
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA lightweight, Li supplementary and lithiophilic interface enables anode-less lithium metal battery prototyping†
Lu
Cheng
,
Jiacheng
Liu
,
Helin
Wang
,
Yuxiang
Guo
,
Ahu
Shao
,
Yunsong
Li
,
Zhiqiao
Wang
,
Yaxin
Zhang
,
Jiawen
Tang
,
Chunwei
Li
and
Yue
Ma
 *
*
State Key Laboratory of Solidification Processing, Center for Nano Energy Materials, School of Materials Science and Engineering, Northwestern Polytechnical University and Shaanxi Joint Laboratory of Graphene (NPU), Xi'an 710072, China. E-mail: mayue04@nwpu.edu.cn
First published on 2nd April 2025
Abstract
Lithium metal batteries (LMBs), while offering exceptional energy density for next-generation energy storage, face inherent challenges such as dendrite growth, non-uniform nucleation, and dynamic interfacial instability that hinder their practical deployment. Herein, a lightweight (0.39 mg cm−2), Li source supplementary and moisture-proof interfacial layer is developed to enable anode-less LMB prototyping. This layer is composed of high-entropy alloys (HEAs) and an in situ grown carbon nanotube (CNT) scaffold, modified with thermally alloyed Li22Sn5 blended with a hydrophobic ethylene-vinyl acetate (EVA) copolymer. The Li22Sn5@EVA composite acts as a moisture-proof cation reservoir, while the HEAs and CNTs synergistically regulate lithium-ion flux and nucleation, promoting uniform lithium deposition and enhancing mechanical stability. The integrated layer (HEA@CNT/Li22Sn5@EVA) enables dendrite-free lithium plating at a high areal capacity of 6 mA h cm−2 and stable cycling in symmetric cells at 2 mA cm−2, even under a 75% depth of discharge. When paired with a LiFePO4 cathode (LFP, 25.53 mg cm−2) in a 145 mA h pouch cell, the prototype achieves a gravimetric energy density of 325.2 W h kg−1 and a power output of 603.5 W kg−1 at an ultralow N/P ratio of 0.22. This interfacial design is broadly applicable to anode-free alkali metal batteries, offering a pathway toward high-energy and high-power energy storage solutions.
Broader contextHigh energy/power density batteries are urgently needed in the electric vehicle and consumer electronics markets. In this study, Cheng et al. created a prototype of an anode-less LMB featuring a lightweight, moisture-proof interface layer and self-supplementary Li sources for bare Cu foil. This offers a novel viewpoint for the optimization strategy of next generation anode-less LMBs. |
Introduction
The ever-increasing demand for consumer electronics and electric vehicles has spurred the development of advanced lithium battery technologies with higher energy densities. The lithium metal anode has attracted significant attention owing to its ultra-high theoretical capacity (3860 mA h g−1)1,2 and lowest electrochemical potential (−3.04 V vs. the standard hydrogen electrode).3 However, uncontrolled lithium deposition leads to dendritic growth, which can penetrate the separator, causing internal short circuits and posing serious safety hazards.4,5 Moreover, the inherently fragile solid electrolyte interphase (SEI) on the lithium metal surface is susceptible to fracture due to significant volume changes during repeated stripping/plating processes, resulting in irregular lithium plating morphology and exacerbated side reactions. Additionally, a portion of the loosely deposited lithium becomes electrically isolated during the stripping process, accumulating as a ‘dead’ lithium layer. This phenomenon not only impairs interfacial Li+ transport but also significantly reduces coulombic efficiency (CE) and accelerates capacity fading during prolonged cycling.6–9Anode-free or anode-less (N/P ratio <0.5) configurations significantly reduce the amount of pre-stored anode materials, offering higher volumetric and gravimetric energy densities.10,11 Furthermore, this configuration simplifies the electrode manufacturing process and reduces production costs. Unfortunately, the practical application of these batteries faces more serious dendrite formation, which can lead to short circuits, rapid depletion of the lithium source, and reduced cycle life.12 To address these challenges, various strategies have been explored, including the design of artificial interphase layers, lithiophilic modifications, and polymer coatings, aimed at improving interfacial stability and regulating Li deposition patterns.13,14 For instance, composite artificial interphase layers,15 nitrides16 and sulfides17–19 have been developed, characterized by high mechanical rigidity and ionic conductivity, to suppress dendrite formation during nucleation.20 However, these interphase layers often exhibit poor adhesion to substrates, leading to delamination during prolonged cycling. Alternatively, the introduction of lithiophilic species to form LixM alloy interphases at the interface effectively reduces the Li nucleation barrier.21–23 Nevertheless, the alloying process inevitably induces volumetric expansion, particle agglomeration, and even electrode pulverization.24–26 To mitigate these issues, 3D architectures have been further introduced to anchor active particles, thereby reducing their migration and detachment during cycling.27,28 However, interphase layers with nucleophilic functional groups may deplete limited cation reservoirs from paired cathodes and lean electrolytes, posing additional challenges to long-term stability.29–31 Consequently, most electrochemical evaluations have been performed in half-cell configurations under low-capacity loadings or in prototype batteries with excessive Li sources (typically > 2×) to compensate for irreversible Li+ loss.32,33 However, these conditions do not fully resolve the challenges of practical applications. Although some substrate designs demonstrate dendrite suppression and reversible plating/stripping in symmetric cells, these evaluations are typically conducted at an ultralow depth of discharge (e.g., less than 20% DOD),34,35 limiting realistic evaluations in full-cell configurations. Furthermore, excessive Li usage compromises the scalability and achievable energy densities of practical battery designs, increasing material and manufacturing costs. Therefore, precise control over lithium utilization—fully leveraging the high-capacity advantages of metallic anodes—remains a critical challenge, requiring further advances in interfacial engineering, electrolyte design, and cell configuration optimization.
Prelithiation, the process of introducing additional active lithium before battery assembly to compensate for lithium loss during the initial charge and subsequent cycles, is widely regarded as an effective strategy for enhancing energy densities and extending cycle life.36 For the traditional high-capacity anodes, Li-containing additives, such as stabilized lithium metal powder (SLMP),37,38 LixSi–Li2O/TiyOz,39 artificial-SEI-coated LixSi or LixSn@Ppy40 with low equilibrium potential, are incorporated during the electrode slurry preparation.41–43 However, the highly reactive nature of these additives makes them incompatible with polar solvents or carbonate electrolytes, causing increased side reactions and challenges such as moisture sensitivity, inhomogeneous lithiation, and stringent environmental adaptability requirements, which often lead to inconsistent performance and higher manufacturing costs.18,26 To design a multifunctional interfacial layer capable of withstanding harsh cycling conditions in anode-less or anode-free configurations, key considerations should include the formation of a mechanically robust SEI, appropriate cation compensation capabilities, high ionic conductivity, chemical stability, and compatibility with roll-to-roll cell assembly processes.41,42,44
Herein, we demonstrate a lightweight (0.39 mg cm−2), moisture-proof interface layer (HEA@CNT/Li22Sn5@EVA) for modulating the Cu foils in anode-less LMB prototypes (N/P = 0.22). This design simultaneously addresses high-throughput Li+ influx, ensuring mechanical stability during high-capacity Li deposition, and providing tailored Li source supplementation. The Li supplementary source is derived from the Li22Sn5 alloy, while the hydrophobic EVA encapsulates the Li containing alloy to ensure environmental adaptability, particularly under conditions of 30% relative humidity. Meanwhile, the carbon-thermal reduction HEAs within the framework of in situ CNTs accommodate high-areal-capacity Li deposition up to 6 mA h cm−2 and at 75% depth of discharge (DOD) under 2 mA cm−2. Furthermore, when the prepared substrate (HEA@CNT/Li22Sn5@EVA) is directly combined with the LFP cathode (25.53 mg cm−2, double sided) in an anode-less configuration (1.5 g A h−1 electrolyte, N/P ratio of 0.22), the 145 mA h pouch-format cell achieves an energy density of 325.2 W h kg−1, an extreme power density of 603.5 W kg−1 and excellent cyclability at the cell level. This multifunctional interfacial design of the Li deposition substrate presents a quantum leap towards the practical use of anode-less, energy/power-dense battery systems.
Results and discussion
As illustrated in the anode-less battery model (Scheme 1a), the deposited Li is highly susceptible to dendrite formation during repeated lithiation/delithiation cycles due to the uneven lithium-ion flux, which compromises both safety and cycling stability. Meanwhile, incomplete Li stripping at the dendrite tips leads to dendrite fragmentation, which becomes electrically isolated, forming dead Li. This dead Li continuously accumulates, resulting in uncontrolled volume expansion and eventually penetrating the separator, causing battery short circuits. In contrast, we have developed an ultra-thin (13.5 μm), lightweight (0.39 mg cm−2), and moisture-tolerant interfacial layer comprising a deep-lithiated alloy (Li22Sn5), HEA@CNT, and a hydrophobic polymer, which effectively regulates Li deposition, suppresses dendrite growth, achieves high CE, ensures stable cycling, and compensates for irreversible Li loss. | ||
| Scheme 1 (a) Schematic illustration of cell prototypes when employing the bare Cu foil or HEA@CNT/Li22Sn5@EVA-Cu substrate paired with the LFP cathode. (b) The HEA@CNT/Li22Sn5@EVA-Cu preparation process of the interface layer. Process I: citric acid molecules are combined with metal cations, vaporized by solvent, and spontaneously ignited to form a crosslinked gel at 360 °C. Process II: growth of CNTs on CVD process and the schematic diagram of HEA@CNT composites. Process III: the one-pot metallurgical process of hot bonding of nano-Sn and molten Li metal. Process IV: the encapsulation of Li22Sn5 in hydrophobic EVA polymers and infiltration in the HEA@CNT scaffold. | ||
The synthesis process of the HEA@CNT skeleton is shown in Scheme 1b (detailed in the Experimental section). In the initial phase, citric acid chelates with Fe(NO3)2·3H2O, Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Al(NO3)2·6H2O and Zn(NO3)2·6H2O in predetermined proportions (Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co:Ni:Al:Zn = 1:1:1:1:1) to form a cross-linked sol–gel network (process I); then, the HEA precursor dry gel was prepared by evaporating the aqueous solution, and the FeCoNiAlZn nanoparticles were reduced by self-combustion at 360 °C. Subsequently, the in situ CNTs were catalysed by the synthesized FeCoNiAlZn nanoparticles to form HEA@CNT composites (process II). At the same time, under the protection of an Ar atmosphere, the chemical molar ratio of nano Sn and Li foil (1:4.4) was mechanically blended, and the deep lithium alloy (Li22Sn5) was prepared by a one-pot metallurgical method (210 °C,10 h, process III). Subsequently, toluene solution HEA@CNT and Li22Sn5 composed of the EVA polymer were directly coated and penetrated onto the HEA@CNT scaffold. After vacuum drying, copper electrode foil covered with HEA@CNT/Li22Sn5@EVA was obtained.
:Co:Ni:Al:Zn = 1:1:1:1:1) to form a cross-linked sol–gel network (process I); then, the HEA precursor dry gel was prepared by evaporating the aqueous solution, and the FeCoNiAlZn nanoparticles were reduced by self-combustion at 360 °C. Subsequently, the in situ CNTs were catalysed by the synthesized FeCoNiAlZn nanoparticles to form HEA@CNT composites (process II). At the same time, under the protection of an Ar atmosphere, the chemical molar ratio of nano Sn and Li foil (1:4.4) was mechanically blended, and the deep lithium alloy (Li22Sn5) was prepared by a one-pot metallurgical method (210 °C,10 h, process III). Subsequently, toluene solution HEA@CNT and Li22Sn5 composed of the EVA polymer were directly coated and penetrated onto the HEA@CNT scaffold. After vacuum drying, copper electrode foil covered with HEA@CNT/Li22Sn5@EVA was obtained.
The field emission scanning electron microscopy (FESEM) image demonstrates the uniform dispersion of alloy particles (Fig. 1a). Due to the superior catalytic effects of transition metal species with unfilled 3d orbitals and the continuous carbon supply from acetylene, the in situ grown CNTs interweave and encapsulate the alloy particles. As illustrated in Fig. 1b–d, the clear lattice fringes at site i indicate the presence of highly crystalline particles within the CNT, with a lattice spacing of 0.15 nm corresponding to the (511) plane of the HEA, suggesting excellent structural stability. Meanwhile, as shown in sites i and ii, the lattice spacing of 0.21 nm corresponds to the (111) plane of the HEA, and a typical graphitic lattice spacing of 0.34 nm is observed adjacent to the alloy particles, demonstrating effective encapsulation of the alloy NPs. The high-angle annular dark-field (HAADF) image of the HEA and the corresponding scanning transmission electron microscopy–energy dispersive X-ray spectroscopy (STEM-EDS) elemental maps confirm the uniform distribution of all elements within the nanoparticles, indicating a homogeneous solid solution without significant element segregation (Fig. 1e). As shown in Fig. S1,† the X-ray diffraction (XRD) pattern of the as-synthesized high-entropy alloy (HEA) nanoparticles clearly demonstrates the presence of crystalline FeCoNiAlZn, as evidenced by distinct diffraction peaks corresponding to this phase, indicating high crystallinity and phase purity. As illustrated in Fig. S2,† based on the Brunauer–Emmett–Teller (BET) surface area (125 m2 g−1) and density functional theory (DFT) pore size range (2–4 nm) analysis, the FeCoNiAlZn@CNT provides ample space to accommodate high areal-capacity Li deposition, while the mesopores facilitate Li-ion transport into the inner regions of the CNTs. For the compositional design rationales of HEA particles, it is necessary to introduce Al and Zn species with lithiophilic properties, as well as to incorporate the Fe, Ni, and Co structural elements in the HEMP for the balanced lattice robustness and electrical conductivity (Fig. S3†).
 | ||
| Fig. 1 (a) FESEM images of the HEA@CNT at low and high magnifications (inset image). (b) TEM image of the HEA@CNT composite. (c and d) HRTEM images of the HEA@CNT composite. Site i: HRTEM image of the representative HEA particle: (111) 0.21 nm and (511) 0.15 nm, and HRTEM of the graphitic carbon nanotubes: graphite (002), 0.33 nm. Site ii: HRTEM image of the representative HEA particle: (111) 0.21 nm and HRTEM of the graphitic carbon nanotubes: graphite (002), 0.34 nm. (d) HAADF-STEM of the HEA@CNT and corresponding STEM-EDS elemental maps of Fe, Co, Zn, Al and Ni signals. | ||
As shown in Fig. 2a, under an argon atmosphere, nano-Sn and Li foil with a chemical molar ratio of 1:4.4 were mechanically mixed, and the deep-lithiated alloy Li22Sn5 was successfully synthesized via a melting method at 210 °C for 10 h. As shown in Fig. 2b and S4a,† the transmission electron microscopy (TEM) image reveals smooth Sn nanoparticles with a uniform morphology and a smooth surface. The selected-area electron diffraction (SAED) pattern of Sn nanoparticles shows three distinct reflection rings corresponding to the (200), (211), and (301) planes, indicating high crystallinity (Fig. 2c). As shown in Fig. 2d, the lattice spacings of 0.149 nm, 0.291 nm, and 0.119 nm are assigned to the (112), (200), and (312) planes of nano-Sn, respectively. Additionally, the size of the as-synthesized Li22Sn5 particles is similar to that of the Sn nanoparticles, retaining a similar morphology (Fig. 2e and S4b†). Meanwhile, as shown in Fig. 2f, the SAED pattern of the as-prepared Li22Sn5 particles shows three distinct reflection rings corresponding to the (511), (640), and (933) planes, confirming the successful formation of the Li22Sn5 alloy. In addition, as shown in Fig. 2g, the peak of nano-Sn disappears, and the diffraction peaks of the as-synthesized Li22Sn5 nanoparticles match the standard card (PDF # 18-753), confirming the formation of the Li22Sn5 alloy. To further validate the alloy formation, X-ray photoelectron spectroscopy (XPS) was performed. As shown in Fig. S2,† the XPS survey spectrum of Li22Sn5 nanoparticles reveals the presence of Sn, Li, and trace amounts of carbon species. After the thermal alloying process (Fig. S5a†), the Li 1s signal shifts significantly to a higher binding energy of 56.4 eV, indicating changes in the chemical environment and further supporting the formation of the Li22Sn5 alloy. Similarly, the Sn 3d peaks observed at around 484.9 eV and 493.3 eV can be assigned to the Li22Sn5 alloy phase, providing additional evidence for successful alloy synthesis (Fig. S5b†).
 | ||
| Fig. 2 (a) One-pot metallurgical process using nano-Sn to alloy thermally with molten Li metal; (b) TEM image of the Sn nanoparticles; (c) SAED pattern of the Sn nanoparticles with an objective aperture as highlighted in the dashed circle in Fig. 3b; (d) HRTEM image of the Sn nanoparticles; (e) TEM image of the Li22Sn5 nanoparticles; (f) SAED pattern of the Li22Sn5 particles with an objective aperture as highlighted in the dashed circle in Fig. 3e; and (g) XRD patterns of the Sn nanoparticle and the as-synthesized Li22Sn5 nanoparticle. | ||
As shown in Fig. 3a, the peel strength of the HEA@CNT/EVA-Cu interface layer increases with the increase of EVA content. In sharp contrast (Fig. 3b), the interface impedance also increases as the EVA content increases. Considering the balance between electrical conductivity and peel strength, the optimal weight ratio of HEA@CNT to EVA is 7:1. The optimal weight ratio of HEA@CNT to EVA was selected as the optimal configuration for subsequent tests. As illustrated in Fig. S6,† the HEA@CNT/Li22Sn5@EVA composite exhibits an initial lithiation process to 0.01 V followed by delithiation to 1 V at 2 mA cm−2 in its first-cycle voltage profiles, while the HEA@CNT/Li22Sn5@EVA-Cu electrode demonstrates a charge capacity of 0.34 mA h cm−2, highlighting its potential as a prelithiation agent to compensate for irreversible Li loss. The initial coulombic efficiency (ICE) of the HEA@CNT/EVA-Cu electrode could obviously be improved from 73.3% in the pristine state to 85.7% and 98.1% as the weight ratio of Li22Sn5 content extended to 10% and 20%, respectively. Moreover, when the content of Li22Sn5 in the electrode increases to 30% by weight, the ICE of the electrode is enhanced to 112.3%, suggesting that excessive Li compensation may potentially lead to metallic Li accumulation within the electrode. As illustrated in Fig. 3d, the EVA encapsulation of the alloy membrane contact angle of the Li22Sn5@EVA membrane (the weight ratio of Li22Sn5 content extends to 10% and 20%) with a water droplet preserved the high contact angle values of ∼89°–70° for 15 s. In contrast, the Li22Sn5@EVA composite membrane with 30% Li22Sn5 (insufficient EVA encapsulation layer) exhibited visible gas bubbles upon water droplet contact, attributed to moisture interaction with exposed Li22Sn5 components. Meanwhile, the carbonate electrolyte (LiPF6 in EC/DEC with 5% FEC and 2% VC) readily penetrated the EVA layer, reducing the contact angle to 0° within 15 s, indicating excellent interfacial wettability. In this regard, considering the balance between ICE and environmental adaptability, the weight ratio of Li22Sn5 content was extended to 20% of the frame, and the optimal weight ratio of Li22Sn5 to HEA@CNT was selected as the optimal configuration for subsequent tests. Fig. 3e and f show the differences in dry-air stability between Li22Sn5 NPs and Li22Sn5@EVA NPs, highlighting the environmental adaptability of the EVA encapsulation. According to the XRD analysis in Fig. 3f, Li22Sn5@EVA remained stable after 3 days of exposure to dry air, demonstrating the protective effect of the EVA layer. In contrast, the LiOH phase of bare Li22Sn5 began to appear after one day of exposure to dry air and dominated after three days (Fig. 3e), indicating significant environmental corrosion. The above analysis demonstrates that the EVA layer effectively protects Li22Sn5 from environmental corrosion, addressing a critical challenge in battery manufacturing. Additionally, the retrievable capacities of the HEA@CNT/Li22Sn5@EVA layer exposed to dry air (∼30% RH) with various durations are shown in Fig. S7 and S8.† As the Li22Sn5@EVA is further incorporated into the HEA@CNT scaffold, the composite interfacial layer exhibits an interconnected architecture with a uniform thickness of ∼13.5 μm (Fig. 3g and h). Correspondingly, the cross-sectional image demonstrates the uniformly distributed Fe, Co, Ni, Al, Zn and Sn signals across the composite layer.
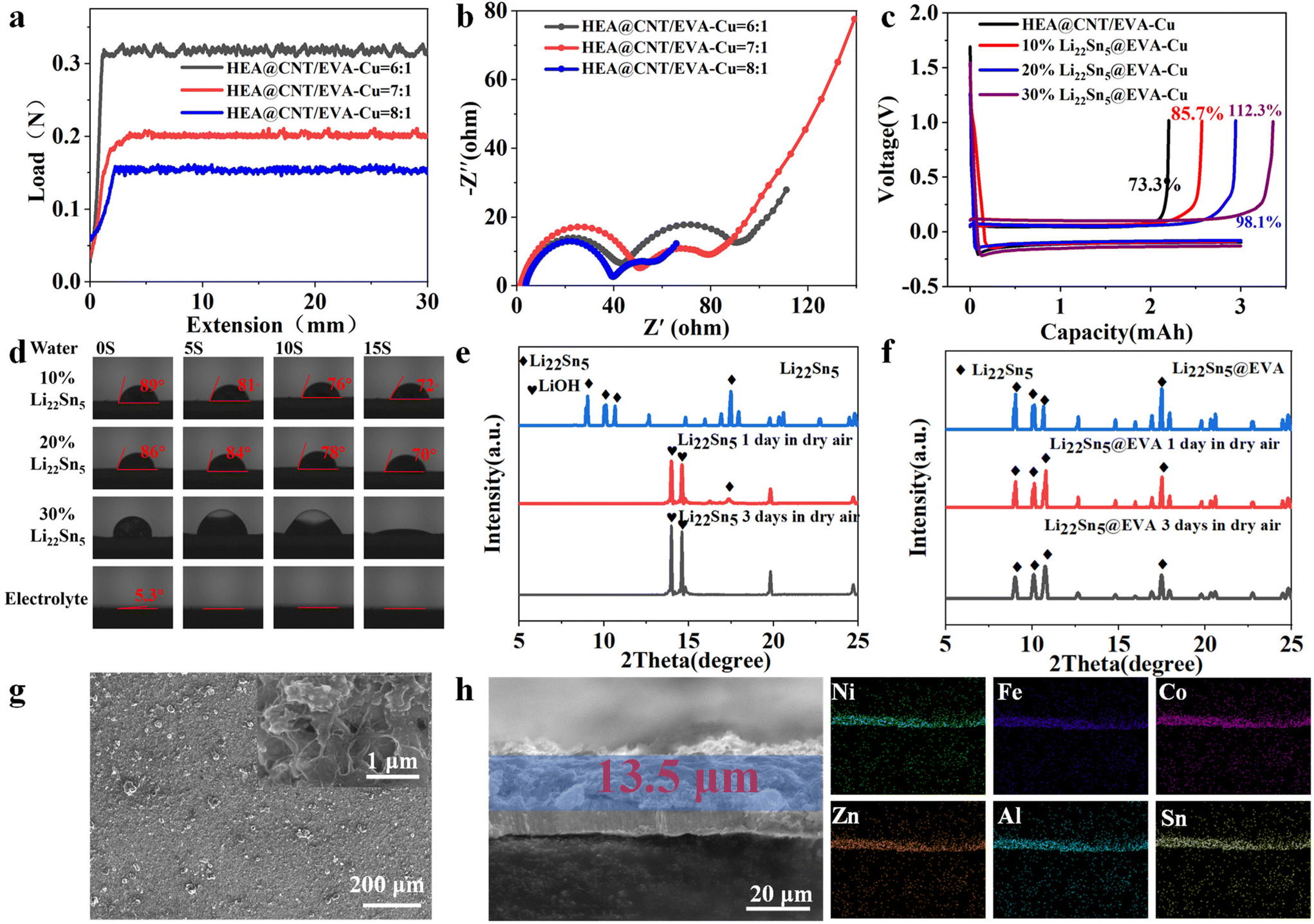 | ||
| Fig. 3 (a) The peel strength of the HEA@CNT/EVA-Cu interface layer varying with the ratio of HEA@CNT to EVA; (b) the Nyquist plot of the HEA@CNT/EVA-Cu interface layer varying with the ratio of HEA@CNT to EVA; (c) voltage profiles of HEA@CNT/EVA-Cu with different amounts of Li22Sn5 scaffold; (d) contact angle evolution of the HEA@CNT/Li22Sn5@EVA-Cu interfacial layer upon the difference in the ratio of Li22Sn5; (e) XRD patterns of Li22Sn5 after exposure to dry air; (f) XRD patterns of Li22Sn5@EVA after exposure to dry air; (g) top-view SEM images of the pristine HEA@CNT/Li22Sn5@EVA-Cu electrode; and (h) cross-sectional SEM image of HEA@CNT/Li22Sn5@EVA-Cu and the corresponding EDS mappings. | ||
To evaluate the synergistic coupling of the HEA@CNT and the Li22Sn5 in the interfacial layer construction for the reversible Li plating process, including the homogeneous nucleation and cation replenishment capability, the half-cell configurations were assembled by casting the HEA@CNT/Li22Sn5@EVA slurry onto the Cu foil as a reference for the Li foil. Fig. 4a and b show the voltage–capacity curves corresponding to HEA@CNT/EVA-Cu and HEA@CNT/Li22Sn5@EVA-Cu half batteries under conditions of 1 mA cm−2 current density and 3 mA h cm−2 deposition, respectively. As shown in Fig. 4a, the HEA@CNT/Li22Sn5@EVA-Cu electrode shows the impressive reversibility of the Li plating/stripping process. The charge-discharge curves at the 50th, 100th and 200th basically coincide, and the capacity change is basically unchanged. In contrast, the capacity of HEA@CNT/EVA-Cu shows sharp fluctuations and a downward trend (Fig. 4b), indicating that Li22Sn5 has made up for the irreversible Li loss. As shown in Fig. 4c, by comparison, the HEA@CNT/Li22Sn5@EVA-Cu electrode shows an average CE value of ∼98.6% for 200 cycles at 1 mA cm−2 and 3 mA h cm−2, which is relatively higher than that of its HEA@CNT/EVA-Cu (∼96.2%) counterpart. Fig. 4d systematically evaluates the interfacial evolution of symmetric cells under high depth-of-discharge (DOD) cycling conditions, a critical parameter for practical applications. Notably, high DOD testing (>50%) better reflects practical application scenarios where full capacity utilization and extended cycle life are simultaneously required. Remarkably, the HEA@CNT/Li22Sn5@EVA-Cu configuration demonstrates exceptional stability even under extreme operation (75% DOD, 6 mA h cm−2), maintaining ultralow voltage polarization (23 mV) over 500 hours without dendrite-induced failure, in stark contrast to the severe voltage fluctuations observed for the bare Cu and HEA@CNT/EVA-Cu substrates. The cycling stability of the symmetric cells was further determined at current densities ranging from 0.5 to 10 mA cm−2 (Fig. 4e). The HEA@CNT/Li22Sn5@EVA-Cu symmetric cell exhibits a critical current density (CCD) value of 10 mA cm−2 with flat voltage plateaus. However, a short-circuit occurred in the symmetric cells of HEA@CNT/CNT-Cu and Cu at 6 mA cm−2 due to the piled-up dendrites and accumulation of dead Li.
 | ||
| Fig. 4 (a) Voltage profiles of HEA@CNT/Li22Sn5@EVA-Cu during the 50th, 100 th and 200th cycles at 1 mA cm−2 with 3 mA h cm−2; (b) voltage profiles of HEA@CNT/EVA during the 50 th, 100 th and 200th cycles at 1 mA cm−2 with 3 mA h cm−2; (c) CE values of the plating/stripping process for various electrodes at 1 mA cm−2 with 3 mA h cm−2; (d) galvanostatic cycling profiles of HEA@CNT/Li22Sn5@EVA-Cu, HEA@CNT/EVA-Cu and Cu foil at 2 mA cm−2 with 6 mA h cm−2. The insets show detailed voltage profiles; (e) rate endurance of the bare Cu, HEA@CNT/EVA-Cu and HEA@CNT/Li22Sn5@EVA-Cu substrates with current densities ranging from 0.5 to 10 mA cm−2. Top-view SEM images of the pristine (f) HEA@CNT/Li22Sn5@EVA-Cu and (j) bare Cu electrode before the Li plating process; top-view SEM images of the (g) HEA@CNT/Li22Sn5@EVA-Cu and (k) bare Cu electrode after the Li plating process with 3 mA h cm−2; top-view SEM images of the (h) HEA@CNT/Li22Sn5@EVA-Cu and (l) bare Cu electrode after the Li plating process with 6 mA h cm−2; top-view SEM images of the (i) HEA@CNT/Li22Sn5@EVA-Cu and (m) bare Cu electrode after the Li stripping process. | ||
To further elucidate the interfacial engineering of HEA@CNT/Li22Sn5@EVA-Cu on the homogenized ion flux, the Li deposition behavior was investigated at the high-capacity deposit loadings (3 mA h cm−2 and 6 mA h cm−2) on the bare Cu and HEA@CNT/Li22Sn5@EVA-Cu electrodes. The pristine HEA@CNT/Li22Sn5@EVA-Cu interfacial layer exhibits an interconnected architecture with a thickness of ∼14.5 μm (Fig. 4f and S9a†). After 3 mA h cm−2 deposition at 1 mA cm−2, the top-view SEM image (Fig. 4g) shows a rather smooth surface without dendrite protrusion from the electrode with a uniform thickness of ∼18.5 μm (Fig. S9b†). In sharp contrast, Li deposits on the bare Cu showed large amounts of visible dendrites (Fig. 4k), which might induce a risk of short circuit of the cell. As the areal capacity accumulated to 6 mA h cm−2, the HEA@CNT/Li22Sn5@EVA-Cu electrode still maintained the smooth surface (Fig. 4h). Meanwhile, the deposits approached ∼26 μm (Fig. S9c†). Notably, the structure of the HEA@CNT/Li22Sn5@EVA-Cu layer could be recovered after the stripping process (Fig. 4i), and the electrode thickness was nearly restored to its original state (Fig. S9d†). In stark contrast, the dendritic Li protruded from the Cu substrate exhibits a drastically increased volume (Fig. 4l); after several complete cycles of the Li plating/stripping process (Fig. 4m), compared with the pristine Cu foil (Fig. 4j), there is a large accumulation of dead Li. During the repeated plating/stripping cycles of the HEA@CNT/Li22Sn5@EVA-Cu electrode, the atomically riveted Zn species of HEA particles as the lithophilic center towards the uniform Li+ influx; while the CNTs were employed as the structural support to reinforce the structural integrity of the interfacial layer. Upon further high-areal-capacity loadings, the HEA@CNT/Li22Sn5@EVA-Cu layer could maintain a uniform distribution of Li ions on the electrode. Upon the reversible Li plating/stripping process, the pristine state of the interfacial layer could be restored without dead Li formation, which largely improved the cation utilization degree as compared to the Cu foil.
To scrutinize the dendrite-free deposition, the Li plating process was monitored in operando through in situ optical microscopy. As demonstrated in Fig. S10a,† the bare Cu gradually developed uneven dendrite growth as the Li deposition was prolonged to 0.5 h. The inhomogeneous deposits were completely stripped with difficulty, leading to compromised reversibility during repeated plating/stripping. Additionally, although HEA@CNT/EVA-Cu exhibits an uneven deposition surface, it is better than that of bare Cu. As the Li deposition extends to 1 h, dendrites gradually form on the HEA@CNT/EVA electrode surface and the electrode surface gradually develops an uneven dendrite growth as the Li deposition is prolonged to 1 h (Fig. S10b†). Encouragingly, the HEA@CNT/Li22Sn5@EVA-Cu exhibited homogenized Li deposition without dendrite protrusions even under high current density and areal capacity loading until 1 h after deposition (Fig. S10c†). The electrochemical impedance spectroscopy (EIS) of the HEMP@RGO-MTL@PH-Cu substrate demonstrated a relatively low and stable resistance after 100 cycles, thereby validating the effectiveness of the lightweight and lithium-compensated interfacial design in enhancing interfacial stability (Fig. S11†).
To further evaluate the compatibility of carbonate-based electrolytes, the full-cell prototype was also assembled by pairing HEA@CNT/Li22Sn5@EVA with the LFP cathode. As shown in Fig. 5a, LFP||Cu exhibits a sharp capacity loss in the carbonate-based electrolyte system due to irreversible Li plating/stripping on the anode side. Similarly, after 100 cycles, the discharge capacity of LFP||HEA@CNT/EVA-Cu decreased from 151.5 mA h g−1 to 15.9 mA h g−1 (Fig. 5b). In sharp contrast, the LFP|| HEA@CNT/Li22Sn5@EVA-Cu cell had a residual capacity of 96.7 mA h g−1 after 100 cycles, which was significantly higher than the LFP||Cu and LFP||HEA@CNT/EVA-Cu full-cells (Fig. 5c). As shown in (Fig. 5d), the LFP||HEA@CNT/Li22Sn5@EVA-Cu full-cell exhibits excellent long-term cycling stability, maintaining a capacity retention rate of 64.8% after 100 cycles. The LFP||Cu full-cell has a capacity retention rate of 60 cycles (a capacity retention rate of 2.8%) or the LFP||HEA@CNT/EVA-Cu full-cell prototype has a capacity retention rate of 100 cycles (a capacity retention rate of 27.9%). In addition, the magnification ratios of LFP||HEA@CNT/Li22Sn5@EVA-Cu at 0.2C, 0.5C, 1C, 3C and 5C were 151.9, 133.1, 105.8, 80.6 and 62.7 mA h g−1, respectively. When the magnification returns to 0.2C, the capacity returns to 138.7 mA h g−1 (Fig. 5e). Note that these rate behaviors are higher than those of the LFP||Cu and LFP||HEA@CNT/EVA-Cu cells at the same current rate. As shown in Fig. 5f, when compared with the LFP||Cu full-cell (0.67 mA h cm−2), LFP||HEA@CNT/Li22Sn5@EVA-Cu showed a capacity retention rate of 64.8% after 100 cycles, while LFP||Cu showed only 22.4%. These results demonstrate that, under the condition of depositing the same amount of metal, the LFP||HEA@CNT/Li22Sn5@EVA-Cu exhibits superior cycling stability.
 | ||
| Fig. 5 (a) Galvanostatic charge/discharge curves of LFP||Cu cell with carbonate-based electrolyte (1.0 M LiPF6 in EC–DEC with 5% FEC and 2% VC); (b) galvanostatic charge/discharge curves of the LFP||HEA@CNT/EVA-Cu cell with a carbonate-based electrolyte; (c) galvanostatic charge/discharge curves of the LFP||HEA@CNT/Li22Sn5@EVA-Cu cell with a carbonate-based electrolyte; (d) cycling stability and CE values of the LFP||Cu, LFP||HEA@CNT/EVA-Cu and LFP|| HEA@CNT/Li22Sn5@EVA-Cu cells at 0.5C; (e) rate performance of the LFP||Cu, LFP||HEA@CNT/EVA-Cu and LFP||HEA@CNT/Li22Sn5@EVA-Cu cells at different rates; and (f) cycling stability values of LFP||HEA@CNT/Li22Sn5@EVA-Cu and LFP||Cu (0.67 mA h cm−2). | ||
To further evaluate the compatibility of the ether-based localized high-concentration electrolyte (LiFSI-1.2DME-3TTE), 145 mA h pouch cell prototypes were also assembled by pairing the HEA@CNT/Li22Sn5@EVA-Cu with the LFP cathode (3.0 mA h cm−2) (Fig. 6a). The detailed specifications of the pouch cells are itemized in Fig. 6b. As shown in Fig. 6c, the LFP||Cu pouch cell exhibits a sharp capacity fading, which could be attributed to irreversible Li plating/stripping on the Cu substrate. Similarly, the discharge capacity of LFP||HEA@CNT/EVA-Cu reduces from 142.9 mA h to 82.1 mA h after 100 cycles (Fig. 6d). In sharp contrast, the LFP||HEA@CNT/Li22Sn5@EVA-Cu model maintained a much higher retrievable capacity of 117.8 mA h after 100 cycles (Fig. 6e). As shown in Fig. 6f, the LFP||HEA@CNT/Li22Sn5@EVA-Cu pouch cell exhibits excellent long-term cycling stability and maintains 95.6% capacity retention after 100 cycles, as compared to the interior capacity retentions of LFP||Cu for 100 cycles (9.3% capacity retention) or the LFP||HEA@CNT/EVA-Cu pouch cell prototype for 100 cycles (capacity retention of 57.7%). Additionally, superior rate capabilities of the LFP||HEA@CNT/Li22Sn5@EVA-Cu are also exhibited, with values of 147.5, 128.3, 110.1, 74.1, and 54.7 mA h obtained at 0.2C, 0.5C, 1C, 3C, and 5C, respectively. A capacity of 136.1 mA h is recovered as the rate returns to 0.2C (Fig. 6g and S12†). Note that these rate behaviors are higher than those of the LFP||HEA@CNT/EVA-Cu and LFP||Cu pouch cells at the same current rates. Fig. 6h demonstrates the Ragone plot of the as-assembled prototype in contrast to other previously reported anode-free battery configurations (based on the total mass of the device level). The LFP||HEA@CNT/Li22Sn5@EVA-Cu pouch cell exhibits a maximum energy density of 325.2 W h kg−1 at 0.2 C, enabling a maximum power output of 603.5 W kg−1 (Tables S1 and S2†).
 | ||
| Fig. 6 (a) The optical image of the 145 mA h anode-less LMB pouch-format configuration. (b) Technological specifications of the 145 mA h pouch cell model. (c) Galvanostatic charge/discharge curves of the LFP||Cu pouch cell with a localized high-concentration electrolyte (LiFSI-1.2DME-3TTE). (d) Galvanostatic charge/discharge curves of the LFP||HEA@CNT/EVA-Cu pouch cell with a localized high-concentration electrolyte. (e) Galvanostatic charge/discharge curves of the LFP||HEA@CNT/Li22Sn5@EVA-Cu pouch cell with a localized high-concentration electrolyte. (f) Cycling stability and CE values of the LFP||Cu, LFP||HEA@CNT/EVA-Cu and LFP||HEA@CNT/Li22Sn5@EVA-Cu models at 0.5C with a localized high-concentration electrolyte. (g) Rate performance of the LFP||Cu, LFP||HEA@CNT/EVA-Cu and LFP||HEA@CNT/Li22Sn5@EVA-Cu models at different rates. (h) Comparison of the energy densities at various power outputs in this work and the performance metrics from the previous studies. | ||
Conclusions
In conclusion, we developed a lightweight (0.39 mg cm−2) and moisture-tolerant interfacial layer for Cu foil to regulate high-throughput metallic deposition across multiple scales, from the Li+ desolvation process to the formation of metallic embryos and the propagation of dendrites. The thermally induced Li22Sn5 alloy, blended with a hydrophobic EVA, acts as a moisture-proof cation reservoir within the scaffold pores, while the in situ grown CNTs, threaded from the interior HEA NPs, reinforce the structural integrity of the framework. This HEA@CNT/Li22Sn5@EVA-Cu design significantly minimizes the risk of dendrite-induced short circuits, enabling the symmetric cell to operate reversibly at 2 mA cm−2 under 75% DOD. When integrated with an LFP cathode in a 145 mA h pouch-format cell, the prototype demonstrates exceptional cycling durability (95.6% capacity retention over 100 cycles) and achieves an energy density of up to 325.2 W h kg−1, at a power output of 603.5 W kg−1. This interfacial modification approach paves the way for anode-less LMB configurations with a maximized Li utilization degree. Future work will focus on extending cycle life with electrolyte formulations and exploring scalable manufacturing processes.Experimental section
Materials
Toluene (99%), poly(vinylidene fluoride-co-hexafluoro propylene) (PVDF), alcohol (99.9%) and N-methyl pyrrolidone (NMP, 99.9%) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Conductive carbon black was purchased from Timcal Ltd. The 1,3-dioxolane (DOL), diethyl carbonate, liquid electrolyte, separator (PP 2500), metallic Li foil and electrode components were supplied by Guangdong Canrd. New Energy Technology Co., Ltd; hard carbon powder was supplied by BTR New Energy Materials Inc.; Sn particles were supplied by Zhengzhou Bak Battery Co., Ltd; and the LiFePO4 (LFP) particles were supplied by BattFlex Technologies Inc. (CATL, China).Preparation of the HEA/CNT nanocomposite
In a typical synthesis, first, all metal nitrate precursors were weighed in equiatomic ratios and dissolved in deionized water. The obtained solution was stirred continuously with a magnetic stirrer for 2 h for homogeneous mixing, and then citric acid was added as the chelating agent in the molar ratio of fuel to oxidant as 0.8:1. Stirring was continued for 2 hours, and ammonia water (NH4OH) was added dropwise to adjust the pH to 7. Second, sols were annealed at 90 °C for 48 h to get the dried gel, and the dried gel was activated in the furnace at 300 °C under an air atmosphere. Finally, the auto-combustion product was mechanically ground and evenly spread on the quartz boat loaded into the tube furnace for the CVD process. In a typical CVD procedure, the obtained precursor was heat-treated in a tube furnace at 5 °C min−1 up to 700 °C in an Ar/H2 mixture (5% H2), and then the reaction was carried out at 700 °C for 30 min using a mixture of acetylene and Ar/H2 at flow rates of 1:9; the resulting product was named FeCoNiAlZn@CNT.
Material characterization
The crystal structure and phase of the materials were analyzed using an X-ray diffractometer (XRD, Bruker D8) with Cu Kα radiation (λ = 1.54056 Å), operating at 40 kV and 30 mA. The ex situ XRD patterns were performed at a step scan of 1.05° and a step time of 25 s. Field emission scanning electron microscopy (FESEM, FEI Nova 450) and transmission electron microscopy (TEM, FEI Talos F200X) with an energy dispersive spectrometer (EDS) were used to characterize the morphology and microstructure of the materials. Raman spectral (HORIBA, France) measurements were carried out to analyze HEMP materials with a 532 nm laser source. Electrochemical impedance spectroscopy was applied in the frequency range of 10 mHz to 100 kHz with a perturbation voltage of 10 mV. X-ray photoelectron spectroscopy (XPS) analysis was applied on a customized XPS system based on the Kratos Axis Supra. The Micromeritics ASAP 2460 system was utilized to measure the specific surface area and porosity of the HEMP materials. The optical contact angle (SL200KB, Kino, USA) was employed to measure the contact angle. The dendrite growth process was observed using in situ optical microscopy (YUESCOPE YM710R).Electrochemical measurements
CR2016 type coin cells were employed in an argon-filled glovebox where both the moisture and oxygen contents were less than 0.1 ppm. The half cells were assembled with Li foil as the counter electrode. The Celgard 2400 membrane was used as the separator. The electrolyte was a mixture of 1.0 M LiTFSI and 0.1 M LiNO3 in 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) with a volume ratio of 1:1 (ether electrolyte system). The duration of lithium plating was fixed at a current density of 1 mA cm−2 with a cycling capacity of 3 mA h cm−2 and then the cut-off potential was controlled as 1.0 V on a Neware CT-4008 battery tester during the stripping process. A symmetric cell was constructed by pairing the prelithiated HEA@CNT/EVA or HEA@CNT/Li22Sn5@EVA electrode (3 mA h cm−2), and the galvanostatic cycling consisting of plating/stripping at 1 mA cm−2 was monitored. For the measurement of pouch cells, the LFP cathode was prepared by mixing LFP, super P and PVDF in NMP onto the Al foil, and then was dried in a vacuum oven at 80 °C for 12 h. Pouch cells were measured in the potential range from 2.5 to 3.8 V at 0.1 C for the first three cycles and a rate of C/2 was applied for subsequent cycles. The carbonate-based electrolyte was a mixture of LiPF6 (1 M) in 1:1 w/w EC/DEC with 5% FEC and 2% VC. The localized high-concentration electrolyte was a mixture of LiFSI-1.2DME-3TTE.
Author contributions
L.C., JC.L. and Y.M. conceived the idea for the project. HL.W. and AH.S. prepared materials. L.C. and JC.L. prepared electrodes and conducted structural characterization studies. YX.G. and ZQ.W. conducted electrochemical measurements. YS.L. performed the operando XRD characterization. JW.T., CW.L., L.C. and Y.M. wrote the manuscript. All authors discussed the results.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts.Acknowledgements
The authors acknowledge the financial support of the National Natural Science Foundation of China (52173229 and 52373229) and the Natural Science Foundation of Shaanxi (2023-JC-JQ-15). Furthermore, the authors also would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for providing several testing instruments.References
- J.-F. Ding, Y.-T. Zhang, R. Xu, R. Zhang, Y. Xiao, S. Zhang, C.-X. Bi, C. Tang, R. Xiang, H. S. Park, Q. Zhang and J.-Q. Huang, Green Energy Environ., 2023, 8, 1509–1530 Search PubMed.
- A. Hu, W. Chen, X. Du, Y. Hu, T. Lei, H. Wang, L. Xue, Y. Li, H. Sun, Y. Yan, J. Long, C. Shu, J. Zhu, B. Li, X. Wang and J. Xiong, Energy Environ. Sci., 2021, 14, 4115–4124 Search PubMed.
- J. Lu, R. Xiong, J. Tian, C. Wang and F. Sun, Nat. Commun., 2023, 14, 2760 Search PubMed.
- V. Sharma, K. Singh and K. Narayanan, Energy Adv., 2024, 3, 1222–1237 RSC.
- Z. Yu, Q. Yang, W. Xue, J. Shen, J. Zhang, S. Zhu, S. Li and Y. Li, Nanoscale, 2023, 15, 4529–4535 RSC.
- S. Zhou, W. Chen, J. Shi, G. Li, F. Pei, S. Liu, W. Ye, L. Xiao, M.-S. Wang, D. Wang, Y. Qiao, L. Huang, G.-L. Xu, H.-G. Liao, J.-F. Chen, K. Amine and S.-G. Sun, Energy Environ. Sci., 2022, 15, 196–205 RSC.
- B. Wu, C. Chen, L. H. J. Raijmakers, J. Liu, D. L. Danilov, R.-A. Eichel and P. H. L. Notten, Energy Storage Mater., 2023, 57, 508–539 CrossRef.
- X. Li, R. Zhu, H. Jiang, Y. Yu, W. Wan, X. Li, C. Wang and Y. Huang, J. Mater. Chem. A, 2022, 10, 11246–11253 RSC.
- Y. Lu, Z. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 Search PubMed.
- Y. Ji, L. Dong, J. Liu, H. Xie, S. Zhong, C. Yang, J. Han and W. He, Energy Environ. Sci., 2024, 17, 4078–4089 Search PubMed.
- K. Jang, H. J. Song, J. B. Park, S. W. Jung and D.-W. Kim, Energy Environ. Sci., 2024, 17, 4622–4633 Search PubMed.
- Q. Cai, X. Qin, K. Lin, Z. Yang, X. Hu, T. Li, F. Kang and B. Li, Nano Lett., 2021, 21, 10252–10259 CrossRef CAS PubMed.
- T. Zhang, H. Lu, J. Yang, Z. Xu, J. Wang, S.-i. Hirano, Y. Guo and C. Liang, ACS Nano, 2020, 14, 5618–5627 Search PubMed.
- J. Gao, C. Chen, Q. Dong, J. Dai, Y. Yao, T. Li, A. Rundlett, R. Wang, C. Wang and L. Hu, Adv. Mater., 2021, 33, 2005305 Search PubMed.
- J. Zhou, B. Hao, M. Peng, L. Zhang, H. Ji, J. Liu, W. Ling, C. Yan and T. Qian, Adv. Energy Mater., 2023, 13, 2204174 Search PubMed.
- R. Zhang, S. Wen, N. Wang, K. Qin, E. Liu, C. Shi and N. Zhao, Adv. Energy Mater., 2018, 8, 1800914 Search PubMed.
- Y. Ouyang, C. Cui, Y. Guo, Y. Wei, T. Zhai and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 25818–25825 CrossRef CAS PubMed.
- X. Liu, T. Zhang, X. Shi, Y. Ma, D. Song, H. Zhang, X. Liu, Y. Wang and L. Zhang, Adv. Sci., 2022, 9, 2104531 Search PubMed.
- M. Liu, Y. Ying, J. Liu, C. Li, R. Hu, J. Liu, H. Huang, A. Zhang, L. Ma, L. Ouyang and M. Zhu, Adv. Energy Mater., 2024, 14, 2403696 Search PubMed.
- H. Wang, Y. Liu, Y. Li and Y. Cui, Electrochem. Energy Rev., 2019, 2, 509–517 CrossRef CAS.
- Q. Wang, B. Liu, Y. Shen, J. Wu, Z. Zhao, C. Zhong and W. Hu, Adv. Sci., 2021, 8, 2101111 Search PubMed.
- X. Li, J. H. Wang, L. Yang, T. Y. Liu, S. Huang, B. Ho, H. Hsueh, J. Chen, L. He, Z. Guo, M. Liu and W. Li, Adv. Mater., 2024, 36, 2409278 CrossRef CAS PubMed.
- N. A. Sahalie, A. A. Assegie, W.-N. Su, Z. T. Wondimkun, B. A. Jote, B. Thirumalraj, C.-J. Huang, Y.-W. Yang and B.-J. Hwang, J. Power Sources, 2019, 437, 226912 Search PubMed.
- Y. Zhao, J. Yan, J. Yu and B. Ding, ACS Nano, 2022, 16, 17891–17910 Search PubMed.
- Y. Sun, J. Li, S. Xu, H. Zhou and S. Guo, Adv. Mater., 2023, 36, 2311687 Search PubMed.
- W. Jia, J. Zhang, L. Zheng, H. Zhou, W. Zou and L. Wang, eScience, 2024, 4, 100266 Search PubMed.
- H. Kang, H. Kang, M. Lyu and E. Cho, EcoMat, 2024, 6, e12498 Search PubMed.
- B. Jagger and M. Pasta, Joule, 2023, 7, 2228–2244 CrossRef CAS.
- P. Du, C. Yuan, X. Cui, K. Zhang, Y. Yu, X. Ren, X. Zhan and S. Gao, J. Mater. Chem. A, 2022, 10, 8424–8431 Search PubMed.
- X. Gao, P. Du, B. Cheng, X. Ren, X. Zhan and L. Zhu, ACS Appl. Mater. Interfaces, 2024, 16, 7327–7337 Search PubMed.
- J. Sun, S. Zhang, J. Li, B. Xie, J. Ma, S. Dong and G. Cui, Adv. Mater., 2023, 35, 2209404 Search PubMed.
- Y. Liu, J. Sun, X. Hu, Y. Li, H. Du, K. Wang, Z. Du, X. Gong, W. Ai and W. Huang, Nano Energy, 2022, 94, 106883 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- P. Zou, C. Wang, J. Qin, R. Zhang and H. L. Xin, Energy Storage Mater., 2023, 58, 176–183 CrossRef.
- K. Lee and J. Sakamoto, Faraday Discuss., 2024, 248, 250–265 RSC.
- S. Chen, Z. Wang, M. Zhang, X. Shi, L. Wang, W. An, Z. Li, F. Pan and L. Yang, Carbon Energy, 2023, 5, e323 CrossRef CAS.
- P. Bärmann, M. Mohrhardt, J. E. Frerichs, M. Helling, A. Kolesnikov, S. Klabunde, S. Nowak, M. R. Hansen, M. Winter and T. Placke, Adv. Energy Mater., 2021, 11, 2100925 CrossRef.
- B. Huang, T. Huang, L. Wan and A. Yu, ACS Sustainable Chem. Eng., 2021, 9, 648–657 CrossRef CAS.
- C. Wang, Y. Han, S. Li, T. Chen, J. Yu and Z. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 12750–12758 CrossRef CAS PubMed.
- S. Li, C. Wang, J. Yu, Y. Han and Z. Lu, Energy Storage Mater., 2019, 20, 7–13 Search PubMed.
- H. Zhang, J. Cheng, H. Liu, D. Li, Z. Zeng, Y. Li, F. Ji, Y. Guo, Y. Wei, S. Zhang, T. Bai, X. Xu, R. Peng, J. Lu and L. Ci, Adv. Energy Mater., 2023, 13, 2300466 CrossRef CAS.
- J. Choi, H. Jeong, J. Jang, A. R. Jeon, I. Kang, M. Kwon, J. Hong and M. Lee, J. Am. Chem. Soc., 2021, 143, 9169–9176 CrossRef CAS PubMed.
- S. Chen, C. Niu, H. Lee, Q. Li, L. Yu, W. Xu, J.-G. Zhang, E. J. Dufek, M. S. Whittingham, S. Meng, J. Xiao and J. Liu, Joule, 2019, 3, 1094–1105 CrossRef CAS.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00042d |
| This journal is © The Royal Society of Chemistry 2025 |