
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sulfur-templated Ni–Ni′ coordination polymer that relies on a polarizable nickel nitrosyl hub†
Manish
Jana
 ,
Michael B.
Hall
* and
Marcetta Y.
Darensbourg
*
,
Michael B.
Hall
* and
Marcetta Y.
Darensbourg
*
Department of Chemistry, Texas A & M University, College Station, Texas 77843, USA. E-mail: hall@science.tamu.edu; marcetta@chem.tamu.edu
First published on 17th December 2024
Abstract
The templating properties of a diaza-nickel-cis-dithiolate towards triphenylphosphine gold(I), yielding a transoid [Ni(N2S2)·2Au(PPh3)] complex (T. A. Pinder, S. K. Montalvo, A. M. Lunsford, C.-H. Hsieh, J. H. Reibenspies and M. Y. Darensbourg, Dalton Trans., 2014, 43, 138–144) suggested that a suitable analogue of d10-Au(I), i.e., {Ni(NO)}10, could generate a tetrahedral nickel node for a [Ni(N2S2)·2Ni(NO)(X)]n coordination polymer. Monomeric precursors, derived from Feltham's [(Ph3P)2Ni(NO)(Cl)] (R. D. Feltham, Inorg. Chem., 1964, 3, 116–119) produced the bidentate/sulfur-chelated [Ni(N2S2)·Ni(NO)(X)] species with loss of PPh3. Exchange of Cl− by azide, N3−, in the {Ni(NO)}10 synthon led to the balance of electrophilicity at Ni(NO) and non-covalent (H-bonding and van der Waals) interactions that stabilized the extended chain of bridging sulfurs, in transoid connectivities, between a square planar NiII and a tetrahedral Ni, the latter within the electronic and spin-delocalized {Ni(NO)}10 system. This study defines a new path that creates coordination polymers using metallodithiolates, the success of which, in this case, depends on the highly polarizable {Ni(NO)}10 unit.
Introduction
Coordination polymers are broadly defined as extended structures based on metal ions linked into an infinite chain, sheet, or three-dimensional construction by bridging ligands.1–4 Once formed, the balance of electronic and steric features/requirements that lead to the thermodynamic preference of observed connections in coordination polymers, may be, in many cases, readily rationalized. However, a priori design principles for a particular set of synthons, are not so easily predicted. Thus, advances in this huge field are typically made from knowledge of serendipitous prior success(es), based on certain building blocks and their fundamental properties.A broad range of molecular compositions and structures are known to result from the ability of metallo-cis-dithiolates (Fig. 1) to capture multiple and numerous exogenous metals at the thiolate sulfurs.5 Various synthetic approaches continue to be developed for entry into hetero bi- and polymetallic complexes, particularly extensive for square-planar, d8 Ni(II)N2S2 derivatives, where the N2S2 platform is contiguous nitrogen and sulfur linked by hydrocarbons. This arrangement places the two thiolates in a propitious position for bidentate ligation to exogeneous metals, including Ni⋯Ni′ arrangements in biology.6 In general, the bonding within [M(η2-μ-SR)2]M′ may be compared to diphosphine-M′ or bipyridine-M′ interactions, as the nucleophilicity of metal-bound thiolate sulfurs has been somewhat deactivated relative to anionic RS−, making the M–S–R similar to neutral P or N donors.5 However, the stereochemistry demanded by the remaining sulfur lone pairs, and the orientation of their orbital lobes in MSR, particularly cis-M(SR)2, provide structural possibilities (as shown in Fig. 1) unavailable to phosphorus or nitrogen donors. Most importantly, they generate stable sulfur-bridged polymetallics.
 | ||
| Fig. 1 Examples of various binding modes of NiN2S2 due to presence of two lone pairs on each sulfur atom.8,9,14 Identifiers taken from deposits in Cambridge Crystallographic Data Base. | ||
The successful linking of NiN2S2 molecules through the attachment of electrophiles to the metal-bound thiolate sulfurs is an obvious strategy that offers an enormous prospect for synthesis of S-based coordination polymers. Most structural platforms involving [M(η2-μ-SR)2]M′ thus far reported are related to bidentate M(η2-μ-SR)2 binding in which the stereochemistry of the [M(η2-μ-SR)2]M′ is governed by the minimization of sulfur lone pair repulsion.7 The cis-configuration generates a butterfly-type hinge at the bridge (Fig. 1, EROPEG).8 However, the placement of two M′ electrophiles in a transoid arrangement relative to the M(μ-SR)2 plane, observed for a bis-[Au(PPh3)]2 derivative (Fig. 1, TINDUR) hints at the opportunity for designing coordination polymers based on NiN2S2 bridges that link metal units in possession of two accessible open sites.9
It is well known that sulfur-based linkers exhibit energetic alignment with transition metal d orbitals, leading to stronger interactions and potentially superior material properties.10 A prominent example from the Anderson group has revealed that a 4Fe4S-based coordination polymer can be used as a heterogeneous, proton–electron coupled transfer mediator during electrocatalysis.11 These highly active and robust cocatalysts can be easily recyclable during the electrocatalysis as compared to the monomeric 4Fe4S clusters.
Here we report a synthon, [Ni(NO)(Cl)(PPh3)2],12 with {Ni(NO)}10 as an electronic analogue of d10 Au+ or {Fe(NO)2}10 (i.e., the total number of electrons in the metal-d orbitals + π* manifold of NO is 10, represented in the Enemark–Feltham (E–F) notation13) which can bind with NiN2S2 similar to the [Ni(N2S2)·Fe(NO)2] complex, EZEXEO in Fig. 1.14 Upon replacement of chloride by azide, the bidentate binding of the NiN2S2 opens up and exposes an open site on the neutral Ni(NO)(N3) unit. This unit is found to serve as a hub for connecting the NiN2S2 linkers in transoid arrangement as seen for the bis-[Au(PPh3)]2 derivative, TINDUR, Fig. 1.9 The successful conversion of the azide derivative into the metallo polymer contrasts to the analogous [Ni(NO)(Cl)(PPh3)2] which is incapable of such development; the latter serves as a basis for understanding the balance of nickel electrophilicity and geometrical preferences. This report defines a new path to construct S-based coordination polymers using available lone pairs of sulfur in metallodithiolate ligands, the success of which is based on the electronically and structurally responsive {Ni(NO)}10 unit.
Results and discussion
Synthesis and characterization
(Caution! All described syntheses of azides result in potential energetic compounds with possible sensitivities towards heat, friction, impact and discharge. Although no incidents occurred during these syntheses personal safety equipment as well as grounded equipment is strongly recommended! Only small quantities (10–20 mg) of material should be used (at a time) during reactions with azide and metal spatulas should never be used.)Relative to phosphines, bipyridines or isonitriles, the sulfur lone pairs in MN2S2 metallodithiolates are of greater nucleophilicity and make them reasonable synthons in the design of sulfur-bridged heterometallics, some of which are directed towards catalysis.15 Reaction of (dadtBz)Ni (dadtBz is N,N′-dibenzyl-3,7-diazanonane-1,9-dithiolate)16 with NiCl2(PPh3)2 did not however produce the expected [(dadtBz)Ni·NiCl2] adduct. Instead, two Cl− ions were displaced, giving the ubiquitous trinickel complex with three square planes arranged in the stair-step configuration, [(dadtBz)Ni·NiII·Ni(dadtBz)]2+, having [NiCl4]2− as an anion, shown in Scheme 1a. The XRD-derived structure of this salt is shown in Fig. S1 and S2 and selected bond angles and lengths are given in Table S1.† Isolation of this complex prompted us to pursue the reported [Ni(NO)(Cl)(PPh3)2] complex as synthon.12
 | ||
| Scheme 1 Synthetic routes to complexes reported in this study. MeOH was used as a solvent for all reactions. Note: complex 2 is the coordination polymer, alternatively referred to as CP2. | ||
The tetrahedral [Fe(CO)2(NO)2] synthon binds with MN2S2 with loss of carbonyl ligands as shown in EZEXEO, Fig. 1.14 We hypothesized that nitric oxide (NO) might be a suitable candidate to accommodate the additional electron density produced from binding of the metallodithiolates while maintaining the geometry of the tetrahedral Ni center of [Ni(NO)(Cl)(PPh3)2]. Accordingly, a stoichiometric amount of the Ni(NO) reagent was added to a methanolic solution of (dadtBz)Ni producing a colour change from brown to dark blue, concomitant with the loss of the ν(NO) absorption of the precursor complex [Ni(NO)(Cl)(PPh3)2] in CH3OH at 1735 cm−1; a new band appeared at ca. 1757 cm−1 (Fig. S3a†). The reaction solution was stirred for an additional hour. Evaporation of the solvent and recrystallization from CH3CN/Et2O yielded complex 1 in 85% yield as a dark green crystalline solid suitable for X-ray diffraction, Scheme 1b. The solid state ν(NO) IR spectrum is shown in Fig. S3b.† Mass spectral analysis of this complex shows m/z = 518.0360 which confirms the presence of the expected neutral complex 1 (Fig. S4 and S5†). Another signal at m/z = 780.1271 was observed, interpreted as due to the presence of a di-nickel complex with one triphenyl phosphine i.e., [(dadtBz)Ni·Ni(NO)(Cl)(PPh3)], analogous to a previously reported gold complex (Fig. 1, identification code: TINDIF).9 Despite several attempts this complex could not be isolated or characterized further. However, a change in the N2S2 ligand backbone led to success.
A [(N2S2)Ni·Ni(NO)(Cl)(PPh3)] complex similar to the Ni–Au TINDIF complex9 was isolated and crystallographically characterized upon changing the Ni synthon from (dadtBz)Ni to (bme-dame)Ni (bme-dame = N,N′-dimethyl-N,N′-bis(2-mercapto-ethyl)-ethylenediamine) during reaction with [Ni(NO)(Cl)(PPh3)2]. The XRD structure of this complex is given in the ESI (Fig. S9 and S10†) and the corresponding mass spectrum is in Fig. S11 and S12.†
In order to remove the metal-bound chloride as well as to see if the structural platform can be expanded from complex 1, reaction with a stoichiometric amount of NaN3 was performed in methanol. Consistent with Feltman's reports, the [Ni(NO)(Cl)(PPh3)2] reacts with NaN3 to form [Ni(NO)(N3)(PPh3)2].17 The FTIR spectrum of the reaction mixture shows absorption bands at ca. 2045 and 1757 cm−1, Fig. S13.† While no shift was observed in the ν(NO) IR band at 1757 cm−1, the appearance of the new absorbance at 2045 cm−1 is taken as convincing evidence of the presence of a metal-bound azide (N3−) in the complex. Removal of methanol and applying crystallization conditions, CH3CN/Et2O, yielded dark greenish-brown coloured needles of complex 2 in 70% yield, Scheme 1c. The solid state IR spectrum of this complex shows ν(NO) and ν(N3) at ca. 1733 cm−1 and 2035 cm−1 (Fig. S14†). Complex 2 can alternatively be obtained upon reaction of (dadtBz)Ni with [Ni(NO)(N3)(PPh3)2] and it can be isolated as crystals from DMF/Et2O solution. X-ray diffraction analysis of the crystals obtained via both routes shows a one-dimensional coordination polymer chain. Detailed crystallographic description of this complex is given below. Once the complex crystalizes as a coordination polymer it becomes insoluble in almost all organic solvents (marginally in DMF). Note: we refer to this product as complex 2 throughout, although it is a coordination polymer.
XRD characterization
The molecular structures of complexes 1, [(dadtBz)Ni·Ni(NO)Cl], and 2, [(dadtBz)Ni· Ni(NO)(N3)]n are shown in Fig. 2 as 50% probability thermal ellipsoid plots; selected metric parameters are tabulated in Table S2.† Complexes 1 and 2 crystalize in the monoclinic P21/c and P21 space groups respectively. The NiN2S2 portion of complex 1, Ni1, is slightly distorted from square planar geometry with τ4 = 0.13. The second Ni center, Ni2, retains a distorted tetrahedral geometry with τ4 = 0.83. The NO molecule bound to Ni2 is almost linear, ∠Ni2–N3–O1 = 175.40°, i.e., an increase in the ∠Ni–N–O bond angle from 152.17° (for monomeric [Ni(NO)(Cl)(PPh3)2]) is observed upon displacement of the PPh3 groups by NiN2S2. A slight increase in the Ni–Cl bond length is also observed (from 2.27 Å in the Ni precursor to 2.31 Å in complex 1) upon coordination with the metallodithiolate ligand. The distance between the Ni centers in complex 1 is 2.778 Å. Based on the sum of the covalent atomic radii of Ni, this Ni–Ni′ distance is beyond bonding. Crystal packing diagrams of this complex along three crystallographic axes are shown in Fig. S8,† and an expanded listing of pertinent metric data is provided in Table S2.†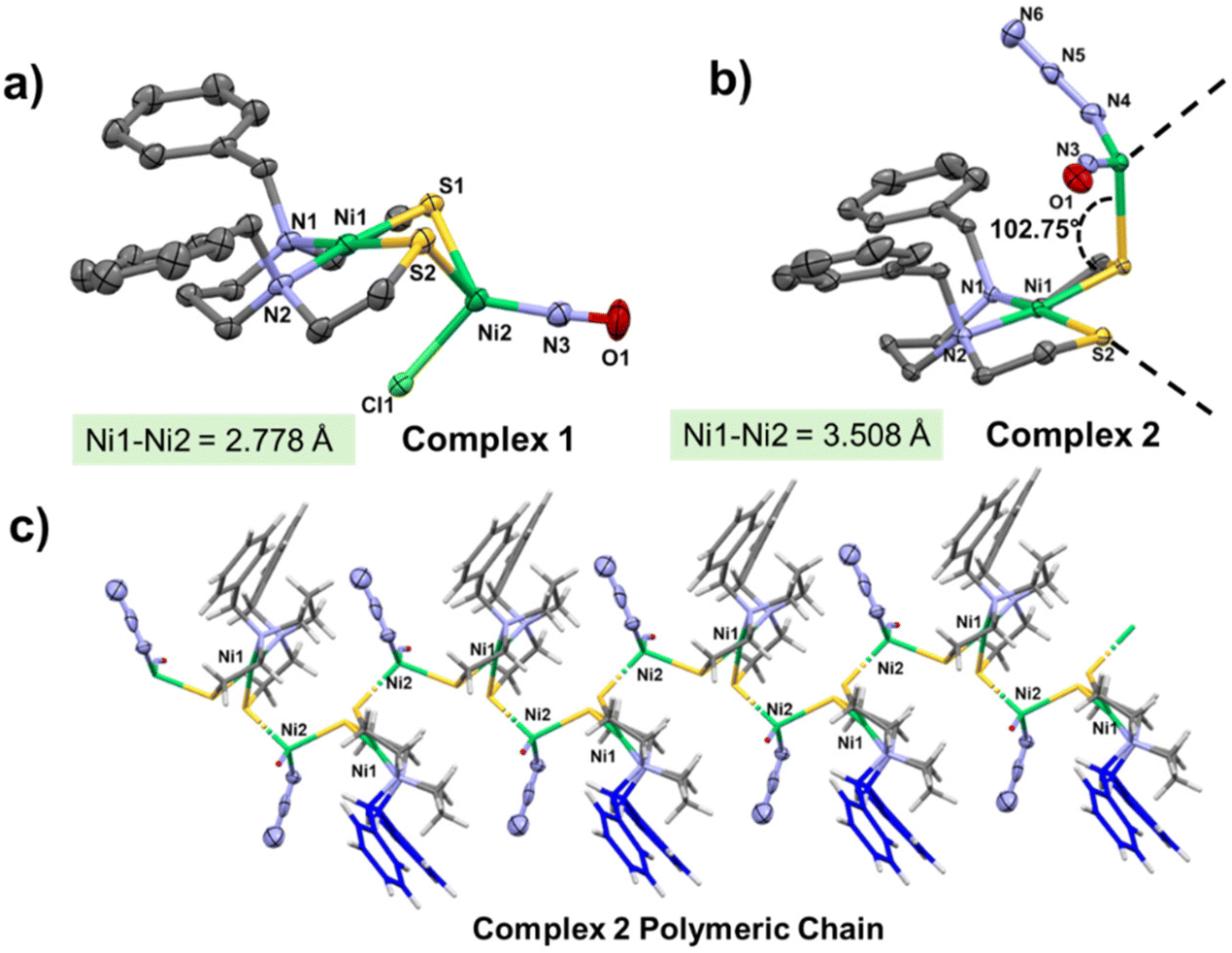 | ||
| Fig. 2 Molecular structures of (a) complex 1 [Ni(dadtBz)Ni(NO)(Cl)] and (b) complex 2 [Ni(dadtBz)Ni(NO)(N3)] (asymmetric unit only is shown) showing 50% probability thermal ellipsoids. H-atoms are omitted for clarity. (c) The 1-D polymeric chain of 2. | ||
Complex 2 crystalizes in the monoclinic P21 space group. It can be crystalized from different solvents such as CH3CN, MeOH as well as DMF. The following discussion refers to the crystals obtained from CH3CN/Et2O. X-ray crystallographic analysis of this complex reveals a one-dimensional coordination polymer structure containing repeating [(dadtBz)Ni·Ni(NO)(N3)] units, Fig. 2c. The unit cell of this species contains two asymmetric units, shown in Figs. S17 and S18.†
The nickel center inside the N2S2 pocket in complex 2 is, as was found for complex 1, in a slightly distorted square planar geometry with τ4 = 0.18; the displacement of the nickel from the mean N2S plane (the plane created by N1N2S2) is 0.015 Å. The other Ni center, Ni2 of the {Ni(NO)(N3)} unit, adopts a distorted tetrahedral geometry with τ4 = 0.84; it is coordinated by S atoms of two distinct metallodithiolate ligands, as well as an NO and an azide, N3−.
The ∠Ni1–S1–Ni2 angle in the CP 2 is 102.75°, in contrast to 76.10° in case of the dinickel complex 1. Upon coordination of the azide unit the metal nitrosyl (∠M–N–O) became significantly bent from 175° (seen for complex 1) to 161° (∠Ni2–N3–O1 = 161.22° for 2). A closer look on the structure of 2 suggests that the oxygen atom of NO molecule is within hydrogen bonding distance (2.5–2.6 Å) from the aliphatic and aromatic protons, Fig. 3d. The distance between the nickel centers increases substantially from 2.77 Å for complex 1, to 3.379 Å and 3.670 Å for the coordination polymer 2.
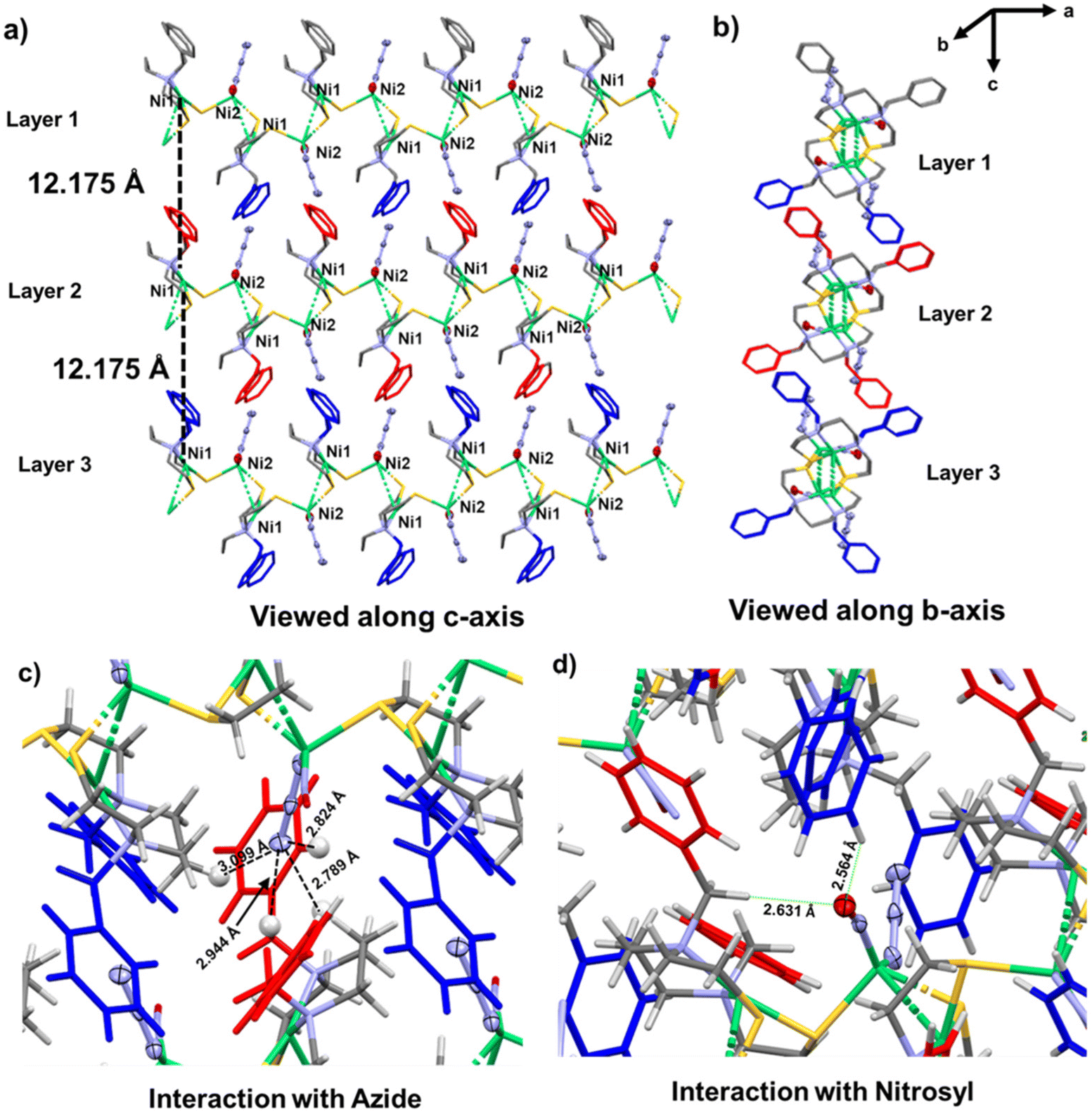 | ||
| Fig. 3 (a) Three layers of complex 2 viewed along c-axis. The distance between the Ni centers in two layers is 12.175 Å. (b) Three layers of complex 2 viewed along b-axis. (c) Zoomed in view of the interaction between the azide group and the aliphatic and aromatic hydrogens. (d) Zoomed in view of the interaction between nitrosyl group and aliphatic and aromatic hydrogens. | ||
We examined the crystal packing of this complex for weak interactions that might assist in stabilizing this polymeric structure. As shown in Fig. 3a, the distance between the nickel centers in two adjacent layers is 12.175 Å (viewed along c-axis). The ‘zoomed-in’ view of the molecular structure shows that the azide group is oriented to indicate hydrogen-bonding interactions with phenyl hydrogens; four contacts are within distances of 2.7–3.1 Å, Fig. 3c.
There appears to be no stabilization by π–π stacking interactions between the aromatic rings18 of the adjacent layers; instead, they are at an angle of ca. 77°, Fig. S19.† The arene planes in the same layer are separated by ca. 6.5 to 6.7 Å, Fig. S20.† In order to evaluate the weak interactions present in the coordination polymer, non-covalent interaction (NCI) analyses were performed on the unit cell of 2, see the Computational section below.
The solid-state structures of 2 obtained from different solvents (CH3CN, MeOH and DMF) find almost identical metric parameters, except for slight differences in the Ni1–Ni2 distances and ∠Ni2–N3–O1 angles, Fig. S22.† The Hirshfeld surface analysis of the crystals obtained from different solvents as well as that of complex 1 are given in the ESI (Fig. S37–40†).
Crystallization from coordinating solvents like MeCN or DMF shows the presence of solvent molecules (CH3CN/DMF) inside the cavities created by stacking of multiple layers side by side, Fig. S22 and S26† (viewed along b-axis).
Bulk purity and thermal stability
To reveal the phase purity of the coordination polymer (complex 2), a PXRD study was carried out on powdered samples within the 2θ range of 5°–40°, Fig. S31.† All significant peaks of the as-isolated powdered complex 2 were noted to be well-matched with the simulated pattern. The PXRD study along with elemental analysis of the coordination polymer confirms its bulk purity and crystallinity.The coordination polymer is found to be stable under ambient conditions for weeks without any loss of NO or azide. Thermogravimetric analysis experiments on crystalline samples of 1 and 2 under N2 environment (Fig. S32†) indicated that complex 1 is stable up to 150 °C, followed by complete structural disruption beyond 150 °C. Complex 2 shows only 6.6 wt% decay up to ca. 141 °C, which can be interpreted as the loss of one mole MeCN per formula unit to yield the non-solvated 2. Complete structural disruption of the coordination polymer was observed after ca. 141 °C, similarly to complex 1.
Redox properties
While the coordination polymer 2 is largely insoluble in CH3CN, its meager solubility in DMF permitted recording of the cyclic voltammograms of complexes, 1 and 2 at 22 °C under Ar atmosphere in DMF containing 0.1 M [n-Bu4N][PF6] (Fig. S33†). Cyclic voltammetry of complex 1 was also attempted in MeCN, Fig. S35.† All values were referenced to internal Fc+/Fc at E1/2 = 0.0 V and were recorded on sweeps initiated in the cathodic direction.Three reduction events were observed in the initial cathodic sweep as shown, Fig. 4. The first irreversible reduction event at ca. −1.55 V for both complexes is assigned to the {Ni(NO)}10/{Ni(NO)}11 couple.19 A second irreversible redox event observed at ca. −1.85 V in DMF is proposed to result from further reduction of the first reduced species. The third reduction event at ca. −2.30 V and its corresponding oxidation at −2.12 V is assigned to the reduction of the Ni2+ center (Ni2+ to Ni1+) housed within the N2S2 ligand.
 | ||
| Fig. 4 Cyclic voltammetry of complex 1 (red) and complex 2 (green) in DMF containing a 0.1 M [nBu4N][PF6] electrolyte and 2.0 mM complex at room temperature. Note that complex 2 has poor solubility in DMF, leading to reduced current intensity during the cyclic voltammetry analysis. | ||
The lower solubility of the coordination polymer in DMF may explain the reduced current intensity in the CV features. Changes upon repeated scans of complex 2 are shown in Fig. S34.† Interestingly, both monomeric and polymeric complexes show almost identical redox behaviour in the cyclic voltammetry experiments which suggests the following: (a) the coordination polymer slightly dissociates in solution to give a monomeric complex similar to complex 1; or (b) the redox-activity of the coordination polymer is similar to that of the monomeric complex. The UV-vis spectra for the monomeric complex as well as the coordination polymer (sparingly soluble in DMF) (Fig. S36†) shows very similar absorption bands and supports the cyclic voltametric result interpreted as the partial dissociation of the coordination polymer in DMF.
Computational section
DFT Calculations were performed for the complex 1 with the TPSS20 and B3LYP21 functionals; the 6-31G(d,p) basis set22 was used for C, H, N, O, S and Cl atoms whereas LANL2DZ basis set23 was used for Ni atoms. The crystal structure of the complex was imported to use as the starting coordinates for the geometry optimization and subsequent vibrational frequency and energy calculations. All species were confirmed to be minimum energy structures by the absence of any imaginary frequencies. Experimentally, the known [Ni(NO)(L3)] complexes have been shown to have a diamagnetic ground state, which are best described as open-shell singlets (OSS) with a HS-Ni(II) center (S = 1) antiferromagnetically coupled to a triplet state NO− (S = 1) ligand.24 Antiferromagnetic coupling for complex 1 was modelled in this manner with the B3LYP functional, and gives an overall spin of Stotal = 0.73. Consistent with experiment, the 1H NMR spectrum of this complex showed broad signals at room temperature (Fig. S6†). As expected, the single determinant wavefunction contains Ms = 0 components from S > 0 spin states. This optimized BS open-shell singlet state is found to be −7.59 kcal mol−1 more stable than the corresponding closed-shell singlet structure. As shown by the wavefunction's spin density in Fig. 5, the majority of the spin is on Ni(II) and the NO− with spin populations of +1.01 and −1.16, respectively. | ||
| Fig. 5 Spin density plot for optimized BS-singlet structure of complex 1. | ||
Interestingly, attempts to optimize the geometry of complex 1 as an open-shell singlet state with the TPSS functional converged to the closed-shell singlet state (Stotal = 0). The calculated structure for both methods are in accord with the experimental crystal structure shown in Fig. S40 and Table S4.†
Parallel calculations for the monomeric form of azide, found as it exists in complex 1, as an OSS in B3LYP and as CSS in the TPSS (Table S4†), shows accumulation of electron density on NO which leads to decrease in the Ni–N–O bond angle. The τ4 value for the Ni2 center decreases from the typical tetrahedral value toward the square planar value as shown in Table S4.† Apparently, these structural changes destabilize the monomeric form of the Ni(N2S2)·Ni(NO)(N3). Experimentally, the Ni–N–O bond is found to be more bent in the polymeric complex 2 than that of monomeric complex 1; nevertheless, the polymer has maintained the strong tetrahedral character of the complex 1 (see Table S2†).
Notably, the Ni–S bond distances are shorter in the polymeric complex than the monomeric complex. We assume this result may contribute to the stabilization of the polymer. In order to examine the non-covalent interactions (NCI) in the coordination polymer, 2, the ground-state geometry of the assemblies found in the solid state was used to evaluate the attractive/repulsive character of the interactions by using Multiwfn to generate NCI Plots.25,26 The Visual Molecular Dynamics (VMD) program was used to visualize the prominent interactions present in the molecule.27 This methodology has been used for coordination polymers to estimate the contributions of a variety of interactions in the solid-state including hydrogen bonding, π–π stacking and other weak van der Waals interactions.28
Since complex 2 is a coordination polymer, for investigations of limited views of units within the CP we terminated the S end with BH3 and the Ni end with NH3. We performed a single point calculation on two asymmetric units of this “molecule” (two layers) which are adjacent to each other. The NCI plot of this construction indicates a prominent CH–π interaction between the aromatic ring of one layer with the aromatic protons of the adjacent layer, Fig. 6. Hydrogen bonding and weak van der Waals interactions as indicated by the green iso-surface, are also observed between the azide group and the aliphatic and aromatic hydrogens (Fig. 6, red circle). The non-covalent interactions (NCI) coupled with strengthening of the Ni–S bonds help to stabilize the azide complex as a coordination polymer.
 | ||
| Fig. 6 (a) NCI index on the unit cell of complex 2 using iso surface value of 0.5 au and the color scale is −0.05 < ρ < 0.05 au. CH–π interactions are shown with arrow and the red circle indicates the CH–N3 interaction. (b) Reduced density gradient plot shows prominent attractive interactions in complex 2. | ||
Conclusion
This synthetic approach to a coordination polymer took advantage of the properties of two synthons:(a) The templating ability of the cis-dithiolates in a NiII(N2S2) complex that leads to the assembly of two strong electrophile/sulfur interactions transoid to each other, linking the NiII with a second nickel imbedded within a polarizable and electroactive {Ni(NO)}10 unit.
(b) The nitrosylated nickel source, with 10 electrons in its “golden” outer orbitals, is responsive to its ligand field. Its ability to electronically deform towards NiII(NO)− is witnessed by shifts in crystallographic parameters. The application of Enemark–Feltham description for the overall electron count accounts for the oxidation state shifts within Ni(NO)10, i.e., [Ni0(NO)+] ↔ [NiI(NO)˙] ↔ [NiII(NO)−], and for the sensitive requirement of the bidentate binding ability of NiN2S2 towards a single metal site, vs. its use as a bidentate bridging ligand that spans coordination sites of two metals.
This electronic deformation or polarization within the delocalized [Ni(NO)]+ unit, i.e., Ni(NO)10, accommodates the alternating assembly of Ni⋯Ni′ in the coordination polymer. While the primary Ni–S–Ni′ bonds in the polymer chains are strong, weak intermolecular interactions, identified in NCI plots, add to the stability of the polymer structure. The role of the substituents on the N2S2 framework must be considered in this regard.
The potential of cis-dithiolates, particularly of nickel, for the assembly of heterobimetallic biomimetics and of catalysts is well developed.15 However, to our knowledge this report is the first to explore and identify a route to coordination polymers based on this background. Given that libraries of MN2S2 exist, there is potential for extensive development to expand the range of S-based coordination polymers and design of materials from their condensation.
Author contributions
Manish Jana: experimental procedures, isolation and characterization (including theoretical calculations) of products, and design of project. M. J. Wrote the first and modified drafts. Michael B. Hall: design of computational approaches and overseer of results, interpretation and presentation. Marcetta Y. Darensbourg: design of project and interpretation/presentation of results. Contributed to writing of manuscript.Data availability
All underlying data are available in the article itself and in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support of the National Science Foundation (MPS CHE 2102159) and the Robert A. Welch Foundation (A-0924) to M. Y. D. for synthesis and characterization of the compounds and (A-0648) to M. B. H for the computational studies. We appreciate the technical assistance and chemical insight of Dr Sarnali Sanfui during preparation of the final draft of the manuscript.References
- K. Biradha, A. Ramanan and J. J. Vittal, Cryst. Growth Des., 2009, 9, 2969–2970 CrossRef.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef PubMed.
- C. S. Diercks, M. J. Kalmutzki, N. J. Diercks and O. M. Yaghi, ACS Cent. Sci., 2018, 4, 1457–1464 CrossRef PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef PubMed.
- (a) J. A. Denny and M. Y. Darensbourg, Chem. Rev., 2015, 115, 5248–5273 CrossRef PubMed; (b) J. A. Denny and M. Y. Darensbourg, Coord. Chem. Rev., 2016, 324, 82–89 CrossRef.
- (a) C. Darnault, A. Volbeda, E. J. Kim, P. Legrand, X. Vernède, P. A. Lindahl and J. C. Fontecilla-Camps, Nat. Struct. Mol. Biol., 2003, 10, 271–279 CrossRef; (b) M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef PubMed; (c) R. E. Treviño and H. S. Shafaat, Curr. Opin. Chem. Biol., 2022, 67, 102110 CrossRef PubMed; (d) H. S. Shafaat, A. C. Manesis and A. Yerbulekova, Acc. Chem. Res., 2023, 56, 984–993 CrossRef PubMed.
- P. Guerrero-Almaraz, M. Quiroz, D. R. Rodriguez, M. Jana, M. B. Hall and M. Y. Darensbourg, ACS Org. Inorg. Au, 2023, 3, 393–402 CrossRef PubMed.
- M. L. Golden, S. P. Jeffery, M. L. Miller, J. H. Reibenspies and M. Y. Darensbourg, Eur. J. Inorg. Chem., 2004, 2004, 231–236 CrossRef.
- T. A. Pinder, S. K. Montalvo, A. M. Lunsford, C.-H. Hsieh, J. H. Reibenspies and M. Y. Darensbourg, Dalton Trans., 2014, 43, 138–144 RSC.
- (a) R. Dong, P. Han, H. Arora, M. Ballabio, M. Karakus, Z. Zhang, C. Shekhar, P. Adler, P. S. Petkov, A. Erbe, S. C. B. Mannsfeld, C. Felser, T. Heine, M. Bonn, X. Feng and E. Cánovas, Nat. Mater., 2018, 17, 1027–1032 CrossRef PubMed; (b) A. J. Clough, N. M. Orchanian, J. M. Skelton, A. J. Neer, S. A. Howard, C. A. Downes, L. F. J. Piper, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2019, 141, 16323–16330 CrossRef PubMed; (c) T. Kusamoto and H. Nishihara, Coord. Chem. Rev., 2019, 380, 419–439 CrossRef; (d) D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef PubMed; (e) M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef PubMed; (f) L. Sun, C. H. Hendon, M. A. Minier, A. Walsh and M. Dincă, J. Am. Chem. Soc., 2015, 137, 6164–6167 CrossRef PubMed; (g) J. A. DeGayner, I.-R. Jeon, L. Sun, M. Dincă and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 4175–4184 CrossRef PubMed; (h) M. Fourmigué, C. E. Uzelmeier, K. Boubekeur, S. L. Bartley and K. R. Dunbar, J. Organomet. Chem., 1997, 529, 343–350 CrossRef; (i) L. Sun, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2013, 135, 8185–8188 CrossRef PubMed; (j) G. Givaja, P. Amo-Ochoa, C. J. Gómez-García and F. Zamora, Chem. Soc. Rev., 2011, 41, 115–147 RSC; (k) J. Xie, J.-N. Boyn, A. S. Filatov, A. J. McNeece, D. A. Mazziotti and J. S. Anderson, Chem. Sci., 2020, 11, 1066–1078 RSC; (l) L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef PubMed.
- (a) N. E. Horwitz, J. Xie, A. S. Filatov, R. J. Papoular, W. E. Shepard, D. Z. Zee, M. P. Grahn, C. Gilder and J. S. Anderson, J. Am. Chem. Soc., 2019, 141, 3940–3951 CrossRef PubMed; (b) N. Jiang, A. Darù, Š. Kunstelj, J. G. Vitillo, M. E. Czaikowski, A. S. Filatov, A. Wuttig, L. Gagliardi and J. S. Anderson, J. Am. Chem. Soc., 2024, 146, 12243–12252 CrossRef PubMed.
- R. D. Feltham, Inorg. Chem., 1964, 3, 116–119 CrossRef.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef.
- P. Ghosh, S. Ding, R. B. Chupik, M. Quiroz, C.-H. Hsieh, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, Chem. Sci., 2017, 8, 8291–8300 RSC.
- (a) C.-H. Hsieh, S. Ding, Ö. F. Erdem, D. J. Crouthers, T. Liu, C. C. L. McCrory, W. Lubitz, C. V. Popescu, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Nat. Commun., 2014, 5, 3684 CrossRef PubMed; (b) S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920–12927 CrossRef PubMed.
- M. Jana, X. Zheng, T. Le, M. Quiroz, P. Guererro-Almaraz, D. J. Darensbourg and M. Y. Darensbourg, Adv. Sci., 2024, 11, 2307113 CrossRef PubMed.
- J. H. Enemark, Inorg. Chem., 1971, 10, 1952–1957 CrossRef.
- D. B. Ninković, J. P. Blagojević Filipović, M. B. Hall, E. N. Brothers and S. D. Zarić, ACS Cent. Sci., 2020, 6, 420–425 CrossRef PubMed.
- S. Kundu, P. N. Phu, P. Ghosh, S. A. Kozimor, J. A. Bertke, S. C. E. Stieber and T. H. Warren, J. Am. Chem. Soc., 2019, 141, 1415–1419 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef; (b) Y. Yang, M. N. Weaver and K. M. Merz Jr., J. Phys. Chem. A, 2009, 113, 9843–9851 CrossRef.
- K. Fujisawa, T. Kataoka, K. Terashima, H. Kurihara, F. de Santis Gonçalves and N. Lehnert, Molecules, 2023, 28, 6206 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
- (a) S. K. Seth, Crystals, 2018, 8, 455 CrossRef; (b) A. Gogoi, A. Das, A. Frontera, A. K. Verma and M. K. Bhattacharyya, Inorg. Chim. Acta, 2019, 493, 1–13 CrossRef; N. A. Ashashi, M. Kumar, R. M. Gomila, A. Frontera, H. N. Sheikh and S. C. Sahoo, J. Mol. Struct., 2022, 1254, 132329 Search PubMed; (c) N. A. Ashashi, M. Kumar, R. M. Gomila, A. Frontera, H. N. Sheikh and S. C. Sahoo, J. Mol. Struct., 2022, 1254, 132329 CrossRef; (d) S. Dutta, S. Chakraborty, M. G. B. Drew, A. Frontera and A. Ghosh, Cryst. Growth Des., 2019, 19, 5819–5828 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2195745, 2368990, 2368992, 2378998, 2379907 and 2380703. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03174a |
| This journal is © The Royal Society of Chemistry 2025 |