DOI:
10.1039/D4DT03020F
(Paper)
Dalton Trans., 2025,
54, 1838-1849
Introduction of substituents for tuning the redox properties of benzoate-bridged paddlewheel diruthenium(II,II) complexes: what does the OH group bring?†
Received
30th October 2024
, Accepted 10th December 2024
First published on 12th December 2024
Abstract
Benzoate-bridged paddlewheel diruthenium(II,II) complexes ([RuII,II2(RnArCO2)4(Lax)2] (Lax = axial ligand); [RuII,II2]) exhibit reversible redox activity involving the oxidized species [RuII,III2]+. The redox activity can be finely tuned over a broad potential range by altering the substituent R on the benzoate-bridging ligand RnArCO2−. The electronic contributions of the substituents R depend on their type and position, as was empirically demonstrated by Hammett for substituents at the meta- and para-positions. However, the substituent effect at the ortho-position is not solely determined by the electronic contribution of R but also by steric hindrance between the o-substituents and adjacent carboxylate groups. Nevertheless, an OH group at the o-position did not provide any steric hindrance, leading to a strong electron-withdrawing effect owing to intramolecular hydrogen bonding between the o-OH group and the adjacent carboxylate group, despite the electron-donating ability of the m- and p-OH groups. The OH group at the o-position induced a significant shift in the redox potential and HOMO energy levels of the [RuII,II2] complexes, thereby stabilizing the [RuII,II2] state. The redox potential and HOMO can be adjusted by introducing additional substituents, such as F, Cl, Me, OMe, and CF3 groups, to cover a wide range, in accordance with an extended Hammett law that considers the contribution of the o-position.
Introduction
Carboxylate-bridged paddlewheel diruthenium(II,II) complexes ([RuII,II2])1 are intriguing entities that act as secondary building units2 for the construction of metal–organic frameworks.3–6 These complexes exhibit reversible redox activity, derived from oxidized [RuII,III2]+, without significant structural change.7,8 [RuII,II2] and [RuII,III2]+ species are both paramagnetic with multiple spin states (S = 1 and 3/2, respectively) and are useful magnetic building units9–12 and they have the potential ability for catalytic activity.13–18 The redox properties and HOMO energy levels of [RuII,II2] can be precisely controlled by chemical modification of the carboxylate ligands.19–22 For example, for the series of [RuII,II2] complexes with substituted benzoate ligands, [RuII,II2(RnArCO2)4(Lax)2] (RnArCO2− = substituted benzoate; Lax = axial ligand), the redox properties and HOMO energy levels can be predicted within the scale of Hammett's law for the congeners substituted at the meta- and para-positions.23–25 However, evaluation of the ortho-substituted congeners is less straightforward because of steric hindrance between the o-substituents and adjacent carboxylate groups, which constrains or weakens electronic conjugation between the aryl ring and carboxylate groups, in addition to the electronic effect of the o-substituents. Thus far, our group has synthesized various benzoate-bridged [RuII,II2] complexes with substituents such as F, Cl, Me, OMe, CF3, and OH, and demonstrated that the redox properties of these complexes follow an extended Hammett's law that includes the pseudo-Hammett constant, σo, obtained empirically by taking into account both the electronic and steric hindrance effects (Table 1),19,20,22 in addition to the common Hammett constants σm and σp for m- and p-substituents, respectively.26–30 Interestingly, the strongly electron-withdrawing nature of the o-OH group was revealed,20 where o-OH exerted a large electron-withdrawing effect, with σo(OH) = 0.667 compared with σo(F) = 0.217 and σo(Cl) = 0.272, although the m- and p-OH groups exhibit electron-donating ability.25 This was first observed for the series of trans-heteroleptic [RuII,II2] complexes, [Ru2{2,6-(CF3)2ArCO2}2{(OH)xArCO2}2(THF)2], where (OH)xArCO2− and 2,6-(CF3)2ArCO2− indicate hydroxy-substituted benzoates and 2,6-(bistrifluoromethyl)benzoate, respectively (Chart 1).22 The o-OH group formed an intramolecular hydrogen bond with the oxygen atom of the bridging carboxylate and o-OH substituent, which not only retained the planarity of the benzoate ligand (associated with the electronic conjugation), but also strongly suppressed the electron-donating ability, consequently lowering the HOMO energy level of the [RuII,II2] unit.22 This effect suggests that introducing additional substitutions such as F, Cl, Me, OMe, and CF3 groups into the m- and/or p-positions in addition to the presence of the o-OH group should adjust the redox potentials and HOMO energy levels over a wide range.
 |
| | Chart 1 Homoleptic [Ru2{RnArCO2}4(THF)2] and heteroleptic [Ru2{2,6-(CF3)2ArCO2}2{RnArCO2}2(THF)2] paddlewheel diruthenium(II,II) complexes described in this study. | |
Table 1 Hammett constants of the relevant substituents
| Substituent |
F |
Cl |
Br |
Me |
OMe |
CF3 |
Ph |
OH |
|
Hammett constants σm and σp were obtained from ref. 24.
Hammett constants σo were taken from ref. 20 and 22.
|
|
para
|
0.062 |
0.227 |
0.232 |
−0.170 |
−0.268 |
0.54 |
0.144 |
−0.370 |
|
meta
|
0.337 |
0.373 |
0.391 |
−0.069 |
0.115 |
0.43 |
0.06 |
0.121 |
|
ortho
|
0.217b |
0.272b |
|
−0.070 |
−0.397 |
0.50 |
|
0.667b |
This study presents 17 new [RuII,II2] complexes with the general formulas (i) [RuII,II2{n-X-4-(OH)ArCO2}4(THF)2] (nX4OH), (ii) [RuII,II2{n-X-2-(OH)ArCO2}4(THF)2] (nX2OH), and (iii) [Ru2{2,6-(CF3)2ArCO2}2{n-X-2-(OH)ArCO2}2(THF)2] (ht-nX2OH), where X indicates a substituent and n is the position of the substituent on the aryl ring; (i) n-X-4-(OH)ArCO2− = 2-chloro-4-hydroxybenzoate, 2Cl4OH; 3-fluoro-4-hydroxybenzoate, 3F4OH; 3-chloro-4-hydroxybenzoate, 3Cl4OH; 3-bromo-4-hydroxybenzoate, 3Br4OH; (ii) n-X-2-(OH)ArCO2− = 3-fluoro-2-hydroxybenzoate, 3F2OH; 4-fluoro-2-hydroxybenzoate, 4F2OH; 4-chloro-2-hydroxybenzoate, 4Cl2OH; 4-methyl-2-hydroxybenzoate, 4Me2OH; 5-chloro-2-hydroxybenzoate, 5Cl2OH; 5-bromo-2-hydroxybenzoate, 5Br2OH; 5-methyl-2-hydroxybenzoate, 5Me2OH; (iii) n-X-2-(OH)ArCO2− = 4-fluoro-2-hydroxybenzoate, ht-4F2OH; 4-chloro-2-hydroxybenzoate, ht-4Cl2OH; 4-methyl-2-hydroxybenzoate, ht-4Me2OH; 4-methoxy-2-hydroxybenzoate, ht-4OMe2OH; 4-trifluoromethyl-2-hydroxybenzoate, ht-4CF32OH; 5-methyl-2-hydroxybenzoate, ht-5Me2OH (Chart 1). Families nX4OH and nX2OH have homoleptic paddlewheel motifs, whereas ht-nX2OH has a trans-heteroleptic paddlewheel motif. The redox activities of these compounds are evaluated via electrochemical measurements in the same medium. Density functional theory (DFT) calculations based on the atomic coordinates of the solid-state structures are used to estimate the HOMO energies of the compounds. Considering the σo(OH) value, all of the complexes adhered to the extended Hammett's law on a unified scale.
Results and discussion
Syntheses and characterization
The present compounds were synthesized using previously reported methods.31,32 The homoleptic [RuII,II2{n-X-4-(OH)ArCO2}4(THF)2] (nX4OH) and [RuII,II2{n-X-2-(OH)ArCO2}4(THF)2] (nX2OH) series were synthesized via ligand substitution of [RuII,III2(OAc)4Cl] (OAc− = acetate), forming [RuII,III2{n-X-4-(OH)ArCO2}4(Cl)] and [RuII,III2{n-X-2-(OH)ArCO2}4(Cl)], respectively, followed by reduction to the [RuII,II2] species of nX4OH and nX2OH using Zn power in THF. The trans-heteroleptic complexes of [Ru2{2,6-(CF3)2ArCO2}2{n-X-2-(OH)ArCO2}2(THF)2] (ht-nX2OH) were synthesized via the ligand substitution reaction of [RuII,III2{n-X-2-(OH)ArCO2}4(Cl)] with 2,6-bis(trifluoromethyl)benzoic acid 2,6-(CF3)2ArCO2H in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometric ratio to produce [RuII,III2{2,6-(CF3)2ArCO2}2{n-X-2-(OH)ArCO2}2(Cl)], followed by reduction to the [RuII,II2] species of ht-nX2OH using Zn powder in THF. All compounds with para-OH substituents contained additional THF molecules as crystallization solvents and proton acceptors of the p-OH group (see Structural Characterization and Elemental Analysis). In the previous study, recrystallization from THF was not successful for [Ru2] with p-OHArCO2− (p-OH; recrystallized from MeOH),30 whereas the use of n-X-4-(OH)ArCO2− (n = 3–5, X = F, Cl, and Br) increased the solubility of the compounds in THF, resulting in successful isolation of single crystals as THF di-adducts. All compounds of nX4OH were isolated as [RuII,II2], although p-OH was obtained as [RuII,III2].30 This is because the halogen group compensates for the electron-donating ability of the p-OH group, stabilizing the low-valence state of [RuII,II2], as discussed hereinafter.
:2 stoichiometric ratio to produce [RuII,III2{2,6-(CF3)2ArCO2}2{n-X-2-(OH)ArCO2}2(Cl)], followed by reduction to the [RuII,II2] species of ht-nX2OH using Zn powder in THF. All compounds with para-OH substituents contained additional THF molecules as crystallization solvents and proton acceptors of the p-OH group (see Structural Characterization and Elemental Analysis). In the previous study, recrystallization from THF was not successful for [Ru2] with p-OHArCO2− (p-OH; recrystallized from MeOH),30 whereas the use of n-X-4-(OH)ArCO2− (n = 3–5, X = F, Cl, and Br) increased the solubility of the compounds in THF, resulting in successful isolation of single crystals as THF di-adducts. All compounds of nX4OH were isolated as [RuII,II2], although p-OH was obtained as [RuII,III2].30 This is because the halogen group compensates for the electron-donating ability of the p-OH group, stabilizing the low-valence state of [RuII,II2], as discussed hereinafter.
The oxidation state of the [Ru2] unit and the electronic configuration of the frontier orbitals formed by Ru–Ru bonding can be easily determined by measuring the magnetic properties of the compounds. The temperature-dependence of the magnetization (M) of the compounds was determined using polycrystalline samples, in the temperature range of 1.8–300 K, by applying a dc field of 0.1 T (= Hdc); dc susceptibility (χ = M/Hdc). The χT vs. T plots are shown in Fig. S1.† The χ–T plots for all compounds were almost identical, gradually increasing upon cooling from 300 K to approximately 100 K and reaching a plateau, followed by an increase at temperatures below approximately 10 K. The χT values of the compounds at 300 K all merged to a narrow range of 0.91–1.16 cm3 K mol−1, and upon cooling, decreased smoothly to fall within the range of 1.1 × 10−2 to 2.4 × 10−1 cm3 K mol−1 at 1.8 K. These χT–T features agree with those for isolated [RuII,II2] complexes with an S = 1 ground state featuring an anisotropic effect due to strong zero-field splitting (ZFS; D ≈ 330–460 K for general [RuII,II2] complexes).33–36 The increase in χ at low temperatures can be ascribed to paramagnetic impurities, such as [RuII,III2]+ species with S = 3/2. Thus, the magnetic data were simulated using a Curie paramagnetic model with S = 1 considering the ZFS, temperature-independent paramagnetism (TIP), and impurities with S = 3/2 (ρ).37 Intermolecular interactions (zJ) have been considered for many magnetically isolated or weakly interacting [RuII,II2] complexes within the framework of the mean-field approximation.38 However, the state zJ = 0 was fixed for all the present compounds because it was not required to obtain an adequate fit. The best fits of the parameters (g = 2.0, fixed) for the compounds are listed in Table S2.† The estimated D values in the range of 337–462 K were consistent with the previously reported values for [RuII,II2] species.33,34 Hence, it was concluded that all complexes had an electron configuration of σ2π24δ2δ*2π*22σ*0 for the Ru–Ru bond frontier orbital sets with degenerate levels of δ* and two π* orbitals.
Structures
All of the compounds were structurally characterized using single-crystal X-ray crystallography (Fig. 1 and Fig. S2†), confirming their paddlewheel features with Ru–Ru bonds in the homoleptic mode for nX4OH and nX2OH and in the trans-heteroleptic mode for ht-nX2OH, with THF as the axial ligand. The crystallographic data are presented in Table S1.† All units had an inversion center at the midpoint of the Ru–Ru bond. Only 4Cl2OH and 4OMe2OH had a mirror plane on the Ru–Ru bond that divided the paddlewheel equally, and a two-fold axis perpendicular to the mirror plane passed through the inversion center, providing high symmetry (C2h), which imposed disorder on the THF molecules, and only one of the four bridged benzoates was crystallographically unique (Fig. 1d and S2d†). The bond lengths around the Ru centers of the compounds are summarized in Table 2. For the series of carboxylate-bridged [Ru2] complexes, the Ru–Oeq (Oeq = equatorial oxygen atoms) bond distance is a good indicator for evaluating the oxidation state (Table 2); these distances are generally in the range of 2.06–2.08 Å and 2.02–2.04 Å for [RuII,II2] and [RuII,III2]+, respectively.1,19,20 The Ru–Oeq bond length varied in the range of 2.057–2.071 Å, indicating that all present compounds feature [RuII,II2]. This is consistent with the magnetic properties described above. The Ru–Ru and Ru–Oax (Oax = axial THF oxygen atom) bond lengths of the complexes were 2.262–2.275 Å and 2.275–2.365 Å, respectively; these lengths were also in the range of [RuII,II2] compounds.1,19,20
 |
| | Fig. 1 ORTEP drawings of 2Cl4OH, (a) 3Br4OH (b), 3F2OH (c), 4Cl2OH (d), 5Cl2OH (e), 5Me2OH (f), ht-4F2OH (g), and ht-4OMe2OH (h); red, gray, green, light-green, purple, and pink represent O, C, F, Cl, Br, and Ru, respectively. The grey bonds represent disordered atomic positions. Displacement ellipsoids are drawn at 50% probability level. The hydrogen atoms and crystallization solvents are omitted for clarity. | |
Table 2 Relevant bond lengths (Å) and angles θ (°) around the Ru centers, where θ represents the dihedral angle between the least-squares planes defined by the phenyl ring of the benzoate ligand and a bridging carboxylate system (atom set Ru2O2C)
|

|
| Compound |
Ru–Ru/Å |
Averaged Ru–Oeq/Å |
Ru–Oax/Å |
Averaged O(o-OH)–Oeq/Å |
θ/° |
Ref. |
| Set-1a |
Set-2b |
|
Phenyl group of C2–C7(8).
Phenyl group of C9(10)–C14(16).
|
|
o-OH
|
2.2681(3) |
2.0650(6) |
2.3098(12) |
2.6035(14) |
8.49 |
7.55 |
30
|
|
2Cl4OH
|
2.2664(16) |
2.057(4) |
2.344(7) |
— |
23.07 |
22.57 |
This work |
|
3F4OH
|
2.2677(3) |
2.0608(8) |
2.3646(16) |
— |
8.14 |
3.91 |
This work |
|
3Cl4OH
|
2.2647(3) |
2.0633(7) |
2.3164(16) |
— |
8.61 |
7.13 |
This work |
|
3Br4OH
|
2.2642(5) |
2.0600(13) |
2.325(2) |
— |
2.99 |
15.57 |
This work |
|
3F2OH
|
2.2718(8) |
2.066(2) |
2.300(4) |
2.595(5) |
5.28 |
2.61 |
This work |
|
4F2OH
|
2.2724(3) |
2.0690(7) |
2.3203(13) |
2.6060(14) |
6.59 |
8.31 |
This work |
|
4Cl2OH
|
2.2698(12) |
2.066(3) |
2.312(6) |
2.583(7) |
3.49 |
— |
This work |
|
4Me2OH
|
2.2669(8) |
2.060(2) |
2.301(4) |
2.570(7) |
4.63 |
— |
This work |
|
5Cl2OH
|
2.2670(3) |
2.0674(7) |
2.3142(14) |
2.6080(14) |
7.92 |
10.05 |
This work |
|
5Br2OH
|
2.2680(4) |
2.0705(10) |
2.318(2) |
2.6175(18) |
6.00 |
8.49 |
This work |
|
5Me2OH
|
2.2620(5) |
2.0608(10) |
2.307(2) |
2.601(3) |
10.50 |
5.60 |
This work |
|
ht-o-OH
|
2.2674(3) |
2.0648(8) |
2.3177(15) |
2.569(3) |
76.70 |
3.38 |
22
|
|
ht-4F2OH
|
2.2702(3) |
2.0680(8) |
2.3245(15) |
2.560(3) |
79.10 |
4.14 |
This work |
|
ht-4Cl2OH
|
2.2658(3) |
2.0659(8) |
2.3178(15) |
2.580(5) |
85.99 |
4.95 |
This work |
|
ht-4Me2OH
|
2.2667(4) |
2.0641(8) |
2.3207(16) |
2.573(3) |
84.81 |
4.91 |
This work |
|
ht-4OMe2OH
|
2.2735(6) |
2.0688(15) |
2.341(2) |
2.565(6) |
79.94 |
2.28 |
This work |
|
ht-4CF32OH
|
2.2664(9) |
2.057(4) |
2.275(4) |
2.603(9) |
69.49 |
24.16 |
This work |
|
ht-5Me2OH
|
2.2752(5) |
2.0708(10) |
2.309(2) |
2.595(5) |
60.74 |
2.83 |
This work |
The dihedral angle (θ) between the least-squares planes composed of two atomic sets (i) the aryl ring of the substituted benzoate ligand and (ii) the bridging carboxyl group and two Ru atoms (i.e., the atom set of CO2Ru2, Chart in Table 2) in the nX2OH series is very small (6.6 ± 2.5°) (Table 2) owing to the formation of intramolecular hydrogen bonds between the o-OH group and carboxylate oxygen, with OOH⋯Oeq (Oeq = carboxylate oxygen atom) distances of 2.56–2.62 Å (Table 2). Notably, positional disorder of the o-OH group was observed in 4Cl2OH and 4Me2OH, with an occupancy of 6:4 and 7:3, respectively, and both o-OH groups formed intramolecular hydrogen bonds. In the ht-nX2OH series, the average θ value for the X-2-(OH)ArCO2− moiety is small (7.2 ± 8.4°), in contrast with the large θ value of 76.7 ± 9.7° for the 2,6-(CF3)2ArCO2− moiety.39 A positional disorder of the o-OH group was also observed in ht-4Cl2OH and ht-4CF32OH, with the occupancy of 9:1 and 6:4, respectively. Interestingly, a minor part of the o-OH group in ht-4Cl2OH did not form an intramolecular hydrogen bond, but rather formed an intermolecular interaction with the CF3 group of an adjacent molecule. Surprisingly, except for 2Cl4OH possessing an o-Cl group (θ = 22.8 ± 0.8°), the value of θ in nX4OH in the absence of o-substituents was relatively small (7.7 ± 4.5°; Table 2),20 which suggests preferential electronic conjugation between the carboxyl group and the aryl ring. The p-OH groups of nX4OH form hydrogen bonds with the crystallization solvent (THF) or adjacent [Ru2] molecules attached to the carboxyl group (Fig. S3†). The effects of the hydrogen bonds can also be observed in the OH stretching vibrations in the IR spectra (Fig. S4†). The OH stretching vibration in nX2OH and ht-nX2OH showed little compound dependency. The OH group peak positions were concentrated in a narrow region (3220 ± 10 cm−1 for nX2OH and 3205 ± 36 cm−1 for ht-nX2OH), and the half-width of the peak also tended to be small. On the other hand, the OH stretching vibration in nX4OH shows compound dependency, with the peak positions distributed over a wide region (3185 ± 137 cm−1), and the half-width of the peak also becomes wide. This may be because the compounds nX2OH and ht-nX2OH form intramolecular hydrogen bonds, making the hydrogen bond properties similar, while the compounds nX4OH form hydrogen bonds with the crystal solvent, making the hydrogen bond properties dependent on the crystal structure.
Electrochemistry in solution
The CVs of the compounds were acquired in N2-saturated THF solutions using n-Bu4N(PF6) as the supporting electrolyte and Ag/Ag+ as the reference electrode (Fig. 2). The electrochemical data are summarized in Table 3, along with some relevant pKa data for the corresponding benzoic acids and the summation of the Hammett constants.24 For all compounds, a reversible, one-electron redox wave with Ic/Ia ≈ 1 was observed, which was assigned to [RuII,II2]/[RuII,III2]+. Notably, the redox potentials (E1/2) of nX2OH (154–327 mV in THF vs. Ag/Ag+) and ht-nX2OH (208–391 mV in THF vs. Ag/Ag+) had relatively high, positive values compared to those of the [RuII,II2] compounds reported previously (Tables S4 and S5†).22,30 The positive shift in the potential was facilitated by substituting halogen groups at the m- and p-positions, in addition to the o-position of the OH group. The p-OH group induced a negative shift of the potential (e.g., −191 mV in THF vs. Ag/Ag+ for p-OH), but the additional substitution of halogen groups at the m-position pushed the potential back to the positive region, providing moderate negative E1/2 values for nX4OH (−136 to −62 mV in THF vs. Ag/Ag+) (Table S4†).30
 |
| | Fig. 2 Cyclic voltammograms of nX4OH (a), nX2OH (b), and ht-nX2OH (c) in THF containing 0.1 M n-Bu4N(PF6) under N2 (mV vs. Ag/Ag+). The ferrocene/ferrocenium couple, Fc/Fc = 213 mV, was observed under the same conditions. | |
Table 3 Electrochemical data for [Ru2(RnArCO2)4(THF)2] and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] measured in THF containing 0.1 M n-Bu4N(PF6) under N2 (mV vs. Ag/Ag+)a and HOMO levels calculated using density functional theory
| Compound |
E
a/mV |
E
c/mV |
E
1/2/mV |
ΔEp/mV |
HOMO energy/eV |
pKa of benzoateb |
∑(xσm + yσp)c |
∑(xσm + yσp + zσo)d |
Ref. |
|
The ferrocene/ferrocenium couple, Fc/Fc+ = 213 mV, was observed under the same conditions described in the Experimental section.
Predicted values were calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02, obtained from the SciFinder-n database.
Hammett constants σm and σp were obtained from ref. 24.
Hammett constants σo were taken from ref. 20 and 22.
|
|
p-OH
|
−134 |
−248 |
−191 |
114 |
(−4.02886) |
4.57 |
−0.370 |
−0.370 |
30
|
|
2Cl4OH
|
−23 |
−249 |
−136 |
226 |
−4.15219 |
3.44 |
−0.370 |
−0.098 |
This work |
|
3F4OH
|
220 |
−417 |
−99 |
636 |
−4.34675 |
4.23 |
−0.033 |
−0.033 |
This work |
|
3Cl4OH
|
114 |
−237 |
−62 |
350 |
−4.30675 |
4.20 |
−0.003 |
−0.003 |
This work |
|
3Br4OH
|
91 |
−237 |
−75 |
331 |
−4.38702 |
4.18 |
0.021 |
0.021 |
This work |
| |
|
o-OH
|
479 |
103 |
291 |
376 |
−4.64689 |
3.01 |
0 |
0.667 |
30
|
|
3F2OH
|
359 |
−52 |
154 |
412 |
−4.97615 |
2.45 |
0.337 |
1.004 |
This work |
|
4F2OH
|
397 |
52 |
225 |
345 |
−4.99220 |
2.85 |
0.062 |
0.729 |
This work |
|
4Cl2OH
|
458 |
94 |
276 |
364 |
−5.00771 |
2.71 |
0.227 |
0.894 |
This work |
|
4Me2OH
|
330 |
55 |
193 |
276 |
−4.43981 |
3.17 |
−0.170 |
0.497 |
This work |
|
5Cl2OH
|
519 |
135 |
327 |
384 |
−5.07629 |
2.64 |
0.373 |
1.040 |
This work |
|
5Br2OH
|
459 |
136 |
298 |
323 |
−5.10894 |
2.61 |
0.391 |
1.058 |
This work |
|
5Me2OH
|
406 |
54 |
230 |
352 |
−4.59982 |
3.30 |
−0.069 |
0.598 |
This work |
| |
|
ht-o-OH
|
491 |
91 |
291 |
232 |
−4.59002 |
2.27/3.01 |
0 |
0.667 |
22
|
|
ht-4F2OH
|
444 |
73 |
258 |
371 |
−4.75982 |
2.27/2.85 |
0.062 |
0.729 |
This work |
|
ht-4Cl2OH
|
484 |
162 |
324 |
322 |
−4.71329 |
2.27/2.71 |
0.227 |
0.894 |
This work |
|
ht-4Me2OH
|
346 |
71 |
208 |
274 |
−4.52852 |
2.27/3.17 |
−0.170 |
0.490 |
This work |
|
ht-4OMe2OH
|
315 |
135 |
225 |
180 |
−4.47900 |
2.27/3.22 |
−0.268 |
0.399 |
This work |
|
ht-4CF32OH
|
470 |
312 |
391 |
158 |
−4.76036 |
2.27/2.45 |
0.540 |
1.207 |
This work |
|
ht-5Me2OH
|
327 |
161 |
244 |
166 |
−4.55002 |
2.27/3.30 |
−0.069 |
0.598 |
This work |
The pKa value of the corresponding benzoic acid (RnArCO2H) is a useful parameter for measuring the electronic effect of the benzoate ligands (RnArCO2−) on the [Ru2] dinuclear center.40Fig. 3 shows the plots of E1/2 as a function of pKa for the [Ru2(RnArCO2)4(THF)2] series and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] reported previously.22 Only the data for the non-o-substituted compounds, except for the o-OH series in this study, were plotted in order to avoid the influence of steric hindrance from the o-substituent in the [Ru2] compounds (2Cl4OH was also excluded from Fig. 3a because it contains an o-Cl group, which demonstrates a good linear relationship for the mono-o-Cl-substituted [Ru2] series (Fig. S5†).20). Although the o-OH group did not produce significant steric hindrance, the nX2OH and ht-nX2OH groups were outside the linear scale created by the [Ru2(RnArCO2)4(THF)2] and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] series, respectively, providing a new group with smaller pKa and positive E1/2 values (Fig. 3). Therefore, the nX2OH and ht-nX2OH groups should be evaluated independently. The relationship is linear depending on the electronic effects of the m- and p-substituents, with slopes similar to those of [Ru2(RnArCO2)4(THF)2] and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2], respectively. The dashed lines in Fig. 3 were drawn by fixing the slope to the red lines for [Ru2(RnArCO2)4(THF)2] and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2]. Conversely, nX4OH (with a p-OH group) has a larger pKa and more negative E1/2, indicating the electron-donating nature of the p-OH group; however, the data for nX4OH followed the linear plot for [Ru2(RnArCO2)4(THF)2] (Fig. 3a).
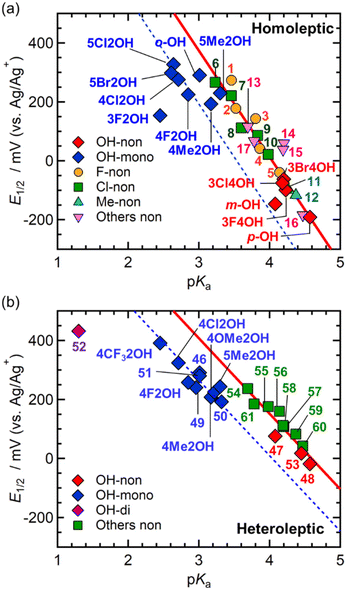 |
| | Fig. 3 Total plot of half-wave redox potential (E1/2) vs. pKa for the corresponding benzoic acids in the homoleptic series [Ru2(RnArCO2)4(THF)2] (a) and heteroleptic series [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2]. The E1/2 values were measured in THF using Ag/Ag+ reference electrode. Data for the compounds without an ortho substituent were chosen for the non-OH series in ref. 20, 21, 26, 27 and 29. Some of the OH–[Ru2] data were obtained from ref. 22 and 30. The solid, red line represents linear least-squares fitting line for non-o-OH substituted series with equations of y = −360.23x + 1454.3 (R2 = 0.8677) for (a) and y = −255.72x + 1174.7 (R2 = 0.8704) for (b). The dashed, blue line represents linear least-squares fitting line for mono-o-OH substituted series as the slope was fixed to that of red, solid line with equations of y = −360.23x + 1273.2 for (a) and y = −255.72x + 1032.5 for (b). Definitions: [Ru2(RnArCO2)4(THF)2], R for 1, 3,4,5-F3; 2, 3,5-F2; 3, 3,4-F2; 4, m-F; 5, p-F; 6, 3,4,5-Cl3; 7, 3,5-Cl2; 8, 3,4-Cl2; 9, m-Cl; 10, p-Cl; 11, m-Me; 12, p-Me; 13, p-CF3; 14, H; 15, p-Ph; 16, p-MeO; 17, p-CHO; [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2], R for 46, o-OH; 47, m-OH; 48, p-OH; 49, 2,3-(OH)2; 50, 2,4-(OH)2; 51, 2,5-(OH)2; 52, 2,6-(OH)2; 53, 3,4-(OH)2; 54, p-CF3; 55, p-Cl; 56, p-F; 57, H; 58, p-Ph; 59, p-Me; 60, p-OMe; 61, p-CHO. | |
As observed previously, the relationship between E1/2 and pKa depends on the number of substituents at the o-position (NSO), which may be due to the alteration of the electronic conjugation between the aryl ring and carboxylate group, due directly to the steric effect of the ortho-substituents with NSO = 1 and 2.19,20 Because no significant steric hindrance was observed in the case of o-OH-substituted benzoate ligands, the discrepancy between the nX2OH and ht-nX2OH groups compared to the non-o-OH-substituted series may be due to another contribution of the electronic effect intrinsically caused by intramolecular o-OH⋯Oeq hydrogen bonding. This electronic effect was evaluated as σo(OH) = 0.667 based on analysis of [Ru2{2,6-(CF3)2ArCO2}2{o-(OH)ArCO2}2(THF)2] (Table 1).22
If all the Hammett constants, including the pseudo-Hammett constants, σo, are considered, the E1/2 values of all [Ru2] compounds should be related in a unified linear plot, that is, through an extended Hammett's law. Fig. 4 shows the plot of E1/2vs. ∑(xσm + yσp + zσo), where x, y, and z represent the number of substituents at the meta-, para-, and ortho-positions, respectively; Fig. 4(a and b) shows the data for the homoleptic and trans-heteroleptic [Ru2] series, respectively. The data for all series are summarized in Table S3,† where the Hammett constants summarized in Table 1 were used.20,22,24 Note that the slopes of the plot for the homoleptic and heteroleptic series are different because the compounds are composed of four and two RnArCO2− ligand blades, respectively (Fig. 4a and b). Linear relationships for the homoleptic and heteroleptic series were observed, with E1/2 (mV) = 362 × ∑(xσm + yσp + zσo) − 60.9 (R2 = 0.918) and E1/2 (mV) = 228 × ∑(xσm + yσp + zσo) + 101 (R2 = 0.938), respectively. These plots indicate that the redox properties of the [Ru2] series can be predicted well in the framework of Hammett analyses; that is, the redox properties can be well-tuned by modifying the substituents of the benzoate ligands. Notably, the electron-withdrawing effect of the o-OH group was significant even in cases where other substituents were present at the m- or p-positions.
 |
| | Fig. 4 Total plot of half-wave redox potential (E1/2) vs. ∑(xσm + yσp + zσo) for homoleptic series of [Ru2(RnArCO2)4(THF)2] (a) and heteroleptic series of [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] (b). The E1/2 values were measured in THF using Ag/Ag+ reference electrode. The data for non-OH compounds were obtained from ref. 20, 21, 26, 27 and 29. Some of the OH–[Ru2] data were obtained from ref. 22 and 30. The solid line represents the linear least-squares fitting line with equations of y = 361.76x − 60.86 (R2 = 0.9177) for (a) and y = 228.21x + 100.77 (R2 = 0.9379) for (b). Definitions: [Ru2(RnArCO2)4(THF)2], R for 18, F5; 19, 2,3,5,6-F4; 20, 2,3,4,5-F4; 21, 2,3,6-F3; 22, 2,4,5-F3; 23, 2,3,4-F3; 24, 2,3,5-F3; 25, 2,4,6-F3; 26, 2,5-F2; 27, 2,3-F2; 28, 2,6-F2; 29, 2,4-F2; 30, o-F; 31, 2,3,4,5-Cl4; 32, 2,3,5-Cl3; 33, 2,3,4-Cl3; 34, 2,4,5-Cl3; 35, 2,5-Cl2; 36, 2,3-Cl2; 37, 2,6-Cl2; 38, 2,4-Cl2; 39, o-Cl; 40, o-Me; 41, 2,4,5-Me3; 42, o-OMe; 43, 4-Cl-2-OMe; 44, 4-F-2-OMe; 45, o-CF3; [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2], R for 62, 2,3,5,6-F4. | |
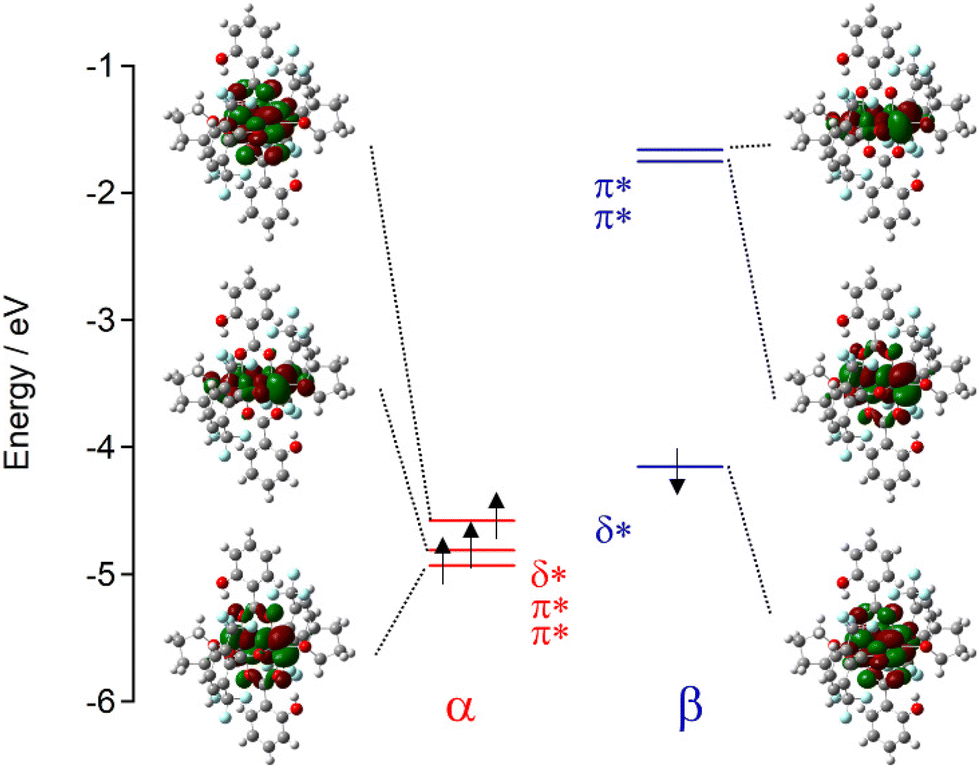
HOMO energy levels
To determine the energy levels of the molecular orbitals (MOs) of the compounds, DFT calculations were performed based on the crystal structures (Fig. 5 and S6 and Table S5†). For all the compounds, the HOMO, which is most likely involved in the [RuII,II2]/[RuII,III2]+ redox couple, was assigned to the δ*(β) frontier orbital.19,20,22,30 The HOMO energy levels of the complexes are listed in Table 3. The estimated HOMO levels of the compounds linearly correlated with pKa (Fig. 6), as observed in the E1/2vs. pKa plots in Fig. 3. The correlations for the nX2OH and ht-nX2OH groups are different from those of the non-o-OH series of [Ru2(RnArCO2)4(THF)2] and [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2]. Consequently, the HOMO energy levels and E1/2 values are strongly correlated in a unified relationship, even for the o-OH series (Fig. 7).19–22,30 The nX4OH compounds have relatively high HOMO energies (−4.387 to −4.152 eV) owing to the strong electron-donating effect of the p-OH group, whereas nX2OH (−5.109 to −4.440 eV) and ht-nX2OH (−4.763 to −4.479 eV) have lower HOMO energies, ascribed to the electron-withdrawing effect of the o-OH group. To evaluate the impact of intramolecular hydrogen bonding, DFT calculations were performed for a model in which the hydrogen atom of the o-OH group was directed outward to interrupt the hydrogen bonding (i.e., the hypothetical model); the atomic coordinates were the same, except for that of H on the o-OH group (Fig. S7† and Tables 4 and S6†).22,30 In these hypothetical non-hydrogen bonding models, the HOMO levels were higher than those of the actual nX2OH (ΔEHOMO ∼ 1 eV) and ht-nX2OH (ΔEHOMO ∼ 0.5 eV), indicating that the intramolecular hydrogen bonding of the o-OH group strongly affects the HOMO levels in the nX2OH and ht-nX2OH series (Table 4).
 |
| | Fig. 5 Frontier orbitals associated with π* and δ* orbitals of 2Cl4OH and corresponding energy levels (eV), where δ* for the β electron corresponds to the HOMO level. | |
 |
| | Fig. 6 Plot of HOMO level vs. pKa for homoleptic series of [Ru2(RnArCO2)4(THF)2] (a) and heteroleptic series of [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] (b). Data for the compounds without an ortho substituent were chosen for the non-OH series from ref. 20, 21, 26, 27 and 29. Some of the OH–[Ru2] data were obtained from ref. 22 and 30. The solid, red line represents the linear least-squares fitting line for non-o-OH substituted series with equations of y = 0.9334x − 8.1874 (R2 = 0.8914) for (a) and y = 0.4963x − 6.5179 (R2 = 0.7966) for (b). The solid, blue line represents the linear least-squares fitting line for mono-o-OH substituted series with equations of y = 0.7495x − 6.9863 (R2 = 0.7613) for (a) and y = 0.3122x − 5.5657 (R2 = 0.7376) for (b). | |
 |
| | Fig. 7 HOMO level vs. half-wave redox potential (E1/2) for homoleptic series of [Ru2(RnArCO2)4(THF)2] (a) and heteroleptic series of [Ru2{2,6-(CF3)2ArCO2}2(RnArCO2)2(THF)2] (b). The E1/2 values were measured in THF using Ag/Ag+ reference electrode. Data for the compounds without an ortho substituent were chosen for the non-OH series from ref. 20, 21, 26, 27 and 29. Some of the OH–[Ru2] data were obtained from ref. 22 and 30. The solid line represents the linear least-squares fitting line with equations of y = (−2.033 × 10−3)x − 4.412 (R2 = 0.7623) for (a) and y = (−1.324 × 10−3)x − 4.291 (R2 = 0.8109) for (b). | |
Table 4 Calculated energy values of the HOMO level of compounds possessing o-OH substituents with or without intramolecular hydrogen bonds, where the values for o-OH and ht-o-OH were referred from ref. 30 and 22, respectively
| Compound |
Actual model with hydrogen bond (eV) |
Hypothetical OH-free model without hydrogen bond (eV) |
|
o-OH
|
−4.64689 |
−3.65231 |
|
3F2OH
|
−4.97615 |
−3.99926 |
|
4F2OH
|
−4.99220 |
−4.00334 |
|
4Cl2OH
|
−5.00771 |
−4.03654 |
|
4Me2OH
|
−4.43981 |
−3.52061 |
|
5Cl2OH
|
−5.07629 |
−4.08688 |
|
5Br2OH
|
−5.10894 |
−4.12579 |
|
5Me2OH
|
−4.59982 |
−3.53857 |
| |
|
ht-o-OH
|
−4.58993 |
−4.07649 |
|
ht-4F2OH
|
−4.75982 |
−4.24416 |
|
ht-4Cl2OH
|
−4.71329 |
−4.21042 |
|
ht-4Me2OH
|
−4.52852 |
−4.02429 |
|
ht-4OMe2OH
|
−4.47900 |
−3.98402 |
|
ht-4CF32OH
|
−4.76036 |
−4.31301 |
|
ht-5Me2OH
|
−4.55002 |
−4.07736 |
Conclusions
Eleven homoleptic complexes (nX2OH and nX4OH) and six heteroleptic [RuII,II2] complexes (ht-nX2OH) were synthesized by introducing substituents such as F, Cl, Me, OMe, and CF3 groups, as well as OH groups, to the bridging benzoate. The o-OH groupacts as a powerful electron-withdrawing group owing to the intramolecular hydrogen bonding between the o-OH group and the oxygen of the bridging carboxyl group, resulting in a pseudo-Hammett constant of σ(OH) = 0.667. The pKa, E1/2, and HOMO levels of [RuII,II2] complexes are critical parameters for evaluating their electronic nature. In a previous report, the E1/2 values and HOMO levels of o-OH-substituted complexes [Ru2{2,6-(CF3)2ArCO2}2{o-OHArCO}2(THF)2] were treated on the same scale as those of non-o-OH complexes; however, this was incorrect. The electronic effect of the o-OH group, associated with intramolecular hydrogen bonding, is significant and intrinsic. Therefore, homoleptic and heteroleptic [RuII,II2] complexes with an o-OH group should be treated differently from the congeners without an o-OH group. We have been gradually expanding the scope of applying this empirical approach to ortho substituents to predict the redox ability of [Ru2] in several previous papers.19,20,22 In the future, as we continue to increase the types of substituents and compounds, we may find out the potential limitations of the applicability of this method, but fortunately, so far, everything has worked well. That is, by adding OH substituents, as well as F, Cl, Me, OMe, and CF3 groups, to the benzoate ligands, the redox properties of the [Ru2] complexes can be widely tuned.
Experimental section
General procedures and materials
Specific synthetic procedures were performed under inert atmosphere (N2) using standard Schlenk-line techniques in a commercial glovebox. All chemicals were of reagent grade and were purchased from commercial sources. The solvents were dried using common drying agents and were distilled under nitrogen atmosphere before use. [RuII,III2(CH3CO2)4Cl] was prepared as reported previously.31
Synthesis of [Ru2(3-F-4-(OH)ArCO2)4(THF)2] (3F4OH), [Ru2(2-Cl-4-(OH)ArCO2)4(THF)2] (2Cl4OH), [Ru2(3-Cl-4-(OH)ArCO2)4(THF)2] (3Cl4OH), [Ru2(3-Br-4-(OH)ArCO2)4(THF)2] (3Br4OH), [Ru2(3-F-2-(OH)ArCO2)4(THF)2] (3F2OH), [Ru2(4-F-2-(OH)ArCO2)4(THF)2] (4F2OH), [Ru2(4-Cl-2-(OH)ArCO2)4(THF)2] (4Cl2OH), [Ru2(4-Me-2-(OH)ArCO2)4(THF)2] (4Me2OH), [Ru2(5-Cl-2-(OH)ArCO2)4(THF)2] (5Cl2OH), [Ru2(5-Br-2-(OH)ArCO2)4(THF)2] (5Br2OH), and [Ru2(5-Me-2-(OH)ArCO2)4(THF)2] (5Me2OH)
The compounds were synthesized in a step-by-step manner via a ligand substitution process involving [RuII,III2]+, followed by reduction to the corresponding [RuII,II2] products;32 the method is similar to that used for previously reported compounds.30 Only the procedure for 3F4OH is described here. [RuII,III2(CH3CO2)4Cl] (711 mg, 1.50 mmol) and 3-fluoro-4-hydroxybenzoic acid (1000 mg, 5.07 mmol) were refluxed in a 1:3 solution of MeOH and H2O (40 mL) for 12 h under aerobic conditions to synthesize [RuII,III2(3-F-4-(OH)ArCO2)4Cl]. The reddish-brown precipitate was collected by filtration, washed with water, and dried in vacuo. Without further purification, a tetrahydrofuran (THF) solution (50 mL) of the crude, red precipitate was stirred with Zn powder (120 mg, 1.84 mmol) for 24 h under nitrogen atmosphere. The brown solution was filtered, and the filtrate was layered with n-hexane and allowed to stand for at least one week, affording 3F4OH as brown crystals in 30% yield. Elemental analysis (%) calculated for C52H64F4O18Ru2 (3F4OH·4THF): C 49.75, H 5.14. Found: C 49.36, H 5.03. IR (KBr): ν(CO2) = 1545, 1391 cm−1. For 2Cl4OH·2THF, yield: 44%. Elemental analysis (%) of C44H48Cl4O16Ru2: C 44.90, H 4.11. Found: C 44.79, H 4.28. IR (KBr): ν(CO2) = 1557, 1392 cm−1. For 3Cl4OH·4THF, yield: 39%. Elemental analysis (%) calculated for C52H64Cl4O18Ru2: C 47.29, H 4.88. Found: C 47.18, H 4.81. IR (KBr): ν(CO2) = 1543, 1384 cm−1. For 3Br4OH·4THF, yield: 51%. Elemental analysis (%) calculated for C52H64Br4O18Ru2: C 41.67, H 4.30. Found: C 41.77, H 4.26. IR (KBr): ν(CO2) = 1548, 1384 cm−1. For 3F2OH, yield: 40%. Elemental analysis (%) calculated for C36H32F4O14Ru2: C 44.72, H 3.34. Found: C 44.70, H 3.46. IR (KBr): ν(CO2) = 1559, 1393 cm−1. For 4F2OH, yield: 27%. Elemental analysis (%) calculated for C36H32F4O14Ru2: C 44.72, H 3.34. Found: C 44.72, H 3.36. IR (KBr): ν(CO2) = 1556, 1450, 1371 cm−1. For 4Cl2OH, yield: 14%. Elemental analysis (%) calculated for C36H32Cl4O14Ru2: C 41.87, H 3.12. Found: C 41.94, H 3.12. IR (KBr): ν(CO2) = 1555, 1438, 1362 cm−1. For 4Me2OH, yield: 63%. Elemental analysis (%) calculated for C40H47O15.5Ru2 (4Me2OH·1.5H2O): C 49.13, H 4.84. Found: C 49.12, H 4.84. IR (KBr): ν(CO2) = 1547, 1434, 1387 cm−1. For 5Cl2OH, yield: 48%. Elemental analysis (%) calculated for C36H32Cl4O14Ru2: C 41.87, H 3.12. Found: C 41.94, H 3.12. IR (KBr): ν(CO2) = 1551, 1440, 1367 cm−1. For 5Br2OH, yield: 37%. Elemental analysis (%) calculated for C36H32Br4O14Ru2: C 35.72, H 2.66. Found: C 35.73, H 2.67. IR (KBr): ν(CO2) = 1551, 1439, 1386 cm−1. For 5Me2OH, yield: 54%. Elemental analysis (%) calculated for C40H46O15Ru2 (5Me2OH·H2O): C 49.58, H 4.79. Found: C 49.64, H 4.65. IR (KBr): ν(CO2) = 1549, 1431, 1367 cm−1.
Synthesis of [Ru2{2,6-(CF3)2ArCO2}2{4-F-2-(OH)ArCO2}2(THF)2] (ht-4F2OH), [Ru2{2,6-(CF3)2ArCO2}2{4-Cl-2-(OH)ArCO2}2(THF)2] (4Cl2OH), [Ru2{2,6-(CF3)2ArCO2}2{4-Me-2-(OH)ArCO2}2(THF)2] (4Me2OH), [Ru2{2,6-(CF3)2ArCO2}2{4-(OMe)-2-(OH)ArCO2}2(THF)2] (4OMe2OH), [Ru2{2,6-(CF3)2ArCO2}2{4-(CF3)-2-(OH)ArCO2}2(THF)2] (4CF32OH), and [Ru2{2,6-(CF3)2ArCO2}2{5-Me-2-(OH)ArCO2}2(THF)2] (5Me2OH)
The compounds were synthesized in a stepwise manner via a ligand substitution process involving [RuII,III2]+, followed by reduction to the corresponding [RuII,II2] products.32 The method is similar to that used for previously reported compounds.22 Only the procedure for ht-4Me2OH is described here. [RuII,III2(CH3CO2)4Cl] (600 mg, 1.27 mmol) and 4-methyl-2-hydroxybenzoic acid (1552 mg, 10.2 mmol) were refluxed in a 1:2 solution of MeOH and H2O (20 mL) for 12 h under aerobic conditions to synthesize [RuII,III2(4-Me-2-(OH)ArCO2)4Cl] as a red precipitate. The resulting [RuII,III2(4-Me-2-(OH)ArCO2)4Cl] (1 mmol, 834 mg) and 2,6-bis(trifluoromethyl)benzoic acid (2 mmol, 516 mg) were refluxed in a 1:3 solution of MeOH and H2O (40 mL) for 12 h under aerobic conditions to obtain [RuII,III2(4-Me-2-(OH)ArCO2)2(2,6-(CF3)2ArCO2)2Cl]. The obtained reddish-brown precipitate was collected via filtration, washed with water, and dried in vacuo. Without further purification, a THF solution (80 mL) of the crude red precipitate was stirred with Zn powder (117 mg, 1.8 mmol) for 48 h at room temperature under nitrogen atmosphere. The brown solution was filtered, and the filtrate was layered with n-hexane and allowed to stand for at least one week, affording ht-4Me2OH as brown crystals in 60% yield. Elemental analysis (%) calculated for C50H52F12O14Ru2 (ht-4Me2OH·2THF): C 45.95, H 4.01. Found: C 46.34, H 3.92. IR (KBr): ν(CO2) = 1579, 1503 cm−1. For ht-4F2OH, yield: 61%. Elemental analysis (%) calculated for C40H30F14O12Ru2: C 41.03, H 2.58. Found: C 41.11, H 2.68. IR (KBr): ν(CO2) = 1572, 1439 cm−1. For ht-4Cl2OH, yield: 59%. Elemental analysis (%) calculated for C40H30Cl2F12O12Ru2: C 39.91, H 2.51. Found: C 39.77, H 2.57. IR (KBr): ν(CO2) = 1581, 1436 cm−1. For ht-4OMe2OH, yield: 60%. Elemental analysis (%) calculated for C44H40F12O14.5Ru2 (ht-4OMe2OH·0.5THF): C 42.93, H 3.28. Found: C 42.68, H 3.29. IR (KBr): ν(CO2) = 1577, 1450 cm−1. For ht-4CF32OH, yield: 61%. Elemental analysis (%) calculated for C44H40F12O14.5Ru2 (ht-4CF32OH·0.5THF): C 42.93, H 3.28. Found: C 42.68, H 3.29. IR (KBr): ν(CO2) = 1577, 1450 cm−1. For ht-5Me2OH, yield: 61%. Elemental analysis (%) calculated for C52H56F12O14.5Ru2 (ht-5Me2OH·2.5THF): C 46.50, H 4.20. Found: C 46.45, H 4.05. IR (KBr): ν(CO2) = 1548, 1484 cm−1.
General physical measurements
Infrared spectra were recorded with KBr disks using a Jasco FT-IR 4200 spectrometer. Magnetic susceptibility measurements were conducted with a Quantum Design SQUID magnetometer (MPMS-XL) by applying a magnetic field of 0.1 T in the temperature range of 1.8–300 K, for which polycrystalline samples embedded in liquid paraffin were used. The experimental data were corrected for the diamagnetic contributions of the sample holder and liquid paraffin using Pascal's constants.41 Cyclic voltammograms (CVs) were recorded in THF with tetra-n-butylammonium hexafluorophosphate (n-Bu4N(PF6), 0.1 M) as the supporting electrolyte, under nitrogen atmosphere, using an electrochemical analyzer (ALS/[H] CH Instruments Model 600A) with a glassy carbon electrode as the working electrode, a Pt counter electrode, and a Ag/AgNO3 reference electrode. First, the CV data were acquired for the solvent containing only the supporting electrolyte. The desired compounds were then added to this solution ([compound] = 1 × 10−3 M), and the CVs were acquired at a scan rate of 0.05 V s−1. Finally, the CV potentials in THF were adjusted using the ferrocene/ferrocenium couple (Fc/Fc+ = 213 mV (ΔE = 91 mV) in THF vs. Ag/Ag+) as an internal standard.
Crystallography
Crystal data were collected on a Rigaku Saturn 724 CCD area detector and a Rigaku Hypix 6000 hybrid photon-counting X-ray detector with multilayer, mirror-monochromated Mo-Kα radiation (λ = 0.71075 Å). The structures were solved using an intrinsic phasing method (SHELXT version 2018/2)42,43 and expanded using Fourier techniques (SHELXL version 2018/3).44 All non-hydrogen atoms were anisotropically refined. Hydrogen atoms were introduced as fixed contributors. Full-matrix least-squares refinements on F2 were based on observed reflections and variable parameters, and converged with unweighted and weighted agreement factors of R1 = ∑||Fo| − |Fc||/∑|Fo| (I > 2.00σ(I)) and wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2 (all data). All calculations were performed using the OLEX2 crystallographic software package.443F4OH, 3Cl4OH, 3Br4OH, and 4F2OH contain a disordered THF, which could be modeled as two distinct parts and optimized for occupancy. 4Cl2OH and 4Me2OH also contained disordered THF imposed by molecular symmetry, whose occupancy was fixed at 0.5. 3F4OH, 4Cl2OH, 4Me2OH, ht-4Cl2OH, and ht-4CF32OH contain positional disorder of the substituent (m-F or o-OH) in the benzoate moiety. ht-4Cl2OH and ht-4Me2OH contain rotational disorder in the o-CF3 group. These disorders were modeled as two distinct parts and were optimized for occupancy. Solvent mask function of OLEX2 software44 was used for 4Cl2OH, 4Me2OH, and ht-5Me2OH, which were reasonably modeled in the presence of a 1.4THF, 2.4THF, and 1THF, respectively, per [Ru2]. The details of the crystal structure analysis are summarized in Table S1.† These data have been deposited as CIFs at the Cambridge Data Centre as supplementary publication notes. CCDC 2394123, 2394124, 2394125, 2394126, 2394127, 2394128, 2394129, 2394130, 2394131, 2394132, 2394133, 2394134, 2394135, 2394136, 2394137, 2394138, and 2394139 for 2Cl4OH, 3F4OH, 3Cl4OH, 3Br4OH, 3F2OH, 4F2OH, 4Cl2OH, 4Me2OH, 5Cl2OH, 5Br2OH, 5Me2OH, ht-4F2OH, ht-4Cl2OH, ht-4Me2OH, ht-4OMe2OH, ht-4CF32OH, and ht-5Me2OH, respectively.† Molecular diagrams were constructed using the VESTA software.45
Computational details
Theoretical ab initio calculations were performed using the DFT formalism, as implemented in Gaussian 16 software,46 employing Beck's three-parameter hybrid functional with the correlation functional of Lee, Yang, and Parr (B3LYP).47 Unrestricted open-shell calculations were performed for molecules containing [Ru2] units. The effective core potential basis function LanL2TZ with polarization (LanL2TZ(f))48–50 was used for Ru atoms, and the 6-31G basis sets with polarization and diffusion functions (6-31+G(d))51–55 were used for C, H, F, Cl, Br, and O atoms, respectively. Spin polarization with SZ = 1 (triplet spin multiplicity) was used for the [Ru2] units in the calculations. The atomic coordinates determined by X-ray crystallography were used in the calculations. In the case of the disordered atoms in the benzoate groups, atoms with major occupancy were used. In the calculations for 4Cl2OH and 4Me2OH, the molecular symmetry must be reduced from C2h to eliminate the structural disorder. C2 and Ci are the candidates for the lower molecular symmetry, and the structures with the latter symmetry were adopted because they have a lower energy, as indicated by DFT calculations.
Author contributions
H. M. conceived the study. Y. W., T. K., and C. I. prepared and characterized the materials. W. K. analyzed the data. W. K. and H. M. supervised the experiments. W. K. prepared the original draft. All authors discussed the results and commented on the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Tables of crystallographic data, magnetic parameters, additional lists of electrochemical data, and HOMO levels of relevant compounds. Figures for magnetic data, ORTEP drawings of several compounds, molecular diagrams highlighting intermolecular hydrogen bonds, plots of E1/2vs. pKa, HOMO levels vs. pKa, and HOMO levels vs. E1/2, and the structural models, which were subjected to DFT calculations to clarify the effect of the intramolecular hydrogen bonds. Computational details are described in the experimental part of the article.
Crystallographic data for 2Cl4OH, 3F4OH, 3Cl4OH, 3Br4OH, 3F2OH, 4F2OH, 4Cl2OH, 4Me2OH, 5Cl2OH, 5Br2OH, 5Me2OH, ht-4F2OH, ht-4Cl2OH, ht-4Me2OH, ht-4OMe2OH, ht-4CF32OH, and ht-5Me2OH have been deposited at the Cambridge Data Centre as supplementary publication as CCDC 2394123, 2394124, 2394125, 2394126, 2394127, 2394128, 2394129, 2394130, 2394131, 2394132, 2394133, 2394134, 2394135, 2394136, 2394137, 2394138, and 2394139, respectively.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (No. 18H05208, 20H00381, 21H01900, 21K18925, 22H00324, and 23K21104) from MEXT, Japan, the GIMRT program, and the E-IMR project, Tohoku University.
References
-
F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds between Metal Atoms, Springer, New York, 3rd edn, 2005 Search PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- O. M. Yaghi, M. ÓKeeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and H. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- R. A. Potyrailo, Chem. Rev., 2016, 116, 11877–11923 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. A. S. Aquino, Coord. Chem. Rev., 2004, 248, 1025–1045 CrossRef CAS.
- M. Mikuriya, D. Yoshioka and M. Handa, Coord. Chem. Rev., 2006, 250, 2194–2211 CrossRef CAS.
- T. E. Vos and J. S. Miller, Angew. Chem., Int. Ed., 2005, 44, 2416–2419 CrossRef CAS PubMed.
-
H. Miyasaka, Hybrid Materials Composed of Organic Radicals and Ru Dimers, in World Scientific Reference on Spin in Organics, ed. Z. V. Vardeny, M. Vohlgenannt and J. S. Miller, World Scientific, Spin in Organics, Materials and Energy, 2018; vol. 4, ch. 4, pp 169–205 Search PubMed.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS PubMed.
- H. Miyasaka, Bull. Chem. Soc. Jpn., 2021, 94, 2929–2955 CrossRef CAS.
- N. Komiya, T. Nakae, H. Sato and T. Naota, Chem. Commun., 2006, 4829–4831 RSC.
- M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207–17216 CrossRef CAS PubMed.
- T. Miyazawa, T. Suzuki, Y. Kumagai, K. Takizawa, T. Kikuchi, S. Kato, A. Onoda, T. Hayashi, Y. Kamei, F. Kamiyama, M. Anada, M. Kojima, T. Yoshino and S. Matsunaga, Nat. Catal., 2020, 3, 851–858 CrossRef CAS.
- K. Makino, Y. Kumagai, T. Yoshino, M. Kojima and S. Matsunaga, Org. Lett., 2023, 25, 3234–3238 CrossRef CAS PubMed.
- J. K. Sailer, J. C. Sharland, J. Bacsa, C. F. Harris, J. F. Berry, D. G. Musaev and H. M. L. Davies, Organometallics, 2023, 42, 2122–2133 CrossRef CAS PubMed.
- C. Itoh, M. Kitada, M. Kondo, S. Masaoka, H. Yoshino, W. Kosaka, Y. Ootani, J. Matsuda, M. Kubo, T. J. Konno and H. Miyasaka, ChemSusChem, 2024, e202400885 CrossRef PubMed.
- H. Miyasaka, N. Motokawa, R. Atsuumi, H. Kamo, Y. Asai and M. Yamashita, Dalton Trans., 2011, 40, 673–682 RSC.
- W. Kosaka, M. Itoh and H. Miyasaka, Dalton Trans., 2015, 44, 8156–8168 RSC.
- Y. Sekine, K. H. Aliyah, T. Shimada, J. Zhang, W. Kosaka and H. Miyasaka, Chem. Lett., 2018, 47, 693–696 CrossRef CAS.
- W. Koska, Y. Watanabe, K. H. Aliyah and H. Miyasaka, Dalton Trans., 2022, 51, 85–94 RSC.
- L. P. Hammet, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
-
M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Interscience, New York, 8th edn, 2020 Search PubMed.
- W. Kosaka, K. Yamagishi, A. Hori, H. Sato, R. Matsuda, S. Kitagawa, M. Takata and H. Miyasaka, J. Am. Chem. Soc., 2013, 135, 18469–18480 CrossRef CAS PubMed.
- W. Kosaka, T. Morita, T. Yokoyama, J. Zhang and H. Miyasaka, Inorg. Chem., 2015, 54, 1518–1527 CrossRef CAS PubMed.
- Y. Sekine, W. Kosaka, H. Kano, C. Dou, T. Yokoyama and H. Miyasaka, Dalton Trans., 2016, 45, 7427–7434 RSC.
- J. Zhang, W. Kosaka, S. Kitagawa, M. Takata and H. Miyasaka, Chem. – Eur. J., 2019, 25, 3020–3031 CrossRef CAS PubMed.
- W. Kosaka, Y. Watanabe, C. Itoh and H. Miyasaka, Chem. Lett., 2022, 51, 731–734 CrossRef CAS.
- R. W. Mitchell, A. Spencer and G. J. Wilkinson, Chem. Soc., Dalton Trans., 1973, 846–854 RSC.
- F. A. Cotton, Y. Kim and A. Yokochi, Inorg. Chim. Acta, 1995, 236, 55–61 CrossRef CAS.
- P. Maldivi, A.-M. Giroud-Godquin, J.-C. Marchon, D. Guillon and A. Skoulios, Chem. Phys. Lett., 1989, 157, 552–555 CrossRef CAS.
- L. Bonnet, F. D. Cukiernik, P. Maldivi, A.-M. Giroud-Godquin and J.-C. Marchon, Chem. Mater., 1994, 6, 31–38 CrossRef CAS.
- E. V. Dikarev, A. S. Filatov, E. Clérac and M. A. Petrukhina, Inorg. Chem., 2006, 45, 744–751 CrossRef CAS PubMed.
- H. Miyasaka, R. Clérac, C. S. Campos-Fernández and K. R. Dunbar, J. Chem. Soc., Dalton Trans., 2001, 858–861 RSC.
- A. Cogne, E. Belorizky, J. Laugier and P. Rey, Inorg. Chem., 1994, 33, 3364–3369 CrossRef CAS.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203–283 CrossRef.
- Among the seven heteroleptic compounds, the θ value of ht-4OMe2OH is exceptionally larger compared to the others (24.16°); if this value is removed, the average θ value becomes much smaller (3.8± 1.2°).
- pKa values are predicted values calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02, which were obtained from SciFinder-n database.
-
E. A. Boudreaux and L. N. Mulay, Theory and Applications of Molecular Paramagnetism, John Wiley and Sons, New York, 1976, p. 491 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- T. Clark, J. Chandrasekhar and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- R. Krishnam, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499–505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of crystallographic data, magnetic parameters, additional lists of electrochemical data, and HOMO levels of relevant compounds. Figures for magnetic data, ORTEP drawings of several compounds, molecular diagrams highlighting intermolecular hydrogen bonds, plots of E1/2vs. pKa, HOMO levels vs. pKa, and HOMO levels vs. E1/2, and the structural models, which were subjected to DFT calculations to clarify the effect of the intramolecular hydrogen bonds. CCDC 2394123–2394139. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03020f |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Yudai
Watanabe
b,
Taku
Kitayama
b,
Chisa
Itoh
b and
Hitoshi
Miyasaka
ab,
Yudai
Watanabe
b,
Taku
Kitayama
b,
Chisa
Itoh
b and
Hitoshi
Miyasaka