes-E-PTZOD-E (Fig. 1D) shows intermediate EDFTH and TSVg values between those of p-PTZOD-E and p-PTZMes-E (Fig. 1C and E). Frontier orbital analyses reveal that the HOMOs of these three alternating copolymers of PTZ and EDOT are relatively evenly distributed along the entire conjugated main chain (Fig. 1D). In contrast, the HOMO of spiro-OMeTAD localizes on only half of the molecular backbone due to the presence of two independent conjugated systems within the spirobifluorene structure (Fig. 1B); if HOMO can delocalize on the entire conjugated skeleton, it may increase the transfer integral of intermolecular hole hopping and reduce the reorganization energy, resulting in a higher hole mobility.
In this study, we synthesized the three target alternating copolymers of PTZ and EDOT via palladium-catalyzed direct arylation polycondensation. Experimental results underscored the significant influence of substituents on the molecular weight of resulting polymers under identical preparation conditions. Furthermore, the HOMO energy level of the organic semiconductor closely correlates with the rate constant for hole extraction from the excited state of FAPbI3 and the hole density generated by air oxidation doping. Notably, both the HOMO energy level and the film morphology of the organic semiconductor significantly affect the efficiency of PSCs. Moreover, different HTLs exhibited marked differences in the 85 °C thermal storage stability of the devices. By utilizing the polymer p-PTZMes-E-PTZOD-E, featuring alternating flexible and rigid substituents, we fabricated PSCs achieving an average efficiency of 25.1%. This performance exceeds that of cells prepared using p-PTZOD-E (19.4%), p-PTZMes-E (23.0%), or spiro-OMeTAD (24.1%) under the same conditions. Importantly, cells based on p-PTZMes-E-PTZOD-E demonstrated excellent stabilities under thermal storage at 85 °C and operation at 45 °C.
2. Results and discussion
2.1. Synthesis and glass transition of alternating copolymers
For the synthesis, purification, and characterization of three alternating copolymers based on PTZ and EDOT, see Section 1.3 of the ESI.† Briefly, 3,7-dibromo-10-mesityl-10H-phenothiazine41 and 3,7-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-10-(2-octyldodecyl)-10H-phenothiazine40 were polycondensed in toluene using the catalytic system developed by Wakioka et al.42 to afford the copolymer p-PTZMes-E-PTZOD-E, featuring alternating flexible and rigid substituents. Under identical conditions, polycondensation of 3,7-dibromo-10-(2-octyldodecyl)-10H-phenothiazine43 (or 3,7-dibromo-10-mesityl-10H-phenothiazine41) with EDOT yielded p-PTZOD-E (or p-PTZMes-E), with only flexible (or rigid) substituent.High-temperature gel permeation chromatography indicated number-average molecular weights of 141 kDa (p-PTZOD-E), 4.9 kDa (p-PTZMes-E), and 152 kDa (p-PTZMes-E-PTZOD-E), relative to polystyrene standards, with polydispersity indices of 2.1, 1.9, and 1.8, respectively. Due to strong interchain interaction, poorly soluble p-PTZMes-E precipitated at a lower degree of polymerization, limiting molecular weight. Intriguingly, p-PTZMes-E-PTZOD-E, with alternating substituents, exhibited a molecular weight and a polydispersity index comparable to p-PTZOD-E.
Glass transition temperature serves as a key metric for evaluating the mechanical properties and operational temperature range of amorphous organic materials. When a material undergoes glass transition, its physical properties, particularly its tensile strength, change significantly. Differential scanning calorimetry revealed glass transition temperature (TDSCg) values of 119 °C (p-PTZOD-E), 200 °C (p-PTZMes-E), and 175 °C (p-PTZMes-E-PTZOD-E) (Fig. S2†). It is well-known that the thermal expansion coefficient of polymers exhibits a smooth step-like change during heating or cooling through the glass transition temperature. Therefore, we performed molecular dynamics simulations of a polymer with each chain having a molecular weight of approximately 10 kDa to calculate specific volumes at various temperatures. Linear fitting of specific volume data in both low- and high-temperature regions allowed us to determine the theoretical glass transition temperature (TSVg), resulting in values of 151 °C, 244 °C, and 211 °C for p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E, respectively (Fig. S3A–C†). It is important to note that due to the rapid cooling rate inherent in molecular dynamics simulations, these TSVg values tend to be higher than the TDSCg values.44 Nevertheless, a robust linear correlation exists between TSVg and TDSCg (Fig. S3D†). Our findings highlight that while incorporating the rigid trimethylphenyl substituent can elevate the glass transition temperature compared to the bulky, flexible 2-octyldodecyl substituent, achieving high molecular weight remains challenging due to solubility constraint. Alternating between these two substituents compromises the glass transition temperature but effectively modulates the molecular weight.
2.2. Energy levels and morphological characteristics of copolymer films
In n-i-p PSCs, the HOMO energy level of a p-type organic semiconductor critically influences two key processes: hole extraction from the photoexcited perovskite and doping via air oxidation. Initially, a solution of polymeric semiconductor was drop-cast onto glassy carbon disk electrodes and allowed to air-dry. Cyclic voltammetry measurements in acetonitrile electrolyte (0.1 M 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide) revealed the electrochemical HOMO energy levels (EECH) of −5.24 eV, −5.12 eV, and −5.20 eV for p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E, respectively (Fig. S4A†).Subsequently, an aqueous solution of PEDOT:PSS and a chlorobenzene solution of organic semiconductor were sequentially spin-coated onto the ITO substrate. Atomic force microscopy imaging demonstrated that spiro-OMeTAD, p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E formed highly smooth amorphous films, with root-mean-square roughness values of 0.35 nm, 0.53 nm, 0.60 nm, and 0.42 nm, respectively (Fig. S5†). Ultraviolet photoelectron spectroscopy measurements of the same samples yielded the HOMO energy levels (EUPSH) of −5.08 eV, −5.00 eV, and −5.05 eV for p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E, respectively (Fig. S6A–C†). Despite discrepancies in absolute HOMO values obtained from density functional theory calculation, cyclic voltammetry, and ultraviolet photoelectron spectrometry, EECH and EUPSH showed strong linear correlations with EDFTH (Fig. S4B and S6D†).
Next, polymeric semiconductors spin-coated onto quartz substrates were analyzed via UV-vis absorption spectrometry to determine their optical bandgaps (Eoptg, Fig. S7†), resulting in values of 2.59 eV, 2.42 eV, and 2.49 eV for p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E, respectively. Combining EUPSH and Eoptg, we determined the LUMO energy levels (EL) of these polymer films using the relation EL = EUPSH + Eoptg. Integrating literature values for the work functions of ITO and gold electrodes, as well as the band edges of tin oxide and FAPbI3,27 we constructed an energy diagram for n-i-p PSCs, as depicted in Fig. 2A.
 | ||
| Fig. 2 (A) Energy diagram of various materials in perovskite solar cells. Numbers below and above the colored bars denote the energy levels of the valence band edge (HOMO) and conduction band edge (LUMO), respectively. Work functions of ITO and Au are also indicated. (B) Schematic depiction of photo-induced electronic processes in an FAPbI3 film covered with a hole transport layer. (C) Semi-logarithmic plot showing the hole extraction rate constant (khe) as a function of driving force (ΔG, the energy difference between the valence band edge of FAPbI3 and the HOMO of the organic semiconductor). The grey solid line represents a nonlinear fit based on Marcus electron transfer theory. (D) Semi-logarithmic plot of hole density (p) versus the HOMO energy level (EUPSH) obtained from ultraviolet photoelectron spectroscopy. The grey solid line indicates a linear fit. | ||
2.3. Hole extraction by copolymers from photoexcited perovskite
As shown in Fig. 2B, upon absorbing photons with energy higher than Eoptg, electrons in the FAPbI3 film are excited from the valence band to the conduction band, simultaneously generating holes in the valence band. Following ultrafast thermal relaxation, these charge carriers undergo either radiative or non-radiative recombination. Concurrently, holes that diffuse to the surface of the FAPbI3 film can be extracted by the HTL. The energy difference (ΔGhe) between the valence band edge of the perovskite and the HOMO of the HTL influences the rate constant (khe) for hole extraction. Hole extraction competes with radiative and non-radiative recombination, thereby determining the hole extraction yield (ϕhe). We evaluated these parameters through time-resolved photoluminescence measurements. For details on sample preparation, measurement, and data analysis, please refer to the ESI.†Upon excitation with a 670 nm pulsed laser, the glass-supported FAPbI3 film with a polystyrene coating exhibits a slow photoluminescence decay at 810 nm, with an amplitude-weighted average lifetime (τ) of 7.3 μs (Fig. S8A and Table S1†). The reciprocal of τ represents the sum of the radiative and non-radiative rate constants. When polystyrene is replaced by one of the aforementioned organic semiconductors, τ significantly decreases. Specifically, the τ values for FAPbI3 films coated with spiro-OMeTAD, p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E are 28 ns, 27 ns, 20 ns, and 24 ns, respectively (Fig. S8B–E and Table S1†). As shown in Fig. 2C, there is a clear correlation between khe and ΔGhe that can be described by Marcus electron transfer theory; the higher the HOMO energy level of the organic semiconductor, the larger the khe. For these four organic semiconductors, the ϕhe values are close to 100% (Table S1†). By comparing the spiro-OMeTAD-based samples studied in our previous work40 and this work, it can be noted that the change of perovskite passivator from 2-phenylethylamine hydroiodide to triphenylmethane-4,4′,4′′-triisocyanate can also result in the difference in khe.
2.4. Hole density, conductivity, mobility, and activation energy of hole conduction in copolymer-based films
For n-i-p type PSCs, the conductivity (σ) of the HTL significantly impacts the cell's internal resistance and efficiency. σ is determined by hole density (p) and hole mobility (μp). Initially, we fabricated metal–insulator–semiconductor (MIS) devices and measured their impedance spectra under varying biases to derive the capacitance of the organic semiconductor layer. Using the Mott–Schottky relationship, we calculated the p values in “pristine” (as-prepared and purified; nominally undoped) organic semiconductors: spiro-OMeTAD (1.3 × 1017 cm−3) < p-PTZOD-E (1.4 × 1017 cm−3) < p-PTZMes-E-PTZOD-E (1.7 × 1017 cm−3) < p-PTZMes-E (2.4 × 1017 cm−3), as shown in Fig. 3A. For “intrinsic” (ideally pure) organic semiconductors, carriers are generated through thermally activated disproportionation reactions; for materials with an Eoptg greater than 2.0 eV, the carrier density is below 107 cm−3.45 Clearly, holes in these “pristine” organic semiconductors must be generated through other mechanisms. The most likely mechanism is oxidative doping by atmospheric oxygen, where oxygen acts as a dopant, marginally oxidizing these high HOMO energy level organic semiconductors.46–48 A small number of neutral organic semiconductor molecules are converted into their corresponding cation radicals, balanced electrostatically by superoxide anions generated from oxygen reduction. Interestingly, the logarithm of p shows a strong linear correlation with the EUPSH of organic semiconductor (Fig. 2D). | ||
| Fig. 3 (A) Relationship between hole density (p) and weight percentage (wt%) of the air oxidation doping promoter TBPH-HFSI. The solid line represents a linear fit. (B) Relationship between electrical conductivity (σ) and TBPH-HFSI wt%. The solid line indicates a power-law fit. (C) Logarithmic plot of hole mobility (μp) versus p. Solid lines serve as guides to the eye. (D) Relationship between activation energy for hole conduction (Ea) and p. The solid line represents a single-exponential decay fit. (E and F) Schematic illustration of hole transport in p-type organic semiconductors via thermal activation hopping. Blue spheres represent holes trapped in deep traps, while red spheres depict holes transported via multiple trapping (cyan) and de-trapping (magenta) processes. | ||
Clearly, the higher the HOMO energy level of an organic semiconductor, the larger the Gibbs free energy (ΔGp) of the air oxidation doping reaction, the higher the equilibrium constant (Kp), and the higher the resulting p; the relationship between ΔGp and Kp is described by ΔGp = −RTln(Kp), where R is the gas constant and T is the temperature in Kelvin. Subsequently, we measured the σ of “pristine” organic semiconductor films using interdigital gold electrodes: p-PTZOD-E (0.01 μS cm−1) < spiro-OMeTAD (0.12 μS cm−1) < p-PTZMes-E-PTZOD-E (0.20 μS cm−1) < p-PTZMes-E (0.23 μS cm−1). Such low σ values fail to meet the PSC requirement for HTLs with low transport resistance.
To enhance the σ, we blended the fluoroalkyl sulfonyl imide salt 4-(tert-butyl)pyridinium 1,1,2,2,3,3-hexafluoropropane-1,3-disulfonimide (TBPH-HFSI) with the organic semiconductors.49 This approach increases the equilibrium constant of the air oxidative doping reaction through the coupling of ion exchange and redox reactions. As shown in Fig. 3B, σ exhibits a power-law increase with the weight percentage (wt%) of TBPH-HFSI. When the wt% of TBPH-HFSI reaches 15%, the σ of the organic semiconductor composite films are: p-PTZOD-E (4.5 μS cm−1) < spiro-OMeTAD (44.4 μS cm−1) < p-PTZMes-E-PTZOD-E (170.7 μS cm−1) < p-PTZMes-E (236.0 μS cm−1). It should be noted that accurately measuring the capacitance of TBPH-HFSI/organic semiconductor composite films using MIS devices is challenging, primarily because the contributions of the silica and polybenzocyclobutene insulating layers to the total capacitance become significant.50 Therefore, we measured the electron paramagnetic resonance (EPR) spectra of the organic semiconductor films (Fig. S9A–D†). As shown in Fig. S9E of the ESI,† the quadratic integral of the EPR signal increases linearly with TBPH-HFSI wt%. By comparing the quadratic integrals of the EPR signals of films with and without TBPH-HFSI, and considering the p values of the “pristine” organic semiconductor films, we estimated the p values of the composite films.8 Following the principles of chemical equilibrium, the p values of the organic semiconductor composite films indeed increase linearly with TBPH-HFSI wt%; the higher the HOMO level, the greater the increase in p (Fig. 3A), which is related to the larger equilibrium constant of the fluoroalkyl sulfonyl imide salt-assisted doping reaction.
After measuring σ and p, the μp was calculated using the equation σ = μpqp, where q is the elementary charge. As shown in Fig. 3C, μp increases gradually with rising p. At the same p level, the μp values for the three PTZ-based alternating copolymers follow the order: p-PTZOD-E < p-PTZMes-E < p-PTZMes-E-PTZOD-E. For p-PTZOD-E, the bulky 2-octyldodecyl side chain on PTZ hinders strong π–π interactions between conjugated backbones, resulting in low μp. In contrast, despite having a lower molecular weight, p-PTZMes-E exhibits higher μp, attributable to more favorable interchain hole transport. The μp of p-PTZMes-E-PTZOD-E is even higher than that of p-PTZMes-E, potentially due to its higher molecular weight favoring intrachain hole transport, as well as interchain connectivity pathways. It is conceivable that in p-PTZMes-E-PTZOD-E, despite half of the PTZ units being substituted with bulky 2-octyldodecyl side chains, strong π–π interactions between PTZMes-EDOT segments may induce a preferential perpendicular local orientation between polymer chains. These interchain π-crossing points might facilitate a mesh-like structure, providing effective pathways for interchain charge transport.51 Alternating flexible and rigid substituents demonstrated here appears to be an effective strategy for enhancing the mobility of polymeric semiconductors. We conducted GIWAXS measurements on p-PTZOD-E, p-PTZMes-E, and p-PTZMes-E-PTZOD-E thin films, all of which exhibited amorphous characteristics, complicating the evaluation of intermolecular π–π interactions. To address this, future studies could employ advanced solid-state nuclear magnetic resonance techniques, integrated with complementary experimental and computational approaches, to better establish structure–property relationships in these complex materials.52
It is well recognized that charge carriers in organic semiconductors exhibit a strong localized character, with the density of states typically following a Gaussian or exponential distribution, and carrier transport occurring via a thermally activated hopping mechanism.53 We measured the σ of the aforementioned films at different temperatures (Fig. S10†), finding that it increases with rising temperature. Using the Arrhenius equation to analyze the temperature-dependent σ, we determined the activation energy (Ea) for hole conduction. The results show that Ea decreases exponentially with increasing p (Fig. 3D).54,55 As illustrated in Fig. 3E, at low p, all holes are trapped in deep traps, resulting in high hopping activation energy and low μp. Conversely, as depicted in Fig. 3F, at high p, deep traps are filled by some holes, leaving the remaining holes in shallow traps, which have low hopping activation energy, leading to high μp.
2.5. Application in PSCs
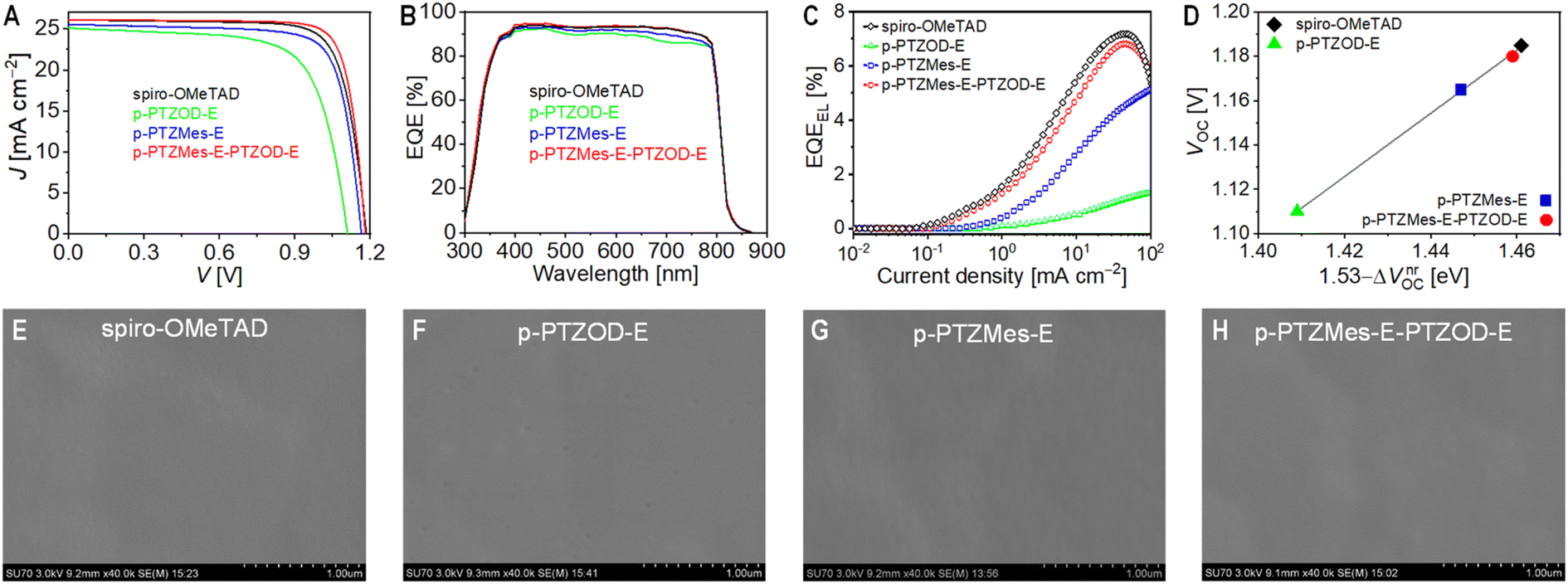 | ||
| Fig. 4 (A) Representative reverse photocurrent density–voltage (J–V) curves of perovskite solar cells employing different hole transport layers under simulated AM1.5G irradiation at 100 mW cm−2. (B) External quantum efficiency (EQE) spectrum. (C) Electroluminescence external quantum efficiency (EQEEL) plotted as a function of current density. (D) Relationship between open-circuit voltage (VOC) and non-radiative recombination VOC loss (ΔVnrOC). The optical bandgap of the perovskite absorber layer is 1.53 eV. The grey solid line indicates a linear fit. (E–H) Scanning electron microscopy images of hole transport layers in perovskite solar cells: (E) spiro-OMeTAD; (F) p-PTZOD-E; (G) p-PTZMes-E; (H) p-PTZMes-E-PTZOD-E. Scale bar: 1 μm. Images were taken after removal of encapsulation materials and the gold electrode. | ||
| Cell | J SC [mA cm−2] | V OC [V] | FF [%] | PCE [%] | J EQESC [mA cm−2] | n | I s [10−18 A] | R s [Ω] | R sh [kΩ] |
|---|---|---|---|---|---|---|---|---|---|
| a J SC: short-circuit photocurrent density; VOC: open-circuit voltage; FF: fill factor; PCE: power conversion efficiency; JEQESC: the integral of the product of the EQE and the photo flux of the AM 1.5G emission (ASTM G173-03) over the wavelength; n: ideality factor; Is: reverse saturation current; Rs: series resistance; Rsh: shunt resistance. The aperture area of metal mask: 0.07 cm2. | |||||||||
| Spiro-OMeTAD | 26.09 | 1.185 | 78.0 | 24.1 | 25.67 | 1.35 | 3.0 | 31.7 | 26.2 |
| p-PTZOD-E | 25.10 | 1.110 | 69.8 | 19.4 | 24.64 | 1.25 | 3.0 | 65.9 | 5.3 |
| p-PTZMes-E | 25.54 | 1.165 | 77.3 | 23.0 | 25.07 | 1.33 | 3.0 | 15.0 | 10.2 |
| p-PTZMes-E-PTZOD-E | 26.09 | 1.180 | 81.6 | 25.1 | 25.65 | 1.35 | 3.0 | 19.4 | 26.2 |
Subsequently, we measured the external quantum efficiencies (EQEs) of the aforementioned cells under monochromatic light irradiation (Fig. 4B). Combining EQE spectra with the standard AM1.5G solar spectrum (ASTM G173-03) allowed us to predict the short-circuit photocurrent density (JEQESC) under AM1.5G conditions. The results revealed a linear correlation between JEQESC and JSC (Fig. S14†). Furthermore, the JEQESC closely matched JSC, indicating minimal mismatch in the solar simulator used for J–V measurements. For the cells with p-PTZMes-E-PTZOD-E and spiro-OMeTAD, EQEs exceeded 90% in the 400–750 nm wavelength range, peaking near 95%. In contrast, cells based on p-PTZOD-E and p-PTZMes-E exhibited decreased EQEs within this range. The decline in EQE intensified with longer wavelengths in the 550–700 nm range, more pronounced in the p-PTZOD-E cell compared to p-PTZMes-E. This EQE deterioration correlates with accelerated recombination of electrons from the perovskite film with holes in the HTLs. The closer proximity of electrons in the perovskite film to the p-PTZMes-E and p-PTZOD-E based HTLs enhances recombination probability, as evidenced by the amplified EQE decay with increasing wavelength. This rapid charge recombination contributes to the observed lower VOC in the J–V measurements.
Next, we measured the electroluminescence external quantum efficiencies (EQEEL) of the aforementioned cells. At an injected current density of 26 mA cm−2, the EQEEL values for the four cells were as follows: spiro-OMeTAD (6.8%) > p-PTZMes-E-PTZOD-E (6.4%) > p-PTZMes-E (3.9%) > p-PTZOD-E (0.9%) (Fig. 4C). The non-radiative open-circuit photovoltage loss (ΔVnrOC) can be calculated using the formula  where kB is the Boltzmann constant, T is the temperature in Kelvin, and q is the elementary charge.56 The ΔVnrOC values for the four cells were: spiro-OMeTAD (69 mV) < p-PTZMes-E-PTZOD-E (71 mV) < p-PTZMes-E (83 mV) < p-PTZOD-E (121 mV). By differentiating the EQE spectrum of the p-PTZMes-E-PTZOD-E cell (Fig. S15†), we estimated the optical bandgap of the FAPbI3-based light absorption layer to be 1.53 eV. Moreover, VOC shows a strong linear correlation with 1.53 − ΔVnrOC (Fig. 4D). The smaller the ΔVnrOC, the higher the VOC.
where kB is the Boltzmann constant, T is the temperature in Kelvin, and q is the elementary charge.56 The ΔVnrOC values for the four cells were: spiro-OMeTAD (69 mV) < p-PTZMes-E-PTZOD-E (71 mV) < p-PTZMes-E (83 mV) < p-PTZOD-E (121 mV). By differentiating the EQE spectrum of the p-PTZMes-E-PTZOD-E cell (Fig. S15†), we estimated the optical bandgap of the FAPbI3-based light absorption layer to be 1.53 eV. Moreover, VOC shows a strong linear correlation with 1.53 − ΔVnrOC (Fig. 4D). The smaller the ΔVnrOC, the higher the VOC.
Additionally, we measured the VOC of the cells under different irradiances (Fig. S16†). By plotting VOC against the logarithm of the irradiance, we calculated the ideality factor (n)57 for each cell: p-PTZOD-E (1.25) < p-PTZMes-E (1.33) < p-PTZMes-E-PTZOD-E (1.35) = spiro-OMeTAD (1.35) (Fig. S17†). The n reflects the inverse of the order of nonlinear charge recombination reactions in the cells, with n values (>1) indicating the presence of localized states in the semiconductors.58 Using the Shockley diode equation59 and these n values, we fitted the J–V curves to obtain the shunt resistance (Rsh), series resistance (Rs), and reverse saturation current (Is) (Table 1). The results indicate that higher Rsh leads to higher VOC, while lower Rs and higher Rsh result in higher FF. Compared to spiro-OMeTAD, the higher FF of the p-PTZMes-E-PTZOD-E cell is attributed to its lower Rs, which is associated with the higher HOMO level of its HTL, leading to faster hole extraction rate and higher conductivity. The lowest FF of the p-PTZOD-E cell is mainly due to its lowest Rsh and highest Rs, the latter being associated with the slowest hole extraction rate and lowest conductivity, stemming from the deepest HOMO energy level of its HTL. Although the HTL of the p-PTZMes-E cell exhibits the fastest hole extraction rate and highest conductivity (highest HOMO level), it shows a moderate FF, mainly due to its low Rsh, which might be related to an excessively high hole density. To confirm this inference, we also prepared the p-PTZMes-E cell with a reduced TBPH-HFSI wt% (10%), which showed slight improvements in JSC (25.72 mA cm−2), VOC (1.170 V), FF (78.5%), and PCE (23.6%).
Next, we removed the encapsulation materials and gold electrode from the PSCs and performed the morphology analysis of the HTLs. Atomic force microscopy images (Fig. S18†) showed that the polycrystalline perovskite thin film had a relatively high root-mean-square roughness of 37 nm. Following the deposition of an HTL, the roughness was significantly reduced, with the values decreasing in the order: p-PTZMes-E (16 nm) > p-PTZMes-E-PTZOD-E (15 nm) > p-PTZOD-E (10 nm) = spiro-OMeTAD (10 nm). Scanning electron microscopy image revealed that on the surface of the perovskite microcrystalline film (Fig. S19†), the HTLs based on spiro-OMeTAD and p-PTZMes-E-PTZOD-E were very intact (Fig. 4E and H), while the HTL based on p-PTZMes-E showed some wrinkles (Fig. 4G). In contrast, the HTL based on p-PTZOD-E exhibited some submicron pits (Fig. 4F). Interestingly, when TBPH-HFSI in the p-PTZOD-E-based HTL was replaced with an equal wt% of 4-(tert-butyl)pyridinium bis(trifluoromethanesulfonyl)imide (TBPH-TFSI),60 the film morphology deteriorated further, showing significant phase separation between TBPH-TFSI and the polymeric semiconductor (Fig. S20A†). Note that TBPH-TFSI and TBPH-HFSI are two different organic salts, with the difference being the chemical structures of the anions. Notably, the “pristine” p-PTZOD-E film exhibited a complete morphology (Fig. S20B†). These comparative experiments suggest that the morphology of the composite film is likely controlled by the miscibility of the organic salt (doping promoter) with the polymeric semiconductor. An incomplete HTL morphology may lead to microscopic contacts between the gold electrode and the perovskite, introducing new charge recombination pathways and resulting in low Rsh.
The above analyses indicated that when designing hole transport materials for n-i-p PSCs, it is necessary to consider the combined quality factors including HOMO energy level, film-forming property, and hole mobility. These physical attributes, as well as doping (hole density and conductivity) control of organic semiconductors, will affect the charge separation and charge recombination of PSCs, ultimately affecting various photovoltaic parameters of the device.
 | ||
| Fig. 5 (A) Power conversion efficiency (PCE) versus time of aging at 85 °C (ISOS-D-2) for solar cells with different hole transport layers. (B) Reverse scan photocurrent density–voltage (J–V) curves of aged cells under 100 mW cm−2 simulated AM1.5G irradiation. (C) External quantum efficiency (EQE) spectra of aged cells. (D) Electronic absorption spectra of cells before and after aging. No clear difference can be observed for the unaged cells, the aged p-PTZMes-E cell, and the aged p-PTZMes-E-PTZOD-E cell. (E–H) Top-view scanning electron microscopy images of the hole transport layers in aged cells: (E) spiro-OMeTAD; (F) p-PTZOD-E; (G) p-PTZMes-E; (H) p-PTZMes-E-PTZOD-E. Scale bar: 1 μm. (I–L) Fluorescence optical microscopy images of the perovskite layers in aged cells: (I) spiro-OMeTAD; (J) p-PTZOD-E; (K) p-PTZMes-E; (L) p-PTZMes-E-PTZOD-E. Scale bar: 5 μm. For panels (D–H), encapsulation material and gold electrode were removed before measurement. For panels (I–L), encapsulation material, gold electrode, and hole transport layer were removed before measurement. Aging conditions: 85 °C for 1000 hours. | ||
Next, we removed the encapsulation materials and gold electrode from the PSCs and measured their UV-vis absorption spectra (Fig. 5D). The results revealed that thermal aging significantly diminished the light absorption of the spiro-OMeTAD and p-PTZOD-E cells, potentially due to perovskite decomposition or phase transformation. In contrast, the light absorption of the p-PTZMes-E and p-PTZMes-E-PTZOD-E cells remained nearly unchanged. Time-resolved photoluminescence measurements (Fig. S22†) showed an increase in τ due to thermal aging, with the extent of increase as follows: p-PTZOD-E (320%) > spiro-OMeTAD (230%) > p-PTZMes-E (120%) > p-PTZMes-E-PTZOD-E (50%). We noticed that the difference in hole extraction rate by a doped organic semiconductor (Section 2.3) and the corresponding “pristine” organic semiconductor in PSC is not significant, because introducing air-doping promoter only increased the hole density from the order of 1017 cm−3 to the order of 1018 cm−3. A further study could be performed on the impact of low concentration doping on the distribution of gap states in these organic semiconductors.61,62 The elevated τ in aged cells may correlate with the formation of wide bandgap species at the perovskite/HTL interface.
Scanning electron microscope images revealed numerous nanopores in the HTLs of aged spiro-OMeTAD and p-PTZOD-E cells (Fig. 5E and F), with larger pores observed in the p-PTZOD-E cell. It is noted that the glass transition temperatures of “pristine” p-PTZOD-E (119 °C) and spiro-OMeTAD (121 °C) are comparable. However, PSCs based on p-PTZOD-E exhibit poorer stability and more pronounced morphological degradation in the hole transport layer, likely due to the already presence of pits in the unaged p-PTZOD-E-based cells. This suggests that both the glass transition temperature and the morphology of the hole transport materials play critical roles in determining the thermal stability of PSCs at 85 °C. It is also worth noting that blending organic salts with organic semiconductors tends to lower the glass transition temperature.7 In stark contrast, the HTLs of the p-PTZMes-E-PTZOD-E and p-PTZMes-E cells maintained their integrity (Fig. 5G and H). Severe degradation of the HTL morphology may result in contacts between the gold electrode and the perovskite, thereby decreasing VOC.
Furthermore, fluorescence microscopy imaging of the perovskite layer after chlorobenzene washing of the HTL showed no discernible patterns in unaged cells (Fig. S23†). After aging, the perovskite layers of spiro-OMeTAD and p-PTZOD-E cells exhibited dense green spots (Fig. 5I and J), likely due to G-band luminescence from PbI2. In contrast, the perovskite layers of p-PTZMes-E and p-PTZMes-E-PTZOD-E cells showed only a few smaller green spots (Fig. 5K and L). Perovskite degradation could diminish light harvesting and induce more structural defects, including deep trap states that reduce charge separation and collection efficiencies, thereby degrading the device's photovoltaic parameters.
 | ||
| Fig. 6 (A–C) Photovoltaic parameters tracked at the maximum power point (MPP) under simulated AM1.5G irradiation at 100 mW cm−2: (A) efficiency at MPP (ηMPP); (B) photocurrent density at MPP (JMPP); (C) photovoltage at MPP (VMPP). (D and E) Series resistance (Rs) and shunt resistance (Rsh) obtained from fitting the J–V curves measured intermittently during MPP tracking. (F) Ion diffusion coefficients (D) in the hole transport layer at 45 °C. | ||
During MPPT, we intermittently measured the J–V curves of the cells and fitted them using the Shockley diode equation59 to extract Rs and Rsh. The time-dependent changes in Rs and Rsh are shown in Fig. 6D and E. Over time, Rs for the spiro-OMeTAD cell exhibited a gradual increase, while Rsh decreased. In contrast, the variations in Rs and Rsh for the p-PTZMes-E-PTZOD-E cell were less pronounced. The reduction in Rsh indicates accelerated charge recombination, likely associated with increased perovskite defects. The increase in Rs may arise from enhanced defects and scattering centers within the perovskite, or possibly from iodide ion migration from the perovskite to the HTL under an electric field, leading to dedoping.63 Molecular dynamics simulations were performed to investigate the diffusion behavior of relevant ions (4-(tert-butyl)pyridinium, TBPH+; 1,1,2,2,3,3-hexafluoropropane-1,3-disulfonimide, HFSI−; formamidinium, FA+; iodide, I−) in both HTLs. These simulations revealed smaller diffusion coefficients for these ions in the p-PTZMes-E-PTZOD-E-based HTL compared to spiro-OMeTAD (Fig. 6F). Suppressing species diffusion within PSCs could effectively regulate defect formation in the perovskite layer and dedoping processes.
3. Conclusions
Through direct arylation polymerization, we synthesized three phenothiazine–ethylenedioxythiophene alternating copolymers featuring varied substituents on phenothiazine: one with 2-octyldodecyl, another with trimethylphenyl, and a third alternating between 2-octyldodecyl and trimethylphenyl groups. This substitution variation led to differences in the HOMO energy levels of the polymeric semiconductors, influencing air oxidation doping and the rate of hole extraction from photoexcited perovskite. These phenothiazine–ethylenedioxythiophene copolymers demonstrated hole density-dependent mobility and conductivity. The combined use of flexible and rigid substituents endowed the polymeric semiconductor composite film with a more uniform surface morphology and enhanced hole mobility. These attributes bolster the efficiency and stability of perovskite solar cells. This work presents a novel strategy for crafting high-performance polymeric semiconductors. While we did not prepare the random copolymers of phenothiazine and ethylenedioxythiophene with flexible 2-octyldodecyl and rigid trimethylphenyl side chains in the present work. It will be interesting to optimize the side chain ratios in the random copolymers.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Conceptualization: P. W. Data curation: B. Z.; Y. C.; L. H. Funding acquisition: Y. Y.; P. W. Investigation: B. Z.; Y. C.; L. H.; N. X.; Y. Y.; J. Z.; Y. Z. Methodology: Y. Z. Project administration: Y. Y. Supervision: N. X. Validation: N. X; J. Z. Visualization: Y. C.; Y. Z. Writing – original draft: Y. C.; Y. Z. Writing – review & editing: N. X.; P. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (2022YFA1204800) and the National Natural Science Foundation of China (No. 22275160, No. 22475187, and No. 52073250) for the financial support. The authors thank Yifan Zhao from the Chemistry Instrumentation Center Zhejiang University for the technical support.References
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H.-B. Kim, S.-I. Mo, Y.-K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Grätzel and D. S. Kim, Science, 2022, 375, 302 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531 CrossRef CAS PubMed.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724 CrossRef CAS PubMed.
- L. He, Y. Zhang, Y. Wei, Y. Cai, J. Zhang and P. Wang, Matter, 2023, 6, 4013 CrossRef CAS.
- X. Zhao, H.-S. Kim, J.-Y. Seo and N.-G. Park, ACS Appl. Mater. Interfaces, 2017, 9, 7148 CrossRef CAS PubMed.
- A. K. Jena, M. Ikegami and T. Miyasaka, ACS Energy Lett., 2017, 2, 1760 CrossRef CAS.
- Y. Wei, Y. Zhang, Y. Ren, B. Zhang, Y. Yuan, J. Zhang and P. Wang, Adv. Funct. Mater., 2023, 33, 2307501 CrossRef CAS.
- Y. Ren, Y. Wei, T. Li, Y. Mu, M. Zhang, Y. Yuan, J. Zhang and P. Wang, Energy Environ. Sci., 2023, 16, 3534 RSC.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418 CrossRef CAS PubMed.
- Z. Zhu, Y. Bai, H. K. H. Lee, C. Mu, T. Zhang, L. Zhang, J. Wang, H. Yan, S. K. So and S. Yang, Adv. Funct. Mater., 2014, 24, 7357 CrossRef CAS.
- S. Ryu, J. H. Noh, N. J. Jeon, Y. C. Kim, W. S. Yang, J. Seo and S. I. Seok, Energy Environ. Sci., 2014, 7, 2614 RSC.
- J. Lee, M. M. Byranvand, G. Kang, S. Y. Son, S. Song, G.-W. Kim and T. Park, J. Am. Chem. Soc., 2017, 139, 12175 CrossRef CAS PubMed.
- L. Zhang, C. Liu, J. Zhang, X. Li, C. Cheng, Y. Tian, A. K.-Y. Jen and B. Xu, Adv. Mater., 2018, 30, 1804028 CrossRef PubMed.
- Y. Kim, E. H. Jung, G. Kim, D. Kim, B. J. Kim and J. Seo, Adv. Energy Mater., 2018, 8, 1801668 CrossRef.
- L. Zhang, C. Liu, X. Wang, Y. Tian, A. K. Y. Jen and B. Xu, Adv. Funct. Mater., 2019, 29, 1904856 CrossRef.
- J. Lee, G.-W. Kim, M. Kim, S. A. Park and T. Park, Adv. Energy Mater., 2020, 10, 1902662 CrossRef CAS.
- G. You, Q. Zhuang, L. Wang, X. Lin, D. Zou, Z. Lin, H. Zhen, W. Zhuang and Q. Ling, Adv. Energy Mater., 2020, 10, 1903146 CrossRef CAS.
- J. Park, S. E. Yoon, J. Lee, D. R. Whang, S. Y. Lee, S. J. Shin, J. M. Han, H. Seo, H. J. Park, J. H. Kim and B.-G. Kim, Adv. Funct. Mater., 2020, 30, 2001560 CrossRef CAS.
- Q. Fu, Z. Xu, X. Tang, T. Liu, X. Dong, X. Zhang, N. Zheng, Z. Xie and Y. Liu, ACS Energy Lett., 2021, 6, 1521 CrossRef CAS.
- H. Opoku, J. H. Lee, J.-J. Lee, H. Ahn and J. W. Jo, ACS Mater. Lett., 2022, 4, 2515 CrossRef CAS.
- Y. Bai, Z. Zhou, Q. Xue, C. Liu, N. Li, H. Tang, J. Zhang, X. Xia, J. Zhang, X. Lu, C. J. Brabec and F. Huang, Adv. Mater., 2022, 34, 2110587 CrossRef CAS PubMed.
- H.-S. Lin, T. Doba, W. Sato, Y. Matsuo, R. Shang and E. Nakamura, Angew. Chem., Int. Ed., 2022, 61, e202203949 CrossRef CAS PubMed.
- Q. Fu, H. Liu, S. Li, T. Zhou, M. Chen, Y. Yang, J. Wang, R. Wang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210356 CrossRef CAS PubMed.
- Q. Fu, X. Tang, H. Liu, R. Wang, T. Liu, Z. Wu, H. Y. Woo, T. Zhou, X. Wan, Y. Chen and Y. Liu, J. Am. Chem. Soc., 2022, 144, 9500 CrossRef CAS PubMed.
- X. Wang, Z. He, H. Chen, L. Yao, C. Li, X. Lin, Z. Zhou, K. Li, W. Wang, W. Cai, Q. Ling and H. Zhen, Adv. Funct. Mater., 2023, 33, 2308435 CrossRef CAS.
- S.-K. Kim, J. Kim, S. Choi, T. Yong, J. Y. Park, G. Lee, S. Han, H. R. You, S. Ko, G. Park, H. Ahn, J. Yang, Y. Kim, B. J. Kim and J. Choi, Adv. Energy Mater., 2023, 13, 2301927 CrossRef CAS.
- Y. Cai, Y. Zhang, J. Zhang, X. Pan, M. R. Andersson and P. Wang, Angew. Chem., Int. Ed., 2024, 63, e202315814 CrossRef CAS PubMed.
- J. J. H. McDowell, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 5 CrossRef.
- J.-M. Raimundo, P. Blanchard, P. Frère, N. Mercier, I. Ledoux-Rak, R. Hierle and J. Roncali, Tetrahedron Lett., 2001, 42, 1507 CrossRef CAS.
- A. K. Mohanakrishan, P. Amaladass and J. A. Clement, Tetrahedron Lett., 2007, 48, 539 CrossRef.
- P. Amaladas, J. A. Clement and A. K. Mohanakrishan, Tetrahedron, 2007, 63, 10363 CrossRef.
- K. Yamazaki, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2013, 34, 69 CrossRef CAS PubMed.
- S. J. Choi, J. Kuwabara and T. Kanbara, ACS Sustainable Chem. Eng., 2013, 1, 878 CrossRef CAS.
- F. Grenier, B. R. Aïch, Y.-Y. Lai, M. Guérette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137 CrossRef CAS.
- S. Narayanan, S. P. Raghunathan, S. Mathew, M. V. M. Kumar, A. Abbas, K. Sreekumar, C. S. Kartha and R. Joseph, Eur. Polym. J., 2015, 64, 157 CrossRef CAS.
- W. Li and T. Michinobu, Polym. Chem., 2016, 7, 3165 RSC.
- Y. Kojima, S. Hayashi and T. Koizumi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1183 CrossRef CAS.
- Y. Zhang, Y. Ren, X. Xie, Y. Wei, L. He, L. Fang, J. Zhang, Y. Yuan and P. Wang, Adv. Funct. Mater., 2022, 32, 2108855 CrossRef CAS.
- Z.-R. Tan, Y.-Q. Xing, J.-Z. Cheng, G. Zhang, Z.-Q. Shen, Y.-J. Zhang, G. Liao, L. Chen and S.-Y. Liu, Chem. Sci., 2022, 13, 1725 RSC.
- Y. Zhang, L. He, Y. Cai, J. Zhang and P. Wang, Angew. Chem., Int. Ed., 2024, 63, e202401605 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-Y. Yu, S.-C. Lin, J. T. Lin, L.-Y. Chen, D.-S. Hsiu, Y. S. Wen, M. M. Lee and S.-S. Sun, J. Mater. Chem. C, 2016, 4, 9499 RSC.
- M. Wakioka, H. Morita, N. Ichihara, M. Saito, I. Osaka and F. Ozawa, Macromolecules, 2020, 53, 158 CrossRef CAS.
- H. Saito, J. Chen, J. Kuwabara, T. Yasuda and T. Kanbara, Polym. Chem., 2017, 8, 3006 RSC.
- Z. Qian, Z. Cao, L. Galuska, S. Zhang, J. Xu and X. Gu, Macromol. Chem. Phys., 2019, 220, 1900062 CrossRef.
- H. Bässler and A. Köhler, Top. Curr. Chem., 2012, 312, 1 Search PubMed.
- T. W. Barrett, H. Wohltjen and A. Snow, Nature, 1983, 301, 694 CrossRef CAS.
- D. M. de Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53 CrossRef CAS.
- C. G. Shuttle, R. Hamilton, J. Nelson, B. C. O'Regan and J. R. Durrant, Adv. Funct. Mater., 2010, 20, 698 CrossRef CAS.
- T. Li, Y. Zhang, M. Ren, Y. Mu, J. Zhang, Y. Yuan, M. Zhang and P. Wang, Angew. Chem., Int. Ed., 2024, 63, e202401604 CrossRef CAS PubMed.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T.-Q. Nguyen, Nat. Mater., 2019, 18, 1327 CrossRef CAS PubMed.
- H. Makki, C. A. Burke and A. Troisi, J. Phys. Chem. Lett., 2023, 14, 8867 CrossRef CAS PubMed.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910 CrossRef CAS.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926 CrossRef CAS PubMed.
- S. Olthof, S. Mehraeen, S. K. Mohapatra, S. Barlow, V. Coropceanu, J.-L. Brédas, S. R. Marder and A. Kahn, Phys. Rev. Lett., 2012, 109, 176601 CrossRef PubMed.
- M. Schwarze, C. Gaul, R. Scholz, F. Bussolotti, A. Hofacker, K. S. Schellhammer, B. Nell, B. D. Naab, Z. Bao, D. Spoltore, K. Vandewal, J. Widmer, S. Kera, N. Ueno, F. Ortmann and K. Leo, Nat. Mater., 2019, 18, 242 CrossRef CAS PubMed.
- F. Li, X. Deng, F. Qi, Z. Li, D. Liu, D. Shen, M. Qin, S. Wu, F. Lin, S.-H. Jang, J. Zhang, X. Lu, D. Lei, C.-S. Lee, Z. Zhu and A. K.-Y. Jen, J. Am. Chem. Soc., 2020, 142, 20134 CrossRef CAS PubMed.
- G. A. H. Wetzelaer, M. Kuik, M. Lenes and P. W. M. Blom, Appl. Phys. Lett., 2011, 99, 153506 CrossRef.
- J. Bisquert and I. Mora-Seró, J. Phys. Chem. Lett., 2010, 1, 450 CrossRef CAS.
- S. Yoo, B. Domercq and B. Kippelen, J. Appl. Phys., 2005, 97, 103706 CrossRef.
- Y. Ren, M. Ren, X. Xie, J. Wang, Y. Cai, Y. Yuan, J. Zhang and P. Wang, Nano Energy, 2021, 81, 105655 CrossRef CAS.
- X. Lin, G. E. Purdum, Y. Zhang, S. Barlow, S. R. Marder, Y.-L. Loo and A. Kahn, Chem. Mater., 2016, 28, 2677 CrossRef CAS.
- A. Fediai, F. Symalla, P. Friederich and W. Wenzel, Nat. Commun., 2019, 10, 4547 CrossRef PubMed.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04998e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |