
Disparity among cyclic alkyl carbonates associated with the cathode–electrolyte interphase at high voltage†
Shuaishuai
Chen
a,
YiHan
Tang
a,
Zhaoxin
Lu
a,
Shun
Wu
a,
Jiliang
Wu
*b,
Zhenlian
Chen
*a and
Deyu
Wang
 *a
*a
aKey Laboratory of Optoelectronic Chemical Materials and Devices, Ministry of Education, School of Optoelectronic Materials & Technology, Jianghan University, Wuhan 430056, China. E-mail: zhenlianchen@jhun.edu.cn; wangdeyu@jhun.edu.cn
bWuhan Zhongyuan Changjiang Technology Development Co. Ltd., Wuhan 430090, China. E-mail: jlwu@whu.edu.cn
First published on 10th June 2024
Abstract
Elevating cut-off voltage is an efficient approach to boost the energy density of Li-ion batteries. However, the failure of conventional carbonate-based electrolyte at high voltage necessitates fundamental understandings of the formation mechanism of the cathode–electrolyte interphase (CEI), especially the contribution from skeleton solvents. In the present work, density functional theory (DFT) calculations reveal the disparity in the productivity and oxidative stability of organic components in the CEI associated with ethylene carbonate (EC), propylene carbonate (PC) and fluoroethylene carbonate (FEC). These two indicators could jointly determine the existence of lithium alkyl carbonates in the CEI, suggested by the species with a m/z of 95 detected with time of flight secondary ion mass spectroscopy (TOF-SIMS). That disparity results in a prominent difference in the capacity retention at high voltage associated with EC, PC and FEC, respectively. These understandings of cyclic carbonates could shed light on bottom-up strategies in electrolyte engineering for the high-voltage electrolyte.
Introduction
Energy density is one of the most important technical indicators of the battery for large-scale energy storage, electric vehicles, and digital products.1 At present, the most practicable way to promote the energy density of lithium-ion batteries is to elevate the cut-off voltage to push the actual capacity to the theoretical limit.2,3 It challenges not only the preparation process of cathode materials, but also the interface stability between the cathode and the electrolyte. One strategy of electrolyte engineering is to ameliorate the precipitation of the conventional electrolyte. Many kinds of functional additives to prevent the side reactions of skeleton solvents have been proposed to promote the electrochemical performance of the lithium-ion battery at high cut-off voltage.4–6 However, interfacial reactions dominating the formation of the CEI, especially the parasitic reaction of skeleton solvents with the cathode and its contribution to the CEI, are still poorly understood, restricting the bottom-up design strategy of electrolyte engineering.The main solvents in state-of-the-art electrolytes comprise linear alkyl carbonates and cyclic alkyl carbonates. Linear alkyl carbonates are easy to oxidatively decompose into species with low weight to be involved in the interfacial reaction on the cathode, leading to products in gas or liquid phases, while for cyclic alkyl carbonates, it has been speculated that they are also decomposed to radicals with low weight to form some types of solid products in the CEI layer, which is similar to the reduction reaction that contributes to organic components of the SEI.7–10 And extensive research had found that ethylene carbonate (EC), the skeleton solvent most widely used in commercial electrolyte for conventional batteries, is poorly compatible with the positive electrode at high voltage. A variety of reaction mechanisms to understand the interfacial reaction of EC on the cathode have been proposed, such as nucleophilic attack, electrophilic attack, dehydrogenation reactions and ring opening reactions.11–13 However, the acceptable working mechanism of EC at high voltage is still lacking.
In contrast, fluoroethylene carbonate (FEC), with one H atom of EC replaced by a F atom, plays an indispensable role in forming LiF contributing to inorganic components and increasing the roughness of both the SEI and CEI. It has also been proposed that LEDCF, a counterpart of LEDC, formed with the reduction reaction on the anode surface.9,14,15 At the cathode–electrolyte interface, when involved in the oxidation reaction, density functional theory (DFT) calculations suggested that the oxidative decomposition path of FEC is similar to that of EC leading to small fragment species.16 Whether FEC only contributes to inorganic LiF in the CEI, or also contributes to organic components, is still unclear.
Propylene carbonate (PC), with one H atom of EC replaced by a methyl group, had been excluded from the skeleton solvent for commercial batteries due to its poor compatibility with graphite associated with the solvation sheath.17 In fact, PC has superior properties including a high dielectric constant, a low melting point, a broad liquid-phase range, a wide electrochemical window, and good compatibility with various transition metal oxide cathode materials.18–20 And it also showed good compatibility with cathodes at high voltage.21 It seems that the long-term “EC–PC disparity” in compatibility with graphite anodes may flip to compatibility with cathodes at high voltage. However, the DFT calculations showed that the HOMO–LUMO energy level as well as both the reduction and oxidation reaction paths do not change significantly with the replacement of the H atom with the methyl group.17,22 This intriguing flip may play a vital role in future electrolyte engineering and demands further fundamental understanding at the molecular level.
In the present work, comparison among the three solvent molecules is carried out with DFT calculations combined with experiments. The calculations on the oxidative decomposition of the solvents indicate that the possibility of the intermediate radicals of PC before the dissociation reaction to form lithium alkyl carbonates is highest and the oxidative stability of the lithium alkyl carbonate associated with FEC is highest. To validate this prediction, the electrochemical performance and detection of CEI composition are carried out on a Li|LiCoO2 battery at a cut-off voltage of 4.3 V and 4.6 V vs. Li/Li+ in electrolytes composed of EC, FEC and PC, respectively, combined with linear carbonates of ethyl methyl carbonate (EMC) and di-methyl carbonate (DMC). Compared with the cut-off voltage of 4.3 V vs. Li/Li+, new species with a m/z of 95, which is most likely LiCO3C2H4−, emerged in the CEI for both PC- and FEC-systems, but not for the EC-system, at the cut-off voltage of 4.6 V. This could explain the rapid capacity fading at the cut-off voltage of 4.6 V vs. Li/Li+ in the EC-system. This work correlates the electrochemical performances and the protection efficiency of the CEI with the productivity as well as oxidative stability of the deposited organic components such as lithium alkyl carbonates, as shown in Fig. 1. The fundamental understanding of the disparity among the three skeleton solvents may shed light on the strategy of electrolyte engineering to boost the energy density of LIBs.
 | ||
| Fig. 1 The schematic of CEI evolution. | ||
Results and discussion
Oxidative decomposition paths contributing to organic components of the CEI
The CEI is generally considered a complex heterogeneous mixture of organic and inorganic compounds.23 For inorganic components such as LiF, Li2CO3 and LixPyFOz, its formation with oxidative decomposition of electrolytes is easily understood and well identified, while for organic components such as lithium alkyl carbonates, the formation and identification are still inconclusive. To understand the disparity in the contribution to organic CEI components from the oxidative decomposition reaction of the three skeleton cyclic alkyl carbonate solvents, reinvestigation on the decomposition path is performed with DFT calculations. The investigation of the oxidation paths in the present work does not focus on the final products with low molecular weight, which have been thoroughly investigated in previous DFT calculations,16,17,24 but on the intermediate state with relatively high molecular weight that could react with other species and ions to form organic components of the CEI. Two kinds of oxidative decomposition process initialized with one electron and one H atom abstracted from the cyclic alkyl carbonates, respectively, are taken into account, and the energy profiles and corresponding images are shown in Fig. 2. | ||
| Fig. 2 Potential energy profile and corresponding images for the oxidative decomposition processes calculated with B3LYP/6-311+G(d,p) methods. (a) For EC+, FEC+, and PC+; (b) for [EC–H], [FEC–H], and [PC–H]. | ||
During the decomposition of the cyclic alkyl carbonates with one electron abstracted, i.e., EC+, FEC+ and PC+, the ring is preferred to be opened with the cleavage of the C–O bond linked with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. When the following dissociation reaction goes through with the cleavage of the C–O bond linked with C–C bond concurrent with H-atom transfer, it leads to gaseous molecule CO2 and the positive radicals OCHCH3+ (acetaldehyde+), OCHCH2F+ (fluoroacetaldehyde+) and H3COCH3+ (acetone+), for EC+, FEC+ and PC+, respectively. This path had been calculated for EC+, but not for FEC+ and PC+, yet.17 And both OCHCH2F+ and H3COCH3+ are more stable than the positive radicals with the same weight generated via other paths.16 For PC+, the energy barrier of the ring-opening is the lowest at about 0.21 eV and of the following dissociation reaction is highest as about 0.29 eV; whereas for EC+, it is the highest at about 0.24 eV for the ring-opening and is the lowest at about 0.20 eV for the dissociation reaction. This suggests that the possibility of forming organic CEI components with the intermediate species during decomposition of PC+ is highest and of EC+ is lowest. Because the carbonate group is dissociated in this kind of intermediate species, it is accessible to form ether-related organic components, and it has to capture other species to form carbonate-related ones.
O. When the following dissociation reaction goes through with the cleavage of the C–O bond linked with C–C bond concurrent with H-atom transfer, it leads to gaseous molecule CO2 and the positive radicals OCHCH3+ (acetaldehyde+), OCHCH2F+ (fluoroacetaldehyde+) and H3COCH3+ (acetone+), for EC+, FEC+ and PC+, respectively. This path had been calculated for EC+, but not for FEC+ and PC+, yet.17 And both OCHCH2F+ and H3COCH3+ are more stable than the positive radicals with the same weight generated via other paths.16 For PC+, the energy barrier of the ring-opening is the lowest at about 0.21 eV and of the following dissociation reaction is highest as about 0.29 eV; whereas for EC+, it is the highest at about 0.24 eV for the ring-opening and is the lowest at about 0.20 eV for the dissociation reaction. This suggests that the possibility of forming organic CEI components with the intermediate species during decomposition of PC+ is highest and of EC+ is lowest. Because the carbonate group is dissociated in this kind of intermediate species, it is accessible to form ether-related organic components, and it has to capture other species to form carbonate-related ones.
It is widely recognized that lithium alkyl carbonates play an important role in the SEI, while their existence in the CEI is still inconclusive. The formation of lithium alkyl carbonates may necessitate the carbonate group being maintained during the ring opening step, i.e., cleavage of the C–O bond linked with the C–C bond rather than the one linked with the CO bond as shown in Fig. 2a. However, the previous calculations reveal that the energy barriers of the former decomposition path are several times of those of the latter one. So, H-abstraction, which has also been proposed to trigger the dissociation of the electrolyte molecules, especially on the oxide cathode surface, is taken as the preliminary of the decomposition shown in Fig. 2b. When the decomposition is initialized with one H atom abstracted from EC and FEC, the ring is preferred to be opened with the cleavage of both C–O bonds linked with the C–C bond and CO bond, directly leading to gaseous CO2 and positive radicals with low weight. However for PC, the ring is preferred to be opened with the cleavage of the C–O bonds linked with the C–C bond, leading to an intermediate radical OCO2CH2C2H3+. Within this species, the carbonate group is maintained and it could be accessible to form lithium alkyl carbonates, unlike the intermediate radicals generated in the oxidative decomposition path with the carbonate group dissociated.
Oxidative stability of organic components
The oxidative stability of organic components plays a critical role in the quality of the CEI. In the present work, the energy levels of the HOMO (the highest occupied molecular orbital) and change in free energy with oxidation (ΔGO) and with H-atom abstraction (ΔGH) are utilized to evaluate the oxidative stability.17,25 It is worth noting that the ether group, aldehyde group, and carbonyl group are less stable than the carbonate group.26,27 The calculated values are collected for the species proposed in previous studies28 as listed in Table S1† and Fig. 3 shows the results for 11 kinds of lithium alkyl carbonates. One can find that the products associated with FEC, especially LFEC and LDFDC, show high oxidative stability, while the products associated with EC and PC show lower oxidative stability. More information about the molecules is shown in Fig. S1, Tables S2 and S3.† | ||
| Fig. 3 The calculated energy level of the HOMO, and the change in free energy with oxidation (ΔGO) and dehydrogenation (ΔGH) for 11 kinds of lithium alkyl carbonates. | ||
The low productivity of carbonate-related organic components with oxidative decomposition reactions combined with the moderate or low oxidative stability of the organic species associated with EC could explain the failure of EC-based electrolyte at high cut-off voltage. And the moderate productivity of carbonate-related organic components combined with the high oxidative stability of the organic species associated with FEC suggests that FEC contributes not only to inorganic but also organic components in the CEI. And the high productivity of carbonate-related organic components associated with two kinds of oxidative decomposition paths combined with the moderate oxidative stability of the organic species associated with PC suggests that the ratio of organic components in the CEI could play a critical role. Recently, a continuum-level model informed by reaction mechanisms obtained from first-principles calculations has been developed to capture the complexities of SEI chemistry and predict the evolution of the SEI.29,30 The calculations on the productivity and oxidative stability of the species could contribute to the simulation at the continuum-level for the CEI.31
Electrochemical evaluation
To investigate the intrinsic electrochemical oxidative decomposition of electrolyte and the oxidative stability of passivation films, linear scanning voltammetry (LSV) is performed with a scan rate of 0.1 mV s−1 between 3 and 6 V vs. Li/Li+ with fresh Super-P electrodes and the cycled Super-P electrode (2.8–4.6 V vs. Li/Li+, 5 cycles), respectively. Through the comparison before and after the cycles shown in Fig. 4, it can be found that the three systems can be stable until the voltage is increased to higher than 5 V vs. Li/Li+. As shown in Fig. 4b and d, the current density at 4.6 V vs. Li/Li+ with the cycled electrodes is a quarter of those of fresh electrodes, indicating that the CEI layers were formed to suppress the electrolyte decomposition. Furthermore, the currents of EC-based systems with used electrodes are still ∼3 times higher than those of the other two systems, indicating the quick consumption of EC-based electrolyte. | ||
| Fig. 4 Linear sweep voltammetry (LSV) curves of electrolytes in Super-P/Li cells at a scan rate of 0.1 mV s−1. (a) Fresh cell, (b) fresh cell at 4.55–4.65 V, (c) cycled cell, and (d) cycled cell at 4.55–4.65 V. | ||
Constant-voltage polarization was also performed for the investigated systems to compare their stability at high potential. In contrast to the fewer variations polarized at 4.3 V vs. Li/Li+, the current densities polarized at 4.6 V vs. Li/Li+ for 10 h show obvious differences, as shown in Fig. S4.† EC-based cells still maintain ∼3.83 μA cm−2, which is ∼3.8 & 2.1 times that of FEC- & PC-based cells. It is worth noting that the leak current for the FEC-based system is alleviated from ∼1.67 to ∼1.03 μA cm−2 as the increase in polarized potential enhanced from 4.3 to 4.6 V vs. Li/Li+. These results indicate that higher amount of EC molecules is required to be decomposed to repair the CEI layer at high potential.
To verify the understanding from the DFT calculations, electrochemical performances of Li|LiCoO2 batteries run with 1 mol per L LiPF6 dissolved in a mixture of EMC & DMC with EC, FEC and PC, respectively, by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in volume, are investigated and shown in Fig. 5 and S5.† The initial discharge capacity and capacity retention for the three systems are very close to each other when run between 2.8 and 4.3 V. The cells with these electrolytes deliver discharge capacities of 157.0, 155.1, & 156.2 mA h g−1 respectively in the first cycle and maintain 69.1%, 79.9% and 74.6% of their initial capacities in the 300th cycle, respectively. The disparity is significant when the systems run between 2.8 and 4.6 V. They present 174.8, 194.8, and 201.4 mA h g−1 respectively in initial cycles. The capacity retention of EC-based sample is significantly degraded to 7.7% in the 300th cycle, in sharp contrast to the high values of 70.2 and 66.1% for FEC- and PC-based systems, respectively. This obvious capacity fading of the EC-based cell could stem from the less stable CEI components, resulting from the low decomposition reaction barrier of the dissociation of EC to gaseous molecules as well as the low oxidative stability of the products deposited on the cathode, as suggested with the DFT calculations. In contrast, LFEC and LDFDC derived from the decomposition of FEC can inhibit the decomposition of FEC thanks to high oxidative stability. And the stabilizing effect of methyl in PC solvents enhances the electrochemical stability of CEI components, which inhibits further decomposition reactions of the electrolyte. Furthermore, the high productivity of lithium alkyl carbonates owing to the high dissociation barrier of PC further improves the effectiveness of forming and repairing the CEI layer.
:1:1 in volume, are investigated and shown in Fig. 5 and S5.† The initial discharge capacity and capacity retention for the three systems are very close to each other when run between 2.8 and 4.3 V. The cells with these electrolytes deliver discharge capacities of 157.0, 155.1, & 156.2 mA h g−1 respectively in the first cycle and maintain 69.1%, 79.9% and 74.6% of their initial capacities in the 300th cycle, respectively. The disparity is significant when the systems run between 2.8 and 4.6 V. They present 174.8, 194.8, and 201.4 mA h g−1 respectively in initial cycles. The capacity retention of EC-based sample is significantly degraded to 7.7% in the 300th cycle, in sharp contrast to the high values of 70.2 and 66.1% for FEC- and PC-based systems, respectively. This obvious capacity fading of the EC-based cell could stem from the less stable CEI components, resulting from the low decomposition reaction barrier of the dissociation of EC to gaseous molecules as well as the low oxidative stability of the products deposited on the cathode, as suggested with the DFT calculations. In contrast, LFEC and LDFDC derived from the decomposition of FEC can inhibit the decomposition of FEC thanks to high oxidative stability. And the stabilizing effect of methyl in PC solvents enhances the electrochemical stability of CEI components, which inhibits further decomposition reactions of the electrolyte. Furthermore, the high productivity of lithium alkyl carbonates owing to the high dissociation barrier of PC further improves the effectiveness of forming and repairing the CEI layer.
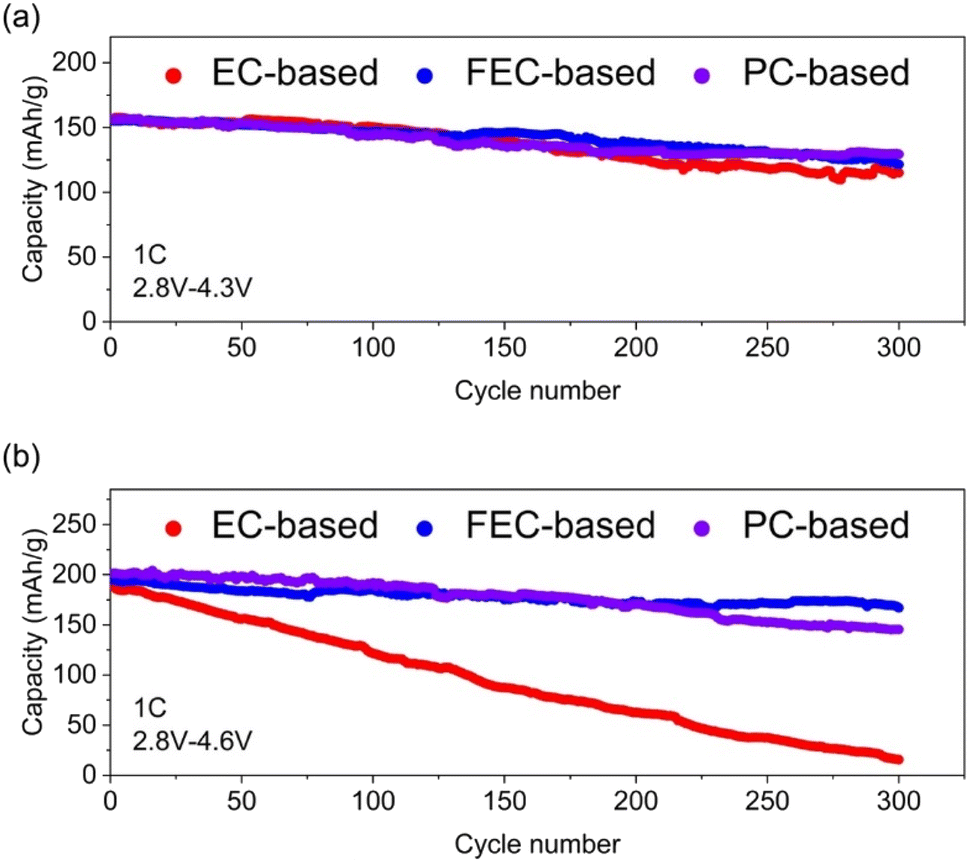 | ||
| Fig. 5 Electrochemical performance of Li/LiCoO2 cells with different electrolytes; the rate is 1C. (a) 2.8–4.3 V vs. Li/Li+ and (b) 2.8–4.6 V vs. Li/Li+. | ||
Mass spectroscopy analysis of CEI components
To understand the role of the CEI components in the electrochemical performances, characterization with time of flight secondary ion mass spectrometry(TOF-SIMS), scanning electron microscopy (SEM), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) are carried out on the surface of cathode LiCoO2 cycled between 2.8 and 4.3/4.6 V vs. Li/Li+ for 5 cycles.For the cathodes cycled in the three kinds of electrolytes between 2.8 and 4.3 V vs. Li/Li+, the species and the evolution of the intensity of the species with etching depth are very close to each other (Fig. S6–S9†). The intensities of the species with m/z 7 and 59 increase rapidly and then very slowly as the etching depth increases. It is reasonable to assign them to Li− and Co−, attributed to the active cathode material. This result also suggests that the cathodes are covered with CEI components. And the species with m/z 17, 26, 45, 67,74, 79, 89, 96 and 105 rapidly decrease in density with etching depth, indicating that these species are associated with the CEI components covered on the cathode surface and can be assigned to OH−, LiF−, LiF2−, LiCO3−, Li2CO3−, PO3−, LiFPO2−, LiPFO3− and Li2PFO3−, respectively.32,33 Except for OH−, the other species are associated with inorganic components Li2CO3 and LixPFyOz.34 The intensity of the other species with low m/z values of 1, 7, 12, 16, and 19 is almost unchanged with etching depth, and these species can be assigned to single elements H, Li, C, O and F, respectively.
When the cathodes are cycled between 2.8 and 4.6 V vs. Li/Li+, the types of species are unchanged for the cathode cycled in EC-based electrolyte, whereas new species with m/z 31 & 95 are detected for cathodes cycled in FEC-based electrolyte and new species with m/z 15, 31 & 95 are detected for cathodes cycled in PC-based electrolytes (Fig. S10–S12†). The species with m/z 15 should correspond to CH3−, the fingerprint of the methyl group in PC molecules. The species with m/z 31 could be associated with OCH3− coming from CH3OCO2Li and/or OP(OCH3)3,35 which show a slow decrease with etching time. And the species with m/z 95 is most likely to be LiOCO2C2H4−, which shows rapid decreases with etching depth. This species should be tightly correlated with the oxidative decomposition of FEC and PC at a high cut-off voltage of 4.6 V vs. Li/Li+. The early MS study also found that the species with a m/z of 95 in the CEI formed in PC-based electrolyte but were absent in EC-based electrolyte.36 This species could be associated with carbonate-related but not ether-related organic CEI components, such as LFEC, LFDC, LDFDC and LPC, which can resist high cut-off voltage as predicted with the aforementioned DFT calculations. And the absence of this species for EC-based systems could be attributed to both the accessibility to be dissociated to fragments with low molecular weight and the low oxidative stability of the carbonate-related CEI components predicted with DFT calculations.
Except for the absence of LiOCO2C2H4− related species, the intensity of the species associated with inorganic CEI components, such as LixPyFOz and Li2CO3, for EC-based systems is also much lower than that for FEC- and PC-based systems. And the intensity of LiF is highest for FEC-based systems and lowest for EC-based systems (cf. the inset overlay in Fig. 6). This is in line with the thickness of the CEI layer observed with SEM and TEM (Fig. 7 and S13†) where the CEI layer in EC-based systems is much thinner than those in FEC- and PC-based systems. Meanwhile, when we compared the TEM images at 4.3 V vs. Li/Li+, as shown in Fig. S14,† it can be seen that there is an amorphous layer on the surface of the particles, the thickness is about 5–10 nm, and the morphology and structure are not significantly different. This is also consistent with the intensity of the peaks assigned to LiF in F 1s spectra (Table S4†). The peak in the C 1s spectrum assigned to O–CO can be attributed to lithium alkyl carbonate products (such as LiOCO2C2H5 and derivatives). It can be seen that the corresponding intensity of EC-based systems is the lowest, suggesting that there are few organic products remaining on the surface, consistent with DFT predictions. It is worth noting that the binding energies associated with –CF2 & C–O in FEC-based electrolyte are higher than those in the other two samples, indicating that FEC and its derivatives are involved in the formation of organic CEI components. Moreover, the signals of Co 2p were observable but lower than those of pristine sample, indicating the existence of the thin CEI layer on the LiCoO2 surface (Fig. S15†), which is consistent with the results observed via TEM.
 | ||
| Fig. 6 TOF-SIMS of the cathode surface cycled between 2.8 and 4.6 V vs. Li/Li+ collected with negative mode and 3D distribution overlay of some fragments. (a) EC-based, (b) FEC-based, and (c) PC-based. (The values of m/z are rounded to an integer.) | ||
 | ||
| Fig. 7 TEM images and XPS spectra of the LiCoO2 electrode after 5 charge/discharge cycles with different electrolytes at 4.6 V vs. Li/Li+. | ||
Conclusion
In the present work, comparative investigations of three cyclic alkyl carbonates on CEI stability have been carried out with DFT calculations combined with experiments. It was found from the DFT calculations that the radicals from PC possess the highest dissociation reaction barrier, whereas FEC-related products have the highest oxidative stability, suggesting that both solvents are beneficial to form a stable CEI layer. The calculations on the oxidative decomposition path of the solvents and the oxidative stability of the possible organic products predicted a higher ratio of organic components in the CEI of PC- and FEC-based systems than that of EC. In the experimental part, it was found that the capacity decayed rapidly at a cut-off voltage of 4.6 V vs. Li/Li+ in EC-systems, as well as high current densities in the tests of linear scanning and constant-voltage polarization. When cycled between 2.8 and 4.6 V vs. Li/Li+, new components emerged in the CEI for both PC- and FEC-systems but not for EC-systems. One important fragment with a m/z of 95 is most likely LiCO3C2H4−, an important fragment associated with the major components in the CEI that play a critical role in the interphase stability. The approaches in this work demonstrated that the fundamental understanding sheds light on electrolyte engineering, as well as developing other critical cutting-edge technologies for lithium batteries.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22179052) and the Excellent Discipline Cultivation Project by JHUN (2023XKZ013).References
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef
.
- O. Borodin, Curr. Opin. Electrochem., 2019, 13, 86–93 CrossRef CAS
- S. Klein, S. van Wickeren, S. Röser, P. Bärmann, K. Borzutzki, B. Heidrich, M. Börner, M. Winter, T. Placke and J. Kasnatscheew, Adv. Energy Mater., 2021, 11, 2003738 CrossRef CAS
- S. Tan, Y. J. Ji, Z. R. Zhang and Y. Yang, ChemPhysChem, 2014, 15, 1956–1969 CrossRef CAS PubMed
- J. Xu, Nano-Micro Lett., 2022, 14, 166 CrossRef CAS PubMed
- M. Zhou, C. Qin, Z. Liu, L. Feng, X. Su, Y. Chen, L. Xia, Y. Xia and Z. Liu, Appl. Surf. Sci., 2017, 403, 260–266 CrossRef CAS
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS PubMed
- E. Björklund, M. Göttlinger, K. Edström, D. Brandell and R. Younesi, ChemElectroChem, 2019, 6, 3429–3436 CrossRef
- Y. Jin, N. H. Kneusels, L. E. Marbella, E. Castillo-Martinez, P. Magusin, R. S. Weatherup, E. Jonsson, T. Liu, S. Paul and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 9854–9867 CrossRef CAS PubMed
- X. Qin, P. B. Balbuena and M. Shao, J. Phys. Chem. C, 2019, 123, 14449–14458 CrossRef CAS
- T. Zhang and E. Paillard, Front. Chem. Sci. Eng., 2018, 12, 577–591 CrossRef CAS
- W. M. Dose, I. Temprano, J. P. Allen, E. Bjorklund, C. A. O'Keefe, W. Li, B. L. Mehdi, R. S. Weatherup, M. F. L. De Volder and C. P. Grey, ACS Appl. Mater. Interfaces, 2022, 14, 13206–13222 CrossRef CAS PubMed
- Y. X. Yao, N. Yao, X. R. Zhou, Z. H. Li, X. Y. Yue, C. Yan and Q. Zhang, Adv. Mater., 2022, 34, e2206448 CrossRef PubMed
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS
- C. Ma, F. Xu and T. Song, ACS Appl. Mater. Interfaces, 2022, 14, 20197–20207 CrossRef CAS PubMed
- L. Xia, B. Tang, L. Yao, K. Wang, A. Cheris, Y. Pan, S. Lee, Y. Xia, G. Z. Chen and Z. Liu, ChemistrySelect, 2017, 2, 7353–7361 CrossRef CAS
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. v. W. Cresce, K. Xu, Q. Li and W. Li, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS
- H. Kaneko, K. Sekine and T. Takamura, J. Power Sources, 2005, 146, 142–145 CrossRef CAS
- B. Wang, H. P. Zhang, L. C. Yang, Q. T. Qu, Y. P. Wu, C. L. Gan and D. L. Zhou, Electrochem. Commun., 2008, 10, 1571–1574 CrossRef CAS
- R. Wagner, S. Brox, J. Kasnatscheew, D. R. Gallus, M. Amereller, I. Cekic-Laskovic and M. Winter, Electrochem. Commun., 2014, 40, 80–83 CrossRef CAS
- H. Q. Pham, E. H. Hwang, Y. G. Kwon and S. W. Song, Chem. Commun., 2019, 55, 1256–1258 RSC
- L. Xing, W. Li, M. Xu, X. Meng and S. Zhao, J. Phys. Chem. B, 2009, 113, 5181–5187 CrossRef CAS
- Sungjemmenla, V. S. K, C. B. Soni, V. Kumar and Z. W. Seh, Energy Technol., 2022, 10, 2200421 CrossRef CAS
- A. S. Wotango, W. N. Su, E. G. Leggesse, A. M. Haregewoin, M. H. Lin, T. A. Zegeye, J. H. Cheng and B. J. Hwang, ACS Appl. Mater. Interfaces, 2017, 9, 2410–2420 CrossRef CAS
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC
- T. Matsushita, K. Dokko and K. Kanamura, J. Power Sources, 2005, 146, 360–364 CrossRef CAS
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Energy Environ. Sci., 2020, 13, 183–199 RSC
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2020, 11, 2003092 CrossRef
- S. C. DeCaluwe, P. J. Weddle, H. Y. Zhu, A. M. Colclasure, W. G. Bessler, G. S. Jackson and R. J. Kee, J. Electrochem. Soc., 2018, 165, E637–E658 CrossRef CAS
- P. J. Weddle, E. W. C. Spotte-Smith, A. Verma, H. D. Patel, K. Fink, B. J. T. de Villers, M. C. Schulze, S. M. Blau, K. A. Smith, K. A. Persson and A. M. Colclasure, Electrochim. Acta, 2023, 468, 143121 CrossRef CAS
- D. Atkins, E. Ayerbe, A. Benayad, F. G. Capone, E. Capria, I. E. Castelli, I. Cekic-Laskovic, R. Ciria, L. Dudy, K. Edström, M. R. Johnson, H. Li, J. M. G. Lastra, M. L. De Souza, V. Meunier, M. Morcrette, H. Reichert, P. Simon, J. P. Rueff, J. Sottmann, W. Wenzel and A. Grimaud, Adv. Energy Mater., 2021, 12, 2102687 CrossRef
- J. Li, W. Li, Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1802957 Search PubMed
- Y. Wu, X. Feng, Z. Ma, L. Gao, Y. Wang, C.-Z. Zhao, D. Ren, M. Yang, C. Xu, L. Wang, X. He, L. Lu and M. Ouyang, eTransportation, 2023, 15, 100216 CrossRef
- H. Sheng, X. H. Meng, D. D. Xiao, M. Fan, W. P. Chen, J. Wan, J. Tang, Y. G. Zou, F. Wang, R. Wen, J. L. Shi and Y. G. Guo, Adv. Mater., 2022, 34, 2108947 CrossRef CAS
- R. Dedryvère, S. Laruelle, S. Grugeon, L. Gireaud, J.-M. Tarascon and D. Gonbeau, J. Electrochem. Soc., 2005, 689–696 CrossRef
- M. Arakawa and J. Yamaki, J. Power Sources, 1995, 54, 250–254 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01759e |
| This journal is © The Royal Society of Chemistry 2024 |