
Single-atom Pd directly anchored on biphenylene: a promising bifunctional electrocatalyst for overall water splitting†
Ting-Ting
Wang
 ,
Yanan
Meng
,
Hai-Cai
Huang
,
Lei
Zhang
* and
Shi-Bo
Cheng
*
,
Yanan
Meng
,
Hai-Cai
Huang
,
Lei
Zhang
* and
Shi-Bo
Cheng
*
School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: shibocheng@sdu.edu.cn; leizhangsd1992@sdu.edu.cn
First published on 19th November 2024
Abstract
The development of bifunctional single-atom catalysts (SACs) for overall water splitting is crucial for clean energy production in the context of sustainable development. Using first-principles calculations, the catalytic capability of different transition metal (TM) atoms supported on biphenylene (Bip) monolayers (TM@Bip, TM = V–Cu, Ru–Ag, and Ir–Au) is comprehensively investigated. Bip can directly anchor TM atoms without engineered vacancies or nitrogen defects. Among the screened SACs, Pd@Bip is found to be an excellent bifunctional catalyst for both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). The overpotentials for the HER and the OER were calculated to be 0.05 and 0.50 V, respectively, which are even superior to the commercialized catalysts like Pt and IrO2. Furthermore, adjusting the d-band center of TM atoms effectively modulates the catalytic activity, and the optimal OER performance of TM–Bip can be achieved with a d-band center of −2.32 eV, which can serve as a principle to design Bip-based SACs. Our findings may serve as a practical theoretical guide for the exploration of effective bifunctional SACs for overall water splitting.
1. Introduction
Electrochemical water splitting is a promising approach for producing hydrogen fuels with renewable electricity, and typically consists of two half-reactions: the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). However, due to the distinct catalytic mechanism for the two half-reactions, HER catalysts often show poor OER efficiency and vice versa. Presently, Pt, Pd, and IrO2/RuO2 are recognized as the most effective catalysts for the HER and OER, respectively.1–3 Nevertheless, the exorbitant cost of noble metals and the additional costs for the preparation of two different electrode materials create economic pressure which limits their large-scale applications. A lot of research effort has also been devoted to developing high-performance catalysts with reduced content and increased utilization efficiency of noble metals. For example, Pd can be alloyed with high-abundance non-noble metals and constructed into various nanostructures like nanospheres, nanorods, nanocubic frames, and nanoparticles.4–7 Pt can be engineered into nanosheets with multifunctional catalytic capacity for the HER, OER, and oxygen reduction reaction (ORR).8 It is even found that iridium dimers (Ir2) hold great promise to promote the dissociation of water in homogeneous and heterogeneous catalytic systems.9Currently, single-atom catalysts (SACs) have emerged as a prominent area of research in the field of electrocatalysis and have been widely used in various electrocatalytic reactions, such as the HER,10 OER,11 CO2 reduction reaction (CO2RR),12 nitrogen reduction reaction (NRR),13 ORR,14 NO reduction reaction (NORR),15 and N2O reduction reaction (N2ORR).16 SACs present numerous advantages over conventional catalysts, including superior catalytic ability, superb selectivity, and tunable structures.17–19 Transition metal (TM) atoms serve as catalytic sites, which uniformly disperse on the substrate surface and consequently enable 100% atomic utilization efficiency. However, the synthesis of SACs remains challenging because the TM atoms tend to aggregate when they display weak interactions with the substrate surface.20 Therefore, it is crucial to select an appropriate substrate to support TM atoms.
In recent years, graphene allotropes have gained attention as promising candidates for anchoring single atom sites, owing to their exceptional durability and abundance in nature.21–27 Pure carbon nanomaterials, with their robust π-interactions in hexagonal networks, are virtually inert, but the addition of TM atoms can dramatically enhance their electrocatalytic properties.28–32 For instance, Senthamaraikannan et al. reported that TM SACs on nitrogen-doped graphene (TM@N3G) exhibit high catalytic abilities for the nitrogen reduction reaction (NRR).33 The Rh@C8N8 monolayer has been theoretically predicted as an effective bifunctional electrocatalyst for the HER and OER.34 Nonetheless, such SACs require embedding TM atoms into the basal plane of graphene via engineered vacancies or nitrogen defects, which makes the experimental synthesis challenging. Exploring novel substrates which can directly anchor TM atoms is still a crucial task for the design of bifunctional SACs.
Recently, Fan et al. synthesized a unique graphene isomer called two-dimensional (2D) biphenylene (Bip) by a dehydrochlorination approach to string 2,5-difluoro-p-phenylenediamine molecules together on the surface of gold.35 Bip consists of tetradentate, hexadentate, and octadentate C-atom rings and exhibits excellent stability, mechanical properties,36–38 and lower thermal conductivity compared to graphene.39,40 Moreover, Bip shows potential applications in optoelectronics and electronics, and as anode materials of electrodes, and catalysts for the ORR.41–47 Despite some preliminary investigations, a more comprehensive understanding of Bip-based SACs is still necessary, particularly regarding bifunctional catalytic capacity for both the OER and the HER, which has not been extensively explored.
In the present work, we constructed TM-supported Bip electrocatalysts (TM@Bip, TM = V–Cu, Ru–Ag, and Ir–Au) and investigated their catalytic activity for the HER and OER based on first-principles calculations. Our results demonstrate that Bip can serve as a great substrate to directly anchor TM atoms due to the low adsorption energies. Notably, Pd@Bip exhibits exceptional catalytic activity for both the HER and the OER, with overpotentials of 0.05 and 0.50 V, respectively. These findings highlight the potential of Pd@Bip as a bifunctional electrocatalyst.
2. Computational details
The Vienna Ab initio Simulation Package (VASP) was used for all calculations in this study.48 The exchange and correlation effects were characterized using the generalized gradient approximation (GGA) approach and the Perdew–Burke–Ernzerhof (PBE) approach.49 A cut-off energy of 450 eV was taken for the plane-wave basis. Moreover, convergence requirements for energy and residual force were set at 10−5 eV and 0.01 eV Å−1, respectively. For the density of states (DOS) calculation, a Monkhorst–Pack K-point mesh of 5 × 5 × 1 was used, while for structure optimization, a 3 × 3 × 1 mesh was employed. Our K-point convergence test suggests that the 3 × 3 × 1 K-point mesh is dense enough for energy-scale calculations (Fig. S1, ESI†). To avoid interactions between the layers, a vacuum of 16 Å was introduced in the z-direction. The van der Waals (vdW) interaction was accounted for using the Grimme correction (DFT-D3).50 In addition, the climbing image nudged elastic band (CI-NEB) method was used to explore reaction energy barriers.51Ab initio molecular dynamics simulations (AIMD) were performed to evaluate the thermal stability of the catalysts. For all computations, solvent effects were taken into account by utilizing the implicit solvent model performed in VASPsol.52 Additional details about the calculations can be found in Note 1 of the ESI.†3. Results and discussion
3.1. Structures of TM@Bip
As shown in Fig. 1a, Bip is composed of tetraene rings that repeat in the x and y directions and belongs to the space group of Pmmm (D2h).39 The pristine supercell has six carbon atoms, which are composed of four α-site and two β-site carbon atoms. In our calculations, the optimized lattice parameters for Bip were determined to be a = 4.52 Å and b = 3.77 Å, which are consistent with previous calculations (a = 4.51 Å and b = 3.76 Å),53 suggesting the reliability of our calculation. The bonds connecting neighboring Cα atoms are found to be 1.46 Å, while those between neighboring Cβ atoms are measured to be 1.45 Å. The length between the Cα and Cβ atoms is determined to be 1.41 Å. As the catalytic capability of a SAC is strongly correlated with the electronic structure of the host,54,55 we first check the electronic structures of pristine Bip, as depicted in Fig. 1b. The pristine Bip reveals metallic behavior, with the states near the Fermi level mainly contributed by the Cα-2p states. As a result, Cα is expected to play a more dominant role when binding with TM atoms.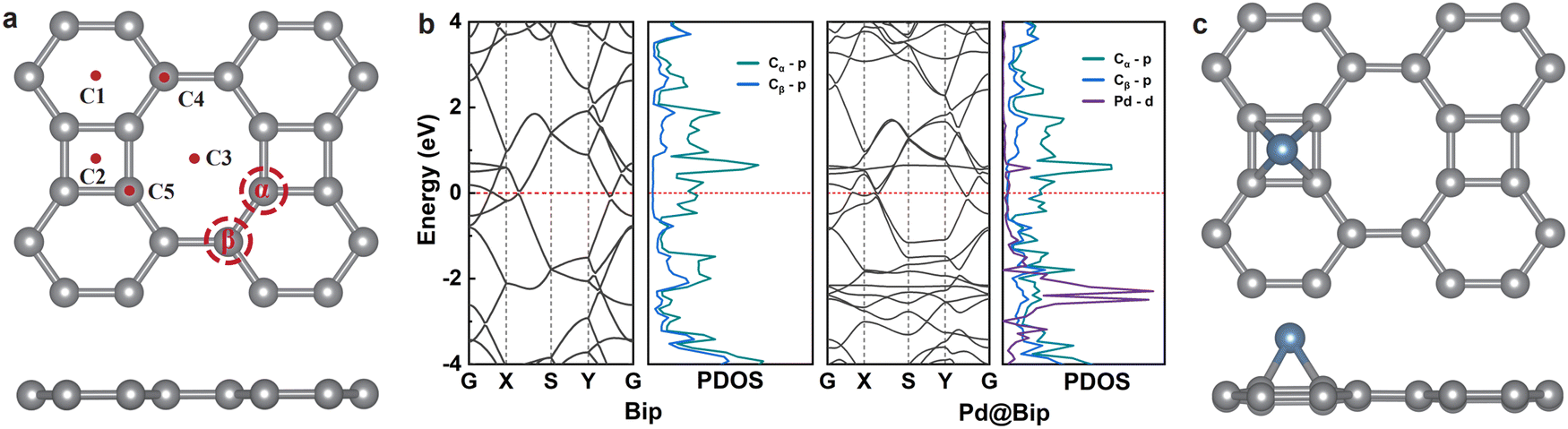 | ||
| Fig. 1 (a) Top and side views of the optimal structure of the pristine Bip. C1, C2, and C3 are the hollow sites of the hexadentate, tetradentate, and octal dentate C-atom rings, respectively; C4 and C5 are the top sites of Cβ and Cα atoms, respectively. (b) The band structures and density of states of Bip and Pd@Bip. (c) Top and side views of Pd@Bip. Pd, blue; C, gray. | ||
As illustrated in Fig. 1a, five adsorption sites are selected to anchor TM atoms on the Bip surface: C1, C2, C3, C4, and C5. The strong interaction between substrates and TM atoms is crucial for SACs,56 and we first checked the adsorption energies (Ead), dissolution potential (Udiss), bond lengths, Bader charges, and magnetic moments of TM atoms on Bip. As summarized in Table S1 (ESI†), the Ead for most TM atoms falls in the range between −4.27 eV and −2.00 eV, demonstrating that Bip can serve as a great substrate to directly anchor TM atoms. The Ead of Fe@Bip and Ag@Bip is positive; thus we exclude them for further discussion in this work. The Udiss is employed to evaluate the electrochemical stability of the catalysts.57 A more positive Udiss demonstrates that the metal atoms can bind strongly with the substrate and can be prevented from dissolution, which indicates a more stable SAC under electrochemical conditions. As presented in Table S1 (ESI†), the Udiss values on most SACs are positive (except V@Bip and Ir@Bip), indicating their electrochemical stability. The bond lengths of TM@Bip fall in the range between 1.97 and 2.25 Å. Only some early transition metals (Cr and Mn) show prominent magnetic moments, while the rest of them are either nonmagnetic or display small magnetic moments around 0.5–0.7μB. It is also observed that the amount of electrons transferred to Bip generally decreases with the increasing nuclear charge within the same period, in line with the sequence of their electron affinity. The optimized configurations for TM@Bip adsorption are depicted in Fig. 1c and Fig. S2 (ESI†). It is evident that TM atoms, such as Cr, exhibit a higher affinity for anchoring at the C1 site, whereas TM atoms, such as Mn, Co, Ni, Ru, and Pd, show a predisposition for the C2 site, and TM atoms like Au tend to adsorb to the C5 site.
The thermal stability of Pd@Bip is checked due to its promising catalytic performance towards both the HER and the OER, as illustrated in the following content of this work. As illustrated in Fig. 2, ab initio molecular dynamics (AIMD) simulations were conducted for Pd@Bip at 300 K and 600 K for 20 ps. Over the 20 ps simulations, no significant energy fluctuations can be found and the atomic structures of Pd@Bip show little distortion, demonstrating its excellent thermal stability. The thermal stability of Pd@graphene (Pd@GR, see Fig. S3, ESI†) is also checked for comparison. As shown in Fig. S4 (ESI†), the Pd atom would migrate on the surface of graphene during AIMD simulations performed at both 300 K and 600 K, indicating that Pd@GR is thermally unstable.
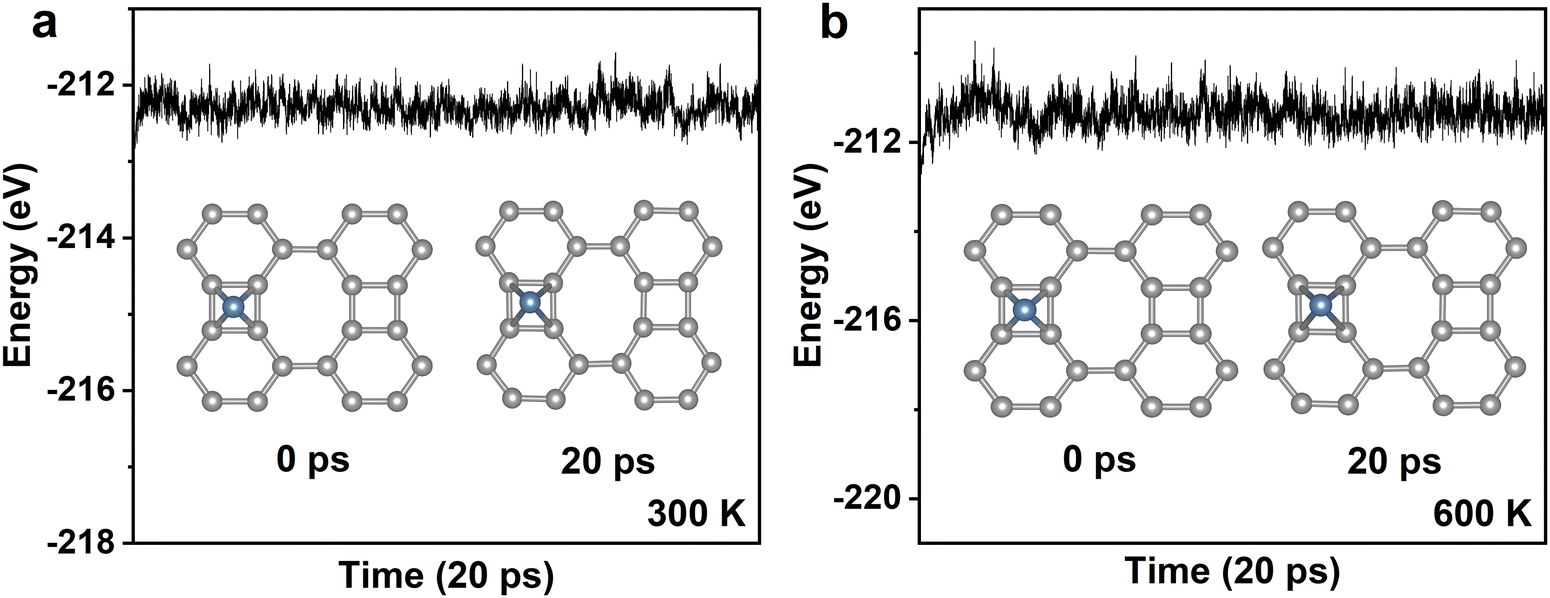 | ||
| Fig. 2 (a) and (b) The total energy change of Pd@Bip at 300 K and 600 K during 20 ps AIMD simulations, respectively. Pd, blue; C, gray. | ||
Such distinct behavior of adatoms can be understood from the migration barriers of Pd on Bip and GR. As shown in Fig. S5 (ESI†), Pd@Bip holds much higher diffusion barriers (0.39 and 0.47 eV) than that of Pd@GR (0.04 eV); hence, Pd anchored on Bip barely moves while the Pd adsorbed on GR migrates among different C–C bridge sites. Our results indicate that Bip can directly anchor TM atoms without engineered vacancies or nitrogen defects, which is usually required for graphene-based SACs.
We also compared the Ead of Pd clusters with different sizes on Bip (Pdn@Bip, n = 2, 3, 4), as shown in Fig. S6 (ESI†). It is found that the adsorption energies of the clusters (−0.58, −0.24, and −0.07 eV per Pd atom for n = 2, 3, and 4, respectively) are significantly larger than the single Pd atom (−2.0 eV per Pd atom). As the size of the Pd clusters increases, the Ead becomes more positive, indicating that the interaction between the Pd clusters and the Bip substrate is considerably weaker compared with a single Pd atom. Consequently, Pd atoms tend to stabilize as a discrete single-atom on Bip, which is highly desired for 2D SACs. In addition, SACs anchored on carbon-based materials like graphene have already been successfully synthesized, as demonstrated by isolated Co atoms anchored on graphene (Co1/G) and Mo-doped holey porous graphene.58,59 Therefore, we believe that the Pd@Bip SAC with great stability can be synthesized using similar synthesis techniques such as atomic layer deposition (ALD), chemical vapor deposition (CVD), and molecular layer deposition (MLD).60
For more structural information for experimentalists, we further simulate scanning tunneling microscopy (STM) images of Pd@Bip. Fig. S7 (ESI†) demonstrates the atomic structure of Pd@Bip and its STM image at a bias voltage of −2.5 V, which mainly corresponds to the electronic states of Pd-d orbitals (as shown in the PDOS, Fig. 1b). The Pd atoms are located above the hollow sites of C4 rings, appearing as much brighter spots than the C atoms in the Bip substrate. Therefore, it is expected that the atomic positions of Pd single atoms should be easily captured by STM.
The density of states (DOS) of TM@Bip offer key insights into their electronic structures and catalytic potentials. As shown in Fig. 1b and Fig. S8 (ESI†), an effective overlap of the C-p and TM-d orbitals was observed, indicating the strong interactions between TM atoms and neighboring C atoms. The d-band theory proposed by Nørskov et al. suggests that the adsorption energy between adsorbates and TM atoms correlates the position of the d-band center. An upward shift of the d-band indicates the less occupied anti-bonding states, leading to the stronger binding energy. The distribution of d-orbitals of a catalyst affects the interaction strength between the reaction intermediate and the catalyst, thus determining the catalytic activity.61–63 As shown in Fig. 1b and Fig. S8 (ESI†), the d-orbitals of Mn, Ni, Pd, Pt, Cu, and Au are located in the deep energy level while those of Cr, Co, Ru, and Rh are situated near the Fermi level. The different d-orbital distributions of these SACs suggest that they will exhibit distinct catalytic capabilities, which is verified by our following free energy calculations and the well-linear relationship between the d-band center and intermediate adsorption energy.
To obtain an in-depth understanding of the significant changes after Pd loading on Bip, we investigated the change in the band structure, as shown in Fig. 1b. The adsorption of Pd lowers the symmetry of Bip and leads to the lifts of degeneracy of the band dispersion near the Fermi level (−1 to 1 eV). A narrow flat band can be found at ∼0.5 eV above the Fermi level, which corresponds to the partly localized Pd-d orbitals. It can also be observed that there are extra bands created within the range from −3 to −2 eV, which are mainly contributed by the Pd-d orbitals. These Pd-d orbitals exhibit hybridization with the C-p orbitals, thus displaying more significant dispersions. The Cα-p orbitals play a more prominent role in the hybridization, which is in line with the bonding geometry between the Pd atom and the surface Cα atoms. Additionally, we show the band structure of Pd@Bip with/without the SOC effect (Fig. S9, ESI†). It can be observed that near the Fermi level, the SOC effect only leads to negligible band splitting, with the energy level nearly unaffected by the inclusion of SOC.
3.2. Catalytic activity towards the HER
The two-step process of the HER involves the adsorption of hydrogen atoms (H*) on the catalyst surface, followed by the release of hydrogen molecules (H2).64 The Gibbs free energy of H* adsorption (ΔGH*) can be used to evaluate the catalytic activity of a catalyst.65,66 A too positive ΔGH* indicates unfavorable adsorption of H*, whereas a too negative ΔGH* represents the unfavorable release of H2. Therefore, an ideal HER catalyst should exhibit moderate adsorption of H* with ΔGH* approaching zero.We first examined the HER performance of the pristine Bip monolayer, in which five different active sites were evaluated: C1, C2, C3, C4, and C5 (Fig. S10, ESI†). According to our calculations, the H* placed at C1, C2, and C3 sites finally moves to the C5 site and H* can only be adsorbed on C4 and C5 sites, with ΔGH* values of 1.28 and 0.42 eV, respectively. Such large ΔGH* values suggest the unfavorable H* adsorption on pristine Bip, rendering it unsuitable for the HER. We further investigated the ΔGH* values for different TM atoms supported on Bip, as depicted in Fig. 3a. The results clearly show a significant decrease of ΔGH* compared to pristine Bip, suggesting the enhanced interaction between H* and the surface. Notably, Pd@Bip, Co@Bip, Rh@Bip, and Ni@Bip exhibit ΔGH* values of 0.05, 0.03, −0.01, and −0.07 eV, respectively, suggesting their excellent HER catalytic performance, which even surpasses that of the widely used Pt (111) catalyst (−0.09 eV).66 The H* adsorption configuration of Pd@Bip is displayed in Fig. S11 (ESI†) as an example, where we found that the H* is adsorbed on the TM atom and tilts to the surface.
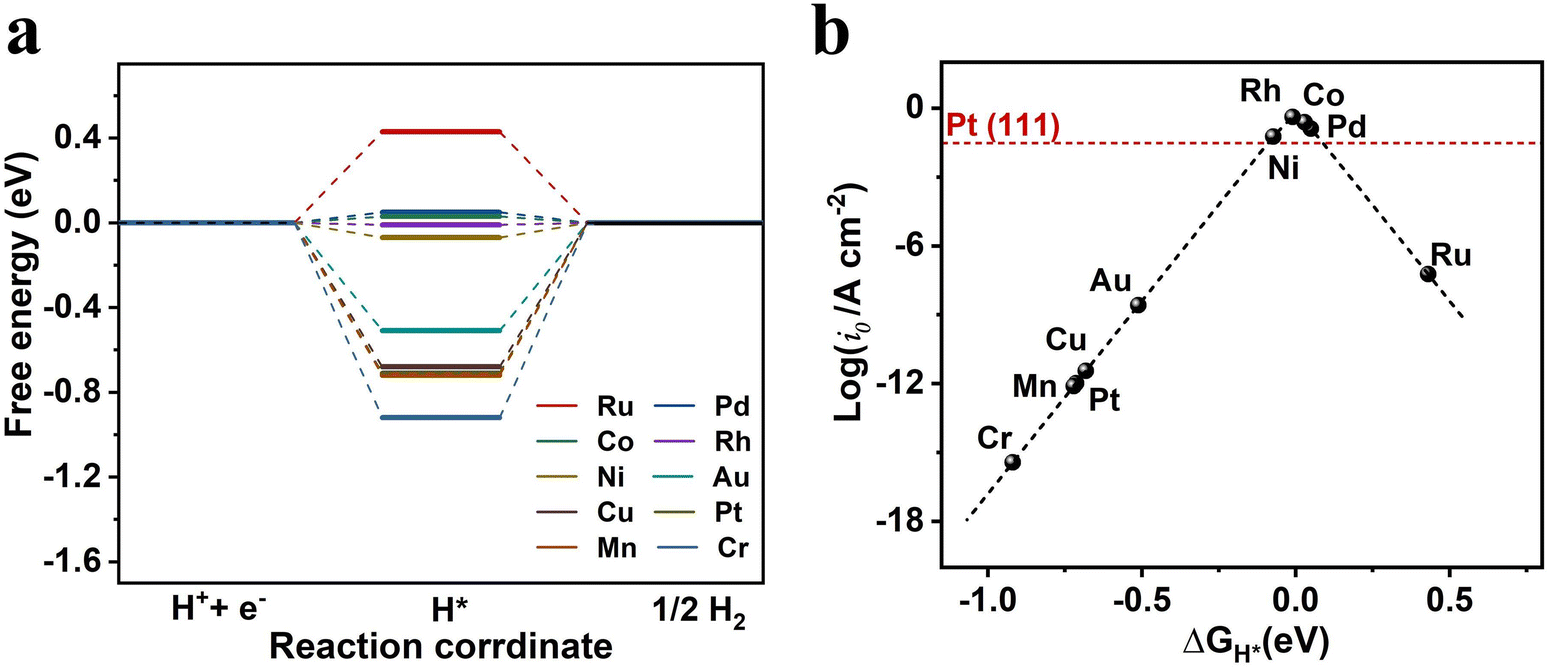 | ||
| Fig. 3 The calculated (a) Gibbs free energy of the HER (ΔGH*) and (b) the HER volcano curve of TM@Bip at 300 K. | ||
The exchange current density (i0) of TM@Bip was then calculated to evaluate the HER catalytic activity directly, as shown in Fig. 3b. The plot in Fig. 3b exhibits a clear volcano shape, with the left side representing strong adsorption of H* and the right side indicating weak adsorption of H*. Notably, the i0 of Pd@Bip, Co@Bip, Rh@Bip, and Ni@Bip locate at the top of the volcano plot, indicating their superior catalytic activity and the moderate binding strength with H*. In contrast, other TM@Bip catalysts exhibit more negative or positive values of i0, attributed to the too strong or weak binding between the catalyst and H*.
The HER after hydrogen adsorption (Volmer reaction) proceeds in acid media following either the Heyrovsky (H+ + H* + e− → H2 + *) or the Tafel (H* + H* → H2 + *) reaction.67 In our simulations, we include one water molecule explicitly when H+ is involved to mimic the hydronium. Fig. 4a and b show the energy barriers of the Heyrovsky and Tafel reactions on Pd@Bip, respectively. It is evident that the energy barrier for the Heyrovsky reaction is only half of that for the Tafel reaction. Therefore, the HER on Pd@Bip favors the Volmer–Heyrovsky mechanism, with the energy barrier comparable to or even smaller than many other catalysts such as 2D B7P2 (0.64 eV), NC-defective 2DPA (0.51 eV), and Ir@NP (0.64 eV).68–70
 | ||
| Fig. 4 The free energy diagram for the (a) Heyrovsky and (b) Tafel reactions on Pd@Bip for the HER at 300 K. Pd, blue; C, gray; O, red; H, pink. | ||
3.3. Catalytic activity towards the OER
The slow kinetics of the OER are widely recognized as a limiting factor in the overall water splitting rate.71,72 Thus, it is essential to screen bifunctional SACs for water splitting, focusing on the OER efficiency. Herein, we investigated the OER catalytic performance of Pd@Bip, Rh@Bip, Ru@Bip, Ni@Bip, Co@Bip, and Au@Bip at different electrode potentials (U), which have demonstrated relatively good HER activity (Fig. 5). As shown in Fig. 5d, the OER of Pd@Bip is energetically uphill at an electrode potential (U) of 0 V. At U = 1.23 V, step (1) becomes downhill, but steps (2), (3), and (4) remain uphill. It is only when the applied potential increases to 1.73 V that all elementary reaction steps become downhill. Consequently, the overpotential (η) is 0.50 V (η![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 1.73−1.23V), with the O* to OOH* transformation being the potential limiting step. The adsorption configurations of O*, OH*, and OOH* for Pd@Bip are displayed in Fig. S11 (ESI†). Similarly, the overpotential of Au@Bip is 0.53 V (η = 1.76–1.23 V), with the OH* to O* transformation as the potential limiting step (Fig. 5a). The overpotential of Pd/Au@Bip is even lower than that of the Ir-based catalyst (0.55 V),2 demonstrating excellent OER catalytic activity.
= 1.73−1.23V), with the O* to OOH* transformation being the potential limiting step. The adsorption configurations of O*, OH*, and OOH* for Pd@Bip are displayed in Fig. S11 (ESI†). Similarly, the overpotential of Au@Bip is 0.53 V (η = 1.76–1.23 V), with the OH* to O* transformation as the potential limiting step (Fig. 5a). The overpotential of Pd/Au@Bip is even lower than that of the Ir-based catalyst (0.55 V),2 demonstrating excellent OER catalytic activity.
 | ||
| Fig. 5 (a)–(f) Gibbs free energy diagrams of the OER processes for Au@Bip, Co@Bip, Ni@Bip, Pd@Bip, Rh@Bip, and Ru@Bip at different electrode potentials (U), respectively. | ||
Interestingly, we found that the OH* species show distinct adsorption behavior on Pd/Au@Bip and the other SACs (Fig. 5). The ΔGOH* values of Pd@Bip and Au@Bip are less negative (−0.04 and −0.03 eV, respectively), whereas those of the other SACs range from −1.32 to −0.60 eV. According to the Sabatier principle, the adsorption of intermediates should neither be excessively strong nor excessively weak.73 For an ideal OER catalyst, the energy barriers for all steps between adjacent intermediate states are 1.23 eV, enabling the OER to occur at its thermodynamic limit with zero overpotential. However, in reality, these energy steps are unevenly distanced, and the overpotential (η) is determined by the largest energy gap between two adjacent steps. In the present work, the strong binding between OH* species and the SAC obstructs the subsequent reactions, which, consequently, leads to the higher overpotentials observed for Rh@Bip, Ru@Bip, Ni@Bip, and Co@Bip (1.07, 1.36, 1.41, and 1.81 V, respectively).
According to the above results, although the HER catalytic performance of Au@Bip is not as good as that of Pt (111), it presents comparable OER catalytic activity with IrO2. Therefore, Au@Bip could be a promising bifunctional catalyst if its catalytic performance can be further regulated. To this end, pH was applied herein since the catalytic performance of the catalysts can be modulated at different pH levels. The Gibbs free energy values of Pd@Bip and Au@Bip for the HER and OER at distinct pH are plotted in Fig. S12 (ESI†). For Pd@Bip, although the OER catalytic performance is improved with the increasing pH, its HER catalytic performance decreases. In contrast, both the HER and OER activities of Au@Bip were enhanced with the increasing pH, attributed to its negative ΔGH* (−0.51 eV) at pH = 0. Under neutral conditions (pH = 7), Au@Bip demonstrates excellent HER and OER properties, making it a promising bifunctional catalyst for water splitting.
The OER pathway on Pd@Bip is also evaluated using NEB calculations (Fig. 6). Firstly, a water molecule adsorbs onto the catalyst and forms an OH* intermediate by losing a proton (H+) and an electron (e−). The Gibbs free energy change for this step is 1.22 eV, with the released proton combining with water to form a hydronium ion (H3O+). Notably, as shown in Fig. 6a, the energy is continuously rising during the decomposition of the H2O molecule (H2O → OH* + H+ + e−). Subsequently, the OH* intermediate undergoes further oxidation, losing another H+ and e− to form an O* intermediate (OH* → O* + H+ + e−), with a Gibbs free energy change of 1.38 eV. In the third step, the O* intermediate reacts with an H2O molecule to generate an OOH* intermediate while releasing H+ and e− (O* + H2O → OOH* + H+ + e−). This step has a Gibbs free energy change of 1.65 eV and is the rate-determining step of the OER, which corresponds to the thermodynamic maximum free energy barrier. Finally, the OOH* intermediate loses H+ and e− to produce an O2 molecule (OOH* → O2* + H+ + e−), with a Gibbs free energy change of 1.43 eV. The maximum energy barrier of Pd@Bip is even superior to other reported catalysts such as CoN3-pyrrole (1.81 eV), Co3@N-TC (1.82 eV), and Fe–NiOOH (1.72eV).28,74,75 Our results verify that the Pd@Bip monolayer exhibits excellent OER catalytic potential in both kinetics and thermodynamics.
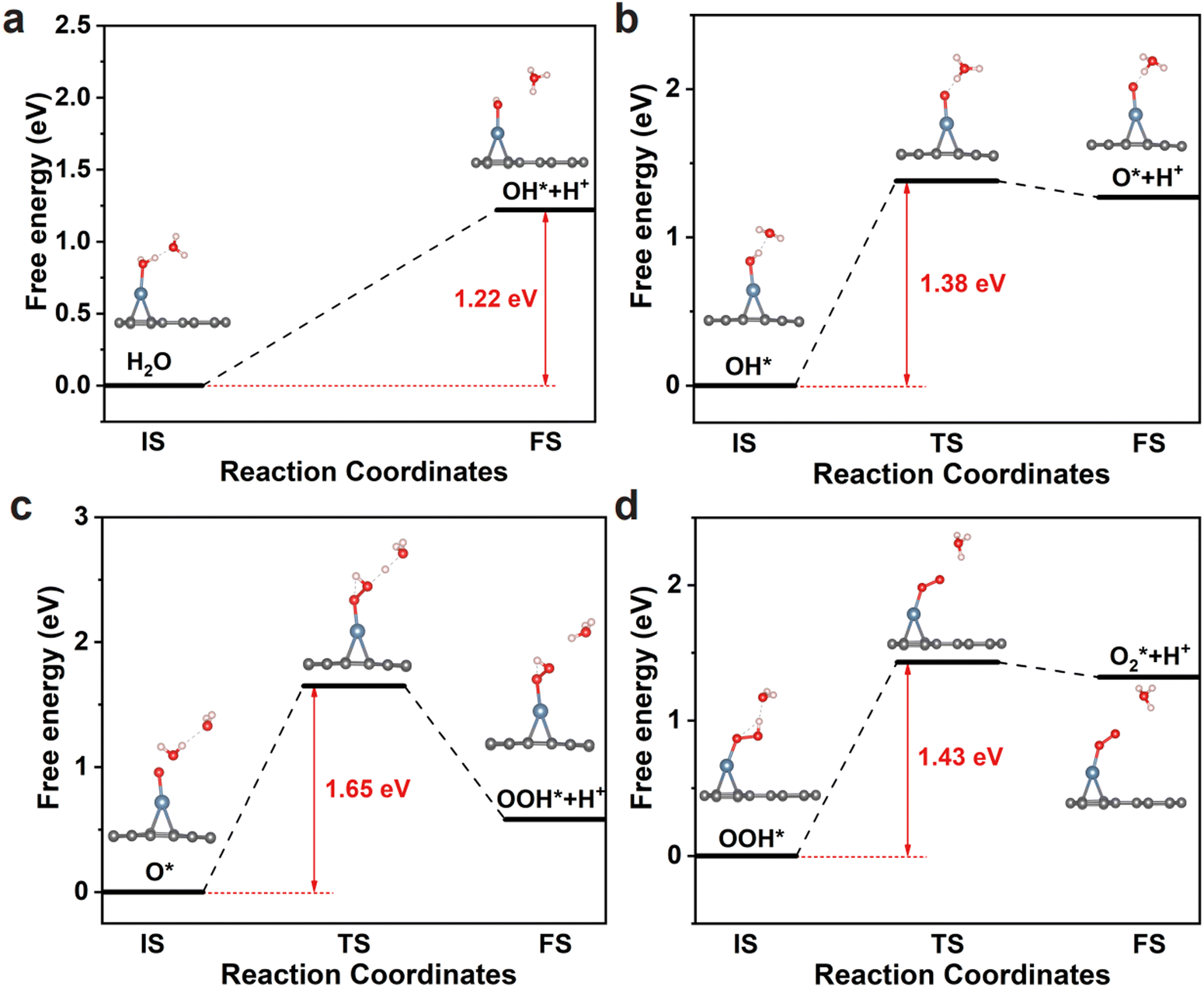 | ||
| Fig. 6 The reaction pathways of Pd@Bip for the OER. Detailed reaction energy profiles of the (a) H2O dissociation, (b) OH* dissociation, (c) OOH* formation, and (d) OOH* dissociation at 300 K. Pd, blue; C, gray; O, red; H, pink. | ||
To understand the distinct ΔGOH* between these SACs, the charge density difference and Bader charges for OH* adsorbed on Pd@Bip are compared with those of Co@Bip, which displays the most negative ΔGOH*. As shown in Fig. 7, the electrons from the TM atoms transfer to the adsorbed OH* and accumulate around the O*. The electron deficiency of TM atoms also attracts a small amount of electrons to the Co@Bip binding regions. Bader charge analysis revealed that Pd and Co atoms transfer 0.47 and 0.73 e− to OH* species, respectively. The smaller charge transfer amounts from Pd to OH* suggest the weaker interaction between OH* groups and the SAC, which leads to the less negative ΔGOH* value of Pd@Bip.76 It is well established that the adsorption characteristics of intermediates strongly influence the catalytic activity of the OER. As shown in Fig. S13 (ESI†), ΔGOH* effectively describes both ΔGO* and ΔGOOH*. To further validate the relationship between ΔGOH* and OER catalytic activity, we examine the connection between ΔGOH* and overpotential in Fig. S13 (ESI†). The results demonstrate a clear negative relationship between ΔGOH* and the overpotential of the OER process. In other words, a more positive value of ΔGOH* is associated with superior OER catalytic performance.
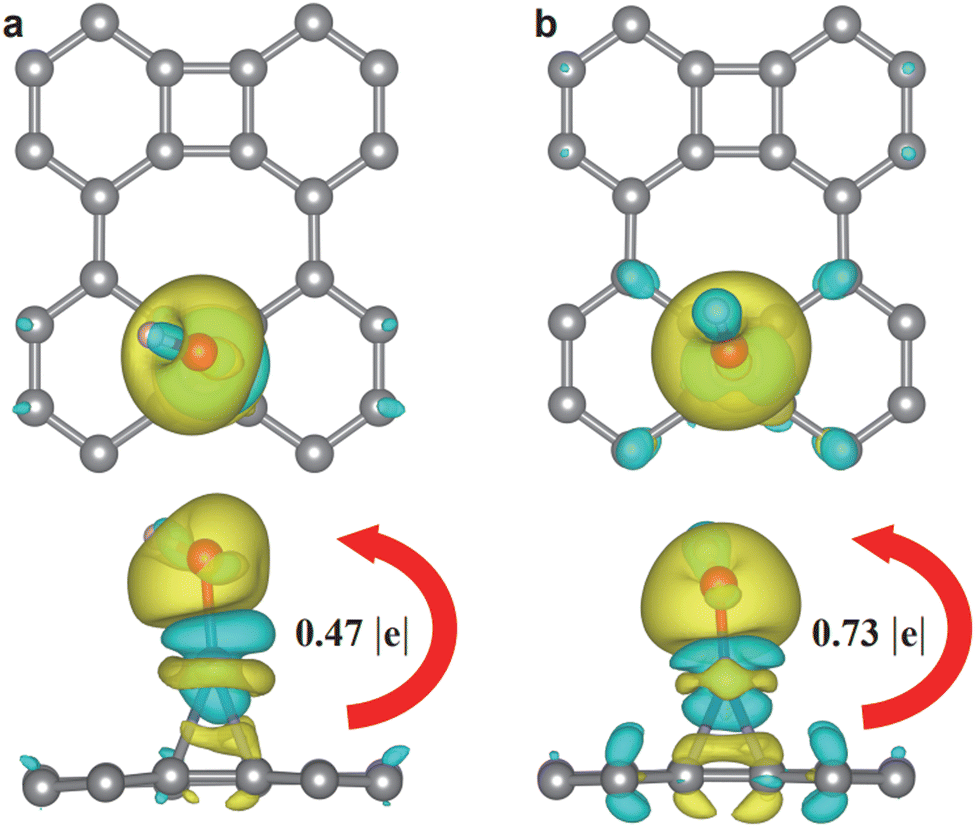 | ||
| Fig. 7 Charge density difference for (a) Pd@Bip and (b) Co@Bip, where the isosurface value is set to be 0.002 e Å−3 and the charge accumulation and depletion areas are shown in yellow and cyan, respectively. | ||
The activity of a SAC is significantly influenced by the d orbitals of the transition metal atoms, which can be captured by the d-band center (εd). In Fig. 8a, we plot the relationships between the εd and ΔGO*, ΔGOH*, and ΔGOOH*, respectively, where a strong linear scaling relationship between εd and the Gibbs free energy changes can be observed. The plots reveal that a lower εd corresponds to weaker interactions between reaction intermediates and TM atoms, which establishes a clear negative correlation between εd and ΔGO*, ΔGOH*, and ΔGOOH*. As the adsorption of intermediates can impact the reaction activity, this linear correlation provides valuable insights for more effective design of catalysts by using εd.
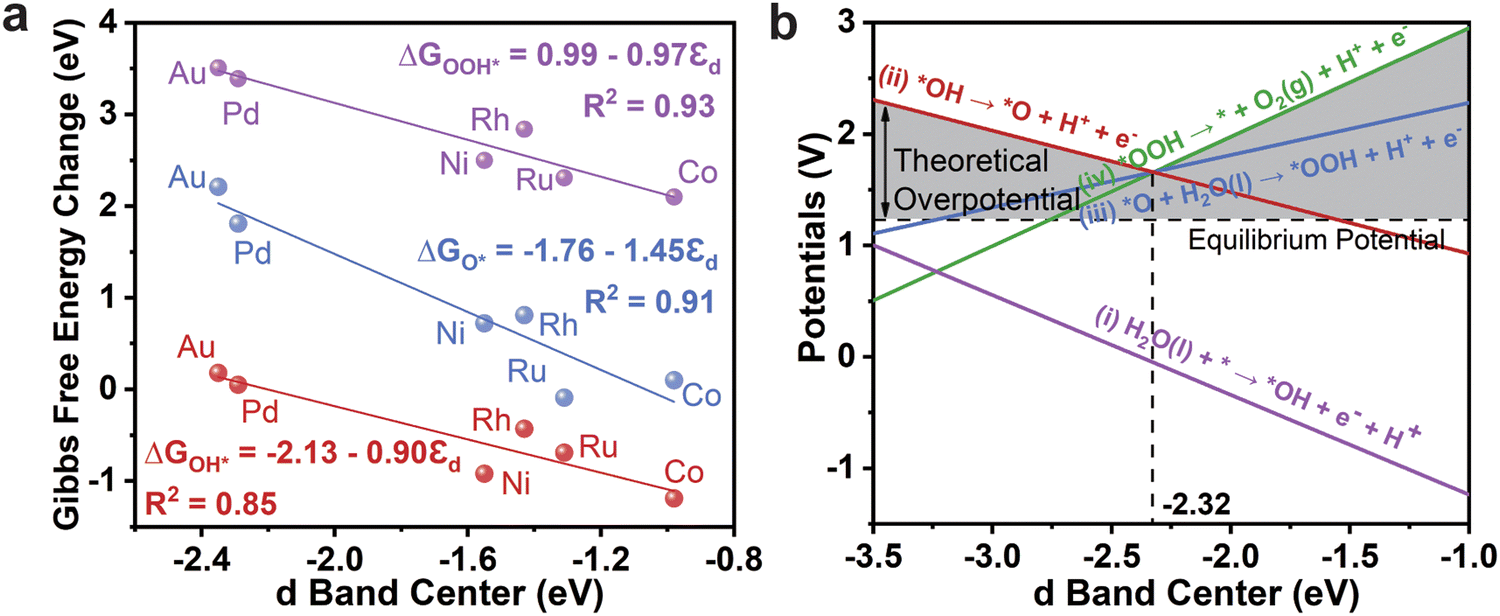 | ||
| Fig. 8 (a) Scaling relationships between three Gibbs free energy changes (T = 300 K) and d-band centers. (b) Potential of each elementary reaction at 300 K as a function of the d-band center, where the horizontal dashed line is the equilibrium potential, the vertical dashed line is the position of the ideal d-band center, and the shaded area is the theoretical overpotential. | ||
As illustrated in Fig. 8b, a clear trend is observed where the theoretical overpotential first decreases and then increases with the elevation of εd, with a turning point at −2.32 eV. When εd is below −2.32 eV, the formation of the O* intermediate (red line) restricts the reaction rate, and the theoretical overpotential decreases with increasing εd. Conversely, when εd exceeds −2.32 eV, the final step in O2 molecule formation (green line) becomes the potential-limiting step, and the theoretical overpotential increases with a higher εd. Generally, a deeper εd will lead to a weaker binding to adsorbed oxygenated intermediates; thus the catalytic activity of Bip-based SACs is initially limited by the too weak adsorption of O*. As the εd exceeds −2.32 eV, the excessive binding between OOH* groups with the SAC then impedes the release of the product (O2 molecules). Based on this analysis, the ideal d-band center for TM@Bip is identified as −2.32 eV, where the OER achieves maximum efficiency. The εd values of Pd@Bip and Au@Bip (−2.29 and −2.35 eV, respectively) closely approach this ideal value; hence, they exhibit the best OER catalytic activity.
4. Conclusions
In conclusion, the HER/OER catalytic activity of TM atoms supported on novel two-dimensional biphenylene monolayers (TM@Bip, TM = V–Cu, Ru–Ag, and Ir–Au) was investigated systematically using DFT calculations. Among these, TM@Bip, Pd@Bip, Co@Bip, Rh@Bip, and Ni@Bip show high HER activity with ΔGH* values of 0.05, 0.03, −0.01, and −0.07 eV, respectively. Meanwhile, Pd@Bip and Au@Bip display excellent catalytic activity for the OER (overpotentials of 0.50 and 0.53 V, respectively), attributed to the moderate interactions between the Pd/Au and adsorbed species. Using εd as a descriptor, we predict that the Bip-based SACs will show the best OER activity when εd reaches −2.32 eV, which is close to the value of Pd@Bip (−2.29 eV). The computational insights affirm Pd@Bip as a highly promising candidate for bifunctional electrocatalysis in both the HER and the OER. Our results may provide useful guidance to synthesize efficient electrocatalysts and hence promote renewable energy production.Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is based upon work supported by the National Natural Science Foundation of China (NSFC) (92161101, 12474274, and 12404107), the Innovation Project of Jinan Science and Technology Bureau (2021GXRC032), the Taishan Scholars Project of Shandong Province (ts201712011), the Instrument Improvement Funds of Shandong University Public Technology Platform (No. ts20220201), the Natural Science Foundation of Shandong Province (ZR2022QB204), and the Shandong Provincial Excellent Young Scientists Fund Program (Overseas) (2024HWYQ-029). The theoretical calculations in this paper have been done on the HPC cloud platform of Shandong University and the HPC platform for theoretical and computational chemistry at the School of Chemistry and Chemical Engineering of Shandong University.References
- Y. Zhu, M. Klingenhof, C. Gao, T. Koketsu, G. Weiser, Y. Pi, S. Liu, L. Sui, J. Hou and J. Li, Nat. Commun., 2024, 15, 1447 CrossRef CAS PubMed.
- S. Kwon, K. A. Stoerzinger, R. Rao, L. Qiao, W. A. Goddard III and Y. Shao-Horn, J. Am. Chem. Soc., 2024, 146, 11719–11725 CrossRef CAS PubMed.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS PubMed.
- B. Huang, Y. Ge, A. Zhang, S. Zhu, B. Chen, G. Li, Q. Yun, Z. Huang, Z. Shi and X. Zhou, Adv. Mater., 2023, 35, 2302233 CrossRef CAS PubMed.
- J.-J. Duan, X.-X. Zheng, H.-J. Niu, J.-J. Feng, Q.-L. Zhang, H. Huang and A.-J. Wang, J. Colloid Interface Sci., 2020, 560, 467–474 CrossRef CAS PubMed.
- H. Guo, M. Li, Y. Yang, R. Luo, W. Liu, F. Zhang, C. Tang, G. Yang and Y. Zhou, Small, 2023, 19, 2207743 CrossRef CAS PubMed.
- B. Salah, A. K. Ipadeola, A. M. Abdullah, A. Ghanem and K. Eid, Int. J. Mol. Sci., 2023, 24, 11832 CrossRef CAS PubMed.
- X. Lv, W. Wei, H. Wang, B. Huang and Y. Dai, Appl. Catal., B, 2019, 255, 117743 CrossRef CAS.
- C. D. Zeinalipour-Yazdi and R. A. van Santen, J. Phys. Chem. A, 2009, 113, 6971–6978 CrossRef CAS.
- B. Jiang, J. Zhu, Z. Xia, J. Lyu, X. Li, L. Zheng, C. Chen, S. Chaemchuen, T. Bu and F. Verpoort, Adv. Mater., 2024, 36, 2310699 CrossRef CAS PubMed.
- F. Jiang, Y. Li and Y. Pan, Adv. Mater., 2024, 36, 2306309 CrossRef CAS PubMed.
- A. O. Ibrahim, W. M. A. W. Daud, M. F. A. Patah, A. Halilu, J. C. Juan and G. Tanimu, Phys. Chem. Chem. Phys., 2024, 26, 10622–10632 RSC.
- J. Wu and Y.-X. Yu, J. Phys. Chem. C, 2022, 126, 12460–12471 CrossRef CAS.
- J. Manopo, P. N. Kusumo, A. Sumboja and Y. Darma, Phys. Chem. Chem. Phys., 2023, 25, 25299–25308 RSC.
- J. Wu and Y.-X. Yu, J. Colloid Interface Sci., 2022, 623, 432–444 CrossRef CAS PubMed.
- R. Gholizadeh and Y.-X. Yu, Appl. Surf. Sci., 2015, 357, 1187–1195 CrossRef CAS.
- H. Zhao, D. Chen, R. Yu, J. Jiao, W. Zeng, J. Zhu, X. Mu, Y. Yao, D. Wu and Y. Zhang, Nano Energy, 2024, 121, 109247 CrossRef CAS.
- X. Mao, W. Gong, Y. Fu, J. Li, X. Wang, A. P. O’Mullane, Y. Xiong and A. Du, J. Am. Chem. Soc., 2023, 145, 21442–21453 CrossRef CAS PubMed.
- P. Mano, S. Namuangruk and K. Takahashi, J. Phys. Chem. C, 2023, 127, 7683–7694 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- F. Yang, X. Zhu, B. Jia, J. Hao, Y. Ma, P. Lu and D. Li, Int. J. Hydrogen Energy, 2024, 51, 195–206 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS.
- Y. Cao, J. Zhao, X. Zhong, G. Zhuang, S. Deng, Z. Wei, Z. Yao and J. Wang, Green Energy Environ., 2021, 6, 846–857 CrossRef CAS.
- J.-Q. Zhou, L. Li, C. Fu, J. Wang, P. Fu, C.-P. Kong, F.-Q. Bai, R. I. Eglitis, H.-X. Zhang and R. Jia, Nanoscale, 2020, 12, 5055–5066 RSC.
- Z. Zhao, J. Hao, B. Jia, X. Zhang, G. Wu, C. Zhang, L. Li, S. Gao, Y. Ma and Y. Li, J. Energy Chem., 2023, 83, 79–89 CrossRef CAS.
- S. Liang, T. Zhang, Y. Zheng, T. Xue, Z. Wang, Q. Wang and H. He, Appl. Catal., B, 2023, 333, 122801 CrossRef CAS.
- X. Zhang, A. Chen, L. Chen and Z. Zhou, Adv. Energy Mater., 2022, 12, 2003841 CrossRef CAS.
- J. Zhu, J. Cao, G. Cai, J. Zhang, W. Zhang, S. Xie, J. Wang, H. Jin, J. Xu and X. Kong, Angew. Chem., Int. Ed., 2023, 62, e202214351 CrossRef CAS PubMed.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen and H. Liu, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed.
- T. G. Senthamaraikannan and D.-H. Lim, Int. J. Hydrogen Energy, 2024, 72, 449–461 CrossRef CAS.
- H. Chen, C. Zhu, C. Wen, M. Wang, M. Zhang, Y. Geng and Z. Su, Appl. Surf. Sci., 2021, 549, 149320 CrossRef CAS.
- Q. Fan, L. Yan, M. W. Tripp, O. Krejčí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert and P. Liljeroth, Science, 2021, 372, 852–856 CrossRef CAS PubMed.
- Y. Luo, C. Ren, Y. Xu, J. Yu, S. Wang and M. Sun, Sci. Rep., 2021, 11, 19008 CrossRef CAS PubMed.
- S. Demirci, T. Gorkan, S. A. Çallıoğlu, V. O. Özçelik, J. V. Barth, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2022, 126, 15491–15500 CrossRef CAS.
- Y. Liao, X. Shi, T. Ouyang, J. Li, C. Zhang, C. Tang, C. He and J. Zhong, J. Phys. Chem. Lett., 2021, 12, 8889–8896 CrossRef CAS PubMed.
- H. P. Veeravenkata and A. Jain, Carbon, 2021, 183, 893–898 CrossRef CAS.
- P. Ying, T. Liang, Y. Du, J. Zhang, X. Zeng and Z. Zhong, Int. J. Heat Mass Transfer, 2022, 183, 122060 CrossRef CAS.
- H. Shen, R. Yang, K. Xie, Z. Yu, Y. Zheng, R. Zhang, L. Chen, B.-R. Wu, W.-S. Su and S. Wang, Phys. Chem. Chem. Phys., 2022, 24, 357–365 RSC.
- T. Liu, Y. Jing and Y. Li, J. Phys. Chem. Lett., 2021, 12, 12230–12234 CrossRef CAS PubMed.
- M. M. Obeid and Q. Sun, Carbon, 2022, 188, 95–103 CrossRef CAS.
- D. Ferguson, D. J. Searles and M. Hankel, ACS Appl. Mater., 2017, 9, 20577–20584 CrossRef CAS PubMed.
- U. Younis, I. Muhammad, Y. Kawazoe and Q. Sun, Appl. Surf. Sci., 2020, 521, 146456 CrossRef CAS.
- L. Xie, T. Chen, X. Dong, G. Liu, H. Li, N. Yang, D. Liu and X. Xiao, ACS Sens., 2023, 8, 3510–3519 CrossRef CAS PubMed.
- S. Mo, J. Seo, S.-K. Son, S. Kim, J.-W. Rhim and H. Lee, Nano Lett., 2024, 24, 4885–4892 CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- T. Han, Y. Liu, X. Lv and F. Li, Phys. Chem. Chem. Phys., 2022, 24, 10712–10716 RSC.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- X. Zhu, Y. Wang, Y. Jing, T. Heine and Y. Li, Mater. Today Adv., 2020, 8, 100091 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. Wu and Y.-X. Yu, Int. J. Hydrogen Energy, 2023, 48, 5961–5975 CrossRef CAS.
- H. Yan, X. Zhao, N. Guo, Z. Lyu, Y. Du, S. Xi, R. Guo, C. Chen, Z. Chen and W. Liu, Nat. Commun., 2018, 9, 3197 CrossRef PubMed.
- P. Du, K. Hu, J. Lyu, H. Li, X. Lin, G. Xie, X. Liu, Y. Ito and H.-J. Qiu, Appl. Catal., B, 2020, 276, 119172 CrossRef CAS.
- Y. Shang, X. Duan, S. Wang, Q. Yue, B. Gao and X. Xu, Chin. Chem. Lett., 2022, 33, 663–673 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- A. Cao and J. K. Nørskov, ACS Catal., 2023, 13, 3456–3462 CrossRef CAS.
- M. Schalenbach, R. Tesch, P. M. Kowalski and R.-A. Eichel, Phys. Chem. Chem. Phys., 2024, 26, 14171–14185 RSC.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- X. Lv, W. Wei, H. Wang, B. Huang and Y. Dai, Appl. Catal., B, 2020, 264, 118521 CrossRef CAS.
- R.-Q. Zhang, T.-H. Lee, B.-D. Yu, C. Stampfl and A. Soon, Phys. Chem. Chem. Phys., 2012, 14, 16552–16557 RSC.
- Y. Wei, F. Gao, H. Huang and G. Jiang, Int. J. Hydrogen Energy, 2022, 47, 8338–8347 CrossRef CAS.
- A. Sharma and B. Chakraborty, Int. J. Hydrogen Energy, 2024, 51, 1306–1313 CrossRef CAS.
- Y. Wang, W. Tian, J. Wan, G. Xiong, H. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2022, 24, 10325–10333 RSC.
- J. Su, R. Ge, Y. Dong, F. Hao and L. Chen, J. Mater. Chem. A, 2018, 6, 14025–14042 RSC.
- L. Xiao, X. Bai, J. Han, T. Tang, S. Chen, H. Qi, C. Hou, F. Bai, Z. Wang and J. Guan, Nano Res., 2024, 17, 2429–2437 CrossRef CAS.
- J. Kari, J. P. Olsen, K. Jensen, S. F. Badino, K. B. Krogh, K. Borch and P. Westh, ACS Catal., 2018, 8, 11966–11972 CrossRef CAS.
- C.-S. Yin, Y. Leng, X. Yang, C.-G. Min and A.-M. Ren, Appl. Surf. Sci., 2024, 643, 158605 CrossRef CAS.
- X. Liu, X. Zhao, S. Cao, M. Xu, Y. Wang, W. Xue and J. Li, Appl. Catal., B, 2023, 331, 122715 CrossRef CAS.
- W. Wang, R. Zhang, H. Chu, Z. Zhan, Q. Huang, Z. Li, X. Wang, F. Bai and W. Zhou, Small, 2024, 2406527 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The calculation details for the HER and OER; the total energies for Pd@Bip using different k-point meshes; the optimized configurations for TM@Bip (TM = Cr–Cu, Ru–Ag and Pt–Au) adsorption; calculated adsorption energies, dissolution potential, nearest neighbor bond lengths, Bader charge and total magnetic moment; the optimized structures and adsorption energies of Pd2–4 clusters anchored on Bip; simulated STM images for Pd@Bip; the optimized configurations for Pd@GR, Pd@GR at 300 K and 600 K during 20 ps AIMD simulations; the potential energy profile of Pd atom diffusion on Bip and GR; the PDOS of TM@Bip; band structures of Pd@Bip (with/without SOC); the possible adsorbed sites of H* on Bip; the adsorption configurations of HER/OER intermediate states for Pd@Bip; the Gibbs free energy change diagram of the HER and OER for Pd@Bip and Au@Bip at different pHs; the relationship between ΔGOH* and ΔGO*, ΔGOOH*, and the overpotential of TM@Bip. See DOI: https://doi.org/10.1039/d4cp03539a |
| This journal is © the Owner Societies 2025 |