
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective glycerol to lactic acid conversion via a tandem effect between platinum and metal oxides with abundant acid groups†
Hui
Luo‡
 *ab,
Mianle
Xu‡
c,
Sihang
Liu
c,
Giulia
Tarantino
a,
Hanzhi
Ye
a,
Hossein
Yadegari
d,
Alain Y.
Li
a,
Ceri
Hammond
a,
Georg
Kastlunger
*c,
Ifan E. L.
Stephens
*d and
Maria-Magdalena
Titirici
*a
*ab,
Mianle
Xu‡
c,
Sihang
Liu
c,
Giulia
Tarantino
a,
Hanzhi
Ye
a,
Hossein
Yadegari
d,
Alain Y.
Li
a,
Ceri
Hammond
a,
Georg
Kastlunger
*c,
Ifan E. L.
Stephens
*d and
Maria-Magdalena
Titirici
*a
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: hui_luo@surrey.ac.uk; m.titirici@imperial.ac.uk
bSchool of Mechanical Engineering Sciences, University of Surrey, Stag Hill Campus, Guildford, GU2 7XH, UK
cCatalysis Theory Center, Department of Physics, Technical University of Denmark (DTU), 2800 Kgs, Lyngby, Denmark. E-mail: geokast@dtu.dk
dDepartment of Materials, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: i.stephens@imperial.ac.uk
First published on 4th November 2024
Abstract
Phasing out petrochemical-based thermoplastics with bio-plastics produced in an energy efficient and environmentally friendly way is of paramount interest. Among them, polylactic acid (PLA) is the flagship with its production accounting for 19% of the entire bioplastics industry. Glycerol electrolysis for producing the monomer lactic acid, while co-generating green H2, represents a promising approach to boost the production of PLA, yet the reaction selectivity has been a bottleneck. Here, we report a combined electrochemical and chemical route using a tandem Pt/C-γ-Al2O3 multicomponent catalyst which can achieve a glycerol-to-lactic acid selectivity of 61.3 ± 1.2%, among the highest performance reported so far. Combining an experimental and computational mechanistic analysis, we suggest that tuning the acidic sites on the catalyst surface is crucial for shifting the reaction towards the dehydration pathway, occurring via dihydroxyacetone intermediate. Within the tandem effect, Pt is the active site to electrochemically catalyze glycerol to dihydroxyacetone and glyceraldehyde, while the γ-Al2O3 provides the required acidic sites for catalyzing dihydroxyacetone to the pyruvaldehyde intermediate, which will then go through Cannizzaro rearrangement, catalyzed by the OH− ions to form lactic acid. This catalytic synergy improves the selectivity towards lactic acid by nearly two-fold. A selectivity descriptor (ΔGGLAD* − ΔGDHA*) from density functional theory calculations was identified, which could be used to screen other materials in further research. Our findings highlight the promise of tandem electrolysis in the development of strategies for selective electrochemical production of high-value commodity chemicals from low value (waste) precursors.
Broader contextUsing renewable energy to produce green hydrogen and chemicals is key for decarbonising the power generation, transport, and chemical industries. Proton exchange membrane water electrolysis is the state-of-the-art technology to generate green hydrogen. However, it has several drawbacks including (1) large thermodynamic potential and sluggish anode kinetics; (2) the use of scarce and expensive iridium as a catalyst; (3) the low economic value of oxygen produced at the anode. Replacing the water oxidation reaction with biomass substrates such as glycerol eliminates the bottlenecks associated with the water oxidation reaction, while enabling green hydrogen production at the cathode at a fraction of the cost. The key to achieve an overall beneficial economic and environmental impact is to selectively produce high-value product on the anode. Herein, we have designed platinum–metal oxide tandem catalyst that can convert glycerol to lactic acid (the precursor for flagship biopolymer poly-lactic acid) at 61.3 ± 1.2% selectivity, while co-produce green hydrogen with an electricity consumption per unit of H2 < 26 kW h kg−1 H2. Our findings highlight the promise of tandem electrolysis in the development of strategies for selective electrochemical production of high-value commodity chemicals from low value (waste) precursors. |
Introduction
The chemical sector consumes vast amounts of oil and gas, accounting for 15% of total primary demand for oil on a volumetric basis and 9% of gas. The use of fossil-fuels as feedstock and energy source, also dictates its unsustainability with direct CO2 emission of 880 Mt in 2018.1 Among all the products in the chemical sector, petrochemical based thermoplastics have an annual production of 222 Mt year−1 in total.2 Globally, these plastics are becoming less attractive to consumers, brands and governments. This shift has made bio-based and bio-degradable plastics a viable, responsible and intelligent alternative to facilitate the transformation towards a zero-emission chemical industry.3Polylactic acid (PLA) is a bio-based polyester and the most utilized degradable bioplastic4 with its production accounting for 18.9% of the entire bioplastics industry.5 Its monomer, lactic acid, is currently prepared via the enzymatic fermentation of sugars, under strict temperature (<313 K) and pH (5–7) conditions, followed by purification through subsequent esterification, distillation and hydrolysis (Fig. 1a).6,7 Although this approach is used by 90% of the lactic acid manufacturers, its low productivity (1–13 g L−1 h−1) and high production cost dictate the low annual growth rate for PLA. Perez-Ramirez and coworkers demonstrated an alternative approach where glycerol is first oxidised to dihydroxyacetone (DHA) and, in a separate batch, the DHA convert into lactic acid at 413 K and 25 bar.6
 | ||
| Fig. 1 (a) Illustrations of three different technique routes to produce lactic acid; (b) Benchmark of literature reported values of electrochemical glycerol conversion to lactic acid. The liquid product distribution towards lactic acid is defined as: mole of lactic acid/mole of total liquid products detected ×100%. Other detectable liquid products include: glyceric acid, tartronic acid, glycolic acid, oxalic acid. (Source of references: Co-DPPE;8 Pt3Au7@Ag;9 Pt-CBAC;10 Pt-CC;11 NixBi1−x;12 Planar Au;13 Au NWs;14,15 Au/Ni(OH)2;14,15 AuPt (15% PtSurf);16 Au/CeO2−x;17 Pt/C-Zeolite;18 hp-PtAu/NF.19 Details in reaction conditions are summarized in Table S1, ESI.† To note the use of liquid product distribution towards lactic acid is for literature comparison. In the discussion below, more standard quantification methods with product yield and faradaic efficiency are used throughout this work. | ||
Glycerol electrolysis to lactic acid could potentially offer a more attractive, continuous route that avoids costs associated with the processing and purification of intermediates (Fig. 1a). Further, being an anodic reaction, this process can be coupled with water reduction, for simultaneous hydrogen production, or other reduction reactions, e.g. CO2 reduction, biomass-derived reduction reactions etc.11,20,21 A life-cycle assessment study demonstrated that electrocatalytic conversion of crude glycerol to lactic acid at 32% liquid product distribution and 100 kg h−1 production rate can result in a 57% reduction in global warming potential compared to the bio- and chemocatalytic processes, when combined with a low-carbon-intensity grid.22 Further emission reduction is possible when the product selectivity towards lactic acid is improved, pointing out the direction for further development. Reports of lactic acid electrosynthesis are limited, as listed in Fig. 1b.8 In most of the cases, the glycerol electrolysis has been performed in a static half-cell configuration rather than a continuous membrane-electrode-assembly (MEA) electrolysis system. However, to maximize the benefits of coupling anode and cathode reactions, the set up must be compatible within a single system with easy product separation and high mass transport, in which case MEA electrolysis cell operating in galvanostatic mode holds greater promise in transferring the research into up-scaling devices than conventional H-cells.23 Besides, to be industrial relevant, the process current density needs to be relatively high (i.e. >200 mA cm−2),24–26 and liquid product distribution towards lactic acid needs to exceed ∼60% to be compatible with the conventional fermentation method.27,28 Reports falling into this area are rare. To date, only Yan et al. reported 80% lactic acid liquid product distribution in their recent works with a two compartment flow cell, yet the method for calculating the glycerol conversion rate and lactic acid selectivity need to be further validated to confirm the high selectivity.14,15 Shi and co-workers also achieved 81% lactic acid liquid product distribution, albiet using a static half cell set up.17 Herein, based on the previous studies, we have chosen to perform our glycerol electrolysis towards lactic acid and H2 co-production in a MEA cell, with a reaction rate of 20 mA cm−2. Although the current density is a magnitude lower than practical industrial scale, the focus here is to achieve high lactic acid yield by designing the electrocatalyst structure and understanding the reaction mechanism. Engineering challenges towards scaling up will later be tackled with strategies such as constructing 3D electrode structure,29 selecting suitable anion-exchange membrane,30,31 investigating in different MEA configurations to minimise electrolyte crossover,32 and engineering the interface microenvironment,33–35 taking inspiration from the electrochemical CO2 reduction community.
Previous studies have shown that the glycerol to lactic acid transformation is a combination of electrochemical deprotonation – heterogeneous dehydration – homogeneous solution phase reaction process, with several intermediate steps involved, as illustrated in red (top) in Fig. 2.16,36,37 Pt-based electrocatalysts are the most active in catalyzing the glycerol deprotonation step towards dihydroxyacetone and glyceraldehyde.38–41 Density functional theory (DFT) calculations have already established the reaction pathways and the factors controlling activity and product selectivity within glycerol (electro-) oxidation on Pt(111) and other metal surfaces.42,43 Subsequently, in a non-electrochemical solution phase step, it is possible to dehydrate DHA to lactic acid.44,45 However, at oxidising potentials on Pt, the dehydration pathway towards lactic acid is less favoured than the competing electrochemical oxidation process towards glyceric acid (shown in blue (bottom) in Fig. 2).16,36,46 Therefore, it is essential to develop an effective strategy to promote the dehydration pathway suppressing the electrochemical oxidation pathway within electrochemical glycerol oxidation.
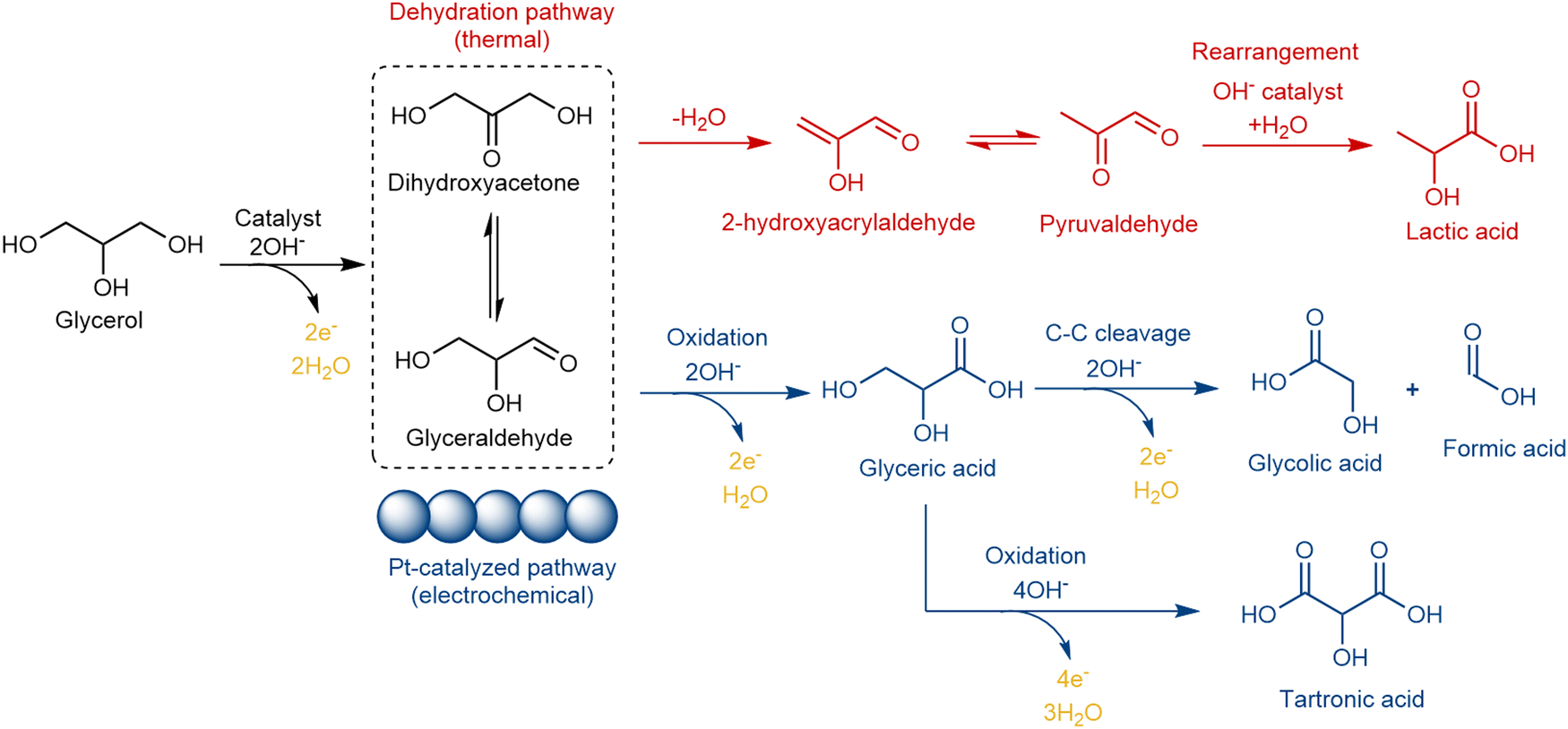 | ||
| Fig. 2 Glycerol dual reaction pathway: dehydration vs. oxidation. Pt based electrocatalysts can effectively catalyze the glycerol → DHA (dihydroxyacetone)/GLAD (glyceraldehyde) step as well as the following oxidation steps.16,36,37 | ||
In thermal (i.e. non electrochemical) heterogeneous catalysis, acidic sites from certain metal oxides, such as TiO2,47 ZrO2,48 Al2O349 and zeolites50–52 can mediate the chemical transformation of DHA to pyruvaldehyde, by coordinating the carbonyl and hydroxyl groups, thus accelerating the keto–enol tautomerization and dehydration of the component.5 When combined with metal catalysts, some metal oxides such as TiO2 can also directly convert glycerol into lactic acid, with the metal providing the dehydrogenation sites to produce DHA, and the transformation to lactic acid taking place on TiO2 surface.53
Taking inspiration from the abovementioned strategies form heterogeneous catalysis, in this paper, we aim to combine electrochemical experiments and DFT simulations to discover tandem electrocatalysts consisting of Pt and metal oxides for improving lactic acid product yield. We first examined the lactic acid product yield % on Pt/C catalyst standalone and employ DFT calculations to identify the reaction limitations. Subsequently, we screened a series of metal oxide materials with different acidic site densities, and designed a multicomponent tandem catalyst system containing Pt sites and metal oxides, to identify the experimental and theoretical descriptor for improving the product yield towards lactic acid leveraging further product optimization attempts.
Results and discussion
Pure Pt catalysts lead to low lactic acid selectivity for glycerol electrooxidation
The glycerol electrolysis was done with a commercial Pt on carbon nanostructured catalyst (Pt/C) mixed with SuperP carbon black (morphology and crystalline structures shown in Fig. S1–S3, ESI†) using a MEA cell as described in the Methods section. We show in Fig. 3 that the Pt/C catalyst can generate lactic acid with a product yield of 32.2 ± 7% and a faradaic efficiency of 21.2 ± 1.3% at a very low cell voltage of ca. 0.6 V. Glyceric acid was detected as the major product (product yield of 38 ± 8% and faradaic efficiency near 50%) and other compounds such as tartronic acid and glycolic acid were detected as minor products, likely formed via further oxidation of glyceric acid, as shown in Fig. 2 and 3. These results indicate that the standalone Pt/C catalyst cannot selectively catalyse the glycerol to lactic acid transformation with high efficiency, due to its high activity towards the competing electrochemical oxidation process. | ||
Fig. 3 Electrolysis data showing the cell voltage change during 1 h experiment under 20 mA cm−2 applied current density. Inserts are the quantified products, defined as  , ,   (top), and (top), and  (bottom), where F is the faradaic constant (96 (bottom), where F is the faradaic constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) and n is the electron transfer number. The electron transfer number used for each compound are as follows: tartronic acid (8); glyceric acid (4); glycolic acid (10/3); lactic acid (2). Values are averaged from three independent measurements. The acronyms in this figure, Fig. 4 and 6 are: Gly: glycerol, GLAD: glyceraldehyde, GA: glyceric acid, TA: tartronic acid, GCA: glycolic acid, LA: lactic acid, FA: formic acid, DHA: dihydroxyacetone, 2-HAC: 2-hydroxyacrylaldehyde, PAH: pyruvaldehyde. 485 C mol−1) and n is the electron transfer number. The electron transfer number used for each compound are as follows: tartronic acid (8); glyceric acid (4); glycolic acid (10/3); lactic acid (2). Values are averaged from three independent measurements. The acronyms in this figure, Fig. 4 and 6 are: Gly: glycerol, GLAD: glyceraldehyde, GA: glyceric acid, TA: tartronic acid, GCA: glycolic acid, LA: lactic acid, FA: formic acid, DHA: dihydroxyacetone, 2-HAC: 2-hydroxyacrylaldehyde, PAH: pyruvaldehyde. | ||
To rationalize the experimental observations on Pt above, we applied DFT to study the competition between the mixed electrochemical & chemical reaction path toward lactic acid and purely electrochemical (EC) pathways to the other products. We have simulated the Pt nanoparticle surfaces on the most stable (111) facet.54 As shown in the free energy diagram in Fig. 4, we find that on a Pt(111) surface at 0.5 V vs. RHE, the most endergonic step of lactic acid production (shown in red in Fig. 4) is the chemical transformation of surface bound 2-hydroxyacrylaldehyde to pyruvaldehyde. On the contrary, the competing electrochemical pathway is virtually exergonic throughout (shown in blue in Fig. 4). The potential response of the latter pathway leads to a takeover of the product via the electrochemical path (shown in blue in Fig. 2 and 4) at increasing overpotentials. However, close to 0 VRHE the limiting potential for both pathways is determined by glycerol electro-oxidation to dihydroxyacetone (*DHA) or glyceraldehyde (*GLAD), respectively, with the formation of *DHA being preferred over *GLAD. The free energy diagrams at different potentials are shown in Fig. S4 (ESI†). Besides, considering the pure electrochemical pathways, some elementary steps (outlined in in Fig. S5, ESI†) exhibit high limiting potentials (up to 0.85 VRHE), as shown in the free energy diagram in Fig. S6a (ESI†), which inhibit the further production of tartronic acid and C–C splitting products at r at the reaction conditions applied in this study. However, the electrochemical reaction proceeds smoothly at 0.5 V vs. RHE, as shown in Fig. S6b (ESI†). Considering the reaction path toward lactic acid (red line in Fig. 4) at 0.5 V vs. RHE, DHA as proton–electron transfer product is quite feasible and the following steps toward lactic acid are non-electrochemical which is potential independent. Therefore, the low selectivity towards lactic acid on pure Pt can be attributed to a combination of limiting electrochemical activity towards DHA and GLAD and the predominance of the electrochemical process towards glyceric acid, tartronic acid and C–C splitting products at elevated overpotentials. To increase lactic acid selectivity, engineering a catalytic system that promotes the dehydration route at moderate electrode potentials is thus required.
 | ||
| Fig. 4 DFT-calculated free energy diagrams of the glycerol oxidation pathways outlined in Fig. 2. on Pt(111) at 0.5 V vs. RHE. The blue line is the purely electrochemical (EC) path and the red line is the Mixed electrochemical and chemical (EC&C) path. The related structures of different pathways are shown at the top and bottom of figure that the main product of purely EC is outlined in blue and the lactic acid is outlined in red. (l): liquid phase, *: adsorption at surface. More details on DFT results are in ESI:† computational details. Color codes for atoms: grey – platinum, white – hydrogen, red – oxygen, dark grey – carbon. | ||
Acidic sites on metal oxides steers lactic acid selectivity
The identified limitations of Pt motivated us to explore new catalyst compositions favouring the non-electrochemical pathway, to improve lactic acid product yield. As mentioned above, previous studies have shown that metal oxides with surface acidic sites can facilitate the DHA to lactic acid transformation, where it is even possible to directly convert glycerol into lactic acid when combined with metal catalysts that drive the first deprotonation step.53 Therefore, we screened several metal oxide materials with different acidic site densities, and designed a multicomponent tandem catalyst system containing Pt sites and metal oxides with abundant acidic sites, in order to combine the electrocatalytic capabilities of Pt to produce DHA from glycerol with the selectivity enhancement of the acid groups on the oxide towards lactic acid.A series of metal oxides with different surface density of acidic sites were tested.55 We fabricated the Pt/C-MOx (MOx (metal oxide) = CeO2, TiO2 and γ-Al2O3) multicomponent electrodes by physically mixing commercial Pt/C (HiSPEC® 9100, Johnson & Matthey) and metal oxide nanopowders (Sigma Aldrich) and spray-coating on carbon paper support, as described previously for Pt/C. The mass ratio between Pt/C and metal oxides as well as the Pt loadings on all electrodes were kept equal as Pt/C, maintained at 1:3 ratio and Pt loading of 0.1 mg cm−2, respectively. The resulting morphology was characterized using electron microscopy, energy-dispersive X-ray spectroscopy (EDS) mapping and XRD. As shown in the TEM and EDS mappings (Fig. 5 and Fig. S2, S3 (ESI†)), in all Pt/C-MOx multicomponent systems, the metal oxide nanoparticles are intimately mixed at the nanoscale, providing good contact for charge and mass transport. From the morphologies and XRD patterns (Fig. S3, ESI†), it can be seen that CeO2 and TiO2 have a more crystalline structure, while Al2O3 is more amorphous.56
 | ||
| Fig. 5 (a) The morphology of Pt/C-Al2O3 multicomponent catalyst: (a) TEM image; (b)–(e) STEM-EDS (b: HAADF-STEM, c: multi-element color mix; d: Al map; e: Pt map). | ||
Electrolysis measurements applying Pt/C-MOx tandem catalysts were carried out under the same conditions as for the Pt/C. All the metal oxides and the carbon black features similar specific surface area (Fig. S9 and Table S2, ESI†), eliminating the effect from significant surface area differences.57Fig. 6a shows the product yield % for each product produced by Pt/C-MOx and Pt/C control. The results show that all Pt/C-MOx exhibit higher product yield towards lactic acid compared to the Pt/C control tested previously. In particular, for Pt/C-Al2O3, the lactic acid product yield % reached 61.3 ± 1.2%, nearly double that of the Pt/C standalone. Although the faradaic efficiency towards glyceric acid in Fig. S8 (ESI†) is still higher than lactic acid, as producing the latter needs only half the number of the electrons, it is obvious that after adding the metal oxides, the partial current density towards lactic acid has significantly improved. The category “Others” take into account all products that are not quantified (carbonate (which is the main component), formate, oxalate, etc.) through HPLC. Since carbonate formation accounts for 12 electrons but only one glycerol gets converted, the proportion represented in FE plot is substantially larger than in product yield. The extended stability of the glycerol electrolysis process was evaluated using the Pt/C-Al2O3 electrode. As shown in Fig. S10 (ESI†), the glycerol electrolysis system exhibits a stable cell voltage below 1 V during 72 h of continuous operation at 20 mA cm−2, corresponding to an electricity consumption per unit of H2 < 26 kW h kg−1 H2 (vs. ∼38 kW h kg−1 H2 for alkaline water electrolyser at similar current density),31 while the lactic acid product yield is maintained above 45%.
 | ||
| Fig. 6 (a) The product yield % for Pt/C-MOx and Pt/C control. Values are averaged from at least three independent measurements. Current density: 20 mA cm−2. Electrolysis duration: 1 h. The corresponding electrolysis data and HPLC chromatograph compound calibration and assignment are shown in Fig. S7 (ESI†). The faradaic efficiency values of each product shown in Fig. S8 (ESI†); (b) experimental NH3-TPD profile on different surfaces: carbon black, TiO2, CeO2 and Al2O3; (c) the correlation between lactic acid product yield % and the acidic surface site density determined by NH3-TPD. (d) The difference in adsorption free energy between GLAD and DHA as a descriptor of experimental results. The DHA and GLAD adsorption structure on (e). (i), (iii) Pt(111) and on (f). (ii), (iv) γ-Al2O3(111), respectively. All relevant structures and energetics in our DFT calculations are provided in ESI,† Section 5, Fig. S12–S14. Color codes: red – oxygen, brown – carbon, silver– platinum, pink – hydrogen, blue – aluminum. | ||
To further validate whether the acidic sites on the metal oxide surfaces play a role, NH3 temperature programmed desorption (NH3-TPD) was used to determine the total acidic site concentration and relative strength of the metal oxide catalysts. The data profile is presented in Fig. 6b, and the acidity of all metal oxides was calculated by integrating the area under the profile curve normalised by the mass of the catalyst. As a benchmark, the surface acidity of pure carbon black was also measured. The values are summarized in Table S2 (ESI†). As Fig. 6c shows, we identified a linear relationship between the lactic acid product yield values and acidic site density of different metal oxides.
DFT calculations were performed to further elucidate why the addition of metal oxides enhances the product yield towards lactic acid by studying the adsorption of reaction intermediates on the metal oxides with respect to Pt.
We have modelled the most stable facets for fluorite-CeO2 and anatase-TiO2 following the XRD results shown in Fig. S3 (ESI†), which suggest predominance of the (111) and (101)-facets respectively. Given γ-Al2O3 amorphous’ nature, which did not even exhibit specific peak in XRD in the combined Pt/γ-Al2O3 system, we adopted the (111)-facet as a pragmatic compromise (see ESI,† Section 5c for more detail). The applied surface structures and the (meta-)stable molecules adsorption in our calculations are shown in Fig. S12 and S13 (ESI†).
As shown in Fig. S14 (ESI†), generally, TiO2 and CeO2 bind the relevant reaction intermediates slightly weaker than Pt. γ-Al2O3, on the other hand, shows strengthened binding throughout. This hints towards γ-Al2O3's enhanced activity, as it can retain DHA and GLAD, thus removing them from the solution phase and preventing further electrochemical oxidation on Pt. The enhanced lactic acid selectivity for the Pt/TiO2 and Pt/CeO2 systems, however, is not evident in this analysis.
We identified that the adsorption free energies of both DHA and GLAD correlate with NH3 adsorption, our probe to titrate the number of acidic sites (cf. Fig. S15, ESI†). Thus, merely increasing the acidic site activity does not fully explain the improved lactic acid yield. The relative adsorption strength of the two key intermediates, on the other hand, shows a clear trend reflecting the identified selectivity behaviour, with experimental lactic acid product yield % correlates linearly with (ΔGGLAD* − ΔGDHA*), the ability of the materials in adsorbing DHA, the precursor of lactic acid, over GLAD (Fig. 6d). We interpret this (ΔGGLAD* − ΔGDHA*). relative behaviour as the ability to preferentially remove DHA from the GLAD–DHA solution equilibrium, leading to further production of DHA in solution at the expense of GLAD. DHA can undergo further reaction on both Pt and the metal oxide surfaces. Thus, DHA is constantly removed from the solution, pushing the solution equilibrium towards producing it even at steady state.
Pure Pt preferably binds GLAD over DHA, while TiO2 and CeO2 adsorb both intermediates with comparable strength and γ-Al2O3 binds DHA stronger. To understand this selectivity descriptor in more detail, we highlight the binding configurations of DHA and GLAD on Pt and Al2O3 in Fig. 6e and f. GLAD tends to chemisorb via its central OH-group on both surfaces. DHA on the other hand is only physisorbed in a flat configuration on Pt, while being chemisorbed via both its terminal OH-groups on the acidic sites of Al2O3. An analogous chemisorbed binding configuration of DHA has been identified on the other studied metal oxide catalysts, as shown in Fig. S13 (ESI†).
Thus, we suggest that while both GLAD and DHA benefit from the acidity of the catalyst binding sites, the chemisorption of DHA on metal oxide catalysts compared to the mere physisorption on Pt is the primary cause in the increase in selectivity and product yield towards lactic acid and motivate the validation of the descriptor (ΔGGLAD* − ΔGDHA*) in future efforts towards lactic acid selectivity improvement.
As a final word of caution, we note that enhanced surface acidity does also lead to a competition of DHA adsorption with *OH formation on the active sites at reaction conditions. We show an evaluation of this deactivation mechanism in Fig. S16–S18 (ESI†). The active sites on both TiO2 and CeO2 have been identified to be *OH and *O free, while γ-Al2O3 exhibited a preferential coverage of 0.4 ML of *OH at reaction conditions. However, the binding of DHA exceeds the formation energy of *OH at the mild oxidative potential applied in this study, indicating its ability to displace *OH. Once, the potential would be raised further (to ∼0.75 V vs. RHE, cf. Fig. S18, ESI†), γ-Al2O3 might be deactivated as *OH's formation is stabilized by more oxidating potentials, in contrast to the adsorption of DHA. Thus, a balance in the surface acidity needs to be targeted which is high enough to exhibit preferential adsorption of DHA over GLAD, while not poisoning the surface with *OH at reaction conditions.
Conclusion
In this work, we have developed a MEA-based tandem catalytic system to boost lactic acid yield via glycerol electrooxidation. The high lactic acid product yield of 61.3% (64% if calculated as liquid product distribution towards lactic acid) at a current density of 20 mA cm−2 was achieved with a Pt/C-γ-Al2O3 multicomponent catalyst that nearly doubled that of Pt/C standalone catalyst. Combining NH3-TPD experiment and first principle calculations, we concluded that the improved yield towards lactic acid is attributed to the high density of surface acidic sites provided by the γ-Al2O3 and their preference for binding DHA, a precursor relevant for the dehydration pathway towards lactic acid, thus preventing further electrochemical oxidation on Pt. ΔGGLAD* − ΔGDHA*, defined as adsorption energy difference between DHA and GLAD, is proposed as a selectivity descriptor, which showed linear relationship to the lactic acid product yield %. Although the research in this field is still at early stage, with current density a magnitude lower than practical industrial scale, the insights gained from this work may bring inspirations to design other electrochemical oxidation reactions for high-value commodity chemical production, facilitating the decarbonisation of the chemical industry. Strategies such as constructing 3D electrode structure, selecting suitable anion-exchange membrane, minimising electrolyte crossover in MEA set up, and engineering the interface microenvironment may be deployed to solve the engineering challenges towards scaling up.Experimental methods
Preparation of electrodes
The anodes were prepared by mixing 3 mg Pt/C 60% catalysts (HiSPEC® 9100, Johnson & Matthey), 9 mg Super P® carbon black (Alfa Aesar) or metal oxides (CeO2, TiO2 or γ-Al2O3) (Sigma-Alrich), 1584 μL ethanol, 4200 μL H2O, and 216 μL Nafion solution (5% w/w). The mixture was then sonicated for 10 min with probe ultrasonicator to obtain a homogeneous slurry. 3 mL of the slurry was spray coated onto 3 × 3 cm2 carbon fiber paper (Freudenberg H23, Fuel Cell Store) to achieve a Pt loading of 0.1 mg cm−2. The cathode used in this reaction was also Pt/C 60% catalyst, prepared with the same spray coating method with a Pt loading of 0.05 mg cm−2. The prepared electrodes were then placed in a vacuum oven and dried at 60 °C overnight.Characterization
HR-TEM images and EDS mappings were obtained by a JOEL-2100F electron microscope at an operating voltage of 200 kV. LEO Gemini 1525 FEGSEM were used for SEM imaging. The XRD patterns were performed using PANanalytical's X’PERTPRO X-ray diffractometer with Ni filtered Cu Kα radiation and an X’Celerator multistrip detector. N2 sorption isotherms were conducted at −196 °C with data collected from pressure range 10−5 to 0.99 in Micromeritics 3Flex system with ∼10 mg of degassed sample (200 °C overnight). NH3-TPD measurements were performed on a homemade system formed by a mass flow controllers (MFC) equipped with a horizontal tube furnace where the sample is placed, connected with an Agilent Technologies 7820A Gas Chromatograph equipped with a thermal conductivity detector. Each analysis was performed packing ∼100 mg of sample into a ¼′′ quartz tube, using two plugs of quartz wool to hold the sample in the center of the tube, which was then placed inside a tubular furnace. Samples were pre-treated in He flow (22.7 mL min−1) at 400 °C for 1 h (ramp rate 10 °C min−1) to remove species initially adsorbed on the surface. NH3 was subsequently adsorbed on to the samples by flowing 0.5 mol% NH3 in He over the sample for 30 minutes at 30 °C at a flow of 38.6 mL min−1. Subsequently, the sample was purged in He flow (29.4 mL min−1) for 45 minutes at 30 °C to remove excess physisorbed NH3. NH3 desorption profiles were collected in He flow (22 mL min−1) using the following program: (i) 10 minutes isotherm at 30 °C, (ii) heating to 400 °C with a ramp rate of 10 °C min−1.Glycerol electrolysis
Commercial Pt/C 60 wt% catalyst was first used to test the glycerol electrolysis towards lactic acid with a Metrohm Autolab PGSTAT204 potentiostat. Electrolysis measurements were carried out using an alkaline anion exchange membrane (AEM) electrolyser cell (Dioxide Materials SKU: 68731) with 5 cm2 geometric surface area. Measurement was done under galvanostatic mode, with constant applied current of 100 mA (20 mA cm−2). The above-mentioned electrodes were used as anodes and cathodes, and the anion-exchange membrane is Fumasep FAA-3-50 (fumatech). Experiments were performed for 1 hour with 1 M glycerol/1 M NaOH as anolyte and 1 M NaOH as catholyte, circulated through the MEA bipolar plate flow channels from the respective electrolyte reservoirs using a dual-channel peristaltic pump (JIHPUMP BT-EA-50) at 50 rpm (corresponding to 50 mL min−1) flow rate. N2 (Zero Grade, BOC) was used as purging gas to the electrolyte reservoirs to maintain an inert atmosphere. The cell temperature was maintained at 60 °C. At the end of the experiments, the anolyte and catholyte in the anode and cathode chambers were sampled and quantified with high performance liquid chromatography (HPLC) respectively, using an Agilent 1260 Infinity II LC System. Different compounds were separated with an Aminex HPX-87H column kept at 65 °C and detected by a refractory index detector at 50 °C. The mobile phase was 5 mM H2SO4 with a flow rate of 0.4 mL min−1, and the injection volume was 1 μL. 200 μL reaction products were collected and acidified with 200 μL 0.55 M H2SO4 solutions.58Computational methods
DFT calculations were performed by using the Vienna ab initio software package (VASP)59 employing the projector-augmented wave method.60 Valence electrons were described with plane waves with cutoff energy up to 500 eV. Methfessel–Paxton smearing with width of 0.2 eV was used. We applied RPBE61 functional and the dispersion correction with Grimme D362 method. Monkhorst–Pack grids63 with dimensions of 3 × 3 × 1 or 3 × 4 × 1, which depends on the surface slab symmetry, were used to sample the Brillouin zone to calculate adsorption energy. The bottom two layers were fixed in the bulk structure whereas the upper layers and adsorbates were allowed to relax in all directions until residual forces were less than 0.01 eV Å−1. Vibrational frequencies of adsorbates were computed using a finite difference method as implemented in VASP (IBRION = 5). For surface Pourbaix Diagram, we used a smaller surface structure to calculate the O* and OH* adsorption phase under different coverage by sampling the surface sites (see ESI,† Section 5e and Fig. S16–S18). The computational hydrogen electrode (CHE)64 was used to calculate the electrode potential dependent reaction energy with proton–electron transfer in the reactant. For convenient handling of all atomic structures, the atomic simulation environment (ASE)65 was used. All computational data, including the adsorption energy of glycerol and each key intermediates, Pourbaix diagram, vibrational calculations, and python analysis scripts are available at https://github.com/CatTheoryDTU/Glycerol_To_LacticAcid.Author contributions
H. L., G. K., I. E. L. S., M. M. T. conceived and designed the experiments. H. L., G. T., H. Y. (Hanzhi Ye), H. Y., A. Y. L. performed the experiments. M. X., S. L., G. K. performed the DFT calculations. H. L., M. X., S. L., G. T., C. H. analysed the data. H. L., M. X., S. L., A. Y. L., G. K., I. E. L. S., M. M. T. co-wrote the paper. All the authors discussed the results and reviewed the manuscript.Data availability
The data that support the findings of this study are available within the main text and the ESI.† All computational data, including the adsorption energy of glycerol and each key intermediates, Pourbaix diagram, vibrational calculations, and python analysis scripts are available at https://github.com/CatTheoryDTU/Glycerol_To_LacticAcid. Data are also available from the corresponding author upon request.Conflicts of interest
There's no conflict of interest to declare.Acknowledgements
The work was supported by a donation from Mr Mark Richardson to the Department of Chemical Engineering at Imperial College London. MX and SL were supported by research grant no. 29450 from Villum Fonden. GK was supported by grant no. 9455, V-Sustain, from Villum Fonden. HY and AYL acknowledges Marie Skłodowska-Curie Individual European Fellowship. CH thanks the support from the Royal Society University Research Fellowship under URF\R\201003 and RF\ERE\210203. IELS acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 866402). MT acknowledges funding from the Royal Academy of Engineering Chair in Emerging Technologies. The authors are most grateful for the guidance and mentoring of Professor Karen Chan in the early stages of this work.References
- International Energy Agency (IEA), Chemicals, ( 2021) Search PubMed.
- P. G. Levi and J. M. Cullen, Mapping Global Flows of Chemicals: From Fossil Fuel Feedstocks to Chemical Products, Environ. Sci. Technol., 2018, 52, 1725–1734 CrossRef CAS.
- European Commission, A Sustainable Bioeconomy for Europe: Strengthening the Connection between Economy, Society and the Environment, 2018 DOI:10.2777/478385.
- K. B. Magdalena Zaborowska, The development of recycling methods for bio-based materials – A challenge in the implementation of a circular economy: a review, Waste Manage. Res., 2022, 1–13, DOI:10.1177/0734242X221105432.
- J. Iglesias, et al., Advances in catalytic routes for the production of carboxylic acids from biomass: a step forward for sustainable polymers, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- M. Morales, et al., Environmental and economic assessment of lactic acid production from glycerol using cascade bio- and chemocatalysis, Energy Environ. Sci., 2015, 8, 558–567 RSC.
- G. M. Lari, et al., Environmental and economical perspectives of a glycerol biorefinery, Energy Environ. Sci., 2018, 11, 1012–1029 RSC.
- C. H. Lam, A. J. Bloomfield and P. T. Anastas, A switchable route to valuable commodity chemicals from glycerol via electrocatalytic oxidation with an earth abundant metal oxidation catalyst, Green Chem., 2017, 19, 1958–1968 RSC.
- Y. Zhou, Y. Shen and J. Xi, Seed-mediated synthesis of PtxAuy@Ag electrocatalysts for the selective oxidation of glycerol, Appl. Catal., B, 2019, 245, 604–612 CrossRef CAS.
- C. S. Lee, et al., Selective electrochemical conversion of glycerol to glycolic acid and lactic acid on a mixed carbon-black activated carbon electrode in a single compartment electrochemical cell, Front. Chem., 2019, 7, 1–11 CrossRef CAS.
- H. Yadegari, et al., Glycerol Oxidation Pairs with Carbon Monoxide Reduction for Low-Voltage Generation of C2 and C3 Product Streams, ACS Energy Lett., 2021, 6, 3538–3544 CrossRef CAS.
- M. S. E. Houache, K. Hughes, R. Safari, G. A. Botton and E. A. Baranova, Modification of Nickel Surfaces by Bismuth: Effect on Electrochemical Activity and Selectivity toward Glycerol, ACS Appl. Mater. Interfaces, 2020, 12, 15095–15107 CrossRef CAS.
- D. Kim, et al., Enhancing Glycerol Conversion and Selectivity toward Glycolic Acid via Precise Nanostructuring of Electrocatalysts, ACS Catal., 2021, 11, 14926–14931 CrossRef CAS.
- Y. Yan, et al., Photoassisted Strategy to Promote Glycerol Electrooxidation to Lactic Acid Coupled with Hydrogen Production, ACS Appl. Mater. Interfaces, 2023, 15, 23265–23275 CrossRef CAS.
- Y. Yan, et al., Electrocatalytic Upcycling of Biomass and Plastic Wastes to Biodegradable Polymer Monomers and Hydrogen Fuel at High Current Densities, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- C. Dai, et al., Electrochemical production of lactic acid from glycerol oxidation catalyzed by AuPt nanoparticles, J. Catal., 2017, 356, 14–21 CrossRef CAS.
- B. Huang, J. Yan, Z. Li, L. Chen and J. Shi, Anode-Electrolyte Interfacial Acidity Regulation Enhances Electrocatalytic Performances of Alcohol Oxidations, Angew. Chem., Int. Ed., 2024, 63, e202409419 CAS.
- M. Aslam, M. Navlani-García, D. Cazorla-Amorós and H. Luo, Efficient and selective glycerol electrolysis for the co-production of lactic acid and hydrogen with multi-component Pt/C-zeolite catalyst, J. Phys.: Mater., 2023, 7, 015002 Search PubMed.
- Y. Li, et al., PtAu alloying-modulated hydroxyl and substrate adsorption for glycerol electrooxidation to C3 products, Energy Environ. Sci., 2024, 17, 4205–4215 RSC.
- H. Luo, et al., Progress and Perspectives in Photo- and Electrochemical-Oxidation of Biomass for Sustainable Chemicals and Hydrogen Production, Adv. Energy Mater., 2021, 2101180, DOI:10.1002/aenm.202101180.
- S. Verma, S. Lu and P. J. A. Kenis, Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- Q. Tu, et al., Electrocatalysis for chemical and fuel production: investigating climate change mitigation potential and economic feasibility, Environ. Sci. Technol., 2021, 55, 3240–3249 CrossRef CAS PubMed.
- S. Cheon, W. J. Kim, D. Y. Kim, Y. Kwon and J. I. Han, Electro-synthesis of Ammonia from Dilute Nitric Oxide on a Gas Diffusion Electrode, ACS Energy Lett., 2022, 7, 958–965 CrossRef CAS.
- J. C. Ehlers, A. A. Feidenhans’l, K. T. Therkildsen and G. O. Larrazábal, Affordable Green Hydrogen from Alkaline Water Electrolysis: Key Research Needs from an Industrial Perspective, ACS Energy Lett., 2023, 8, 1502–1509, DOI:10.1021/acsenergylett.2c02897.
- P. De Luna, et al., What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- D. Segets, C. Andronescu and U. P. Apfel, Accelerating CO2 electrochemical conversion towards industrial implementation, Nat. Commun., 2023, 14, 7950 CrossRef CAS.
- F. Bonk, J. R. Bastidas-Oyanedel, A. F. Yousef, J. E. Schmidt and F. Bonk, Exploring the selective lactic acid production from food waste in uncontrolled pH mixed culture fermentations using different reactor configurations, Bioresour. Technol., 2017, 238, 416–424 CrossRef CAS PubMed.
- Y. Wang, et al., Chemical synthesis of lactic acid from cellulose catalysed by lead(II) ions in water, Nat. Commun., 2013, 4, 2141 CrossRef.
- S. Suter and S. Haussener, Optimizing mesostructured silver catalysts for selective carbon dioxide conversion into fuels, Energy Environ. Sci., 2019, 12, 1668–1678 RSC.
- C. A. Giron Rodriguez, et al., Influence of Headgroups in Ethylene-Tetrafluoroethylene-Based Radiation-Grafted Anion Exchange Membranes for CO2 Electrolysis, ACS Sustainable Chem. Eng., 2023, 11, 1508–1517 CrossRef CAS.
- Z. Liu, et al., The effect of membrane on an alkaline water electrolyzer, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- C. Dai, et al., Suppressing product crossover and C–C bond cleavage in a glycerol membrane electrode assembly reformer, Energy Environ. Sci., 2024, 17, 6350–6359 RSC.
- Y. Lum and J. W. Ager, Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu, Energy Environ. Sci., 2018, 11, 2935–2944 RSC.
- J. Gu, et al., Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 5, 268–276 CrossRef CAS.
- F. Pelayo García de Arquer, et al., CO2 Electrolysis to Multicarbon Products at Activities Greater than 1 A cm−2, Science, 2020, 367, 661–666 CrossRef.
- R. K. P. Purushothaman, et al., An efficient one pot conversion of glycerol to lactic acid using bimetallic gold-platinum catalysts on a nanocrystalline CeO2 support, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS.
- A. C. Brix, et al., Electrocatalytic Oxidation of Glycerol Using Solid-State Synthesised Nickel Boride: Impact of Key Electrolysis Parameters on Product Selectivity, ChemElectroChem, 2021, 8, 2336–2342 CrossRef CAS.
- T. Li and D. Harrington, Overview of glycerol electrooxidation mechanisms on Pt, Pd and Au, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS.
- Z. Yu, X. Yue, J. Fan and Q. Xiang, Crystalline Intramolecular Ternary Carbon Nitride Homojunction for Photocatalytic Hydrogen Evolution, ACS Catal., 2022, 12, 6345–6358 CrossRef CAS.
- M. Valter, E. C. Dos Santos, L. G. M. Pettersson and A. Hellman, Selectivity of the first two glycerol dehydrogenation steps determined using scaling relationships, ACS Catal., 2021, 11, 3487–3497 CrossRef CAS.
- M. Valter, et al., Electrooxidation of Glycerol on Gold in Acidic Medium: A Combined Experimental and DFT Study, J. Phys. Chem. C, 2018, 122, 10489–10494 CrossRef CAS.
- B. Liu and J. Greeley, Decomposition Pathways of Glycerol via C–H, O–H, and C–C Bond Scission on Pt(111): A Density Functional Theory Study, J. Phys. Chem. C, 2011, 115, 19702–19709 CrossRef CAS.
- M. Valter, E. C. dos Santos, L. G. M. Pettersson and A. Hellman, Partial Electrooxidation of Glycerol on Close-Packed Transition Metal Surfaces: Insights from First-Principles Calculations, J. Phys. Chem. C, 2020, 124, 17907–17915 CrossRef CAS.
- B. Liu and J. Greeley, Decomposition pathways of glycerol via C–H, O–H, and C–C bond scission on Pt(111): a density functional theory study, J. Phys. Chem. C, 2011, 115, 19702–19709 CrossRef CAS.
- H. Yan, et al., Selective oxidation of glycerol to carboxylic acids on Pt(111) in base-free medium: a periodic density functional theory investigation, Appl. Surf. Sci., 2019, 497, 143661 CrossRef CAS.
- Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Efficient synthesis of lactic acid by aerobic oxidation of glycerol on Au–Pt/TiO2 catalysts, Chem. – Eur. J., 2010, 16, 7368–7371 CrossRef CAS.
- K. M. A. Santos, E. M. Albuquerque, L. E. P. Borges and M. A. Fraga, Discussing Lewis and BrØnsted acidity on continuous pyruvaldehyde Cannizzaro reaction to lactic acid over solid catalysts, Mol. Catal., 2018, 458, 198–205 CrossRef CAS.
- E. M. Albuquerque, L. E. P. Borges, M. A. Fraga and C. Sievers, Relationship between Acid–Base Properties and the Activity of ZrO2-Based Catalysts for the Cannizzaro Reaction of Pyruvaldehyde to Lactic Acid, ChemCatChem, 2017, 9, 2675–2683 CrossRef CAS.
- E. M. Albuquerque, L. E. P. Borges and M. A. Fraga, Lactic acid production from aqueous-phase selective oxidation of hydroxyacetone, J. Mol. Catal. A: Chem., 2015, 400, 64–70 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Conversion of Sugars to Lactic Acid Derivatives Using Heterogeneous Zeotype Catalysts, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- P. P. Pescarmona, et al., Zeolite-catalysed conversion of C3 sugars to alkyl lactates, Green Chem., 2010, 12, 1083–1089 RSC.
- R. M. West, et al., Zeolite H-USY for the production of lactic acid and methyl lactate from C3-sugars, J. Catal., 2010, 269, 122–130 CrossRef CAS.
- D. Chu, H. Zhou and Z. Luo, CrOx decoration on Fe/TiO2 with tunable and stable oxygen vacancies for selective oxidation of glycerol to lactic acid, New J. Chem., 2022, 46, 18744–18750 RSC.
- L. Vitos, A. V. Ruban, H. L. Skriver and J. Kollár, The surface energy of metals, Surf. Sci., 1998, 411, 186–202 CrossRef CAS.
- K. I. Shimizu, K. Kon, K. Shimura and S. S. M. A. Hakim, Acceptor-free dehydrogenation of secondary alcohols by heterogeneous cooperative catalysis between Ni nanoparticles and acid-base sites of alumina supports, J. Catal., 2013, 300, 242–250 CrossRef CAS.
- J. Gu, J. Wang and J. Leszczynski, Structure and Energetics of (111) Surface of γ-Al2O3: Insights from DFT Including Periodic Boundary Approach, ACS Omega, 2018, 3, 1881–1888 CrossRef CAS.
- C. Chen, et al., Cu–Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- H. Luo, et al., Role of Ni in PtNi Bimetallic Electrocatalysts for Hydrogen and Value-Added Chemicals Coproduction via Glycerol Electrooxidation, ACS Catal., 2022, 14492–14506, DOI:10.1021/acscatal.2c03907.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. K. Nørskov, et al., Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- A. Hjorth Larsen, et al., The atomic simulation environment—a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00236a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |