DOI:
10.1039/D4TA00460D
(Paper)
J. Mater. Chem. A, 2024,
12, 15240-15253
Food-waste-derived hydrochar to a low-cost p-type semiconducting photocatalyst (Zn–Al@HC): multifunctional role in real wastewater treatment and environmental sustainability†
Received
20th January 2024
, Accepted 11th March 2024
First published on 13th March 2024
Abstract
The present investigation demonstrates the effectiveness of a novel Zn–Al@HC photocatalyst supported by anaerobically digested food waste-derived hydrochar (HC) for the photocatalytic activation of peroxydisulphate (PDS) and abatement of salicylic acid (SA) in the UV/PDS/Zn–Al@HC system. The morphological and photo-electrochemical characterization of Zn–Al@HC supported the formation of ZnAl2O4 crystals and the contraction of band gap energy from 3.80 eV (ZnAl2O4) to 2.36 eV (Zn–Al@HC) by the inclusion of HC (1.86 eV) as a carbon-based support material. Moreover, a remarkable reduction in recombination of photogenerated e−/h+ was witnessed in response to UV irradiation in the UV/PDS/Zn–Al@HC system for SA degradation. Under optimal operating conditions, the proposed treatment system achieved 94.45 ± 0.65% degradation of SA. Furthermore, the radical scavenging test revealed the dominance of hydroxyl radical followed by sulphate radicals in the UV/PDS/Zn–Al@HC system, and a potential mechanism was deduced. Cyclic voltammetry for 20 repetitive cycles in a harsh medium affirms the photo-stability of Zn–Al@HC, even under real environmental conditions. The energy-economic valuation of the as-synthesized photocatalyst unveiled the total operating cost and electrical energy per order of UV/PDS/Zn–Al@HC to be 0.1536 $ per m−3 order−1 and 0.0130 kW h m−3 order−1, respectively. Thus, the system proves to be a sustainable and economical option for the management of waste and remediation of chemicals of emerging concern, which can be implemented as future large-scale technology for wastewater treatment.
1. Introduction
Salicylic acid (SA) is a chemical of emerging concern (CEC) that is widely used as a raw material in the manufacturing of cosmetics, analgesics, antipyretics, anti-inflammatory drugs such as aspirin, painkillers, and other drugs. Recalcitrant organic SA is highly toxic, endocrine disrupting, non-biodegradable, and bioaccumulates in nature; hence, wastewater containing SA needs to be treated before being discharged into the aquatic system.1,2 Recently, the photocatalysis-based advanced oxidation process (AOP) has received significant attention for CEC remediation because of its non-toxicity, low cost, better performance, complete mineralization, high stability, and potential for utilizing solar energy.3,4 The mechanism of photocatalytic degradation involves two stages, including the onsite generation of free radicals (either hydroxyl (˙OH) or sulphate (SO4˙−)) and the reaction of generated radicals with target contaminants leading to their partial or complete mineralisation.5 However, SO4˙−-based AOPs are dominating the present research area as the standard redox potential of SO4˙− (2.5–3.1 eV) is higher than that of ˙OH (1.9–2.7 eV), it has a longer half-life, can non-selectively target the organic pollutants, and SO4˙−-based AOPs are more compatible in a wider pH range.6 These radicals can be generated via the activation of either peroxydisulphate (PDS) or peroxymonosulphate (PMS). The most environment-friendly approach for persulphate activation is UV irradiation, which directly breaks down the chemical bonds of contaminants. Furthermore, UV light can also aid the disintegration of peroxide bonds in the oxidants, which releases free radicals for contaminant degradation.7
The heterogeneous semiconductor catalyst ZnO is prominently used to assist the photodegradation process due to its low cost and similar photocatalytic activity to the well-established TiO2 photocatalyst.8 Moreover, ZnO possesses a broad energy band gap of 3.4 eV with a high exciton binding energy (approx. 60 meV), making it suitable for absorbing UV irradiation as well as high-energy photons.9,10 However, the high resistivity of ZnO, low visible light absorption (compared to UV), and faster recombination of photogenerated electrons and holes (e−–h+) are responsible for its limited application. The optical and photocatalytic properties of ZnO can be upgraded by incorporating a group III element, e.g., aluminium (Al), which is non-toxic, thermally stable, brings changes in lattice parameters, and induces surface structural defects. Generally, the surface defects improve the photocatalytic activity owing to the reduction in the recombination of charge carriers.11
Furthermore, the e−–h+ recombination can be retarded by the construction of heterojunction composites using organic and inorganic materials, which facilitate PDS activation via direct e− transfer. For instance, heterojunction photocatalysts PDINH/MIL-88A(Fe) and Fe3(PO4)2·8H2O (vivianite) were reported to effectively activate PDS through a similar synergistic effect and therefore, successfully participated in CEC elimination.12,13 In this direction, incorporation of a carbon-based support material such as hydrochar (HC) provides better stability and recyclability to the semiconductor-based photocatalysts.14 Compared to commercially available carbon materials, i.e., activated carbon or biochar (BC), the synthesis of HC is less energy intensive and it is compatible with moisture-containing precursors (e.g., digestate) as well. Moreover, the HC-based catalyst was also reported to have a significant impact on oxidant (such as PDS, PMS) and hydrogen peroxide (H2O2) activation to generate reactive radicals in AOPs.15 As a step towards an economic and sustainable catalyst synthesis, researchers are utilizing sludge/digestate as the precursor for HC. For instance, Wei and the co-researcher utilized iron-modified sewage sludge-derived HC as a catalyst for PDS activation and achieved 99.72% degradation efficiency for tetracycline (antibiotic).
To the extent of our understanding, this is the first investigation that utilised anaerobically digested food waste (AD-FW) derived HC modified with Zn–Al precursor for the synthesis of a photocatalyst (Zn–Al@HC). Further, its application in a UV/PDS system for SA degradation has been demonstrated. The detailed characterization (morphological, structural, optical, and photo-electrochemical) of Zn–Al@HC and optimization of operating parameters were performed. Furthermore, the effect of anions, versatile aqueous matrix, and catalytic mechanism during the UV/PDS/Zn–Al@HC process, and the mineralisation pathway for SA degradation are also proposed based on the radical scavenging experiments. Importantly, the energy-cost analysis emphasizes the potential of Zn–Al@HC as a low-cost, highly efficacious, and sustainable photocatalyst for SA degradation in the future.
2. Experimental analysis
2.1. Reagents, chemicals, and Zn–Al@HC synthesis approach
For the present investigation, chemicals such as sodium persulphate (Na2S2O8), sodium chloride (NaCl), sodium fluoride (NaF), sodium phosphate (Na3PO4), sodium carbonate (Na2CO3), sodium nitrate (NaNO3), zinc oxide (ZnO), and aluminium oxide (Al2O3) of laboratory grade were procured from Merck, India, whereas SA was procured from Sigma Aldrich (C7H6O3, >99% pure). Additional reagents used during the experiments were tert-butyl alcohol (TBA; C4H10O, Advent Chembio Pvt. Ltd, 99%), p-benzoquinone (BQ; ≥99.5%), methanol (MeOH; CH3OH, Merck, 99%), 5,5-dimethyl-1-pyrroline N-oxide (DMPO, Sigma Aldrich, >99%), and ethylenediaminetetraacetate disodium salt dihydrate (Na2-EDTA; 99%). Carbon tetrachloride (CCl4, 99%) was procured from Universal Fine Chemicals, India. The photocatalyst was synthesized using the co-precipitation technique16 from the anaerobic digestate of food waste, as described further in Text S1.†
2.2. Physicochemical, morphological, and electrochemical characterization
Pristine HC (210 °C, 4 h) and the photocatalyst Zn–Al@HC (210 °C, 4 h) were characterized for physicochemical and morphological characteristics as described in detail (Text S2†). Furthermore, the absorption bands and photocatalytic activity of the prepared novel catalyst were determined using the UV-vis diffuse reflectance spectra (DRS) and chronoamperometry (CA). Photoluminescence (PL) spectroscopy was carried out using a fluorescence spectrophotometer (Cary Eclipse, Agilent Technologies, USA) to analyse the charge carrier separation efficacy of the as-synthesized catalyst. The PL spectroscopy was conducted in the wavelength range of 200 nm to 600 nm with an excitation wavelength of 350 nm at ambient temperature. A similar protocol for the PL characterization was followed by Li et al. and Ghosh et al.17,18 Additional electrochemical analysis including Cyclic Voltammetry (CV), Linear Sweep Voltammetry (LSV), Mott–Schottky (M–S) analysis, and Electrochemical Impedance Spectroscopy (EIS) were conducted both under UV irradiation and in the dark to determine the photocurrent response.
2.3. UV/PDS/Zn–Al@HC photocatalytic degradation and optimization
Photocatalytic degradation experiments was conducted as per the method mentioned in Text S3.† After 30 min of the initial adsorption test, the system was exposed to a source of UV radiation (11 W, Phillips, India) for 120 min. The sample was collected at regular intervals of 20 min and monitored using a UV-visible spectrophotometer at a wavelength of 297 nm.19 The UV/PDS/Zn–Al@HC photocatalytic degradation was performed to investigate the effect of different anions, including chloride (Cl−), nitrate (NO3−), fluoride (F−), carbonate (CO32−), and phosphate (PO43−) at variable concentrations, i.e., 0 to 15 mM. Later, a comparative analysis of the SA degradation efficiency in three different water matrices, i.e., distilled water, tap water, and wastewater treatment plant (WWTP) effluent (procured from the Indian Institute of Technology campus, Kharagpur, West Bengal, India) was performed and evaluated under the optimized conditions to affirm its real field scalability. Furthermore, the residual sulphate and metal ion concentration in the final UV/PDS/Zn–Al@HC effluent was determined using Ion Chromatography (IC, Eco-IC, Metrohm, Switzerland) to assess the downstream potent hazards of the UV/PDS/Zn–Al@HC effluent.
2.4. Toxicity and reusability evaluation
To determine the toxic effects of untreated SA samples and UV/PDS/Zn–Al@HC treated SA samples, phytotoxicity assessment on Vigna radiata was conducted. The assessment was performed in a Petri dish where five seeds were sown and watered (5 ml each day) for twelve days with both treated and untreated water separately. The results were evaluated in terms of root and shoot length on the last day. Furthermore, Zn–Al@HC was recovered using a vacuum filtration process from the treated effluent of the UV/PDS/Zn–Al@HC system and dried in an oven at 60 °C to evaluate the reusability capacity (conducted in triplicate) of the as-synthesised catalyst.
3. Results and discussion
3.1. Morpho-chemical characterization and properties of Zn–Al@HC
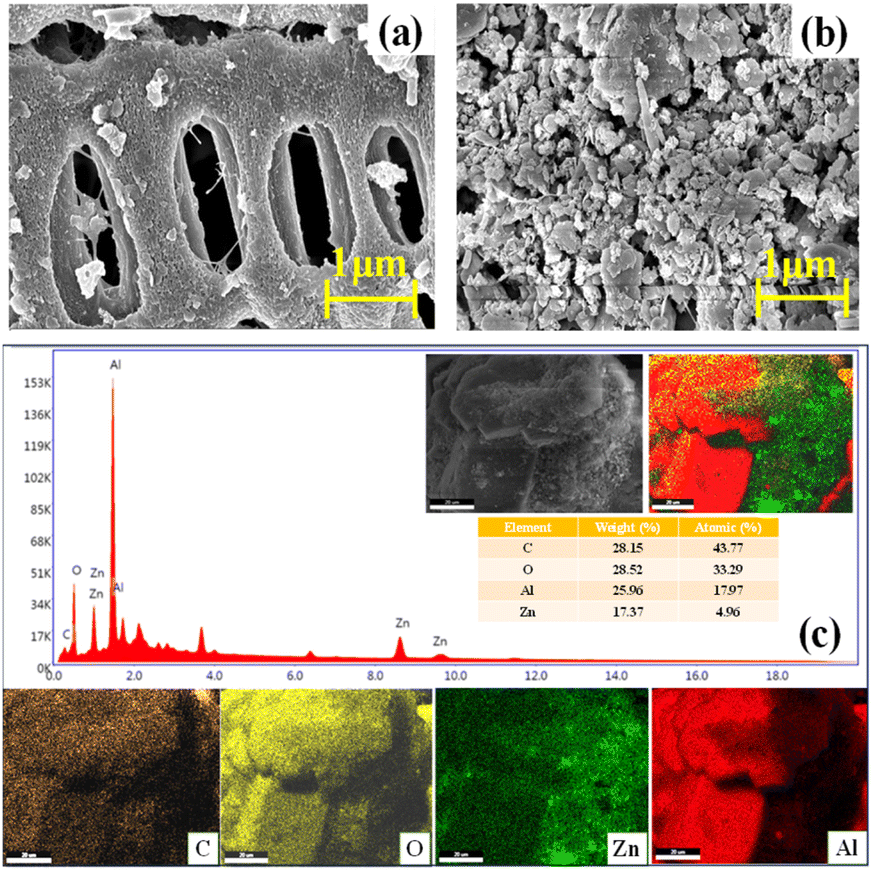
The scanning electron microscopy (SEM) micrograph of the pristine HC derived from AD-FW revealed the existence of fibrous and highly porous skeleton-like morphology (Fig. 1a). The porosity of the HC verified its ability to function as the best support material for the Zn–Al composite. However, the texture of pristine HC was destroyed after Zn–Al activation and resulted in more porous microspheres in the Zn–Al@HC. Moreover, the SEM images of Zn–Al@HC showed the appearance of white ultrafine particulates of ZnAl2O4 on the surface of HC and a great effect on the textural properties of the HC (Fig. 1b).20 The cross-linking between the small microspheres to form large microspheres suggests the better occurrence of polymerization and condensation reaction during the HTC of AD-FW in the presence of Zn–Al.21 Additionally, it supports the harvesting of UV irradiation due to increased surface area and multiple scattering of light within the porous framework. Furthermore, the elemental composition was assessed using Energy Dispersive Spectroscopy (EDS), which revealed the uniform distribution of each element within the Zn–Al@HC photocatalyst (Fig. 1c).
 |
| | Fig. 1 (a and b) SEM interpretation of pristine HC and the as-synthesized Zn–Al@HC photocatalyst, (c) EDS spectrum of Zn–Al@HC (with the corresponding elemental mapping of C, O, Zn, and Al, respectively). | |
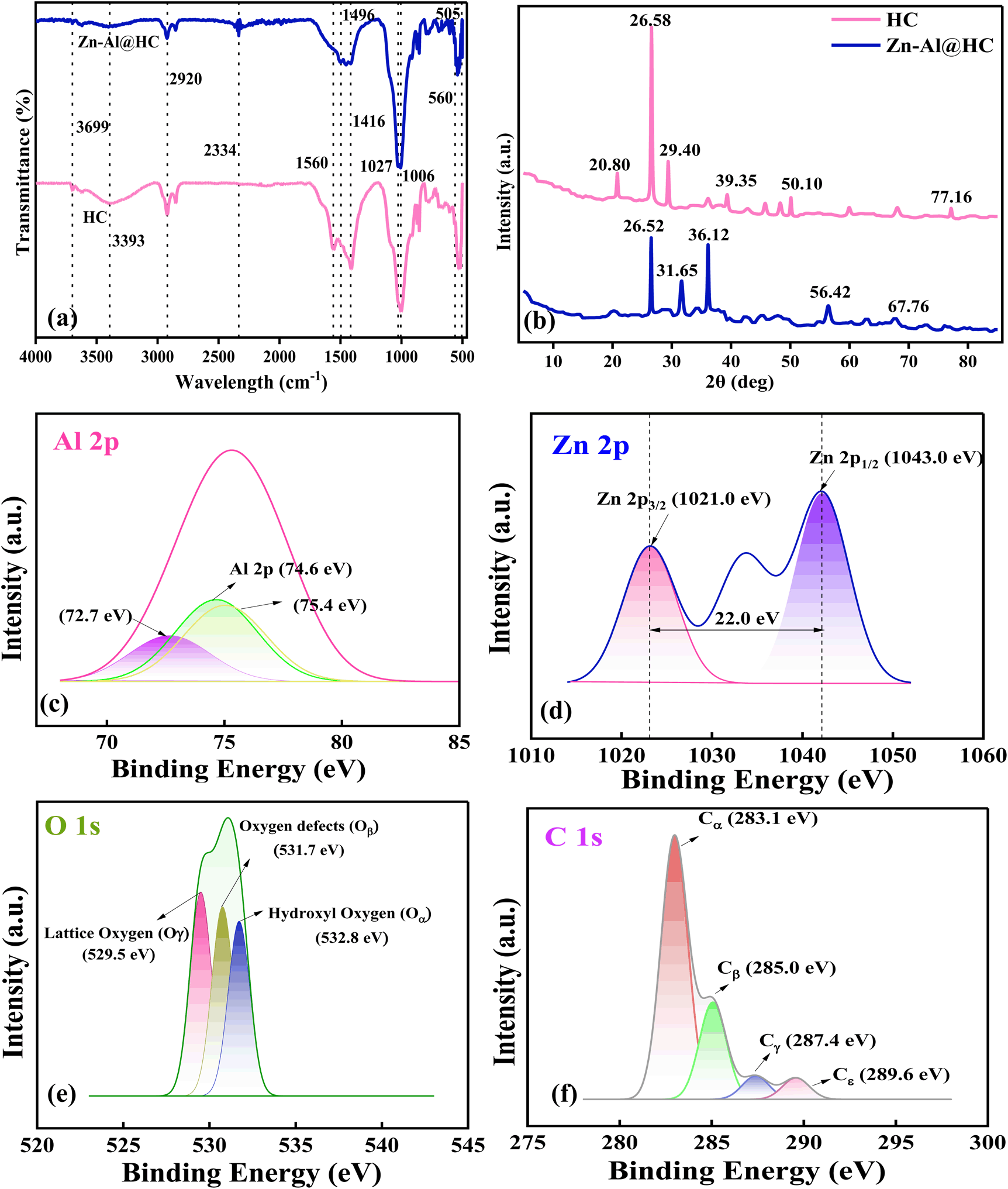
The Fourier transform infrared spectroscopy (FTIR) spectra of pristine HC and Zn–Al@HC have been presented in Fig. 2a, which suggested the dominance of the oxygen functional groups, such as aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of conjugated quinones and ketones in the wavelength between 1500 cm−1 and 1600 cm−1. Both the samples exhibited common broad bands between 3699 cm−1 and 2920 cm−1 due to the –OH stretching of the interlayer water molecule. The formation of a hydrogen bond is responsible for the broadening of the band. However, the absence of –OH stretching mode in Zn–Al@HC represented the hydrophobic nature of the prepared photocatalyst Zn–Al@HC.22 The original structure of HC remains intact after Zn–Al@HC composite formation. In the FTIR spectra, the bands around 560 cm−1 and 680 cm−1 represent the vibration of Zn–O bonds at the tetrahedral site, whereas the small peaks around 505 cm−1 could be attributed to the Al–O bonds in the octahedral site of the ZnAl2O4 spinel.23 The visible band around 700–450 cm−1 can be related to the lattice vibration modes of O–Zn–O.24
O stretching of conjugated quinones and ketones in the wavelength between 1500 cm−1 and 1600 cm−1. Both the samples exhibited common broad bands between 3699 cm−1 and 2920 cm−1 due to the –OH stretching of the interlayer water molecule. The formation of a hydrogen bond is responsible for the broadening of the band. However, the absence of –OH stretching mode in Zn–Al@HC represented the hydrophobic nature of the prepared photocatalyst Zn–Al@HC.22 The original structure of HC remains intact after Zn–Al@HC composite formation. In the FTIR spectra, the bands around 560 cm−1 and 680 cm−1 represent the vibration of Zn–O bonds at the tetrahedral site, whereas the small peaks around 505 cm−1 could be attributed to the Al–O bonds in the octahedral site of the ZnAl2O4 spinel.23 The visible band around 700–450 cm−1 can be related to the lattice vibration modes of O–Zn–O.24
 |
| | Fig. 2 (a) FTIR, (b) XRD of pristine HC and the Zn–Al@HC photocatalyst, and (c–f) XPS spectra of the Zn–Al@HC photo-catalyst (Al 2p, Zn 2p, C 1s, O 1s). | |
The X-ray diffraction (XRD) characterization of the pristine HC and as-synthesized Zn–Al@HC photocatalyst is represented in Fig. 2b. In accordance with the XRD pattern, differentiated peaks (2θ) at 20.80°, 26.58°, 29.40°, 39.35°, 50.10°, and 77.16° were evident for the pristine HC. The presence of peaks between 10° and 35° reflects the amorphous nature of the carbon-based support material.25 Interestingly, the diffraction peak around 26.5° in both HC and Zn–Al@HC showed a high degree of graphitization. However, the peaks at 20.8° and 29.40° correspond to inorganic impurities SiO2 and CaCO3 in the pristine HC, which were absent after Zn–Al activation of HC.26 Furthermore, a decrease in the intensity of the SiO2 peak was observed with the addition of the Zn–Al composite during the synthesis, which might be due to carbon gradually covering the crystal plane of SiO2.27
For the synthesized Zn–Al@HC photocatalyst, the XRD pattern obtained at 31.65° [220], 36.12° [311], and 56.42° [422], (JCPDS no: 00-005-0669) affirmed the formation of ZnAl2O4 spinel phase in the photocatalyst supported by the carbonaceous structure, which follows the previous investigation conducted on the BC-ZnAl2O4 composite28 and Zn/Al LDHs@C.29 Note that the decrease in crystallization degree and improvement in the amorphous sites in the Zn–Al@HC photocatalyst can cause structural defects and impact both the photo-induced e−/h+ pairs and the efficiency of resulting charge carrier separation. This phenomenon will be further supported by CA and EIS results in the following sections.
X-ray Photoelectron Spectroscopy (XPS) was carried out to further confirm the elemental concoction and surface electronic state of Zn–Al@HC. The XPS region of Al 2p is located in the range of 72–75 eV (Fig. 2c), which illustrates the presence of Al3+ on the plane of the photocatalyst. As per the literature, the Al 2p peak corresponds to Al3+, i.e., aluminium with oxidation number 3+. Also, the results were consistent with the assigned peak of 74.3 eV in Fe2O3/Zn–Al LDH by Li et al. (2020).30 Moreover, the Zn 2p spectrum (Fig. 2d) of Zn–Al@HC at 1021.0 eV and 1043.0 eV belongs to the two components Zn 2p1/2 and Zn 2p3/2, respectively, due to spin–orbit splitting. The difference in binding energies between these components is 22 eV, which supports the XRD results of ZnAl2O4 formation in the synthesized composite and a clear transition from Zn to Zn2+ state of the metal.9,31
Herein, from Fig. 2e, four C peaks were observed, which could be attributed to the oxygen-based functional groups of the HC-based photocatalyst (Zn–Al@HC). The XPS spectrum of Cα at 283.1 eV corresponds to two-coordinated carbon atoms (such as aliphatic/aromatic carbon groups; C–H, C–C).32 Furthermore, the peaks at 285.0 eV (Cβ), 287.4 eV (Cγ), and 289.6 eV (Cε) reflect the presence of hydroxyl/ether/C–O, quinone/carbonyl/CO, and carboxylic/ester/O– CO groups. Additionally, Cβ affirms the presence of C in a graphitic environment, whereas the deconvolution at Cγ supports the formation of C–N bonds in the synthesized photocatalyst.33 Both graphitic C and C–N are responsible for enhanced photocatalytic activity due to improved charge absorption properties and the synergy between their crystallographic and electronic structure.34
The high resolution spectra of O 1s in Zn–Al@HC are evident in Fig. 2f and are fitted into three peaks at 529.5 eV, 531.7 eV, and 532.8 eV, which correspond to lattice oxygen (Oγ), oxygen defects (Oβ), and adsorbed water molecules on the surface (Oα).35 The cumulative effects of the chemical bonding state and photogenerated e− transfer are responsible for the outstanding photocatalytic properties of the synthesized Zn–Al@HC. Moreover, Brunauer–Emmet–Teller (BET) surface analysis (BEL, Japan, Inc.) demonstrated a surface area of 19.51 m2 g−1 and pore volume of 0.032 cm3 g−1 of the as-synthesized Zn–Al@HC catalyst, which are relatively higher than those of the HC (14.27 m2 g−1, 0.016 cm3 g−1). In addition, the adsorption–desorption curves of both HC and Zn–Al@HC exhibit a Type IV isotherm (Fig. S1a and b†) with an H3 hysteresis loop and a pore radius of 4.57 nm and 1.57 nm, which mainly suggest the existence of a mesoporous structure with capillary condensation that tentatively contributes to radical generation by activating PDS in the UV/PDS/Zn–Al@HC system.36
3.2. Photoelectrochemical characterization and stability of Zn–Al@HC
3.2.1. UV-vis diffuse reflectance spectra (DRS) (Kubelka–Munk's plot).
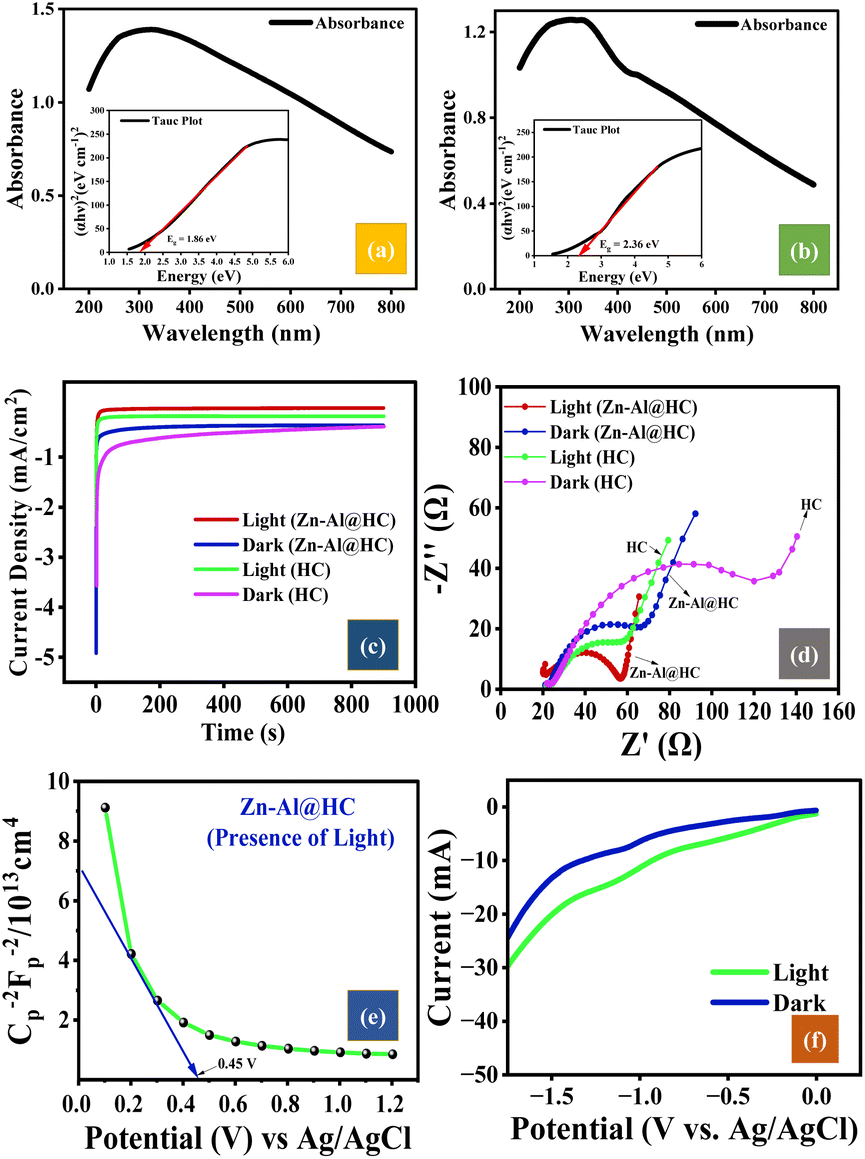
UV-vis DRS was performed to evaluate the optical properties of the synthesized photocatalyst Zn–Al@HC. The photocatalytic reactions are highly dependent on the light absorption by the material and the migration of light-induced e−–h+.37 The strong absorption band in the UV-visible region for pristine HC is visible in Fig. 3a, while Zn–Al@HC has a strong absorption band illustrating bathochromic shifts compared to HC Fig. 3b. The absorption edge of Zn–Al@HC is located at approximately 370 nm in line with the previously reported value of 390 nm by Ramani et al. and Suwanboon et al. for zinc oxide and g-C3N4/ZnAl2O4 nanoparticles, respectively.38,39 This result indicates the prominent optical properties of Zn–Al@HC in the UV region.40
 |
| | Fig. 3 (a and b) UV-DRS of pristine HC and Zn–Al@HC, (c) chronoamperometric analysis of HC and Zn–Al@HC (UV illuminated and dark system), (d) Nyquist plot for HC and Zn–Al@HC under UV and dark conditions, and (e) Mott–Schottky plot under UV light, (f) LSV curves under UV illuminated and dark conditions. | |
The energy band gap value (Eg) of the sample was estimated from extrapolation of the Tauc plot of (αhv)2vs. photon energy (hv) to zero based on the Kubelka–Munk formula (eqn (1)).
where
α is the absorption coefficient (6.626 × 10
−34 J s),
h is Plank's constant,
v is the frequency of light,
A is the band tailing parameter, and
n is the optical transition coefficient, respectively.
As per the literature, ZnO, Al2O3, and ZnAl2O4 have wide energy band gaps of 3.4 eV, 7.1 eV, and 3.8 eV, respectively,41 which were significantly reduced by the doping of Zn–Al on the HC surface having a band gap of 1.86 eV (Fig. 3a). The resultant band gap of the synthesized composite Zn–Al@HC was found to be 2.36 eV (Fig. 3b) and predicted to exhibit enhanced photocatalytic activity. The results were similar to those obtained in the ZnO–BC composite photocatalyst having a band gap of 2.97 eV, higher than that of pristine BC (1.52 eV) but lower than that of ZnO (3.06 eV). The ZnO–BC photocatalyst was reported to achieve 98.5% degradation of ciprofloxacin in 240 min.42
The reduction in the band gap of Zn–Al@HC as compared to ZnAl2O4 might be due to the structural defects (as affirmed by FTIR and XPS analysis) and interaction with the carbon content of HC. Structural defects improved the interaction of the electric surface charge of metal oxides in the Zn–Al@HC composite and modified the e−–h+ separation process or radical generation under UV irradiation.42 Thus, the synthesized composite has a better ability to absorb more UV-visible light, which enhances the photocatalytic degradation of SA. Similarly, other HC-based photocatalysts have been reported to effectively degrade emerging contaminants as shown in Table S1.†
3.2.2. Photo-induced chronoamperometry.
Chronoamperometry analysis for both pristine HC and as-synthesized Zn–Al@HC was conducted in a typical three-electrode system for 960 s and at an applied potential of −1 V. The trends of photocurrent obtained under both UV illumination and dark conditions are presented in Fig. 3c. The chronoamperometric inferences are consistent with the EIS results, representing the better stability and photocurrent density of Zn–Al@HC compared to HC. The photocurrent plots for Zn–Al@HC and HC illustrated an average photocurrent density of −0.15 mA cm−2 and −0.58 mA cm−2. Additionally, the photocurrent density decreases nearly 5.6 times in the dark conditions. Furthermore, the photocurrent response of Zn–Al@HC under UV illumination remained more stable and reversible for a longer duration. This might be attributed to the activation of Zn–Al@HC in the presence of UV irradiation to produce e−–h+ charge carriers and lower e−–h+ recombination.43 Thus, Zn–Al@HC proved to be a highly potent photocatalyst for contaminant degradation under UV exposure.
3.2.3. Nyquist plots.
To investigate the charge transfer resistance of the photocatalyst, EIS was conducted. As stipulated in Nyquist plots (Fig. 3d), the semicircle arc of Zn–Al@HC (under light exposure conditions) is smaller as compared to that in the dark and pristine HC. The Nyquist plots also corroborate the formation of a heterojunction and reduce the resistance for charge carrier migration in the Zn–Al@HC photocatalyst as compared to the pristine HC. Additionally, a remarkable reduction in the recombination of photogenerated e−/h+ was witnessed in response to UV irradiation in the UV/PDS/Zn–Al@HC system for SA degradation. The UV-DRS analysis affirmed the formation of a heterojunction between HC and Zn–Al composite and lessened the resistance for charge carrier migration and the analogous recombination rate.44
3.2.4. Mott–Schottky (M–S) analysis.
The M–S analysis was conducted using 0.05 M Na2SO4 solution in the presence of optimum 0.01 M PDS at 1000 Hz frequency in the dark conditions and under a constant AC potential. The M–S analysis is generally carried out to identify the semiconducting nature (i.e., p-type (−ve slope) or n-type (+ve slope)) of the Zn–Al@HC photocatalyst and carrier charge density. Zn–Al@HC shows the −ve (in Fig. 3e) slope confirming the p-type semiconducting properties, which was also supported by Liu et al.45 From the M–S plot, the flat band potential (Efb) of Zn–Al@HC was found to be 0.45 eV vs. Ag/AgCl. The Efb at the standard electrode potential of Ag/AgCl (E0Ag/AgCl ∼0.198 eV at 25 °C) was converted to Efbvs. normal hydrogen electrode potential (ENHE) using eqn (2). The pH value of Na2SO4 and PDS solution was found to be 6.97.| | | ENHE = EAg/AgCl (at pH 6.9) − 0.059 (7 – pH of the electrolyte) + E0Ag/AgCl | (2) |
For Zn–Al@HC, Efb is found to be equal to 0.646 eV vs. NHE from the above eqn (2). Furthermore, to examine the photocatalytic mechanism of Zn–Al@HC, the potential of the valence band (EVB) and conduction band (ECB) should be determined using eqn (3) and (4).
where X is the absolute electronegativity of Zn–Al@HC (∼5.34 eV),
E0 is the free e
− energy on the H-scale (∼4.50 eV) and
Eg is the energy band gap of Zn–Al@HC (∼2.36 eV from the Kubelka–Munk plot).
For the as-synthesized photocatalyst, X and Eg are approximately equal to 5.34 eV and 2.36 eV. Thus, the calculated EVB and ECB for Zn–Al@HC are about 2.02 eV and −0.34 eV, respectively. As per the literature, the potential for superoxide radicals O2˙−/O2 is −0.33 eV vs. NHE, ˙OH/H2O is +1.99 eV vs. NHE, and ˙OH/OH− is +2.40 eV vs. NHE.46 The ECB for Zn–Al@HC is −0.34 eV, which is more negative than the O2˙−/O2 potential, therefore, the UV/PDS/Zn–Al@HC system has greater tendency towards the generation of O2˙−, which might later transform into ˙OH.47 Additionally, the photogenerated h+ of EVB are sufficient to perform either the photolysis of H2O2 by 2 e− reduction of O2 (i.e., O2/H2O2 ∼ +0.682 eV) or oxidation of hydroxyl ion (OH−) and H2O to generate ˙OH (i.e., ˙OH/H2O ∼ +1.99 eV, OH−/H2O ∼ +1.23 eV) imperative to the formation of ˙OH.48 Moreover, the photocatalyst has the oxidative selectivity to produce H2O2 (H2O2/H2O ∼ +1.76 eV) and further release ˙OH (H2O2/˙OH ∼ +0.73 eV).49,50 Hence, the UV/PDS/Zn–Al@HC system has both the active species O2˙− and ˙OH besides the photogenerated holes. The domination of ˙OH was also established via a radical scavenging experiment, therefore corroborating with the aforementioned hypothesis, which is elaborated in Section 3.6. Another co-existent radical is PDS (S2O82−/S2O8˙−) having a redox potential of +2.01 eV, however, it is incapable of producing SO4˙− (2.5–3.1 eV) directly.51
3.2.5. Electrochemical/voltammetry analysis.
Additionally, CV and LSV were also executed under UV illumination and the dark condition for the synthesized photocatalyst (Zn–Al@HC). The analyses were performed at a scan rate of 0.05 V s−1 in the presence of 50 mM Na2SO4 electrolyte with platinum and Ag/AgCl as counter and reference electrodes, respectively. As stipulated in LSV curves (Fig. 3f), Zn–Al@HC showed lower response without UV, whereas it manifested a higher cathodic photocurrent of 30.12 mA (1.3 times higher) in the presence of UV. The electrochemical evaluation of the as-synthesized catalysts for photocatalytic activity indicated the easier charge migration in Zn–Al@HC, thereby greatly enhancing the photogenerated e−–h+ in the conduction and valence bands.
Furthermore, after 20 cycles of CV under ideal experimental conditions (i.e., in the absence of PDS), the results revealed a much higher and more stable current response of Zn–Al@HC, which affirms its high reusability and recyclability. In addition, profound oxidation and reduction peaks were also observed at 1.5 V and −1.0 V, respectively, which signify the enhanced redox properties of the as-synthesised catalyst (Fig. 4a). On performing the same 20 cycles CV under harsh operating conditions (under 0.01 M PDS, 50 mg L−1 SA, 50 mM Na2SO4), a current drop of 26.5% was observed under UV-exposed conditions, which was much less compared to that in the dark (Fig. 4b). This is attributed to the high photostability and high photoelectrochemical response of Zn–Al@HC under a UV-exposed system, supporting the results obtained from LSV and CA analysis.
 |
| | Fig. 4 (a) 20 Cycles CV under ideal operating conditions, (b) 20 cycles CV for Zn–Al@HC (real environmental conditions; both UV illuminated and dark systems). | |
3.2.6. Photoluminescence analysis.
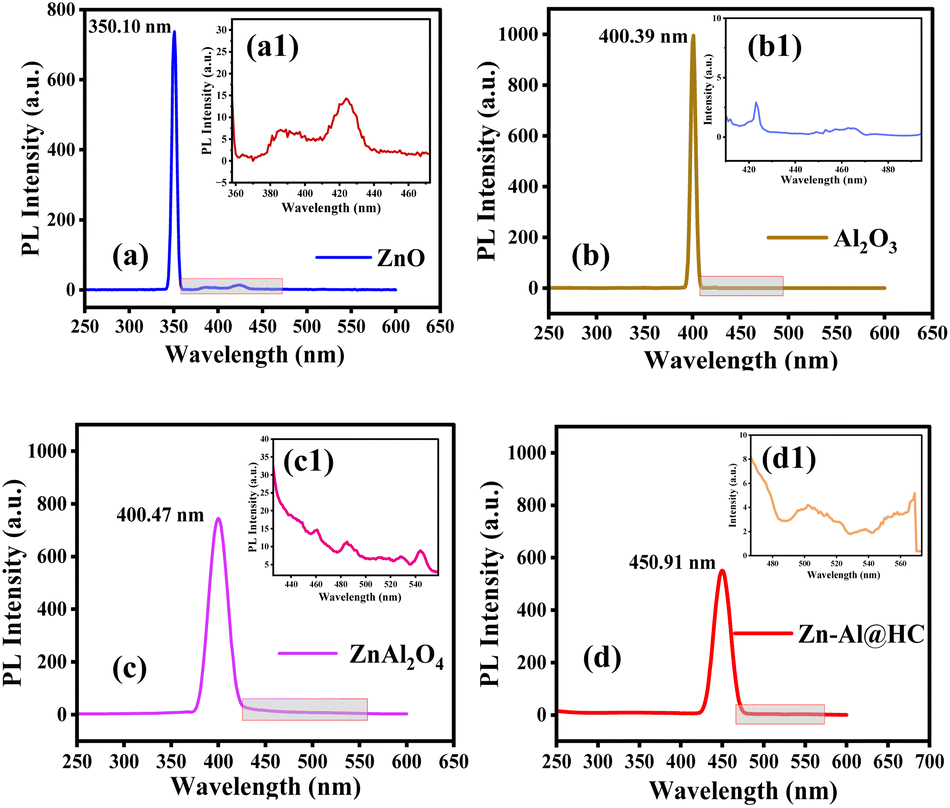
From the results of PL spectra (Fig. 5a–d), it was observed that ZnO exhibited a high intensity peak in the UV region at 350.10 nm, whereas Al2O3 exhibited a higher peak with a slight wavelength shift in the UV-vis region at 400.39 nm, which shows the lower photocatalytic activity of Al2O3. However, the intensity of the peak reduced in the case of ZnAl2O4 crystals at 400.47 nm, which was minimum in the case of as-synthesized Zn–Al@HC (at 450.91 nm), illustrating the superior photocatalytic properties of the as-synthesized catalyst. This suggests that the added HC may function as an electron trap, allowing the ZnAl2O4 electrons to be attracted to the HC when exposed to UV light.52 Furthermore, it speeds up the migration and separation of electron–hole pairs, suppresses the charge-carrier recombination, and increases the effectiveness of Zn–Al@HC for the SA degradation process.17,53 Furthermore, a red shift as observed in the sub-spectra (Fig. 5a1–d1) emphasizes the compatibility of Zn–Al@HC for visible light simulation, which also extends its applicability for the solar-photo degradation process.
 |
| | Fig. 5 PL spectra and sub-spectra of (a and a1) pure ZnO, (b and b1) pure Al2O3, (c and c1) co-precipitated ZnAl2O4, (d and d1) and as-synthesized Zn–Al@HC photocatalyst. | |
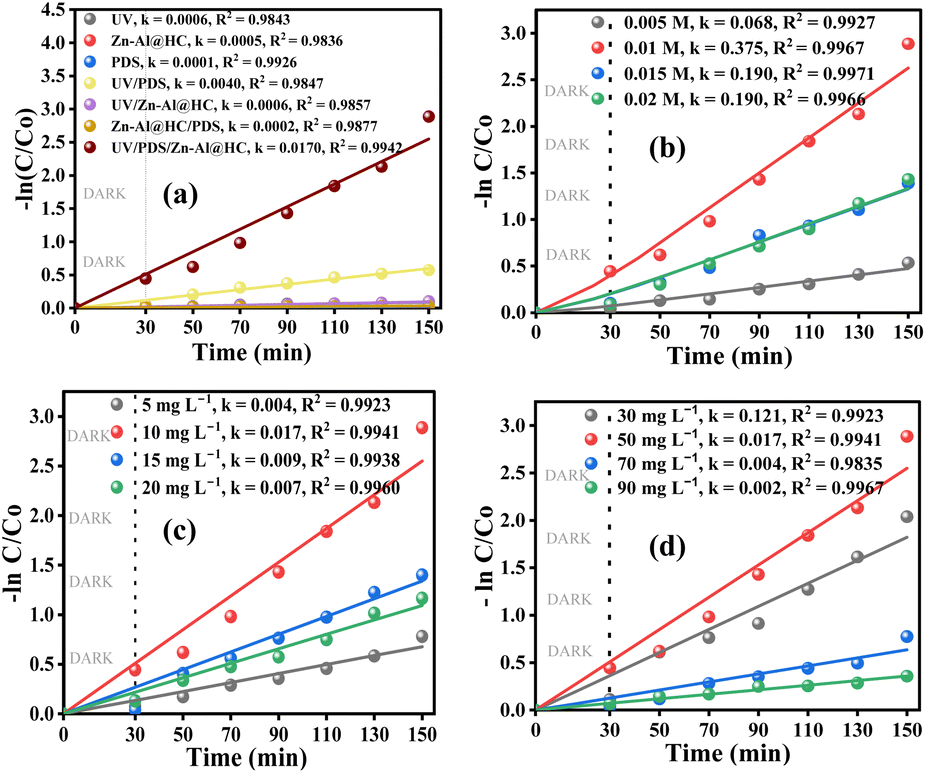
3.3. UV/PDS/Zn–Al@HC degradation of SA and control experiments
The photocatalytic performance of Zn–Al@HC as the PDS activator in SA degradation was evaluated under UV light. As illustrated in Fig. 6a, the control UV irradiation has minimal SA degradation efficiency, indicating that the contribution of UV light alone is negligible. Meanwhile, SA was barely degraded in 150 min (including 30 min of dark study) in the case of control Zn–Al@HC and control PDS. The negligible degradation in the presence of PDS alone might be due to its limited oxidation potential (2.01 V). Moreover, Zn–Al@HC had a less than 10% (i.e., 9.34 ± 0.32%) efficiency of SA degradation under UV irradiation alone, which might be due to extensive recombination of charge carriers in the case of the pure catalyst. Without UV exposure, Zn–Al@HC and PDS together were unable to degrade SA, manifesting the significance of UV light in the system. Interestingly, an improvement in SA degradation efficiency to 43.35 ± 0.15% was observed after the introduction of UV into the PDS system, thus indicating the activation of PDS in the presence of UV light; however, the degradation efficiency is still less than 50% (Fig. S2a†). After exposing the Zn–Al@HC-based photocatalyst to the UV/PDS system, outstanding photocatalytic activity and enhancement in the SA degradation (up to 94.45 ± 0.65%) were noticed. As for the SA solution, the photocatalytic degradation process followed the pseudo-first-order kinetics as indicated in eqn (5).| |  | (5) |
where k represents the degradation rate constant (min−1), Co is the initial concentration of SA (mg L−1), and Ct is the final concentration of the SA after irradiation for time t (min), respectively.
 |
| | Fig. 6 (a) Photocatalytic degradation of SA and kinetics involved under different (b) PDS dosages, (c) Zn–Al@HC dosages, and (d) pollutant concentrations. (pH0 of 7.0, temperature of 27 °C, Zn–Al@HC of 10 mg L−1, UV irradiation of 11 μW cm−2, SA concentration of 50 mg L−1, and PDS of 0.01 M.) | |
These results emphasized the existence of a synergistic effect between PDS and Zn–Al@HC (PDS activation effect of Zn–Al@HC) under UV light. Additionally, the PDS entrapped the conduction band photoelectrons, prevented e−–h+ recombination, and generated SO4˙− and ˙OH with high oxidizing ability. The mechanism behind the role of Zn–Al@HC in the radical generation is illustrated in eqn (6)–(9).
| | | Zn–Al@HC + hv → Zn–Al@HC (h+ + e−) | (6) |
| | | S2O82− + e− → SO4˙− + SO4˙− | (7) |
| | | SO4˙− + H2O → ˙OH + SO42− + h+ | (8) |
| | | h+/ SO4˙−/˙OH + SA → intermediates → CO2 + H2O | (9) |
3.3.1. Effect of peroxydisulphate (oxidizer) dosage.
In the heterogeneous PDS photocatalytic system, PDS dose is directly correlated with the number of reactive radicals generated. Fig. 6b stipulates the effect of PDS concentration on SA degradation in the UV/PDS/Zn–Al@HC system. On increasing the PDS concentration from 0.005 to 0.01 M, an increment in degradation efficiency from 41.38 ± 0.50% to 94.45 ± 0.65% (Fig. S2b†) was observed under constant Zn–Al@HC (10 mg L−1) and UV conditions. The increment might be due to greater generation of reactive radicals for the higher dose of PDS. However, when PDS dosage was further increased from 0.015 to 0.02 M the SA degradation rate was significantly decreased. For a particular dose of photocatalyst, e− and h+ are constant, hence, the excessive PDS concentration might be responsible for the side reactions of PDS with the available SO4˙− radicals (eqn (10) and (11)).54 These side reactions decrease the number of reactive radicals for the oxidation of SA, therefore the optimal PDS concentration of 0.01 M was selected and used for further experiments.| | | SO4˙− + S2O82− → SO42− + S2O8˙− | (10) |
| | | SO4˙− + SO4˙− → S2O82− | (11) |
3.3.2. Effect of Zn–Al@HC (catalyst) dosage.
The effect of Zn–Al@HC dosage on the catalytic degradation of SA (50 mg L−1) in the UV/PDS/Zn–Al@HC system was investigated at 0.01 M PDS dose (Fig. 6c). On decreasing the Zn–Al@HC dosage from 20 mg L−1 to 15 mg L−1 and 10 mg L−1, an increasing trend in SA degradation efficiency, i.e., 68.81 ± 1.50%, 75.35 ± 1.35%, and 94.45 ± 0.65% was witnessed (Fig. S2c†). This might be due to the agglomeration of particles and high opacity of the aqueous solution at a high concentration of the catalyst, which hinders UV penetration through the solution.55 However, when the Zn–Al@HC dose was further reduced to 5 mg L−1 from 10 mg L−1, the SA removal was drastically decreased to 54.20 ± 0.50% from 94.45 ± 0.65%. The reason for the sudden decrease is attributed to the lack of active sites for PDS activation and SA elimination under a low dosage of Zn–Al@HC.56
3.3.3. Effect of initial SA (pollutant) concentration.
The effect of the initial concentration of pollutant on the degradation capability of the Zn–Al@HC photocatalyst was evaluated at the optimum catalyst and PDS dose of 10 mg L−1 and 0.01 M, respectively. Despite the low concentration of the model pollutant SA found in real wastewater, to check the performance efficiency of the UV/PDS/Zn–Al@HC system, higher concentrations of SA were chosen in this investigation. The results in Fig. S2d† revealed the decrement in degradation efficiency with increasing SA concentration beyond 50 mg L−1. Based on the scenario, the results of Fig. 6d can be interpreted as that at a fixed concentration of Zn–Al@HC, PDS, and irradiation intensity, the number of reactive radicals generated (SO4˙− and ˙OH) at the catalyst surface can be assumed to be constant. Thus, the lower degradation at higher concentrations of SA might be due to the insufficient generation of reactive radicals and the lesser interaction between the reactive species and the pollutant molecules.57 Also, the availability of large pollutant molecules interferes with the UV (photons) absorption effectively, thereby resulting in a decrease in degradation efficiency with an increase in the initial concentration of SA.58 However, examining the degradation efficiency at a concentration of 30 mg L−1 also resulted in a slightly lower degradation rate, which might be due to incompetent exposure to radicals with the SA molecule. Thus, 50 mg L−1 can be considered the optimal SA concentration for the effective working of the catalyst in the UV/PDS system.
3.3.4. Effect of co-existing ions.
Halide ions (Cl− and F−) are often found in surface water, groundwater, or seawater in the concentration range of 0.1–500 mg L−1. These halide ions undergo photo excitation on UV exposure and produce radicals that selectively degrade pollutants. Additionally, these ions also act as ˙OH scavengers, thereby decreasing the ˙OH radical-mediated degradation of SA.59 A correlation between the added Cl− ion and SA degradation rate was obtained for the experiment (Fig. S3a†). When the Cl− concentration increased from 5 to 15 mM, the SA degradation was slightly enhanced from 52.24 ± 0.66% to 66.87 ± 0.45%, which might be due to the interaction between the SO4˙− and Cl− to produce the Cl˙ radical with a high redox potential of 2.40 V and degrade SA further. However, the overall SA degradation rate decreased from 0.017 ± 0.001 min−1 to 0.004 ± 0.0002 min−1 in the presence of Cl−, which is attributed to the reaction between the Cl− and ˙OH (2.80 V) that produced comparatively fewer active oxidants such as ClOH˙−, Cl˙, and Cl2˙− (eqn (12)–(16)), thus lowering the degradation efficiency in the UV/PDS/Zn–Al@HC system on Cl− exposure.
A similar trend was also observed earlier for acetaminophen and trichloroethylene degradation by the UV/PDS system,60,61 whereas the presence of F− ion enhanced the SA degradation rate maximum up to 0.025 min−1 at a F− concentration of 15 mM (removal efficiency of 97.29 ± 0.55%). However, the results showed an inhibitory effect of F− at a concentration of 10 mM. The SA degradation efficiency was 67.75 ± 0.50% for 5 mM and 84.55 ± 0.61% for 10 mM concentration of F− ion (Fig. S3b†). Hence, F− ion had a two-sided effect (both positive and negative) on the SA degradation in the UV/PDS/Zn–Al@HC system.
| | | Cl− + ˙OH/SO4˙− → ClOH˙−/SO42−/Cl˙ | (12) |
| | | Cl˙ + H2O → ClOH˙− + H+ | (14) |
| | | 2Cl− + Zn–Al@HC (hVB+) → Cl2 | (15) |
| | | S2O82− + 2Cl− → 2SO42− + Cl2 | (16) |
The effect of NO3− on the photocatalytic degradation of SA is provided in Fig. S3c.† The addition of a low dosage of NO3− had an inhibitory effect on the SA degradation and decreased the degradation efficiency of SA. On comparing with the NO3− free system having an SA degradation rate of 0.017 min−1 (NO3− = 0 mM), the degradation rate reduced to 0.0074 min−1 (for 5 mM), 0.0071 min−1 (for 10 mM), and 0.009 min−1 (for 15 mM). The negative effect of NO3− is attributed to the radical scavenging properties of NO3−, which reacts with the ˙OH and the h+ (eqn (17) and (18)). The results obtained are consistent with the previous investigation performed on the photocatalytic degradation of carbamazepine.62
| | | NO3− + ˙OH → NO3˙ + OH− | (18) |
A slight suppression in the SA degradation was observed (Fig. S3d†) in the case of CO32− addition, possibly due to the conversion of CO32− to HCO3− on reaction with water (eqn (19)). Both CO32− and HCO3− compete for the radical species ˙OH and SO4˙− to produce less reactive radical moieties CO3˙− and HCO3˙.63 Previous investigations reported the same trend concerning the CO32− ion on the carbamazepine degradation in the thermally activated PDS system.64
| | | CO32− + H2O → HCO3− + OH− | (19) |
In the presence of phosphate ions in the aqueous phase, an accelerating effect on SA degradation was noticed. Enhanced degradation efficiencies of 95.03 ± 0.84%, 99.95 ± 0.02%, and 99.96 ± 0.01% were observed at 5, 10, and 15 mM of PO43−, respectively (Fig. S3e†). This can be attributed to the adsorption of PO43− on the Zn–Al@HC surface which promotes e−–h+ separation via inducing an electrostatic field on the Zn–Al@HC surface, therefore enhancing the photocatalytic degradation of SA.59 As shown in Fig. S4a–e,† the effect of anions on the UV/PDS/Zn–Al@HC revealed that Cl− had the most inhibitory effects on SA degradation in the concentration range of 5–15 mM, followed by NO3−, CO32−, F−, and PO43− (Cl− > NO3− > CO32− > F− > PO43−).
3.3.5. Effect of an aqueous matrix and residual sulphate.
The synchronous decay of SA by the UV/PDS/Zn–Al@HC system was evaluated (Fig. S5a and b†) in three different types of aqueous matrixes including distilled water, tap water, and secondary treated sewage (WWTP effluent). The results are thoroughly explained in Text S4.†
3.4. Identification of the predominant radical species
To elucidate the effect of different radical species involved in the UV/PDS/Zn–Al@HC system on SA degradation, a scavenging experiment using TBA and MeOH was conducted. MeOH could rapidly react with both ˙OH (k = 9.7 × 108 M−1 s−1) and SO4˙− (k = 1.1 × 107 M−1 s−1); whereas, TBA could scavenge ˙OH radical with the reaction rate constant of k = 6.0 × 108 M−1 s−1, but not SO4˙− having k = 4.0 × 105 M−1 s−1.65 The addition of TBA at the concentration of 50 mM to the UV/PDS/Zn–Al@HC system decreased the degradation efficiency from 94.45 ± 0.65% to 41.22 ± 0.15%, which shows the dominancy of ˙OH in the SA degradation (Fig. 7a and b).
 |
| | Fig. 7 (a) Scavenging effect of TBA, MeOH, Na2-EDTA, CCl4, and BQ on SA decay and (b) kinetics in the UV/PDS/Zn–Al@HC system. | |
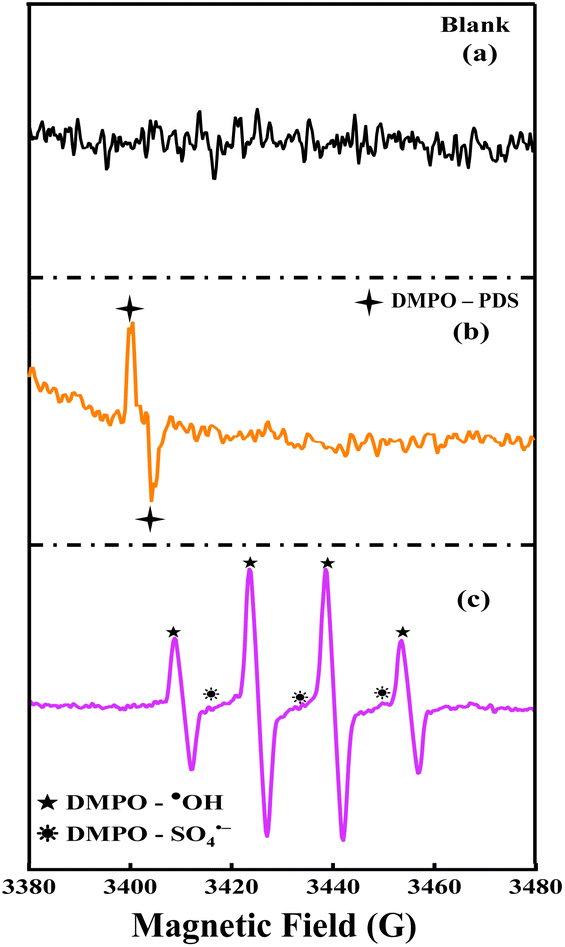
The SA degradation in the TBA exposed system might be mainly due to the photolysis and activity of SO4˙− in the system. Furthermore, the exposure of MeOH at the same concentration lowers the degradation rate to 25 ± 0.45%, confirming the co-existence and significant contribution of SO4˙− in the UV/PDS/Zn–Al@HC system. In addition, BQ, Na2-EDTA, and CCl4 were employed to identify the O2˙−, h+, and e−, respectively.66 Furthermore, in the presence of BQ, Na2-EDTA, and CCl4, SA degradation was suppressed to 68.15%, 71.48%, and 66.35% in 150 min, respectively. Therefore, the aforementioned results confirmed that both ˙OH and SO4˙− generated by stimulating PDS in the presence of Zn–Al@HC and UV light played vital roles in the degradation of SA (eqn (7) and 8); however, the role of O2˙−, h+, and e− also cannot be neglected. Overall, ˙OH was identified as the principal radical responsible for the abatement of SA in the UV/PDS/Zn–Al@HC system. To validate the involvement of reactive oxygen species in the process, an EPR analysis was performed at room temperature using a 100 mM solution of the DMPO spin-trapping agent. DMPO reacts with short-lived radicals generated in the Zn–Al@HC/PDS system to form stable adducts, which have extended lifespans (eqn (20)–(22)). Clearly, Fig. 8a does not show any peak in the spectrum, thereby revealing no formation of reactive oxygen species in the blank solution, whereas the EPR spectrum of pure PDS revealed sharp peaks (indicated by a star mark) in the spectra of the DMPO–PDS system, which is attributed to the nitroxide radical (5,5-dimethylpyrrolidone-(2)-oxyl-(1); DMPOX) formed as a result of the oxidation of DMPO (Fig. 8b). This observation was consistent with the results of control experiments. However, the distinctly higher peaks of DMPO-˙OH with wave intensity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1, displayed by the Zn–Al@HC/PDS system, signify the accelerated activity of the catalyst towards ˙OH generation (Fig. 8c),67 while the minor peaks of DMPO-SO4˙− with the wave intensity of 1:1:1 support the co-existence of the SO4˙− radical.67 Hence, the major role of ˙OH was affirmed by both the scavenging and EPR experiments.
:2:2:1, displayed by the Zn–Al@HC/PDS system, signify the accelerated activity of the catalyst towards ˙OH generation (Fig. 8c),67 while the minor peaks of DMPO-SO4˙− with the wave intensity of 1:1:1 support the co-existence of the SO4˙− radical.67 Hence, the major role of ˙OH was affirmed by both the scavenging and EPR experiments.
| | | DMPO + SO4˙− → DMPO-SO4˙− | (20) |
| | | DMPO-SO4˙−+ H2O → DMPO-HO˙ + h+ + SO42− | (21) |
 |
| | Fig. 8 EPR (a), blank (b), DMPO-PDS (c), DMPO-˙OH/SO4˙− for the PDS activated Zn–Al@HC photocatalyst. | |
3.5. Catalytic mechanism involved in the use of the UV/PDS/Zn–Al@HC system
Based on the scavenging experiment and detected free radicals, the mechanism involved in the UV/PDS/Zn–Al@HC system for activating PDS by Zn–Al@HC and further SA degradation is proposed in Fig. 9. Initially, the adsorption mechanism is caused by the capturing of both SA and PDS on the Zn–Al@HC surface and generation of photogenerated e− and h+ (eqn (23) and (24)). However, the process of photocatalytic degradation occurred both on the catalyst surface and aqueous media simultaneously. During the process, the dominant ˙OH radical can be produced in several ways, including the reaction of photogenerated h+ with the OH− and H2O molecules (eqn (26)–(28)), the reaction between O2˙− and H2O molecules (eqn (25); (29) and (30)), and the reaction of SO4˙− with OH− and H2O molecules according to eqn (31) and (32), and eqn (8).42,68
 |
| | Fig. 9 Proposed photocatalytic mechanism of Zn–Al@HC for reactive radical generation, PDS activation, and SA degradation. | |
Furthermore, the photocatalyst Zn–Al@HC tentatively contributed to the generation of in situ H2O2 which further releases ˙OH (eqn (33)–(35)) for contaminant degradation confirming the abundance of ˙OH during the EPR.
| | | ZnAl2O4 (eCB−) + ZnAl2O4 (hVB+) → 2ZnAl2O4 (e−–h+) | (23) |
| | | Zn–Al@HC (eCB−) → HC (e−) − ZnAl2O4 | (24) |
| | | Zn–Al@HC (eCB−) + O2 → O2˙− | (29) |
| | | O2˙− + 2H2O → H2O2 + 2OH− → 2H2O2 + e− → 2˙OH + 2OH− | (30) |
| | | SO4˙− + OH− → ˙OH + SO42− | (31) |
| | | ˙OH/SO4˙− + SA → CO2 + H2O + Degradation products | (32) |
| | | 2H2O → H2O2 + 2 (H+ + e−) | (33) |
On the other hand, the probable generation of SO4˙− can take place via either photodecomposition of PDS by UV light (eqn (36)), or activation of PDS by the HC-based catalyst via an e− transfer mechanism (eqn (37)–(39)), or reaction between PDS and photogenerated e− (eqn (7)). Both the generated radicals attack the SA molecules and photocatalytically degrade to intermediates and then to CO2 and H2O on further oxidation (eqn (32)).69
| | | S2O82− + HC surface–COOH → SO4˙− + HSO4− + HC surface–COO˙ | (37) |
| | | S2O82− + HC surface–OH → SO4˙− + HSO4− + HC surface–O˙ | (38) |
| | | S2O82− + HC surface–CO → SO4˙− + SO42− + HC surface–CO˙ | (39) |
3.6. Mineralisation sequence, evolution of oxidation products, and total organic carbon removal during SA degradation in the UV/PDS/Zn–Al@HC system
From the inference of the Liquid Chromatograph Mass-Spectroscopy (LC-MS) analysis (as shown in Fig. S6†) of the initial and final treated sample taken under optimum conditions, a plausible reaction pathway for the SA mineralization and intermediate formation is proposed in Fig. 10. As per the results, oxidative degradation of SA started with the attack of ˙OH radical on SA, leading to the formation of polyhydroxylated benzene derivatives, such as 2,3-dihydroxybenzoic, 2,5-dihydroxybenzoic, and 2,6-dihydroxybenzoic acids.70 Simultaneously, the decarboxylation of SA resulted in the formation of catechol and it further oxidized to form 1,2-benzoquinone.71 The cleavage of the benzene ring of 1,2-benzoquinone in the aqueous medium gives rise to aliphatic carboxylic acids, such as muconic acid. Maleic acid and oxalic acids are formed by the addition of ˙OH to the double bond of the muconic acid. Then maleic acid was oxidized to malic acid and further decomposed to acetic acid and CO2. On the other hand, the generated oxalic acid transformed into formic acid and then to CO2.72 The p-position of the CO groups initiated an attack on 1,2-benzoquinone, leading to the formation of H2O2 intermediates. These intermediates subsequently converted into 2-hydroxy-1,4-benzoquinone through the release of water. 2-Hydroxy-1,4-benzoquinone was oxidized to fumaric acid, caffeic acid, glutaconic acid, and other small molecules such as glucose and fructose.73
 |
| | Fig. 10 Proposed degradation pathway for the photocatalytic degradation of SA by the Zn–Al@HC photocatalyst in the UV/PDS system. | |
Furthermore, 2,3-dihydroxybenzoic acid was oxidized to oxoacetic acid and ethane-1,1,2- tricarboxylic acid. These compounds are further converted to ethanoic acid and propanoic acid. Then, propanoic acid was oxidized to carbonic and hydroxyl acetic acid. Subsequently, hydroxyl acetic acid was again converted to ethanoic and carbonic acids, which directly mineralised to CO2 and H2O.74 The degradation products and intermediates formed with their respective m/z values are listed in Table S2.†
Despite the high SA degradation rate obtained in the present experiment, SA was not completely mineralized to CO2 and H2O. There might be the formation of the above discussed organic by-products, which might be toxic to aquatic and terrestrial systems. To better evaluate the final organic concentration, the total organic carbon (TOC) was determined at each designated sampling time for the system having an initial concentration of SA = 50 mg L−1, PDS = 0.01 M, and Zn–Al@HC = 10 mg L−1. Results showed a drastic decrease in TOC content, and a maximum of 85.21 ± 1.25% of TOC removal efficiency was achieved after 360 min. Similar results were obtained by Shinde et al. and Garza-Campos et al. with a TOC abatement efficiency of 88.9% and 87%, respectively, during the photocatalytic degradation of SA by deploying TiO2/AlFe2O3 as the photocatalyst and coupling a solar photoelectron-Fenton-solar heterogeneous photocatalysis system.75,76
Finally, phytotoxicity evaluation was performed against Vigna radiata because of its rapid germination, low-cost, and highly sensitive nature to scrutinize the toxicity of the treated sample (containing transitional intermediates). The results showed significant growth of roots up to 1 ± 0.5 cm and shoots up to 6 ± 0.9 cm in the treated sample that contained 2.7 mg L−1 of SA after eliminating 94.45 ± 0.65% of SA under optimum conditions for twelve days (Fig. 11a and b). However, no growth of Vigna radiata was observed on exposure to the untreated sample having 50 mg L−1 of SA. The results emphasised the degradation of SA into less toxic products during the UV/PDS/Zn–Al@HC treatment and intermediates (hydroxylated compounds) have a low inhibitory effect and the phytotoxicity decreased. The past investigations discovered that the untreated SA solution hindered the germination and lengthening of the root and shoot of Vigna radiata and Vicia faba.77,78 Additionally, the results demonstrated that SA concentrations of 3.95 and 4.16 mg L−1 remained after the electrocoagulation, and electro-Fenton treatments showed significant germination and elongation of seeds.77,78 Thus, the treated effluent can be reliably utilized for irrigation, gardening, and other recreational purposes.
 |
| | Fig. 11 Evaluation of Vigna radiata germination on exposure to (a) treated and (b) untreated wastewater sample; (c) reusability assessment of Zn–Al@HC. | |
Furthermore, the reusability assessment of Zn–Al@HC revealed the outstanding performance of the catalyst for 10 continuous cycles (Fig. 11c) under optimized conditions, which was also demonstrated by the 20 cycles CV mentioned earlier. Results of the reusability test revealed that the SA degradation dropped to 88 ± 0.5% after the 9th successive cycle from 94.45 ± 0.65% (1st cycle), with maximum Al and Zn leaching of 1.2 ± 0.2 mg L−1 and 0.6 ± 0.1 mg L−1, respectively, which is significantly less than the United States, Environmental Protection Agency (US-EPA) guidelines (Al <5.0 mg L−1, and Zn <2.0 mg L−1) for irrigation,79,80 thus revealing the excellent potency of the catalyst even after 9 cycles of reuse. After usage, Zn–Al@HC was regenerated using alkali EtOH to enhance the sustainability of the catalyst, and nearly 90 ± 0.5% SA degradation efficiency was achieved after regeneration.
3.7. Cost and performance assessment
The photodegradation of SA by one order of magnitude in 1 m3 of wastewater in the photochemical process utilizes a significant amount of electrical energy and oxidants, thereby raising the total operational cost of the UV/PDS system. Thus, electrical energy per order (EEUV), total oxidant consumed (ox), and their corresponding CostUV and CostOX for batch reactions can be estimated using eqn (40)–(43), respectively.| |  | (40) |
| | | CostUV ($ per m−3) = EEUV (kW h m−3 order−1) × electricity cost ($ per kW h−1) | (41) |
| |  | (42) |
The total operating cost (Costtotal) of the system can be estimated using eqn (43).
| | | Costtotal ($ per m−3) = CostUV + CostOX | (43) |
Herein, EEUV is the electrical energy per order (kW h m−3 order−1), I represents the light intensity input to the photochemical system (11 μW cm−2), t signifies the reaction duration (150 min), V is the volume of liquid in the system (1 L), A is the surface area of water receiving the UV irradiation (cm2), and C0 and Ct are the initial and ultimate amounts of SA (mg L−1), respectively. The cost of electricity and PDS were considered to be $ 0.1 per kW h and $ 0.064 per kg, respectively, as per Indian market cost.81
Under the optimal experimental conditions of 50 mg L−1 of SA, 10 mg L−1 of Zn–Al@HC, PDS dosage of 0.01 M, and UV irradiation of 11 μW cm−2, UV/PDS/Zn–Al@HC consumed 0.013 kW h m−3 order−1 of EEUV, which was about 345, 2223, and 6686 folds lower than those of UV/PDS, Zn–Al@HC/UV, and Zn–Al@HC/PDS, respectively (Table 1). Similarly, the Costtotal of UV/PDS/Zn–Al@HC was estimated at 0.1536 $ per m−3 order−1 to achieve 94.45 ± 0.65% degradation of SA. The lowered EEUV and Costtotal of UV/PDS/Zn–Al@HC shows that higher pollutant elimination can result in lower energy consumption and operating costs.
Table 1 Cost assessment of UV/PDS/Zn–Al@HC, UV/PDS, Zn–Al@HC/PDS, and Zn–Al@HC/UV for the degradation of SA
| Degradation process |
UV/PDS/Zn–Al@HC |
UV/PDS |
Zn–Al@HC/UV |
Zn–Al@HC/PDS |
| EEUV (kW h m−3 order−1) |
0.0130 |
4.4879 |
28.8975 |
86.9246 |
| CostUV ($ per m−3) |
0.0013 |
0.4488 |
3.4677 |
8.6925 |
| Costox ($ per m−3) |
0.1523 |
0.1523 |
0.1523 |
0.1523 |
| Costtotal ($ per m−3) |
0.1536 |
0.6011 |
3.6200 |
8.8448 |
| Costtotal (Rs per m−3) |
12.6828 |
49.6235 |
298.8338 |
730.1381 |
An earlier investigation on the degradation of SA using an electro-Fenton system consumed 0.171 kW h m−3 order−1 of EEo and 0.22 $ m−3 to obtain 91.68% degradation of 50 mg L−1 of SA.77 Similarly, another investigation demonstrated the degradation of 50 mg L−1 of SA by 69.5% within 100 min at a pH of 3.0, requiring 11.91 kW h m−3 of electrical energy in the peroxi-coagulation process.2 Thus, compared to the earlier investigations, UV/PDS/Zn–Al@HC required considerably less electrical energy and operating cost, making it a more cost-effective treatment as an alternative for treating SA-laden wastewater. Few other investigations have also adopted different cost-effective methods to treat SA, which are summarised in Table S3.†
4. Conclusions
The present research claimed the efficacious valorisation of AD-FW using HTC for the synthesis of an economic and highly efficient Zn–Al-based photocatalyst. The novel composite of a Zn and Al precursor with the HC resulted in enhanced photocatalytic and electrochemical properties of the synthesized catalyst (Zn–Al@HC) compared to the pristine HC. Zn–Al@HC decomposed PDS to reactive radicals via UV irradiation, and established about 94.45 ± 0.65% degradation of the model pollutant SA in the setup under optimum operating conditions (Zn–Al@HC = 10 mg L−1, PDS = 0.01 M, SA = 50 mg L−1). An increment in the catalyst and PDS dosage had a positive correlation with the SA degradation rate. Despite the scavenging effect of anions Cl−, NO3−, and CO32−, SA degradation efficiencies of 66.87 ± 0.45%, 74.67 ± 0.60%, and 88.64 ± 0.10% were achieved, which highlighted the potent applicability of Zn–Al@HC under harsh conditions. In contrast, the exposure to anions F− and PO43− showed excellent SA degradation with more than 99% efficiency. Moreover, Zn–Al@HC showed satisfactory performance in real wastewater with an SA abatement efficiency of 78.19 ± 0.10%. Furthermore, the catalyst was found to be highly stable as demonstrated by 20 cycles of CV analysis. The EEUV and Costtotal of the UV/PDS/Zn–Al@HC system were 0.0130 kW h m−3 order−1 and 0.1536 $ per m−3 order−1, which evidenced the cost-effectiveness of the UV/PDS/Zn–Al@HC system compared to the other reported photocatalytic degradation systems for SA. Thus, the suggested treatment system can be implemented at the pilot or field-scale at the tertiary stage in WWTPs and can be explored for other organic CECs in the future.
5. Environmental implications
SA is extensively utilized in the production of pharmaceuticals, cosmetics, antipyretics, and painkillers. However, it causes environmental hazards due to its high toxicity, endocrine disrupting properties, non-biodegradability, and tendency to accumulate in the ecosystem. Conventional WWTPs of the industrial sector struggle to eliminate CECs from contaminated water due to their non-biodegradable nature. To address this challenge, photocatalysis systems have emerged as promising advanced tertiary treatment methods and can be added as a downstream process to any biological unit in conventional WWTPs. Photocatalysis systems offer advantages such as high degradation rates, operational simplicity, and shorter processing times. In this context, deriving photocatalysts from AD-FW can solve multiple environmental issues, such as inappropriate FW management, AD management, less waste to landfill, carbon emission and severe health hazards due to the unscientific disposal of hazardous waste.
Similarly, in an attempt to extend the scope of the procedure, metals (Zn and Al) used for photocatalyst synthesis at a large scale could also be derived from industrial waste including electroplating residues, zinc batteries, spent aluminium cans, etc. targeting the other solid waste stream (E-waste). Hence, the present novel material Zn–Al@HC photocatalyst could be a game-changing solution to reduce environmental pollution, support the concept of circular economy (Fig. 12), and promote environmental sustainability at a relatively lower cost.
 |
| | Fig. 12 Proposed field scale Zn–Al@HC implementation scheme for pollution abatement, reducing energy requirement, and environmental sustainability. | |
5.1. Novelty statement
For the first time, an AD-FW derived photocatalyst (Zn–Al@HC) synthesized using a greener approach (one-pot method, lower energy requirements, and waste-derived precursor) is utilized for remediation of SA from wastewater. Also, process optimization under diverse operating conditions and real environment application is thoroughly discussed in the present investigation, i.e., unique from previous investigations. Additionally, waste-derived catalysts cut back the cost of utilizing commercial expensive catalysts and can offer the photocatalysis technology at an affordable cost and enhance the sustainability.
Author contributions
Shraddha Yadav: conceptualization, formal analysis, investigation, methodology, writing – original draft; writing – review & editing. Monali Priyadarshini: validation, visualization, writing – original draft, data curation, writing – review & editing, investigation. Azhan Ahmad: software, visualization, writing – original draft, writing –review & editing. Makarand M. Ghangrekar: funding acquisition, project administration, resources, supervision, validation, writing – review & editing. Brajesh K. Dubey: resources, supervision, validation, writing – review & editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The current research work is funded by a research grant from the Department of Science and Technology, Government of India (File no.: DST/TMD (EWO)/OWUIS-2018/RS-10).
Notes and references
- Y. Liu, Y. Sun and Z. Peng, Desalination, 2022, 537, 115866 CrossRef CAS.
- İ. Y. Köktaş and Ö. Gökkuş, Chemosphere, 2022, 293, 133566 CrossRef PubMed.
- A. Adak, I. Das, B. Mondal, S. Koner, P. Datta and L. Blaney, Emerging Contam., 2019, 5, 53–60 CrossRef.
- A. Huang, D. Zhi and Y. Zhou, Environ. Pollut., 2021, 286, 117310 CrossRef CAS PubMed.
- M. Priyadarshini, I. Das, M. M. Ghangrekar and L. Blaney, J. Environ. Manage., 2022, 316, 115295 CrossRef CAS PubMed.
- O. K. Türk, G. Adalar, S. Yazici Guvenc, E. Can-Güven, G. Varank and A. Demir, Environ. Sci. Pollut. Res., 2023, 30, 869–883 CrossRef PubMed.
- H. Xu, T. Zhang, D. Wang, D. Cai, S. Chen, H. Wang, S. Shu and Y. Zhu, Sep. Purif. Technol., 2022, 300, 121833 CrossRef CAS.
- V. M. Daskalaki, M. Antoniadou, G. Li Puma, D. I. Kondarides and P. Lianos, Environ. Sci. Technol., 2010, 44, 7200–7205 CrossRef CAS PubMed.
- M. Mitra, A. Ghosh, A. Mondal, K. Kargupta, S. Ganguly and D. Banerjee, Appl. Surf. Sci., 2017, 402, 418–428 CrossRef CAS.
- F. Wang, Y. Liu, M. Cao, B. Zhou, H. Chen, R. Yuan, S. Liu and B. Xing, Environ. Sci. Technol., 2023, 57, 3691–3702 CrossRef CAS PubMed.
- U. P. S. Gahlaut, V. Kumar and Y. C. Goswami, Phys. E, 2020, 117, 113792 CrossRef CAS.
- X. H. Yi, H. Ji, C. C. Wang, Y. Li, Y. H. Li, C. Zhao, A. Wang, H. Fu, P. Wang, X. Zhao and W. Liu, Appl. Catal., B, 2021, 293, 120229 CrossRef CAS.
- X. H. Yi, T. Y. Wang, H. Y. Chu, Y. Gao, C. C. Wang, Y. J. Li, L. Chen, P. Wang, H. Fu, C. Zhao and W. Liu, Chem. Eng. J., 2022, 449, 137784 CrossRef CAS.
- S. Yadav, A. Ahmad, M. Priyadarshini, B. K. Dubey and M. Ghangrekar, FlatChem, 2024, 44, 100623 CrossRef CAS.
- J. Wei, Y. Liu, Y. Zhu and J. Li, Chemosphere, 2020, 247, 125854 CrossRef CAS PubMed.
- U. M. García-Pérez, A. Martínez-De La Cruz and J. Peral, Electrochim. Acta, 2012, 81, 227–232 CrossRef.
- X. Li, T. Wan, J. Qiu, H. Wei, F. Qin, Y. Wang, Y. Liao, Z. Huang and X. Tan, Appl. Catal., B, 2017, 217, 591–602 CrossRef CAS.
- U. Ghosh and A. Pal, J. Water Process Eng., 2022, 50, 103315 CrossRef.
- S. Krishnan and A. Shriwastav, Environ. Res., 2023, 216, 114568 CrossRef CAS PubMed.
- A. Kamal, U. Haroon, H. Manghwar, K. H. Alamer, I. M. Alsudays, A. T. Althobaiti, A. Iqbal, M. Akbar, M. Anar and M. Nazish, Molecules, 2022, 27, 5333 CrossRef CAS PubMed.
- H. B. Sharma and B. K. Dubey, Waste Manage., 2020, 118, 521–533 CrossRef CAS PubMed.
- S. Soltani, U. Rashid, R. Yunus and Y. H. Taufiq-Yap, Fuel, 2016, 178, 253–262 CrossRef CAS.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Mater. Sci. Eng., B, 2018, 227, 136–144 CrossRef CAS.
- A. Elhalil, R. Elmoubarki, A. Machrouhi, M. Sadiq, M. Abdennouri, S. Qourzal and N. Barka, J. Environ. Chem. Eng., 2017, 5, 3719–3726 CrossRef CAS.
- Z. Huang, L. Shi, Y. Muhammad and L. Li, J. Colloid Interface Sci., 2021, 586, 423–432 CrossRef CAS PubMed.
- L. Yang, H. Wang, J. Sun, Y. Xu, P. Li, Y. Xu and S. Wu, IET Renew. Power Gener., 2021, 15, 1397–1407 CrossRef.
- Z. Chen, H. Jia, Y. Guo, Y. Li and Z. Liu, Environ. Res., 2022, 208, 112695 CrossRef CAS PubMed.
- S. Siara, C. Elvis, R. Harishkumar and P. Velayudhaperumal Chellam, Mater. Res. Bull., 2022, 145, 111530 CrossRef CAS.
- S. Masoumi and A. K. Dalai, J. Cleaner Prod., 2020, 263, 121427 CrossRef CAS.
- Q. Li, G. Wei, Y. Yang, Z. Li, L. Zhang and Q. Huang, J. Hazard. Mater., 2020, 394, 122566 CrossRef CAS PubMed.
- P. Zhang, R. Y. Hong, Q. Chen and W. G. Feng, Powder Technol., 2014, 253, 360–367 CrossRef CAS.
- X. Zhu, Y. Liu, F. Qian, Z. Lei, Z. Zhang, S. Zhang, J. Chen and Z. J. Ren, Environ. Sci. Technol., 2017, 51, 10756–10764 CrossRef CAS PubMed.
- J. T. Titantah and D. Lamoen, Diam. Relat. Mater., 2007, 16, 581–588 CrossRef CAS.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- Z. Zhang, Z. Hua, J. Lang, Y. Song, Q. Zhang, Q. Han, H. Fan, M. Gao, X. Li and J. Yang, CrystEngComm, 2019, 21, 4607–4619 RSC.
- S. Biswas, H. Siddiqi, B. C. Meikap, T. K. Sen and M. Khiadani, Water, Air, Soil Pollut., 2020, 231(3) CAS.
- S. Sutar, S. Otari and J. Jadhav, Chemosphere, 2022, 287, 132200 CrossRef CAS PubMed.
- M. Ramani, S. Ponnusamy and C. Muthamizhchelvan, Opt. Mater., 2012, 34, 817–820 CrossRef CAS.
- S. Suwanboon, P. Amornpitoksuk, T. Rattana and C. Randorn, Ceram. Int., 2020, 46, 21958–21977 CrossRef CAS.
- K. V. Kumar and S. D. Bhavani, East Eur. J. Phys., 2023, 2023, 355–362 CrossRef.
- K. Guan, P. Zhou, J. Zhang and L. Zhu, J. Mol. Struct., 2020, 1203, 127425 CrossRef CAS.
- M. Amir, T. Fazal, J. Iqbal, A. A. Din, A. Ahmed, A. Ali, A. Razzaq, Z. Ali, M. S. U. Rehman and Y. K. Park, J. Ind. Eng. Chem., 2022, 115, 171–182 CrossRef CAS.
- N. Iqbal, J. Photochem. Photobiol., A, 2022, 427, 113813 CrossRef CAS.
- D. Roy, S. Neogi and S. De, Chem. Eng. J., 2022, 430, 133069 CrossRef CAS.
- J. Liu, J. Ma, X. Du, F. Han, S. Shi and G. Yang, J. Alloys Compd., 2021, 862, 158017 CrossRef CAS.
- A. K. Dey, S. R. Mishra and M. Ahmaruzzaman, Environ. Sci. Pollut. Res., 2023, 30, 53887–53903 CrossRef CAS PubMed.
- X. An, Y. Chen, M. Ao, Y. Jin, L. Zhan, B. Yu, Z. Wu and P. Jiang, Chem. Eng. J., 2022, 435, 135087 CrossRef CAS.
- D. C. Madu, S. M. Islam, H. Pan and C. J. Barile, J. Mater. Chem. C, 2021, 9, 6297–6307 RSC.
- Y. Nosaka and A. Nosaka, ACS Energy Lett., 2016, 1, 356–359 CrossRef CAS.
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Chem, 2021, 7, 38–63 CAS.
- A. Fernandes, M. J. Nunes, A. S. Rodrigues, M. J. Pacheco, L. Ciríaco and A. Lopes, Molecules, 2021, 26, 4821 CrossRef CAS PubMed.
- Y. Liang, Z. Zeng, J. Yang, G. Yang and Y. Han, Colloids Surf., A, 2021, 624, 126796 CrossRef CAS.
- Z. Guo, M. Deng, H. Wang, X. Xiang, C. Zhang and B. Wang, J. Phys. Chem. Solids, 2024, 185, 111729 CrossRef CAS.
- B. Sun, H. Li, Q. Wei, S. Xue, A. Zhou and X. Yue, Sep. Purif. Technol., 2022, 301, 122039 CrossRef CAS.
- H. Heidarpour, M. Padervand, M. Soltanieh and M. Vossoughi, Chem. Eng. Res. Des., 2020, 153, 709–720 CrossRef CAS.
- R. Li, H. Hu, Y. Ma, X. Liu, L. Zhang, S. Zhou, B. Deng, H. Lin and H. Zhang, J. Cleaner Prod., 2020, 276, 124246 CrossRef CAS.
- D. Lambropoulou, E. Evgenidou, V. Saliverou, C. Kosma and I. Konstantinou, J. Hazard. Mater., 2017, 323, 513–526 CrossRef CAS PubMed.
- S. Bekkouche, S. Merouani, O. Hamdaoui and M. Bouhelassa, J. Photochem. Photobiol., A, 2017, 345, 80–91 CrossRef CAS.
- A. Tufail, W. E. Price and F. I. Hai, Membranes, 2020, 10, 1–15 CrossRef PubMed.
- C. Tan, N. Gao, S. Zhou, Y. Xiao and Z. Zhuang, Chem. Eng. J., 2014, 253, 229–236 CrossRef CAS.
- C. Liang, Z. S. Wang and N. Mohanty, Sci. Total Environ., 2006, 370, 271–277 CrossRef CAS PubMed.
- X. Gao, Q. Guo, G. Tang, W. Peng, Y. Luo and D. He, J. Water Reuse Desalin., 2019, 9, 301–309 CrossRef CAS.
- S. Dhaka, R. Kumar, S. hun Lee, M. B. Kurade and B. H. Jeon, J. Cleaner Prod., 2018, 180, 505–513 CrossRef CAS.
- J. Deng, Y. Shao, N. Gao, Y. Deng, S. Zhou and X. Hu, Chem. Eng. J., 2013, 228, 765–771 CrossRef CAS.
- C. Tan, D. Fu, N. Gao, Q. Qin, Y. Xu and H. Xiang, J. Photochem. Photobiol., A, 2017, 332, 406–412 CrossRef CAS.
- M. Abdelbaky, A. M. Abdelghany, A. H. Oraby, E. M. Abdelrazek and M. M. Rashad, J. Inorg. Organomet. Polym. Mater., 2023, 33, 3304–3314 CrossRef CAS.
- S. Ding, J. Wan, Y. Wang, Z. Yan and Y. Ma, Chem. Eng. J., 2021, 422, 130406 CrossRef CAS.
- M. Sabri, A. Habibi-Yangjeh and S. Vadivel, Mater. Chem. Phys., 2020, 239, 121988 CrossRef CAS.
- A. A. Babaei, M. Golshan and B. Kakavandi, Process Saf. Environ. Prot., 2021, 149, 35–47 CrossRef CAS.
- E. Guinea, C. Arias, P. L. Cabot, J. A. Garrido, R. M. Rodríguez, F. Centellas and E. Brillas, Water Res., 2008, 42, 499–511 CrossRef CAS PubMed.
- N. Rabaaoui and M. S. Allagui, J. Hazard. Mater., 2012, 243, 187–192 CrossRef CAS PubMed.
- J. Wang, M. Xu, X. Liang, Y. Zhang, D. Yang, L. Pan, W. Fang, C. G. Zhu and F. Wang, Sep. Purif. Technol., 2021, 263, 118368 CrossRef CAS.
- D. Mukherjee, A. K. Ray and S. Barghi, Processes, 2016, 4, 13 CrossRef.
- S. J. George, R. Gandhimathi, P. V. Nidheesh and S. T. Ramesh, Clean: Soil, Air, Water, 2014, 42, 1701–1711 CAS.
- S. S. Shinde, C. H. Bhosale and K. Y. Rajpure, J. Mol. Catal. A: Chem., 2011, 347, 65–72 CrossRef CAS.
- B. Garza-Campos, E. Brillas, A. Hernández-Ramírez, A. El-Ghenymy, J. L. Guzmán-Mar and E. J. Ruiz-Ruiz, J. Hazard. Mater., 2016, 319, 34–42 CrossRef CAS PubMed.
- M. Priyadarshini, A. Ahmad and M. M. Ghangrekar, Environ. Pollut., 2023, 322, 121242 CrossRef CAS PubMed.
- A. Ahmad, M. Priyadarshini, I. Das, M. M. Ghangrekar and R. Y. Surampalli, J. Environ. Chem. Eng., 2023, 11, 109049 CrossRef CAS.
-
M. of E. and P. British Columbia, Aluminum Water Quality Guidelines – Wildlife , Livestock Watering , and Irrigation (Reformatted Guideline from 1988), Ministry of Water,Land and Resource Stewardship, 1988 Search PubMed.
-
M. of E. L. and P. British Columbia, Ambient Water Quality Guidelines for Zinc. Water Management Branch Ministry of Environment and Climate Change Strategy, 1999 Search PubMed.
- Y. Zhang, Y. Xiao, Y. Zhong and T. T. Lim, Chem. Eng. J., 2019, 372, 420–428 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Brajesh K.
Dubey
b
*ab and
Brajesh K.
Dubey
b