
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantum control of photoion circular dichroism using orthogonal laser beams†
Jason B.
Greenwood
 * and
Leah
Donnelly
* and
Leah
Donnelly
Centre for Light-Matter Interactions, School of Mathematics and Physics, Queen's University Belfast, Belfast, UK. E-mail: j.greenwood@qub.ac.uk
First published on 7th May 2025
Abstract
The circular dichroism manifested in the photoionization yields of the chiral molecule fenchone has been investigated using two laser beams of different wavelengths intersecting at 90 degrees. This allowed different two-colour ionization schemes to be investigated so that the circular dichroism contributions of each step in the multiphoton ionization process could be deduced. The results showed that the circular dichroism generated by a beam at a wavelength of 260 nm could be strongly influenced by the polarization state of a 520 nm beam propagating in a perpendicular direction. The asymmetry in the ion yields was found to change depending on whether the 520 nm beam was left or right circularly polarized, and if it was linear polarized in a direction parallel or perpendicular to the propagation direction of the 260 nm beam. The control exhibited by the direction of the linearly polarized light is attributed to an orientation dependence of the circular dichroism due to selective excitation of the isotropic ensemble by the 520 nm beam. By contrast, when both beams were circularly polarized, the dependence on the polarization direction of the 520 nm pulse is ascribed to either the interference between different ionization pathways or excitation of a chiral vibronic wavepacket in the molecule. These results are the first demonstration of chiroptical quantum control of total ion yields, showing in principle that all-optical enantio-sensitive chemical processing may be possible in the future.
Introduction
Chirality is a critical property of the building blocks of Life on Earth. Chiral enantiomers are mirrored pairs of molecules that can’t be superimposed on one another by rotations and translations alone. They have identical physical properties but, in nature, the amino acids and sugars found in proteins and DNA have almost exclusively one handedness. The reason for and origin of this homochirality remains one of the great unsolved scientific questions.1–3Chiroptical phenomena have been observed and studied for more than 200 years. In the 19th century this included observations of optical rotation (OR) by Biot in 1815,4 Pasteur's recognition in 1848 that OR had a geometrical origin,5 and Cotton's discovery of circular dichroism (CD) in 1895.6 OR and CD are related phenomena arising from differences in the real and imaginary components of the refractive index of a material or solution for left and right circularly polarized light. These phenomena are extensively used for chiral analysis and as tools for investigating the structural conformation of complex molecules such as proteins. However, the small differences obtained for the different polarization states limits further applications.
The relative size of the CD can be quantifiedby a dimensionless asymmetry parameter g which is defined as
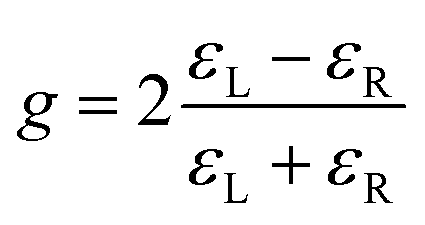
Since the start of this century, a number of new chiroptical phenomena have been discovered with intrinsically larger asymmetries which could allow more sensitive enantiomeric differentiation. These include microwave three wave mixing,7–10 Coulomb explosion imaging,11–13 photoion circular dichroism (PICD)14–31 and photoelectron circular dichroism (PECD).32–52
PICD is due to differences in ion yields produced from LCP and RCP photoionization of the molecule, usually through multiphoton ionization. The polarization dependence emerges from the circular dichroism present in the transitions involved in the ionization process, which could be one photon or multiphoton transitions between electronic states of the molecule or its molecular cation. Therefore, PICD is closely related to conventional CD and is quantified in a similar way

PICD values can be enhanced for cold molecules in the gas phase if a narrowband laser picks out specific vibrational states in the excitation.23,27 In addition to the parent molecular ion, yields of fragment ions can also be measured. These can demonstrate different PICD values as production of different ions may involve different excitation pathways, such as further photon absorption by the molecular ion.
Multiphoton PICD measurements have also been found to substantially change depending on whether nanosecond or femtosecond laser pulses are used. The broadband nature of femtosecond pulses means a greater number of electronic and vibrational states will be populated and the average of these contributions tends to produce lower PICD values. For example, the g-value for resonant excitation of the n → π* transition in 3-MCP was found to be 27% at 324 nm with a nanosecond laser16 and was even higher when the gas was supersonically cooled so that specific vibrational states were excited.23 In contrast, lower g values have been found for femtosecond pulses which reduced further as the pulse length was shortened.17 As fs laser pulses are more intense, saturation of the ion yields may also contribute to a reduction in the PICD values, although a very recent study found no intensity dependence.53
All of these PICD studies have been undertaken in the gas phase with the molecules randomly orientated. However, if the molecules have a fixed orientation, then the circular dichroism needs to be described by a tensor rather than a simple numerical value, which arises from the coupling between electric dipole and the magnetic dipole or electric quadrupole moments of the transition.54 The first investigations of anisotropic circular dichroism in isolated molecules have been recently investigated via PICD measurements.55 Using multi-coincidence ion fragment imaging of a methane-like chiral molecule, strong field multiple ionization was used to identify the spatial orientation of each molecule. By correlating different orientations to the ion yields, it was shown that the PICD could be enhanced by two orders of magnitude compared to the isotropically averaged value. At lower intensities with methyl oxirane, a subset of the molecular orientations was selected by measuring the momentum of one ionic fragment relative to the laser propagation direction, yielding a factor of 5 enhancement in the PICD. Similar anisotropy in the PICD was also identified by Jeong et al.56 using a supersonically cooled gas jet and a narrow linewidth nanosecond laser, to obtain PICD values across the S0–S1 band origin of pseudoephedrine and styrene oxide. This allowed the contributions from the P, R, and Q rotational bands to be identified, each of which correspond to different molecular orientations due to the selection rules.
This potential to control and enhance PICD contributions could open up possible applications. As well as providing faster and more sensitive chiral analysis, strong enantio-dependent interactions could lead to all-optical chiral purification methods via selective photolysis. This would also enable ultrafast pump–probe PICD studies of molecular dynamics in the same way that strong asymmetries observed in time-resolved PECD are starting to show remarkable potential.36,57,58
There are different approaches through which quantum control of population transfer in a chiral molecule could be used to obtain stronger PICD yields. By combining laser pulses of different colours, different quantum pathways to the ion continuum could be controlled by the relative magnitude, phase and polarization of the individual pulses. Near-perfect state-specific enantiomeric enrichment of rotational states in a chiral molecule has already been achieved using tailored microwave pulses.59 Similar control of electronic states is no doubt more challenging but potential schemes are being investigated theoretically.60
Excitation of non-stationary states also provides a way in which the ionization yield could be controlled. Chiral electronic wavepackets can be created from a superposition of states excited with a short, circularly polarized pulse, known as photoexcitation circular dichroism.61 This has been observed in the anisotropic photoelectron angular distributions produced from ionization of the wavepacket by a linearly polarized photon. For similar asymmetries to be manifested in the total ion yield, the ionization process would need to probe the asymmetry of the wavepacket, for example by having the polarizations of the excitation and ionization pulses co-rotating or counter-rotating relative to the chiral wavepacket.
Ultimately, if PICD asymmetries are to reach values similar to those found with PECD, then the chiroptical interaction needs to be manifested directly through electric dipole interactions. This new field of research was launched in a seminal paper which proposed the creation of a locally chiral electric field configuration.62 Such fields map out a 3D chiral shape, so that the chiroptical interactions can be manifested within the dipole approximation. The present paper, which is an initial step towards this goal, describes how perpendicular laser beams with different colours and polarization have been used to investigate how PICD asymmetries can be controlled.
Experimental
Time of flight mass spectrometer
A schematic diagram of the apparatus used for the experiments is shown in Fig. 1. This was a vertically orientated time-of-flight (TOF) mass spectrometer which has been used in previous studies.63–66 The ion acceleration region consisted of a flat repeller plate and an extraction aperture which held one end of a cylindrical tube with a resistive coating on its inner surface. The other end of the tube was grounded so a uniform electric field was produced along its length. Ionization was achieved by two perpendicular laser beams which intersected on the axis of the TOF between the repeller and extraction plates. An effusive gas jet of fenchone was passed through a hole in the repeller plate and along the TOF axis to intersect with the laser beams. The chamber had a base pressure of 2 × 10−8 mbar which rose to 10−6–10−5 mbar when the gas jet was operational. | ||
| Fig. 1 Time-of-flight mass spectrometer used for the measurements; G – gas jet; R – repeller plate; E – extraction plate; C – cylinder with resistive inner coating; A – aperture. The gas jet was intersected orthogonally by 520 nm and 260 nm laser pulses with each beam slightly off focus. Different polarization combinations were used, for example in this figure the 260 nm pulses are circularly polarized and 520 nm pulses linearly polarized in the horizontal direction so that its electric field is perpendicular to the plane of polarization of the 260 nm pulses. | ||
The potentials on the repeller and extraction plates were set to generate a fields of 3 × 105 V m−1 in the interaction region and 6 × 104 V m−1 inside the cylinder. This unusual configuration, which does not give the optimum mass resolution, was used to de-focus the ions generated outside the laser beams’ intersection volume, causing them to be stopped by an aperture in the flight tube. In this way the signal arising from the joint interaction of the two laser beams was enhanced relative to ions produced outside this volume.
A Photonis MiniTOF microchannel detector with an 8 mm active diameter was placed at the end of a 50 cm flight tube. Pulses from the MiniTOF passed through an ET Enterprises AD8 amplifier-discriminator to produce 17 ns TTL pulses. These pulses were counted in digital counters, each of which were gated with timing windows corresponding to ion masses of interest. With this setup a mass resolution of m/Δm ≈ 200 was achieved. During data acquisition the total count rate was maintained below 40 kHz for a laser repetition rate of 200 kHz.
Laser and optics
A Spectra Physics Spirit 1040-8-HE laser system was used for the measurements. The Ytterbium doped laser crystal generated a 1040 nm fundamental wavelength which for the present experiments was doubled internally to produce 520 nm, 300 fs pulses at a repetition rate of 200 kHz. A beamsplitter divided the pulses with 70% directed into a beamline with a Mach–Zehender delay stage. These 15 μJ pulses were then focussed by a 30 cm focal length lens into the interaction region. In the other beamline the pulses were frequency doubled in a BBO crystal to produce 260 nm, 0.35 μJ pulses and focussed into the TOF with a 20 cm focal length lens.For the present measurements, the beams intersected slightly off focus to maximise the proportion of the ions obtained from the two-beam interaction relative to that obtained from the sum of each individual beam. The widths of both beams at the overlap were measured by recording the signal as the 520 nm beam was translated vertically through the 260 nm beam. In this way, the intensities of the 520 nm and 260 nm pulses were estimated at 1 × 1012 and 5 × 1010 W cm−2 respectively.
To control the polarization, each beam passed through a half-waveplate followed by a quarter-waveplate. The half-waveplates were rotated to change between left- and right- circular polarization (LCP, RCP) and linear polarization in the vertical or horizontal direction (VLP, HLP). When one beam was HLP and the other circularly polarised, the linear polarization was perpendicular to the circular polarization plane (as shown in Fig. 1), while VLP was parallel to the polarization plane. Using the rotating waveplate method,67 the degree of circular polarization (Stokes parameter S3) for all the waveplate angles which were used to produce circular polarization, was between 97% and 99% for both beams.
Data acquisition
The nomenclature used to describe asymmetries in ion yields arising from circular dichroism varies in the literature. It has been presented as circular dichroism in the ion yield (CDIY), photoion circular dichroism (PICD) or the g-value itself is used. Given that the g-value refers to the circular dichroism for a single transition between two specific states, in this paper we have used PICD for the asymmetry in the ion yields which can be accumulated from several transitions, each with their own g-value.To obtain PICD values, two separate measurements of the ion yield for LCP and RCP must be made. If these are measured sequentially, this can introduce errors due to fluctuations in target density, laser parameters or imperfections in the retarder being used to create the circular polarization. In the present measurements these errors were minimised by the high stability of the laser output (0.06% rms), by rapidly switching between polarization states (every few seconds), and by reversing the order in which the data was acquired. Previous PICD experiments have used achiral reference molecules and/or retro-reflected pulses which generate dual peaks in the TOF spectrum to reduce these errors.24,29
Imperfections in the waveplates mean that there are small variations in the magnitude of the circular polarization in oppositely circulating pulses. As the ionization process is non-linear this can lead to sizeable systematic biases in the ion yields. To minimise this systematic error and any errors due to drift in the experimental conditions, IL and IR yields were acquired by rotating the half-waveplate to an angle corresponding to LCP followed immediately by a move to the nearest angle corresponding to RCP. This was done for all other angles of the half waveplate which correspond to LCP/RCP pairs and then repeated but in reverse order with the yield from RCP measured before LCP. The PICD values were then calculated for each of these pairs and a mean and standard error calculated. This acquisition ‘rotation’ was repeated multiple times and an overall weighted mean and standard error obtained. This protocol reduced the systematic error due to the waveplate imperfections but small systematic biases (b) remained. The measured PICD(m) and true PICD(t) values were related by
| PICD(m) = e.PICD(m) + b |
The enantiomers of fenchone, 1S,4R (+) and 1R,4S (−), were used for the present study. By definition, the true PICD values for the enantiomers are equal and opposite, PICD+(t) = −PICD−(t). For our measurements we eliminated the systematic bias by measuring both enantiomers under identical experimental conditions and taking an average of their difference.
| PICD+(m) − PICD−(m) = e+PICD+(t) − e−PICD−(t) |

All the PICD values presented in this paper correspond to the true value of the 1S,4R (+) enantiomer, PICD+(t), with the uncertainties presented to one standard error in the mean. The samples used for the measurements were obtained from Merck and used without further purification. The supplier specified enantiomer excesses for each sample, were e+ = 0.98 and e− = 0.80.
To validate this new experimental apparatus and acquisition methodology, a PICD value averaged over all ion masses of –0.25 ± 0.03% was measured for (+)-alpha pinene using 520 nm pulses. This was in good agreement with a value of –0.29 ± 0.02% obtained from the total electron yield measured with a different instrument normally used to study PECD.68
Results and discussion
Fenchone has been used extensively for studies of chiroptical phenomena such as photoelectron circular dichroism and photoexcitation circular dichroism. Its bicyclic monoterpenoid structure limits conformational change and its electronic structure has been well-investigated experimentally and theoretically.56,69 Most previous resonant multiphoton ionization studies have focused on excitation of 3s and 3p Rydberg states, accessible with two 400 nm photons,34,36,38,44,48,70–72 but the wavelengths used in the present study provide access to the first excited π* state (A). This n–π* transition from the ground state (X) is electric dipole forbidden, so its single photon excitation cross section is small.73Previous measurements of ultraviolet absorption coefficients and circular dichroism for gas phase fenchone,73 show that the X–A excitation cross section peaks around 4.2 eV (296 nm) and is at least a factor of 100 less than for the higher lying bands. The g-value of 5% is considerably larger than that normally found for the electronic circular dichroism obtained from absorption of one photon. Measurements of the PICD by Loge and Boesl18 at 295 nm using a ns laser matched the magnitude and sign of this asymmetry.
Absorption of one 260 nm (4.78 eV) photon or two 520 nm (2.39 eV) photons used in the present study excite much higher rovibrational states within the A-band and would have a further order of magnitude lower transition probability than at the peak of the band. The adiabatic and vertical ionization energies of the molecule have been measured at 8.5 eV and 8.6 eV respectively,74,75 meaning that a minimum of four 520 nm or two 260 nm photons are required for ionization. The weakness of the X–A transition and our use of femtosecond pulses with high intensity, means that some of the ionization could be proceeding non-resonantly despite energetic overlap with the A band. A schematic energy level diagram for fenchone and possible transitions for both wavelengths is shown in Fig. 2.
 | ||
| Fig. 2 Fenchone energy level diagram and ionization schemes for the 520 nm and 260 nm laser pulses. X and X′ are the ground states of the fenchone neutral and cation, A is the π* first excited state of the neutral, with the D band also shown. The generation of fragments by absorption of 2 × 520 nm photons or 1 × 260 nm photon is only indicative and is more likely to proceed if further photons are absorbed. | ||
Mass spectra
Mass spectra obtained from ionization of fenchone are shown in Fig. 3. The spectra are dominated by the parent molecular ion at mass 152. The main fragments with masses of 81 and 69 are enhanced more significantly than the parent ion when both beams are present. These fragments can only be produced from the break-up of the cyclic structure involving the cleavage of two bonds. The positive charge ends up on one or other of the fragments with the neutral fragment picking up an additional H atom in the dissociation process. Breakage of these two bonds would require the absorption of at least two further 520 nm photons (or one 260 nm) beyond the ionization limit.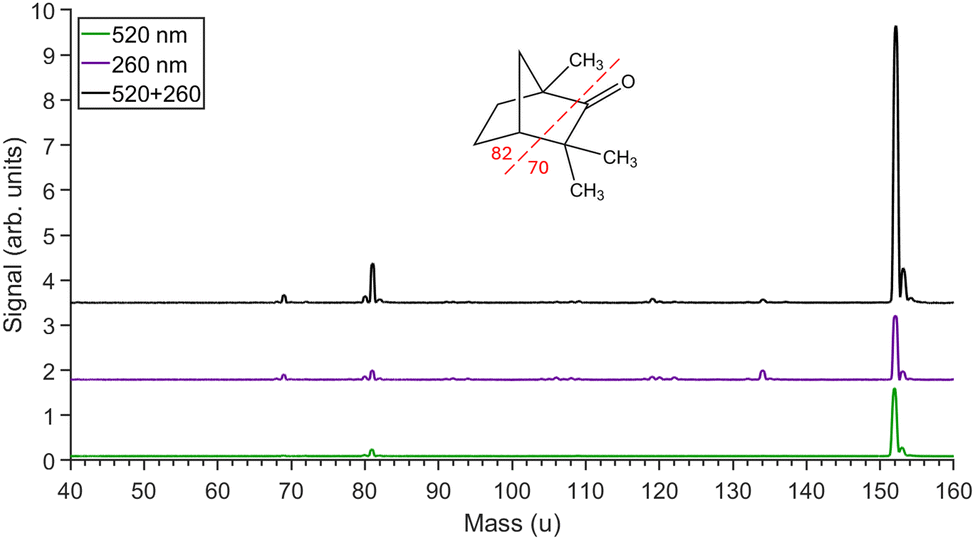 | ||
| Fig. 3 Mass spectra obtained for circularly 520 nm pulses only (green, bottom), 260 nm pulses only (purple, middle) and for both pulses (black, top) when circularly polarized. The fenchone parent molecular ion peak is at 152 u with the main fragment ions at 81 u and 69 u. The inset shows the structure of fenchone indicating which bonds must be broken to generate the main fragment ions observed in the spectrum. Minor peaks between 81 u and 152 u are due to aromatic contaminants left over from previous experiments. | ||
To optimize the temporal overlap of the beams, the yields of each main peak observed in the mass spectrum were recorded as a function of the delay of the 520 nm pulses relative to the 260 nm pulses (Fig. 4). The 650 fs width of the peak is not reflective of the pulse lengths of the individual beams as this is also determined by the off-focus spatial extent of the orthogonal beams. There is evidence of a decay process lasting about 300 fs in the pump–probe delay curves, particularly for the 81 u fragment. This indicates that the A state is being populated and is a key staging post for absorption of the additional photons needed to break up the ring structure. The observed decay is probably due to intra-vibrational redistribution from highly excited levels within the A band.
 | ||
| Fig. 4 Signal rates obtained as a function of the delay between RCP 260 nm pulses and VLP 520 nm pulses. The parent molecular (152 u) and fragment ions (81 u, 69 u, each scaled by a factor of 4) count rates are plotted. Positive delays correspond to the 520 nm pulse arriving after the 260 nm pulse. Each ion has been fitted with a function (black curves) comprising a Gaussian distribution (red), an error function with an exponential decay of lifetime τ (purple), and a second error function with no decay (blue). These individual components are shown for the 81 u mass from which a lifetime of about 300 fs was obtained. | ||
One-colour PICD
PICD values produced from each individual laser beam, plotted in Fig. 5, have magnitudes in the range 0.1–0.3%. Although the PICD was previously measured at 5% for 295 nm,73 our laser has a larger bandwidth, higher intensity and longer wavelength. As the X–A cross section at 260 nm is particularly weak, some of the ion signal could also be due to non-resonant ionization. | ||
| Fig. 5 PICD measurements for the major (+)fenchone cations detected in the mass spectrum for each individual laser colour; 520 nm – green circles; 260 nm – purple squares. The underlying mass spectrum is also plotted (not to scale). | ||
It is noticeable that the PICD values for the fragments are different from the parent ions. For 260 nm they are 0.15% more positive, while for 520 nm only, the 81 u fragment is 0.4% higher which results in a change of sign compared to the parent ion. This indicates that there are contributions to the PICD at both wavelengths resulting from transitions in the molecular ion due to the need for above threshold absorption of photons to produce fragment ions.
Two-colour PICD
Two-colour PICD results were taken at a time delay corresponding to the maximum enhancement in the parent ion signal (see Fig. 4). The enhancement in the ion yield above the background level varied depending on the polarization states of the individual pulses. The weakest enhancement was found when one pulse was circular while the other one was HLP so that it was perpendicular to the plane of the other pulse's polarization. The two-colour signal enhancement was a factor of 2–3 greater than the background for the parent ion, and 3–5 for the fragment ions.Background contributions generated from ionization by each individual pulse were subtracted from the signal by measuring ion yields at a negative time delay corresponding to the 520 nm pulse arriving about 2 ps before the 260 nm pulse. In this way the true two-colour signal was extracted. In all the two-colour measurements, the PICD signal was obtained from the difference in the LCP and RCP signal from one of the beams while the polarization of the other beam was kept fixed.
Fig. 6 shows the two-colour PICD values for the differential absorption of LCP vs. RCP for 520 nm pulses obtained while the 260 nm polarization was fixed at HLP, VLP, LCP or RCP. Comparison with the PICD obtained using only 520 nm pulses shows that no matter the polarization of the 260 nm pulses, the two-colour results mirror the one-colour results and are slightly more positive (≈0.15%).
 | ||
| Fig. 6 (a) Two-colour PICD of (+)fenchone derived from the differences in ion yields from changing the 520 nm polarization between LCP and RCP while the polarization of the 260 nm is fixed at: HLP – red diamond; VLP – blue diamond; LCP – black cross; RCP – magenta cross. Plotted for comparison are the one-colour 520 nm only PICD values – green circles. Note that for PICD values corresponding to the same ion mass, the points have been slightly offset in mass for easier visual comparison. (b) The one-colour (520 nm) PICD ionization scheme with the individual circular dichroism contributions for each excitation step, as derived from the data, are presented as percentage g-values. (c) The probable two-colour ionization scheme with the corresponding polarization states used for each beam. | ||
From the data in Fig. 5 and 6(a) the likely excitation route via two-colour absorption, and the individual g-values for the contributing transitions, could be deduced as shown in Fig. 6(b) and (c). If the excitation of the A state (the excitation step) is mainly due to the 260 nm pulse, then ionization from the A state (the ionization step) by the 520 nm pulse generates no contribution to the PICD as the parent ion PICD is close to 0%. There is also a relatively large circular dichroism (about +0.4%) from absorption in the molecular ion by 520 nm photons which results in production of the mass 81 fragment (the fragmentation step). This is less clear for the 69 fragment due to poorer statistics. Comparison between the two-colour and one-colour results also indicates that X–A transitions by 2 × 520 nm absorption must be contributing −0.15% to the overall PICD in the 520 nm only results.
When two-colour PICD is obtained with the 260 nm pulse being switched between LCP and RCP, more interesting results are found. To help understand the relative contributions to the PICD from the 260 nm pulses, measurements were taken with the 260 nm pulse at an intensity 5 times higher than that used for all the other results. This meant that the excitation step was dominated by the 260 nm excitation and hence the ionization step must have proceeded by absorption of 520 nm photons for this wavelength to contribute to the two-colour signal. As the 520 nm beam was linearly polarised, this ionization step could not contribute to the PICD and any asymmetry must arise from 260 nm X–A excitation.
Fig. 7 shows that under these two-colour conditions the PICD was very small, indicating that since the 520 nm polarization is linear for the ionization step, there is a −0.1% PICD contribution from the X–A excitation step by a 260 nm photon. This means that the positive PICD values found in the one-colour 260 nm measurements must arise from the ionization step, contributing around +0.25% as summarised in Fig. 7(b).
 | ||
| Fig. 7 (a) Two-colour PICD of (+)fenchone derived from the differences in ion yields from changing the 260 nm polarization between LCP and RCP while the polarization of the 520 nm is fixed at: HLP – red diamond; VLP – blue diamond. Plotted for comparison are the one-colour 260 nm only PICD values – purple squares. Unlike all the other results in this paper, the intensity of the 260 nm was increased to 2.5 × 1011 W cm−2 meaning X–A excitation step was being dominated by absorption of one 260 nm photon. (b) the one-colour (260 nm) PICD ionization scheme with the individual circular dichroism contributions for each excitation step, as derived from the data, are presented as percentage g-values. (c) the two-colour ionization schemes for 520 nm VLP and HLP. | ||
The fact that there is small but significant circular dichroism in the ionization step by 260 nm pulses is contrary to the prevailing view that any such contributions are minimal. As there can be multiple transitions to continuum states, one might expect the overall contribution to tend to average to zero. There has only been one previous direct measurement of CD for ionization from an excited state.21 The g-value for this transition was found to be small compared to the excitation from the ground state, being less than 0.3%. Kröner has theoretically investigated the resonant and non-resonant CD contributions to ion yields via stationary and non-stationary states in methyl oxirane and indicated that there can be a small contributions to PICD values from the ionization step.76
The existence of non-zero CD in the ionization step by 260 nm provides an opportunity to investigate how this might be influenced by the way in which the A-state is prepared. Using the same polarization scheme as in Fig. 7 but with lower intensity 260 nm pulses, the pulses which primarily contribute to the excitation and ionization steps were reversed. Fig. 8(c) and (d) shows that the A-state was then predominantly populated by absorption of two 520 nm linearly polarized photons while the ionization step proceeded by absorption of one circularly polarized 260 nm photon. Fig. 8(a) shows that in this case the PICD was strongly dependent on the direction of the 520 nm linear polarization. Compared to the one-colour 260 nm measurements, for both parent and fragment ions, the two-colour PICD results became more positive when the 520 nm polarization was parallel to the 260 nm polarization plane and more negative when perpendicular. Since this change between VLP and HLP corresponds to 4 standard errors in the mean (for masses 152 and 81), we can say with high confidence that spatial orientation of the ensemble following the excitation step strongly influences the circular dichroism in the ionization step and the fragmentation step.
 | ||
| Fig. 8 (a) Two-colour PICD of (+)fenchone derived from the differences in ion yields from changing the 260 nm polarization between LCP and RCP while the polarization of the 520 nm is fixed at: HLP – red diamond; VLP – blue diamond. Plotted for comparison are the one-colour 260 nm only PICD values – purple squares. These polarizations were identical to those used in Fig. 7 but with a reduced 260 nm intensity of 5 x 1010 W cm−2. This means that the X–A excitation step was primarily through excitation by the 520 nm pulses. (b) and (c) the dominant two-colour ionization schemes for 520 nm VLP and HLP respectively showing percentage g-values which have been modified by the polarization direction used for the excitation step. | ||
When the 520 nm pulse polarization was switched from linear to circular polarization, this produced a substantial change in the ionization probability (about a factor of 3 when using 520 nm only). This meant that while the excitation step was more probable when the polarization of the 520 nm pulses were linear (Fig. 8(c)). When circularly polarized the excitation rate was more balanced with that obtained from the 260 nm pulses. In this scenario there is the possibility that when both beams are circular, the circular dichroism could be influenced by interference between the two excitation/ionization pathways shown in Fig. 9(b). Our PICD measurements under these conditions (Fig. 9(a)) show a statistically significant difference in the two-colour 260 nm PICD depending on whether the 520 nm pulse is fixed at either LCP or RCP (for masses 152 and 81).
 | ||
| Fig. 9 (a) Two-colour PICD of (+) fenchone derived from the differences in ion yields from changing the 260 nm polarization between LCP and RCP while the polarization of the 520 nm is fixed at: LCP – black cross; RCP – magneta cross. Plotted for comparison are the one-colour 260 nm only PICD values – purple squares. (b) and (c) the two-colour ionization pathways where for each measurement the 520 nm polarization was fixed at LCP and RCP respectively. | ||
Alternatively, it could be that the excitation step creates a chiral vibronic wavepacket which leads to the ionization step being dependent on the polarization (LCP v RCP). This photoexcitation circular dichroism (PXCD) has been previously observed via the photoelectron angular distribution,61 but has yet to be shown as a way of controlling of integrated ion yields.
Conclusions
In conclusion, we have re-purposed a time-of-flight mass spectrometer to probe chiral molecular structure using orthogonally intersecting laser pulses. By overlapping visible (520 nm) and ultraviolet (260 nm) femtosecond pulses in space and time, we have extracted contributions from two-colour photoion circular dichroism with uncertainties of 0.1% or less. This has allowed us to show how the relative ion yields can be controlled beyond the inherent circular dichroism present from interactions with a single colour pulse. By exciting an intermediate state with linearly polarised 520 nm light, a spatially orientated ensemble of excited molecules was created which substantially modifies the circular dichroism produced from the subsequent ionization of this state by a 260 nm photon.Control of the ion yields was also demonstrated when both beams were circularly polarized. In this case there can be no orientation dependence of the circular dichroism as preferential excitation of molecules with transition moments in the polarization plane of the exciting pulse does not change following a switch in the circulation direction. Instead, interference between one-photon and two-photon pathways could be occurring, or a chiral wavepacket is being generated which influences the PICD obtained from ionization of this state. The latter would be the first evidence of photoexcitation circular dichroism observed via ion yields.
Ultimately, while these exciting results demonstrate quantum control of chiral ionization processes, the small asymmetries generated through electronic circular dichroism make the experiments lengthy and produce only modest changes in the photoionization and photofragmentation yields. This is because circular dichroism is not manifested in the dipole approximation and relies on the interference between the electric dipole and magnetic dipole (or higher) moments of the transition. However, the recent interest in generating locally chiral light for interactions within the dipole approximation implies that more extensive chiroptical control is possible. For instance, if much larger PICD asymmetries can be generated, then all-optical enantiomeric purification could be achieved by separation of the ions using external fields or via selective photo-destruction of one of the enantiomers.
Creating locally chiral fields requires the overlap of multi-colour, non-collinear beams. However, due to phase differences between the intersecting beams, the handedness of the locally chiral pulses changes across the interaction region and any differences in the net yields tend to average to zero. For a successful experimental realisation, the chiral pulses must also be globally chiral so that the handedness is invariant as the relative phase between non-collinear beams changes. Ayuso et al.62 have already identified a scheme to achieve this using two beams each consisting of collinear 1ω/2ω pulses.
Ye et al.77 have proposed another scheme using one collinear 1ω/2ω beam where both pulses are linearly polarized in the same direction and are intersected perpendicularly with a circularly polarised 1ω beam. This configuration is attractive as the shape of the electric field configuration would be constant across the interaction volume, with phase changes only causing a rotation of the electric field about the direction of the 1ω beam. The present instrument has been designed so that this chiral pulse configuration can be implemented for future experiments. This offers the exciting possibility of observing and controlling more dramatic PICD asymmetries within the dipole approximation.
Author contributions
LD was involved in setting up the experiment, acquiring and analysing the data, and editing several drafts of the manuscript. JBG developed the concept, set up the experimental apparatus and the data acquisition system, and supervised the experimental work and analysis. JBG wrote the original draft of paper and subsequent versions.Data availability
The numerical data which is graphically displayed in the paper is tabulated in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Ian Williams, Yann Mairesse, Valerie Blanchet, Bernard Pons and David Ayuso for their valuable discussions which have contributed to the development of this experiment and interpretation of the results. This work has been supported by the Royal Society's Paul Instrument Fund (PI170043) and International Exchange Scheme (IES\R1\241228), and from Queen's University's Agility Plus fund.Notes and references
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2019, 11, a032540 CrossRef CAS PubMed.
- Y. Chen and W. Ma, PLoS Comput. Biol., 2020, 16, e1007592 CrossRef CAS PubMed.
- Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436–3476 RSC.
- R. Bentley, Perspect. Biol. Med., 1995, 38, 188–229 CrossRef CAS PubMed.
- G. Vantomme and J. Crassous, Chirality, 2021, 33, 597–601 CrossRef CAS PubMed.
- P. Laur, Compr. Chiropt. Spectrosc., 2012, 2, 1–35 Search PubMed.
- M. D. Marshall, H. O. Leung, S. R. Domingos, A. Krin, M. Schnell, N. A. Seifert, Y. Xu and W. Jager, Phys. Chem. Chem. Phys., 2022, 24, 28495–28505 RSC.
- D. Patterson, M. Schnell and J. M. Doyle, Nature, 2013, 497, 475–477 CrossRef CAS PubMed.
- V. A. Shubert, D. Schmitz, D. Patterson, J. M. Doyle and M. Schnell, Angew. Chem., Int. Ed., 2014, 53, 1152–1155 CrossRef CAS PubMed.
- A. K. Patterson, L. H. El-Qarra and D. K. Smith, Chem. Commun., 2022, 58, 3941–3944 RSC.
- M. Pitzer, R. Berger, J. Stohner, R. Dorner and M. Schoffler, Chimia, 2018, 72, 384–388 CrossRef CAS PubMed.
- M. Pitzer, K. Fehre, M. Kunitski, T. Jahnke, L. Schmidt, H. Schmidt-Bocking, R. Dorner and M. Schoffler, J. Visualized Exp., 2017, 126, 56062 Search PubMed.
- M. Pitzer, M. Kunitski, A. S. Johnson, T. Jahnke, H. Sann, F. Sturm, L. P. Schmidt, H. Schmidt-Bocking, R. Dorner, J. Stohner, J. Kiedrowski, M. Reggelin, S. Marquardt, A. Schiesser, R. Berger and M. S. Schoffler, Science, 2013, 341, 1096–1100 CrossRef CAS PubMed.
- U. Boesl von Grafenstein and A. Bornschlegl, ChemPhysChem, 2006, 7, 2085–2087 CrossRef PubMed.
- H. G. Breunig, G. Urbasch, P. Horsch, J. Cordes, U. Koert and K. M. Weitzel, ChemPhysChem, 2009, 10, 1199–1202 CrossRef CAS PubMed.
- C. Loge, A. Bornschlegl and U. Boesl, Anal. Bioanal. Chem., 2009, 395, 1631–1639 CrossRef CAS PubMed.
- P. Horsch, G. Urbasch, K. M. Weitzel and D. Kroner, Phys. Chem. Chem. Phys., 2011, 13, 2378–2386 RSC.
- C. Loge and U. Boesl, ChemPhysChem, 2011, 12, 1940–1947 CrossRef CAS PubMed.
- P. Horsch, G. Urbasch and K. M. Weitzel, Chirality, 2012, 24, 684–690 CrossRef CAS PubMed.
- C. Loge and U. Boesl, ChemPhysChem, 2012, 13, 4218–4223 CrossRef CAS PubMed.
- C. Loge and U. Boesl, Phys. Chem. Chem. Phys., 2012, 14, 11981–11989 RSC.
- U. Boesl, A. Bornschlegl, C. Loge and K. Titze, Anal. Bioanal. Chem., 2013, 405, 6913–6924 CrossRef CAS PubMed.
- K. Titze, T. Zollitsch, U. Heiz and U. Boesl, ChemPhysChem, 2014, 15, 2762–2767 CrossRef CAS PubMed.
- U. Boesl and A. Kartouzian, Annu. Rev. Anal. Chem., 2016, 9, 343–364 CrossRef CAS PubMed.
- A. Hong, H. Jang, C. Jeong, M. C. Choi, J. Heo and N. J. Kim, J. Phys. Chem. Lett., 2016, 7, 4385–4390 CrossRef CAS PubMed.
- J. Lepelmeier, K. Titze, A. Kartouzian, U. Boesl and U. Heiz, ChemPhysChem, 2016, 17, 4052–4058 CrossRef CAS PubMed.
- J. Lepelmeier, J. L. Alonso-Gomez, F. Mortaheb, U. Boesl, U. Heiz and A. Kartouzian, Phys. Chem. Chem. Phys., 2017, 19, 21297–21303 RSC.
- C. S. Lehmann and K. M. Weitzel, Phys. Chem. Chem. Phys., 2020, 22, 13707–13712 RSC.
- T. Ring, C. Witte, S. Vasudevan, S. Das, S. T. Ranecky, H. Lee, N. Ladda, A. Senftleben, H. Braun and T. Baumert, Rev. Sci. Instrum., 2021, 92, 033001 CrossRef CAS PubMed.
- C. Jeong, H. J. Eun, J. Yun and N. J. Kim, J. Phys. Chem. A, 2022, 126, 4295–4299 CrossRef CAS PubMed.
- C. S. Lehmann, D. Botros and K. M. Weitzel, Phys. Chem. Chem. Phys., 2022, 24, 15904–15911 RSC.
- N. Bowering, T. Lischke, B. Schmidtke, N. Muller, T. Khalil and U. Heinzmann, Phys. Rev. Lett., 2001, 86, 1187–1190 CrossRef CAS PubMed.
- L. Nahon, G. A. Garcia, C. J. Harding, E. Mikajlo and I. Powis, J. Chem. Phys., 2006, 125, 114309 CrossRef PubMed.
- C. Lux, M. Wollenhaupt, T. Bolze, Q. Liang, J. Kohler, C. Sarpe and T. Baumert, Angew. Chem., Int. Ed., 2012, 51, 5001–5005 CrossRef CAS PubMed.
- C. S. Lehmann, N. B. Ram, I. Powis and M. H. Janssen, J. Chem. Phys., 2013, 139, 234307 CrossRef PubMed.
- S. Beaulieu, A. Comby, B. Fabre, D. Descamps, A. Ferre, G. Garcia, R. Geneaux, F. Legare, L. Nahon, S. Petit, T. Ruchon, B. Pons, V. Blanchet and Y. Mairesse, Faraday Discuss., 2016, 194, 325–348 RSC.
- A. Comby, S. Beaulieu, M. Boggio-Pasqua, D. Descamps, F. Legare, L. Nahon, S. Petit, B. Pons, B. Fabre, Y. Mairesse and V. Blanchet, J. Phys. Chem. Lett., 2016, 7, 4514–4519 CrossRef CAS PubMed.
- A. Kastner, C. Lux, T. Ring, S. Zullighoven, C. Sarpe, A. Senftleben and T. Baumert, ChemPhysChem, 2016, 17, 1119–1122 CrossRef CAS PubMed.
- S. Beaulieu, A. Comby, D. Descamps, S. Petit, F. Legare, B. Fabre, V. Blanchet and Y. Mairesse, J. Chem. Phys., 2018, 149, 134301 CrossRef CAS PubMed.
- A. Comby, E. Bloch, C. M. M. Bond, D. Descamps, J. Miles, S. Petit, S. Rozen, J. B. Greenwood, V. Blanchet and Y. Mairesse, Nat. Commun., 2018, 9, 5212 CrossRef CAS PubMed.
- C. M. M. Bond and J. B. Greenwood, J. Phys.: Conf. Ser., 2019, 1289, 012027 CrossRef CAS.
- A. Comby, C. M. M. Bond, E. Bloch, D. Descamps, B. Fabre, S. Petit, Y. Mairesse, J. B. Greenwood and V. Blanchet, Chirality, 2020, 32, 1225–1233 CrossRef CAS PubMed.
- A. Kastner, G. Koumarianou, P. Glodic, P. C. Samartzis, N. Ladda, S. T. Ranecky, T. Ring, S. Vasudevan, C. Witte, H. Braun, H. G. Lee, A. Senftleben, R. Berger, G. B. Park, T. Schafer and T. Baumert, Phys. Chem. Chem. Phys., 2020, 22, 7404–7411 RSC.
- V. Blanchet, D. Descamps, S. Petit, Y. Mairesse, B. Pons and B. Fabre, Phys. Chem. Chem. Phys., 2021, 23, 25612–25628 RSC.
- K. Fehre, S. Eckart, M. Kunitski, C. Janke, D. Trabert, M. Hofmann, J. Rist, M. Weller, A. Hartung, L. P. H. Schmidt, T. Jahnke, H. Braun, T. Baumert, J. Stohner, P. V. Demekhin, M. S. Schoffler and R. Dorner, Phys. Rev. Lett., 2021, 126, 083201 CrossRef CAS PubMed.
- G. Nalin, K. Fehre, F. Trinter, N. M. Novikovskiy, N. Anders, D. Trabert, S. Grundmann, M. Kircher, A. Khan, R. Tomar, M. Hofmann, M. Waitz, I. Vela-Perez, G. Kastirke, J. Siebert, D. Tsitsonis, H. Fukuzawa, K. Ueda, J. B. Williams, D. Kargin, M. Maurer, C. Kustner-Wetekam, L. Marder, J. Viehmann, A. Knie, T. Jahnke, M. Ilchen, R. Dorner, R. Pietschnig, P. V. Demekhin and M. S. Schoffler, Phys. Chem. Chem. Phys., 2021, 23, 17248–17258 RSC.
- P. Kruger, J. H. Both, U. Linne, F. Chirot and K. M. Weitzel, J. Phys. Chem. Lett., 2022, 13, 6110–6116 CrossRef PubMed.
- J. Miles, D. Fernandes, A. Young, C. M. M. Bond, S. W. Crane, O. Ghafur, D. Townsend, J. Sa and J. B. Greenwood, Anal. Chim. Acta, 2017, 984, 134–139 CrossRef CAS PubMed.
- C. Sparling, A. Ruget, N. Kotsina, J. Leach and D. Townsend, ChemPhysChem, 2021, 22, 76–82 CrossRef CAS PubMed.
- C. Sparling, S. W. Crane, L. Ireland, R. Anderson, O. Ghafur, J. B. Greenwood and D. Townsend, Phys. Chem. Chem. Phys., 2023, 25, 6009–6015 RSC.
- I. Powis, Chirality, 2008, 20, 961–968 CrossRef CAS PubMed.
- I. Powis, Adv. Chem. Phys., 2008, 138, 267–329 CrossRef CAS.
- S. Das, J. Ghosh, S. Vasudevan, S. T. Ranecky, T. Rosen, N. Ladda, H.-G. Lee, T.-J. Stehling, F. Westmeier, J. Mikosch, A. Senftleben, T. Baumert and H. Braun, Phys. Chem. Chem. Phys., 2025, 25, 8043–8051 RSC.
- T. Bondo Pedersen and A. E. Hansen, Chem. Phys. Lett., 1995, 246, 1–8 CrossRef.
- K. Fehre, N. M. Novikovskiy, S. Grundmann, G. Kastirke, S. Eckart, F. Trinter, J. Rist, A. Hartung, D. Trabert, C. Janke, M. Pitzer, S. Zeller, F. Wiegandt, M. Weller, M. Kircher, G. Nalin, M. Hofmann, L. P. H. Schmidt, A. Knie, A. Hans, L. Ben Ltaief, A. Ehresmann, R. Berger, H. Fukuzawa, K. Ueda, H. Schmidt-Bocking, J. B. Williams, T. Jahnke, R. Dorner, P. V. Demekhin and M. S. Schoffler, Phys. Chem. Chem. Phys., 2022, 24, 26458–26465 RSC.
- C. Jeong, J. Yun, J. Heo and N. J. Kim, Phys. Chem. Chem. Phys., 2023, 25, 22644–22649 RSC.
- V. Wanie, E. Bloch, E. P. Mansson, L. Colaizzi, S. Ryabchuk, K. Saraswathula, A. F. Ordonez, D. Ayuso, O. Smirnova, A. Trabattoni, V. Blanchet, N. Ben Amor, M. C. Heitz, Y. Mairesse, B. Pons and F. Calegari, Nature, 2024, 630, 109–115 CrossRef CAS PubMed.
- V. Svoboda, N. B. Ram, D. Baykusheva, D. Zindel, M. D. J. Waters, B. Spenger, M. Ochsner, H. Herburger, J. Stohner and H. J. Wörner, Sci. Adv., 2022, 8, eabq2811 CrossRef CAS PubMed.
- J. Lee, E. Abdiha, B. G. Sartakov, G. Meijer and S. Eibenberger-Arias, Nat. Commun., 2024, 15, 7441 CrossRef CAS PubMed.
- M. Mondelo-Martell, D. Basilewitsch, H. Braun, C. P. Koch and D. M. Reich, Phys. Chem. Chem. Phys., 2022, 24, 9286–9297 RSC.
- S. Beaulieu, A. Comby, D. Descamps, B. Fabre, G. A. Garcia, R. Géneaux, A. G. Harvey, F. Légaré, Z. Mašín, L. Nahon, A. F. Ordonez, S. Petit, B. Pons, Y. Mairesse, O. Smirnova and V. Blanchet, Nat. Phys., 2018, 14, 484–489 Search PubMed.
- D. Ayuso, O. Neufeld, A. F. Ordonez, P. Decleva, G. Lerner, O. Cohen, M. Ivanov and O. Smirnova, Nat. Photonics, 2019, 13, 866–871 CrossRef CAS.
- L. Belshaw, F. Calegari, M. J. Duffy, A. Trabattoni, L. Poletto, M. Nisoli and J. B. Greenwood, J. Phys. Chem. Lett., 2012, 3, 3751–3754 CrossRef CAS PubMed.
- E. P. Mansson, S. Latini, F. Covito, V. Wanie, M. Galli, E. Perfetto, G. Stefanucci, H. Hubener, U. De Giovannini, M. C. Castrovilli, A. Trabattoni, F. Frassetto, L. Poletto, J. B. Greenwood, F. Legare, M. Nisoli, A. Rubio and F. Calegari, Commun. Chem., 2021, 4, 73 CrossRef CAS PubMed.
- E. P. Mansson, S. De Camillis, M. C. Castrovilli, M. Galli, M. Nisoli, F. Calegari and J. B. Greenwood, Phys. Chem. Chem. Phys., 2017, 19, 19815–19821 RSC.
- F. Calegari, D. Ayuso, A. Trabattoni, L. Belshaw, S. De Camillis, S. Anumula, F. Frassetto, L. Poletto, A. Palacios, P. Decleva, J. B. Greenwood, F. Martin and M. Nisoli, Science, 2014, 346, 336–339 CrossRef CAS PubMed.
- B. Schaefer, E. Collett, R. Smyth, D. Barrett and B. Fraher, Am. J. Phys., 2007, 75, 163–168 CrossRef.
- J. B. Greenwood and I. D. Williams, Phys. Chem. Chem. Phys., 2023, 25, 16238–16245 RSC.
- S. Beauvarlet, E. Bloch, D. Rajak, D. Descamps, B. Fabre, S. Petit, B. Pons, Y. Mairesse and V. Blanchet, Phys. Chem. Chem. Phys., 2022, 24, 6415–6427 RSC.
- C. Lux, M. Wollenhaupt, C. Sarpe and T. Baumert, ChemPhysChem, 2015, 16, 115–137 CrossRef CAS PubMed.
- H. G. Lee, S. T. Ranecky, S. Vasudevan, N. Ladda, T. Rosen, S. Das, J. Ghosh, H. Braun, D. M. Reich, A. Senftleben and T. Baumert, Phys. Chem. Chem. Phys., 2022, 24, 27483–27494 RSC.
- A. Kastner, T. Ring, B. C. Kruger, G. B. Park, T. Schafer, A. Senftleben and T. Baumert, J. Chem. Phys., 2017, 147, 013926 CrossRef PubMed.
- F. Pulm, J. Schramm, J. Hormes, S. Grimme and S. D. Peyerimhoff, Chem. Phys., 1997, 224, 143–155 CrossRef CAS.
- D. P. Singh, N. De Oliveira, G. A. Garcia, A. Vredenborg and I. Powis, ChemPhysChem, 2020, 21, 2468–2483 CrossRef CAS PubMed.
- I. Powis, C. J. Harding, G. A. Garcia and L. Nahon, ChemPhysChem, 2008, 9, 475–483 CrossRef CAS PubMed.
- D. Kroner, Phys. Chem. Chem. Phys., 2015, 17, 19643–19655 RSC.
- Y. S. Chong Ye, L. Fu and X. Zhang, 2022, arXiv, 2210.07023v2, https://arxiv.org/abs/2210.07023v2.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp01127b |
| This journal is © the Owner Societies 2025 |