
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrated CO2 capture and electrochemical conversion: coupled effects of transport, kinetics and thermodynamics in the direct reduction of captured-CO2 adducts†
Avishek
Banerjee
 and
Carlos G.
Morales-Guio
*
and
Carlos G.
Morales-Guio
*
Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, USA. E-mail: moralesguio@ucla.edu
First published on 8th January 2025
Abstract
Upgrading anthropogenic CO2 from concentrated point sources or directly from the atmosphere is a valuable approach in closing the carbon cycle. Existing processes capture the CO2, concentrate it into pure gas streams, transport it, and then convert it into fuels and chemicals in a separate process plant. This sequential approach results in higher energy and operating costs which can be reduced by integrating the capture and conversion steps to directly reduce the captured CO2-bound adduct to value-added products. The direct reduction of the captured CO2-bound adduct is called the captured-CO2 reduction reaction (c-CO2RR). Understanding of c-CO2RR has been obscured by the higher intrinsic complexity of the system. The CO2 capture media is a complex space of several buffer reactions that allow the co-existence of different carbon species in solution depending on CO2 loading, temperature, pressure, and pH. In order to design improved capture agents and catalysts for integrated CO2 capture and conversion, it is essential to identify the carbon source and the primary factors influencing product formation on a c-CO2RR catalyst. This review delineates the strategies to determine the active carbon species for integrated CO2 capture and conversion systems. Furthermore, it summarizes the fundamental applications of mass transport, thermodynamics, and kinetics across various c-CO2RR scenarios.
 Avishek Banerjee | Avishek Banerjee received his BTech in Chemical Engineering from National Institute of Technology Durgapur, India. He then pursued his MSc at McGill University in Montreal, Canada, where he worked with non-thermal plasma for synthesis of valuable fuels and chemicals. In 2022, he joined UCLA as a PhD student in the Department of Chemical and Biomolecular Engineering. His research focuses on the electrocatalytic approaches of integrated CO2 capture and conversion, with an emphasis on understanding the roles of transport, thermodynamics and kinetic phenomena in these processes. |
 Carlos G. Morales-Guio | Carlos G. Morales-Guio is an Assistant Professor of Chemical and Biomolecular Engineering at UCLA. He received his BEng degree in Chemical Engineering from Osaka University and his MS and PhD in Chemistry and Chemical Engineering from EPFL. He then moved to California as a postdoctoral research fellow at Stanford University. His lab at UCLA is interested in understanding engineering principles relevant to the scale-up of electrocatalytic processes. The lab focuses on understanding the fundamentals of mass, charge and heat transport at the electrode/electrolyte interface, reactor design, process intensification and process automation to guide the development of sustainable energy applications. |
Broader contextExisting processes for CO2 capture and conversion start by separating the CO2 from the effluent of a point source or the atmosphere, and then this concentrated CO2 stream is used as the feedstock for a catalytic conversion step in a separate unit. The CO2 capture step involves multiple thermal and pressure cycles that result in energy losses and increased operating costs. Recently, a one-step process called reactive capture of CO2 has been proposed in which CO2 in its captured form (c-CO2) is directly transformed into value-added products while circumventing the CO2 release step. The research on the electrochemical c-CO2 reduction reaction (c-CO2RR) is just starting, but it is already evident that the complexity of the system pushes the limits of what is known in science and engineering. Understanding individual phenomena of CO2 capture and CO2 conversion is not enough. When capture and conversion are integrated, new phenomena emerge that are unique to the performance of the capture step in the presence of a catalyst, and to the performance of catalysis in the presence of the capture agent. Thus, it is of critical urgency that all researchers entering this field learn the fundamentals of transport, thermodynamics, and kinetics that link CO2 capture and conversion. This review covers the latest developments in c-CO2RR research and our viewpoint on the challenges and future opportunities that exist in this space as the field matures. |
1 Introduction
CO2 contributes to almost 79% of total greenhouse gas emissions and is considered the major cause of global warming.1 The development of technologies that contribute to the reduction of CO2 emissions and its direct capture from the atmosphere is more urgent than ever before. Global emissions of CO2 are estimated to be approximately 35 billion tons per year, with approximately 2 billion tons per year attributed to the chemical industry.2,3 Despite the increasing adoption of electrification for heating in various chemical manufacturing processes, burning of fossil fuels for heating is just a fraction of the CO2 emissions and there remains a pressing need to source carbon atoms for chemicals from sources different from fossil fuels. As the demand for chemicals continues to grow, technologies that can upgrade the anthropogenic CO2 from concentrated point sources and the atmosphere are needed to close the carbon cycle. Although the use of renewable electricity to capture and convert CO2 into value-added chemicals offers a pathway to decarbonization, significant challenges persist in terms of energy intensity and cost compared to conventional methods. Overcoming these hurdles is crucial to making these processes industrially viable.4Existing processes capture CO2, concentrate it in pure gas streams, compress CO2, and then convert it into fuels and chemicals, all in separate reaction units (top panel in Fig. 1). This sequential multistep approach results in higher energy and operating cost, arising from the inefficiency of the CO2 release and capture media regeneration step before CO2 compression. CO2 release from direct air capture (DAC) processes have an estimated energy cost between 3400 and 8810 kJ per kg CO2 for solid and liquid sorbent, while the CO2 compression step along with the transport of gas requires an energy of 324–432 kJ per kg of CO2.5–10 Recently, a one-step route has been proposed where CO2 capture and conversion are integrated into one process that circumvents the inefficient CO2 release step, by directly reducing the captured CO2-bound adduct to value-added chemicals (bottom panel in Fig. 1). The elimination of the inefficient temperature and pressure swing cycles during CO2 capture and conversion can potentially reduce 50% of the total energy cost. The gains, however, must occur on both processes: (1) the CO2 capture, and (2) the electrochemical captured-CO2 reduction reaction (c-CO2RR) steps. This requires that the yield and energy efficiency of the c-CO2RR electrolyzer is comparable to current state of the art CO2 electrolyzers.11 In principle, integrated CO2 capture and conversion has the potential to be more efficient and less expensive than the conventional approach relying on CO2 electrolyzer technologies for the CO2 reduction reaction (CO2RR). However, a long road remains ahead to bring c-CO2RR electrolyzer technologies in par with today's CO2RR electrolyzers.
 | ||
| Fig. 1 Illustration showing the conventional CO2RR pathway (shown by the green path) and the c-CO2RR pathway (shown by the orange path). The CO2RR pathway is a multistep process comprising additional energy costs from the CO2 release, compression, and transport steps before the CO2 reduction reaction can take place, whereas the integrated c-CO2RR is a one-step process that could potentially reduce energy and operating costs. Furthermore, both the pathways are interrelated to the fundamental principles of thermodynamics, transport, and kinetics which influences the product formation. | ||
CO2 capture technologies are much more mature than the reduction step in c-CO2RR. Many studies have been conducted on solvent-based capture technologies with amines or carbonates. However, preliminary work suggest that amines might not be appropriate for c-CO2RR due to their strong interactions with the catalyst surfaces and the enhanced hydrogen evolution reaction.12 Alternative capture agents like phenoxides,13 alkoxides,12 and amino acids14 could indeed result in renewed interest in the development of capture agents specifically designed for integration with c-CO2RR, beyond those being commercialized today. Further optimization of the capture step is guaranteed once we find promising catalysts for c-CO2RR and as we start to extract capture agent design rules for integrated carbon capture and conversion.
Integrated CO2 capture and conversion systems are complex, with several carbon species co-existing in the reactors. Identifying the source of carbon while quantifying products, and elucidating the key factors that affect the activity and selectivity of a catalyst, is important. Although c-CO2RR has been studied in the context of electrochemical,15–19 thermal20–23 and biological systems,24–27 this review focuses solely on the electrochemical route. In recent years, researchers have reported evidence of electrochemically reducing the captured CO2-bound adduct to products.28 However, significant challenges still remain in understanding the transformation of the captured CO2-bound adduct. In the electrolyte solution the vapor–liquid equilibrium, ion-speciation, and reaction equilibriums dynamically varies as a function of the temperature, pressure, and pH. These factors are further governed by the fundamental principles of mass transport, thermodynamics, and kinetics, which when not carefully investigated can convolute the way we interpret the data collected from c-CO2RR.
In the field of electrocatalysis, there is a consensus within the scientific community regarding the importance of transport, thermodynamics, and kinetics; however their full integration into scientific studies remain somewhat limited. Thermodynamics determines the feasibility of a process by delineating the minimum energy requirements and equilibrium conditions. It also links variations in local concentration, temperature, pressure, and electric field of an electrochemical system to changes in the electrochemical potential. Thermodynamics further determine the direction of mass and energy fluxes in and out of a control volume, which is linked to spacial and temporal differences in local concentration, temperature, and pressure. Transport properties determine the scalability of a process by giving insights into technically feasible energy and mass fluxes, which in turn will be linked to reactions in multidimensional space and time coordinates. Transport phenomena are used to describe processes of diffusion, convection, and migration of species in electrochemical cells. Different reactors have different hydrodynamics, resulting in different transport properties. By and large, the electrocatalysis community has failed to recognize that we measure experimentally reactor kinetics and not reaction kinetics, slowing the progress of the field. Thus, understanding of mass, heat, and charge transport is necessary to decouple reaction kinetics from reactor kinetics, and this is also true for c-CO2RR systems. Thermodynamics, transport, and kinetics cannot be disentangled because of their interdependence. In order to overcome the challenges of successfully scaling-up electrochemical systems, it is necessary to measure, model and control the coupling of thermodynamics, transport, and kinetics even when studying the most basic electrochemical system at a bench scale (Fig. 1).
Most reviews and perspectives focuses on the different amine capture agents and discusses scalability challenges that could arise from c-CO2RR, while comparing the value proposition of c-CO2RR with traditional CO2RR.11,29–34 In contrast, our review is meant to serve as an introduction to the field of c-CO2RR for chemists, material scientists, and engineers who although might have learned the various concepts of transport, thermodynamics, and kinetics separately, have not seen how these three concepts come together at different length and time scales in c-CO2RR. This review will further aim to lay out some guidelines on the processes and factors that influence product selectivity and that should be considered while studying and reporting data for c-CO2RR systems. It also includes the latest breakthrough results and the wider implications for the scientific community, and provides the tools necessary for understanding the fundamentals of electrochemical c-CO2RR.
2 Understanding vapor–liquid equilibrium and ion speciation under c-CO2RR conditions
The different species present in a solution filled with a CO2-bound capture agent can be probed by understanding the vapor–liquid equilibrium of the system. When a capture agent is dissolved in a solution, and then it is put in contact with a gas phase containing CO2, free and dissolved CO2 will always be present in solution due to the equilibrium reactions between the gas and liquid phases. The amount of dissolved CO2 is furthermore a function of CO2 loading, pH, temperature and pressure of the system. Industrially, amines are currently the leading CO2 sorbents, and an entire field of science and engineering has been developed around the optimization of CO2 absorption capacities of different amines.35–39 This section dives deeper into CO2 capture with amines used primarily in post-combustion processes along with insights into other groups of capture agents, including hydroxyls used for direct air capture (DAC),40–44 and the untapped group of alkoxides and phenoxides that is recently gaining interest.13,452.1 Chemical reaction processes
The CO2 absorption process into different capture solutions is governed by different equilibrium reactions, allowing different species to coexist in the system.For amine capture solutions, it is known that the species present are RNH2, RNH3+, RNHCOO−, H3O+, OH−, HCO3−, CO32−, CO2, and H2O. R represents an alkyl/alkanol group and can belong to a primary, secondary or tertiary family of amines. Primary and secondary amines have a higher basicity than water, and thus CO2 being the Lewis acid attacks the amine to undergo a zwitterion mechanism to form carbamates. Initially, two amines are consumed for each CO2 molecule absorbed, generating an ammonium carbamate. However, if the CO2 loading is increased and the amine is almost depleted, then the ammonium carbamate decomposition reaction starts occurring to form ammonium bicarbonate and carbonate species.46 Similarly in the presence of more sterically hindered amines, like tertiary amines, water comparatively becomes a stronger Lewis base and can directly assist in the formation of ammonium bicarbonate with the amines.47 The following chemical reactions describe the formation of different species.37,38
Water ionization:
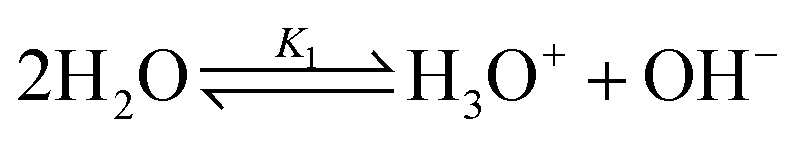 | (1) |
CO2 reaction with water:
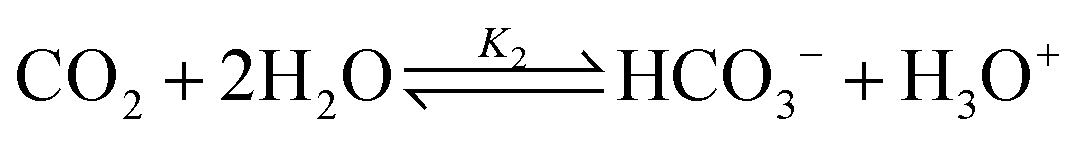 | (2) |
dissociation of bicarbonate ion:
 | (3) |
zwitterion formation from RNH2 and CO2 reaction:
 | (4) |
carbamate formation by deprotonation of zwitterion:
 | (5) |
 | (6) |
 | (7) |
carbamate hydrolysis:
 | (8) |
carbamate consumption:
 | (9) |
de-protonation of amine:
 | (10) |
bicarbonate formation:
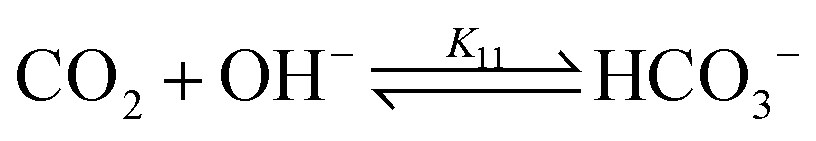 | (11) |
The equilibrium constants of these reaction, K1–K11, can subsequently be used to understand the speciation as a function of pH, temperature and CO2 loading. In the case of primary and secondary amines, carbamate is the main species formed when the CO2 is bound to the amine, while tertiary amines follow a base-catalyzed hydration mechanism producing only bicarbonate ions and no carbamate, which would essentially mean that reactions (4)–(9) would not occur in the presence of tertiary amines.
Similarly, hydroxyls commonly used for DAC, form bicarbonate and carbonate species in the solution. The different species that are known to co-exist are H3O+, OH−, HCO3−, CO32−, CO2, and H2O, i.e., reactions (1)–(3), and (11) would only occur in such systems, and it reduces the number of buffer reactions compared to amine capture agents. In CO2RR, we are largely concerned only with the equilibrium reactions between CO2 and bicarbonates. It should thus be evident for researchers working with CO2RR that c-CO2RR represents a step-up in complexity when amines and other capture agents are involved.
Other capture agents belonging to the alkoxide family have also been recently investigated for c-CO2RR. Kowalski et al., recently investigated potassium methoxide as a CO2 capture agent, and observed that the methoxide ions require alcohols (other non-aqueous medium) as the solvent medium to capture CO2 and form methyl carbonate. In an aqueous medium, the OH− present in the reaction primarily behave as the capture agent forming HCO3− and CO32−, limiting the methylcarbonate production pathway. In the presence of alkoxide ions, similar reactions were described to understand the capture mechanism.13
Alcohol ionization:
 | (12) |
CO2 reaction with alkoxides:
 | (13) |
In the alkali metal alkoxide systems, the speciation looks fairly simple with only species being the ROH, RO−, ROCOO−, CO2, and H+ in addition to the alkali metal cation. The R could represent a primary, secondary, tertiary or an aromatic alcohol group.
The equilibrium equations shown here only describe the vapor–liquid equilibrium, while a full description of the system needs to add the solid components and all the reactions that lead to precipitation of species as well as dissolution of the solids in equilibrium. For simplicity, solid phases will be not discussed here but these need to be considered for any practical system.
2.2 CO2 loading effect on speciation
The concentration distribution in the bulk of the solution is a function of the CO2 loading when the temperature and pressure is constant. From the different equilibrium constants of the reactions shown in Section 2.1, vapor–liquid equilibrium models have been developed to predict the CO2 absorption in different amines. Fig. 2 shows a typical chemical speciation distribution at different CO2 loadings in amines. The CO2 loading (mol mol−1) is defined as the ratio of mol of CO2 present (dissolved + captured) to the mol of capture agent present in the solution. The concentration of CO32− does not vary significantly, especially for primary and secondary amines, when the CO2 loading is varied between 0–1 mol mol−1. At low CO2 loadings, it is typically observed that all the CO2 dissolved in the solution gets converted to carbamate as denoted by reactions (5)–(7). CO2 loadings of 0.5 mol mol−1 usually give the highest carbamate concentration, which could be also rationalized from the stoichiometry of the reaction where 2 amine molecules are required to capture 1 CO2 molecule. Through the predicted equilibrium concentration of different species as a function of CO2 loading, it has been shown that the carbamate consumption, as denoted by reaction (8), and CO2 hydrolysis starts to simultaneously occur at around 0.3 mol mol−1. When a CO2 loading of 1 mol mol−1 is reached the speciation is mostly dominated by bicarbonate and free CO2 present in the system, with little to no carbamate. Thus for electrochemical c-CO2RR, it is necessary to probe the loading of the systems to understand the speciation and also it is beneficial to be at a loading <0.5 mol mol−1 if the intention is to reduce the carbamate species within the system. However in case of higher loadings, if small amounts of carbamate present near the active site of the catalyst is reduced, it is possible that the bicarbonate can equilibrate back to carbamate. The speed of this equilibration will vary and would depend on the pKa and the equilibrium constants of the different amines. For comparison, CO2 loadings cycle typically between 0.3 and 0.7 during the thermal regeneration of amine capture solutions, where the low loadings belong to the CO2-lean capture solution that is fed to the absorber unit and the high loading belong to the CO2-rich amine streams that leave the bottom of the absorber.38,48 | ||
| Fig. 2 VLE models showing equilibrium concentration of different species in liquid-phase as a function of loading in (a) 2 M MEA at 301 K which is a primary amine (lines represent model and dots represent experimental data points from Liu et al.)49 (b) 2 M DEA at 301 K which is a secondary amine (lines represent model and dots represent experimental data points from Liu et al.)49 (c) 2 M MDEA at 298 K which is a tertiary amine (lines represent model and dots and hollow dots represent experimental data points from Derks et al. and Jakobsen et al. respectively)50,51 Reproduced with permission from ref. 48, copyright 2024 American Institute of Chemical Engineers. | ||
Although most studies just look at speciation considering all amines behave similarly, but there lies differences in how these vary between primary, secondary and tertiary amines. As can be seen from Fig. 2, the VLE models of a primary, secondary and tertiary amine shows that the equilibrium concentration of the different species are different as a function of loading when the sterics of the amines change. Also, even between the same group of amines, the speciation curves could be different, which mainly arises from the different equilibrium constants of carbamate consumption and amine deprotonation in aqueous systems, K9 and K10. For instance, between monoethanolamine (MEA) and diethanolamine (DEA), at 0.5 mol mol−1 of loading the carbamate concentration is slightly higher in MEA than in DEA (Fig. 2a and b). From the equilibrium constant values, it was determined that the carbamate consumption (reaction (9)) is slightly faster for DEA compared to MEA which is responsible for the difference. Also, at loading of 1 mol mol−1 slightly lower concentrations of RNH3+ and HCO3− was observed compared to MEA. The pKa of DEA is lower than MEA and so it acts as a weaker Lewis base leading to weak binding of CO2 to the amine. This leads to faster deprotonation of DEA allowing the carbamate consumption reaction to accelerate (reaction (9)). For tertiary amines like methyldiethanolamine (MDEA), the dominant species that forms is bicarbonate and thus the CO2 loading is equivalent to bicarbonate formation with small amounts of CO32−. This small amount of CO32− arises from the high pKa of MDEA that accelerates the deprotonation of the bicarbonate (Fig. 2c).
When hydroxyls are used as capture agents, the main species present in the system is HCO3−, CO32− and dissolved CO2. It is understood that at the beginning of CO2 loading the main reaction is the formation of HCO3− with CO32− forming at low CO2 loadings, when the pH of the solution is highly alkaline.45
Similarly alkoxide capture agents can be modeled to understand the speciation at different CO2 loadings. Considering the stoichiometry of alkoxides, it requires 1 molecule of alkoxide to capture 1 molecule of CO2, which means the highest concentration of alkylcarbonate will form at CO2 loading of 1 mol mol−1. Also, the bonding characteristics of CO2 on alkoxides is similar to a deprotonated amino group. Recently, Kowalski et al. studied the loading of CO2 in a methoxide capture agent in methanol solvent. Experimentally the mass change was measured while flowing pure CO2 in 5 wt% methoxide and they observed that the change in mass was higher in the presence of methoxide in the methanol solvent compared to when no methoxide was present in the methanol. They further experimentally calculated the CO2 loading to be 1.09 mol mol−1 which is similar to the expected stoichiometric loading (Fig. 3).12
 | ||
| Fig. 3 Comparison of mass change from CO2 loading in (a) mass change during CO2 absorption in 100 mL of pure methanol. The net mass change is approx. 0.7 g of CO2 after subtracting the CH3OH evaporation amount measured under the flow of Ar and the mass change observed during CO2 flowing into the methanol solvent. (b) Mass change during capture of CO2 in 100 mL of 5 wt% CH3OK in CH3OH. CO2 forms CH3OCOO− when reacted with the methoxide ions and the mass change for this process is about 3.5 g. This measurement is also an experimental verification of the fact that the solubility of CO2 is different in water and in methanol. The CO2 loading, defined as the ratio of mol CO2 captured and dissolved to the mol CH3OK present, was calculated to be 1.09 mol mol−1 from (b).12 Reproduced with permission from ref. 12, copyright 2024 American Chemical Society. | ||
2.3 pH effects on chemical speciation
The pH of the solution affects the equilibrium species distribution in the capture solution. As CO2 is an acidic gas, its capture is favored in more alkaline conditions. The concentration of the different species when plotted as a function of pH shows that at very low pH the species concentration is dominated by free CO2 and HCO3−, at intermediate pH the species concentration is dominated by the carbamate, and at very high pH the dominant species is the amine capture agent in its initial form. In MEA the highest carbamate concentration is achieved at a pH of 9.3 whereas for DEA it is at a pH of 8.85, which also corresponds closely to the pKa of these amines which are 9.45 and 8.88 at 298 K respectively.52 The faster rates of carbamate consumption and CO2 hydrolysis in DEA can also be realized from Fig. 4a and b, where it can be seen that the HCO3− and free CO2 shoots up slightly quicker at lower pH compared to MEA. The pH is also a function of the CO2 loading, and typically it decreases as more CO2 is captured within the system, giving rise to the different speciation at lower pH. Also, the partial pressure of CO2 in equilibrium with the solution is higher at higher temperatures. This translates into a higher release rate at higher temperatures resulting in a more alkaline solution, indicating temperature as an important parameter in regulating the pH. Also, the partial pressure of CO2 increases by several orders of magnitude at a low pH, which is attributed to the presence of a high concentration of free CO2 in the bulk of the solution. Thus in general it can be concluded that for aqueous systems a more alkaline pH is favorable for CO2 capture, while a more acidic pH favors the release of CO2 from the CO2 captured adduct. Consequently, pH-swing for release of CO2 from the captured adduct is currently being explored as an alternative to temperature-swing or pressure-swing cycles.53–56 Similar conclusions can be drawn for alkaline CO2 capture systems where hydroxyls are used as the capture agent. In such systems, a low pH is accompanied by the formation of carbonic acid H2CO3, a neutral pH consists of HCO3−, while a high pH forms CO32− in the system.57 | ||
| Fig. 4 Equilibrium speciation as a function of pH at 298 K for (a) 2 M MEA (primary amine), (b) 2 M DEA (secondary amine) and (c) 2 M MDEA (tertiary amine) obtained from VLE models. It can be seen that at a low pH, dissolved CO2 has a very high concentration along with high concentration of HCO3−. Reproduced with permission from ref. 48, copyright 2024 American Institute of Chemical Engineers. | ||
Although the vapor–liquid equilibrium models can be leveraged to predict the speciation as a function of pH in the aqueous systems, for alkoxide/phenoxide capture agents, the solvent medium is primarily non-aqueous. Thus, it becomes difficult to define pH under such conditions and find equivalent speciation plots. However, Zhang et al. reported that the CO2 binding strength to different alkoxides followed a linear scaling relation (LSR), and more interestingly it lied on the same LSR as the amines, which suggests that the binding of CO2 is solely a function of the pKa of the capture agents (Fig. 5). Furthermore, the findings of their investigation can be extended to conclude that the equilibrium constants which vary between different amines for developing the VLE models are mainly a characteristic of the changing pKa of the amines.13
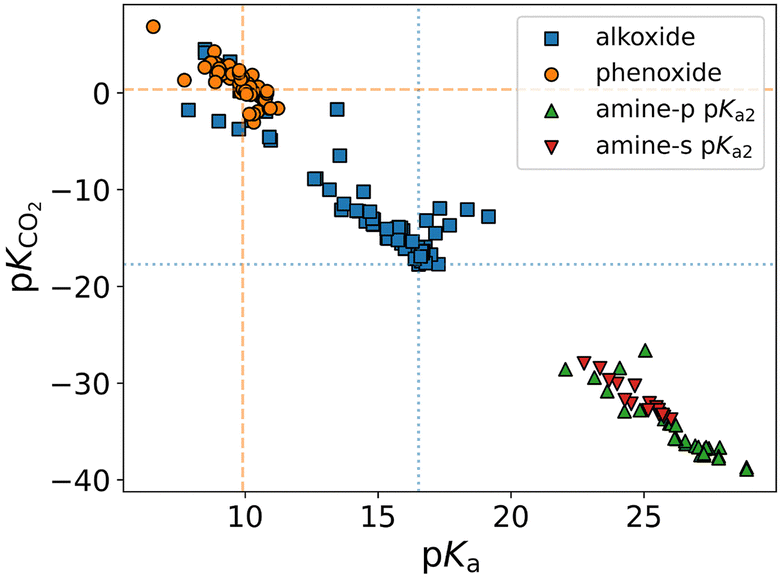 | ||
| Fig. 5 DFT calculated values of CO2 binding constant (pKCO2) as a function of pKa for different amines and alkoxides. The orange dashed line and the blue dotted line represent the data points of phenoxide and methoxide, respectively.13 Reproduced with permission from ref. 13, copyright 2022 National Academy of Sciences. | ||
2.4 Temperature effects on equilibrium concentrations
The equilibrium species concentration of capture solutions as a function of temperature can be probed to investigate its influence on c-CO2RR. The equilibrium constants of all the reactions stated in Section 2.1 are a function of the enthalpy (ΔH) and temperature (T) by eqn (14). | (14) |
For amine capture solutions, at low CO2 loading the RNHCOO− concentration increases with temperature, and reaches a maximum value at 0.5 mol mol−1 loading. As the loading is increased more, it has been observed that the RNHCOO− concentration declines. This decline accelerates at higher temperatures which could be explained by Le Chatelier's principle, where higher temperatures shift the equilibrium to the left, which in amine capture solutions shift the equilibrium for reactions (5)–(7) towards the formation of the zwitterionic intermediate to ultimately form dissolved CO2 and RNH2, as shown in reaction (4). Furthermore, carbamate consumption reaction becomes more dominant at loadings >0.5 mol mol−1, as K9 is negatively related to temperature denoting the process is exothermic. In addition, the temperature was shown to have an effect on the concentration of dissolved CO2. As the loading increased, the concentration of dissolved CO2 was increased, and it increased further at higher temperatures. At higher loadings, it is rationalized that the amine capture agent is completely consumed and then the CO2 undergoes hydrolysis to form bicarbonates in the system. Also, as CO2 hydrolysis is an exothermic reaction, it facilitates the release of more CO2 at higher temperatures. Consequently, when the partial pressures of CO2 is compared to the CO2 loading with the use of the temperature dependent Henry's constant, it is observed that higher temperature results in a higher partial pressure of CO2, implying lower solubility in the solution (Fig. 6). When comparing primary and secondary amines, DEA shows a lower partial pressure of CO2 compared to MEA, which again arises because of the faster carbamate consumption and CO2 hydrolysis in secondary amines. Also for MDEA which is a tertiary amine, the effect of temperature on partial pressure of CO2 had little effect compared to MEA and DEA (Fig. 6c).
 | ||
| Fig. 6 Partial pressure of CO2 as a function of CO2 loading and temperature for (a) 2.5 M MEA (primary amine) where solid lines are predicted from VLE models and hollow dots are experimental data points from Lee et al.,58 (b) 3.33 M DEA (secondary amine) where solid lines are predicted from VLE models and hollow dots are experimental data points from Bottinger et al.,59 and (c) 2 M MDEA (tertiary amine) where solid lines are predicted from VLE models and hollow dots and solid dots are experimental data points from Sulaiman et al. and Jou et al.39,60 Reproduced from with permission ref. 48, copyright 2024 American Institute of Chemical Engineers. | ||
For hydroxyl capture agents, the partial pressure of CO2 as a function of loading is only governed by CO2 hydrolysis and HCO3− formation, reactions (1)–(3). The partial pressures at similar conditions of pressure and temperature are usually higher for such systems implying more free dissolved CO2 present in the solution.
For alkoxide capture agents, the partial pressure of CO2 is one order of magnitude lower than amine/hydroxyl capture agents. This difference arises mainly because of the higher Henry's constant when using methanol as solvent and also a higher solubility of CO2 in methanol.
Temperature has a significant effect on the chemical speciation of the system and can be captured in the vapor–liquid equilibrium model used while studying c-CO2RR systems. At high temperature conditions, the CO2-bound adduct can release free CO2 and regenerate the capture agent. This is known as temperature-swing capture agent regeneration. In conventional carbon capture and utilization processes, the capture step is followed by a high-temperature capture agent regeneration process while simultaneously releasing a concentrated stream of CO2 for further storage or upgrading. Similarly, in c-CO2RR, the captured CO2-adduct will decompose at higher temperatures leading to low c-CO2RR performance, however, consequently higher temperatures will release more free CO2 in the system that can compete for the active sites of the catalyst for further reduction. Banerjee et al., in their study showed that by increasing the temperature of the capture solution during electrolysis, they can achieve a higher partial current density of CO using silver catalyst. They concluded that this behavior is mainly due to the release of free CO2 in the system as they increase the temperature from 20 °C to 40 °C.48 In such cases, at higher temperatures the performance of c-CO2RR can appear to be improved, but this is not to be confused with the direct reduction of the CO2-bound adduct. Therefore, understanding the vapor–liquid equilibrium effects discussed throughout Section 2 will be important in identifying the active carbon species getting reduced as the field of c-CO2RR evolves.
3 Identifying the source of carbon in c-CO2RR
The VLE models can provide a clear understanding of the speciation and its variation with the pH, temperature and CO2 loading. Leverick et al. quantified the speciation of amines with different pKa through NMR analysis to verify the speciation with the VLE-models and identify the source of carbon. There have been three main approaches that have been undertaken to identify the active carbon species which are by leveraging characterization techniques to correlate with the VLE-models of different amines, continuum modeling, and correlating the two-film theory with reaction transport model. The last method provides a robust way for quick screening of different capture agents, especially if VLE models are not easily accessible. This section goes through each method in detail.3.1 Characterization techniques coupled with VLE-models
Recently, the most detailed study to identify the source of carbon in c-CO2RR systems with the VLE-models have been performed by Leverick et al. using amine capture agents. Using a H-cell reactor with an Ag catalyst, they investigated the faradaic efficiency (F.E.) of CO in the presence of amines with varying pKa and different propensity to form carbamates. They reported that at pH > 10 the F.E. CO is <0.1% and the F.E. CO increases to >20% when the pH is decreased, and it is ubiquitous to all the amines tested (Fig. 7c). To rationalize this behaviour they correlated the experimental data to the VLE-models, and explained that the carbamate concentration decreases at lower pH but the concentration of HCO3− and dissolved CO2 increases, which also increases the CO2 partial pressure (Fig. 7a and b). The changes in carbamate concentration were further tracked with 13C NMR along with the different species present in the solution. Furthermore, they showed that the partial current density of CO (jCO) had a first-order dependence with the partial pressure of CO2 verifying that the source of carbon is the dissolved CO2 in the solution and not the carbamate itself. In cases, where they did not see a linear increase of jCO with decreasing pH, they discussed the possibility of the different amines having different activity towards HER, which could suppress the production of CO.61 | ||
| Fig. 7 (a) Concentration of carbamate from NMR vs. pH for 2 M KCl + 2 M amine with DMAE (square), AMP (right-pointing triangle), 2A1P (left-pointing triangle), and MEA (circle). (b) Concentration of bicarbonate from NMR vs. pH. (c) FE of CO (filled symbols) and H2 (open symbols) production vs. pH from potentiostatic measurements at −1.30 vs. SHE-iR.61 Reproduced with permission from ref. 61, copyright 2023 American Chemical Society. (d) jCO at −1.1 V vs. normal hydrogen electrode (NHE) of Ni–N/C and cAg for the c-CO2RR in CO2-absorbed MEA (1–5 M) solutions with purging Ar.15 Reproduced with permission from ref. 15, copyright 2022 Royal Society of Chemistry. | ||
Similar studies were performed by Kim et al. using a membrane electrode assembly with a single atom Ni catalyst and a Ag foil as catalyst. With both catalysts, they probed the source of carbon for the produced CO by changing the concentration of the carbamate in the system and also by changing the temperature of the reactor. They also quantified the carbamate concentration in the system using 13C NMR and observed a zero-order dependence on the F.E. CO with the carbamate concentration, meaning the carbamate did not act as the active species for reduction (Fig. 7d). They further observed an increase in the F.E. CO as the temperature increased, which they attributed to more dissolved CO2 released in the system at higher temperature.15
Thus, VLE models can be an important source to identify the active species. Changing the CO2 loading, temperature, pH, and pressure of the system affects the species distribution and thus the VLE models in conjunction with the multi-variable dataset can rationalize the results to provide a basis for identification. In c-CO2RR studies, keeping track of these parameters can help in understanding how the amount of dissolved CO2 changes in the system, and these changes can be correlated with the changing faradaic efficiency/partial current density of the products. If there were to be any direct reduction of the CO2-bound capture agent, the total partial current density of the reduced product should have a dependence on the carbamate concentration of the solution.
3.2 Continuum models
Safipour et al. investigated CO2RR with MEA solution in a flow-cell configuration to investigate the species getting reduced. By increasing the concentration of MEA they observed an increase in HER activity and reduction in CO production activity. To understand this effect, they developed a 1D continuum model to determine the concentration distribution of different species within the electrical double layer. Assuming concentration dependent Tafel kinetics, the electrode reactions were modelled. It was found that the CO2 concentration at the surface of the electrode is lower in the presence of amines compared to conventional CO2RR, which was responsible for the lower CO2 conversion rates. The high activity for HER was related to the presence of high concentration of MEAH+ at the surface which can act as a proton source. While a higher concentration of carbamate was observed to be present at the surface, the lower partial current density of CO compared to amine free electrolytes ruled out the possibility of any carbamate reduction (Fig. 8).17 | ||
| Fig. 8 Simulated boundary-layer concentration profiles at an applied potential of −1.5 V vs. SHE, where the zero coordinate corresponds to the Ag cathode surface. (a) Comparison of the dissolved CO2 concentration profile in 0.1 M KHCO3 electrolyte with and without 0.2 M MEA additive. (b) MEA, MEAH+, and carbamate concentration profiles in 0.1 M KHCO3 + 0.2 M MEA electrolyte, showing the depletion of MEAH+ and accumulation of MEA and carbamate relative to their respective bulk values. Carbamate concentration is elevated due to the reaction of locally generated MEA with aqueous CO2, causing the result in (a).17 Reproduced with permission from ref. 17, copyright 2023 American Chemical Society. | ||
Thus in different c-CO2RR settings, a similar approach can be taken to estimate the captured CO2-bound adduct concentration at the surface of the electrode and compare it with the partial current density of the reduced product to estimate if there is any CO2-bound capture agent reduction. Physics-based continuum models is therefore a key tool in establishing connections between device level performance metrics (like current density and product selectivity), electrode characteristics, and operational parameters. In c-CO2RR systems, continuum models can provide information regarding polarization behaviour, transport of products and reactants, and limitations in operational lifespan, which when complemented with experimental investigations can expedite the development of c-CO2RR technologies.62
3.3 Two-film theory combined with reaction transport model
Recent investigations by Shen et al. and Kowalski et al. used the rotating cylinder electrode (RCE) setup to study the activity for c-CO2RR.16 Shen et al. looked into different mass transport regimes by changing the rotation speed and observed that the partial current density of CO is linearly dependent with the partial pressure of CO2. They further developed a model for the partial current density of CO under limited mass transport regime of dissolved CO2 to correlate jCO to the amount of dissolved CO2 in the system. Kowalski et al. used the same model to study the activity of c-CO2RR and compared methoxide capture agents to hydroxyls and amines. They also reported dissolved CO2 as the primary species being reduced in c-CO2RR. This model is robust and unique where it measures the partial pressure of CO2 from the headspace in operando to identify the active species getting reduced and allows for quick screening of different activity descriptors. The two main parameters that are relevant to understand the model are (i) the mass film transfer coefficient at the gas–liquid interface and (ii) the mass film transfer coefficient at the liquid–electrode interface.16| CO2(g) ⇌ CO2(dissolved) | (15) |
 | (16) |
The Henry's constant, HCO2 is a function of the temperature, pressure and also the solvent that is used in the system. Kowalski et al. in their study also pointed out that when looking at c-CO2RR systems under different capture agents such as amines and alkoxides, the solubility of CO2 is different in the different solvents and this changes the Henry's constant value to be used in eqn (16). The difference in solubilities in different media must be taken into account while investigating c-CO2RR. This can be achieved through the use of VLE models and multi-scale models.
| Sh = 0.204Re0.59Sc0.33 | (17) |
| JCO2,max = km,CO2CCO2,bulk | (18) |
| jCOmax = 2FJCO2,max | (19) |
 | ||
| Fig. 9 Left panel: Experimental and transport model-based maximum partial current density for the reduction of dissolved CO2 in a solution at equilibrium with a partial pressure of CO2 determined using a gas chromatogram. Experiments containing amines were conducted using 0.099 M KClO4 + 0.001 M KOH. All the experiments were conducted using an electrodeposited Ag catalyst as the working electrode, a Pt counter electrode, and a Ag/AgCl reference electrode. Right panel: Applied potential versus partial CO current density for experimental data in the left panel.16 Reproduced with permission from ref. 16, copyright 2023 Elsevier. | ||
4 Reactor and catalyst design for c-CO2RR
Reactor and catalyst design have been extensively studied in the past for CO2RR, but there is not much consensus on how these parameters play a role in c-CO2RR. Indeed, many strategies have been suggested to improve CO2RR performance. However, the community has now started to recognize that electronic modifications (d-band center tuning), alloying, and interface engineering alone are not sufficient to deliver a scalable CO2RR technology. Jang et al. has recently shown that transport alone has a larger impact in product selectivity during CO2RR on Cu than interface engineering (cation effects Cs+, K+, Na+etc.) or the predominant surface termination (i.e. comparison of pc-Cu, Cu(111), Cu(100), and Cu(751)).64 Much of what has been claimed as catalytic enhancements are not maintained when the electrode areas are increased, indicating that the catalyst has a limited effect on the performance of the CO2RR electrolyzer.65,66 The explanations based on morphology control, d-band center tuning, and interface engineering often emerge as an explanation to an experimental observation in a restricted set of experimental conditions. Reproducibility across labs working on CO2RR is an issue.67 We note that this is not limited to CO2RR, but that this is indeed a widespread issue in electrocatalysis.68 Our purposeful reduced discussion of catalyst optimization strategies in this review originates from our interest in discussing at depth what we consider has been phenomena ignored in the field, that is, the role of transport in electrocatalysis for c-CO2RR applications. Nevertheless, in order to provide a balanced discussion, other optimization strategies that do not focus on the role of transport are also discussed in Section 4.2. Additionally, in this section, we reflect on the effect that reactor geometry has on product selectivity where often different reactors result into different product distribution when using the same catalyst, suggesting reaction kinetics and reactor kinetics are convoluted in most electrocatalytic studies.In this section, we will dive deeper into these discussions to understand how much knowledge from CO2RR can be translated to c-CO2RR, if at all.
4.1 Role of catalyst morphology and geometry
In the past, a bulk of research for CO2RR has mostly focused on selecting catalyst based on their electronic properties and DFT calculations of binding energies of different intermediates. Two main descriptors for CO2RR has been the binding energies of *CO and *H,69,70 but complexities arising from the presence of capture agents raises questions on whether CO2RR descriptors could be translated to c-CO2RR. Recently, Choi et al., in their work showed that catalyst in the presence of amine capture agents can undergo corrosion, so binding energies of the amine to the surface is an important parameter when looking into c-CO2RR.71Early works by Hori et al. was conducted in an H-cell configuration where the CO2 dissolved in the solution is reduced to products.72 This happens in the two-phase solid–liquid interface. Hori broadly divided metals into two categories; (i) CO producing metals like Au, Ag, Cu, and (ii) HCOO− producing metals like Sn, Pb, In, Cd, Hg. To operate at higher current densities reactor configurations were later modified to gas-diffusion electrolyzers which operate at the three-phase boundary of solid–gas–liquid. However, with new reactor geometries researchers did not observe changes in selectivity of the catalysts, meaning the intrinsic activity of these catalysts at the two-phase boundary and the three-phase boundary remained the same. Although early reports suggest that CO2RR catalysts do not work well in c-CO2RR systems, there is still value in working with these catalysts to see if there is a suitable catalyst that can activate the CO2-bound captured complex to make reduction products. The idea is that if a suitable catalyst is identified then there is value in developing a new reactor design like the gas-diffusion electrodes to operate at higher current densities, however it is still likely that in the case of c-CO2RR the reaction will still occur at the two-phase boundary where the catalyst will be in contact with the liquid as in the H-cells.
Although most studies have focused on catalyst activity, recent studies have shown that catalyst morphology and geometry also plays an important role in determining product distribution and tuning them could increase the efficiency of electrochemical systems. For CO2RR, several studies have already demonstrated that morphology of the catalyst plays a role to increase faradaic efficiency however for direct reduction of the captured CO2-bound adduct, there are additional buffer reactions occurring at the interface which makes it harder to delineate the mechanisms. In this section, we will look into the direct reduction of the CO2-bound capture agents from a catalyst perspective, where the two most important things is determining the active species at the surface of the electrode and investigating the effect of the geometry of catalyst on the electroreduction of these CO2-bound capture agents.
The transport of species from the bulk of the electrode to the catalyst surface (external mass transport) and from the catalyst surface to inside the pores (internal mass transport) both influences the product distribution in CO2RR and simultaneously can influence c-CO2RR. Studies that have used porous catalysts to increase the CO2 conversion argued that a porous structure by means of internal mass transport can increase the OH− concentration within the pores and can suitably create a local CO2 environment which increases the CO2 utilization. The pH also plays an important role as a high pH reduces HER by reducing the concentration of protons.73,74 In 1989 Hori et al. showed that a locally high pH at the electrode–electrolyte interface can facilitate C–C coupling to form multicarbon products.75 A highly porous electrode have been argued to enhance this effect creating a very high pH within the pores by means of internal mass transport. Thus studies have looked into different mesoporous, microporous and nanoporous structure for CO2RR. Macropores can shorted the ion diffusion distance by increasing the buffer-volume of the reactions, while mesopores facilitate mass transport properties and micropores help in increasing the active surface area.76
Tuning the morphology of the catalyst can thus affect the local electronic structure by altering the grain boundaries, edges, facets, and corners. This can modify the pH, mass transport, adsorption/desorption kinetics, and local CO2 concentration, at different scales. Different morphologies including nanocavities, mesopores, nanocages, crystalline porous frameworks and porous thin films have been studies in the past for CO2RR.76
Ma et al. using Cu nanowires showed that the structure of the catalyst can regulate the pH causing the changes in the internal transport of the HCO3− and OH− species in and out of the pores, which altered the selectivity leading to more preferred CO dimerization pathway (Fig. 10a).77 Zhuang et al. showed that a Cu catalyst structure with nanocavity can promote the C–C coupling between the C2 and C1 intermediates to form C3 products.78 They observed that a nanocavity could concentrate C2 species inside the cavities through steric confinement, increasing the C3 production inside the cavities (Fig. 10b). Mesoporous structures, mainly carbon-based, were also investigated for CO2RR, as larger surface areas could potentially increase the active sites.79–81 Although pristine carbon is known to be electrochemically neutral, chemical doping using N, B, F, and S atoms have been used to break the neutrality to generate active sites for CO2RR.82,83 Hursan et al. showed that morphological attributes like porosity and pore size can have an affect on the performance of N-doped catalysts on CO2RR.84 They observed that the nonporous N-doped catalyst was the least hydrophobic and produced the highest HER, compared to the porous films which enhanced CO2RR performance. To describe this behavior, they correlated wettability with surface energy. Changes in wetting properties influences the dynamics of gas bubble formation on the electrodes. The non-porous samples can result in large bubbles that can block the active sites of the catalyst for CO2 reduction. They highlight that the release of gas bubbles which are dependent on the wetting properties of the catalyst can bring about changes in mass transport leading to changes in the catalytic current densities. Hydrophobic surfaces enhance gas bubble diffusion and release. Other reports also suggest that the surface hydrophobicity can diminish the catalyst-water contact inducing a high concentration of OH− near the electrode, increasing the local pH and decreasing HER85 (Fig. 10c). The wetting properties of a material are usually determined by the interfacial interaction forces between a solid and the fluid. These van der Waals forces, which are dependent on polarity, dipole moment, structures, and charging states, develop at the solid–liquid interface. The net surface energy then determines the wetting properties of the material.85,86 It has thus been observed that higher surface energy generally leads to more hydrophilic surfaces. However, during electrochemistry, these forces will act under an electric field which can further modify the interaction of these forces. Different studies have reported that the electrowetting properties of electrodes can be adjusted by tuning (i) the dielectric properties of the electrode and electrolyte and (ii) choosing a catalyst PZC such that there is no excess-charge at the solid–liquid interface.85 Therefore, wetting properties have potential to further enhance the performance of c-CO2RR and must be explored during the development of c-CO2RR technologies. Hierarchical porosity have also been investigated by Ni et al. where they used an F-doped hierarchical nanocage structure and compared it to F-doped solid carbon and F-doped carbon with mesopores. They observed that the hierarchical structure possessed the largest surface area among all and also the highest F.E and current density for CO, which was rationalized by increased mass transport and low diffusion resistance from the hierarchical structure (Fig. 10d).87 Dutta et al. showed that porous thin films, like Cu foam, have a difference in product selectivity where they make more C2H4 and C2H6 compared to CO in flat Cu. The catalytic difference was attributed to the presence of (100) facet in porous electrodes which have an intrinsic activity for C–C coupling (Fig. 10e).88 Existing knowledge on CO2RR can thus provide a basis on the activity of catalysts with different geometries for c-CO2RR and should be further investigated to understand how the presence of amines or other capture agents might alter the product distribution.
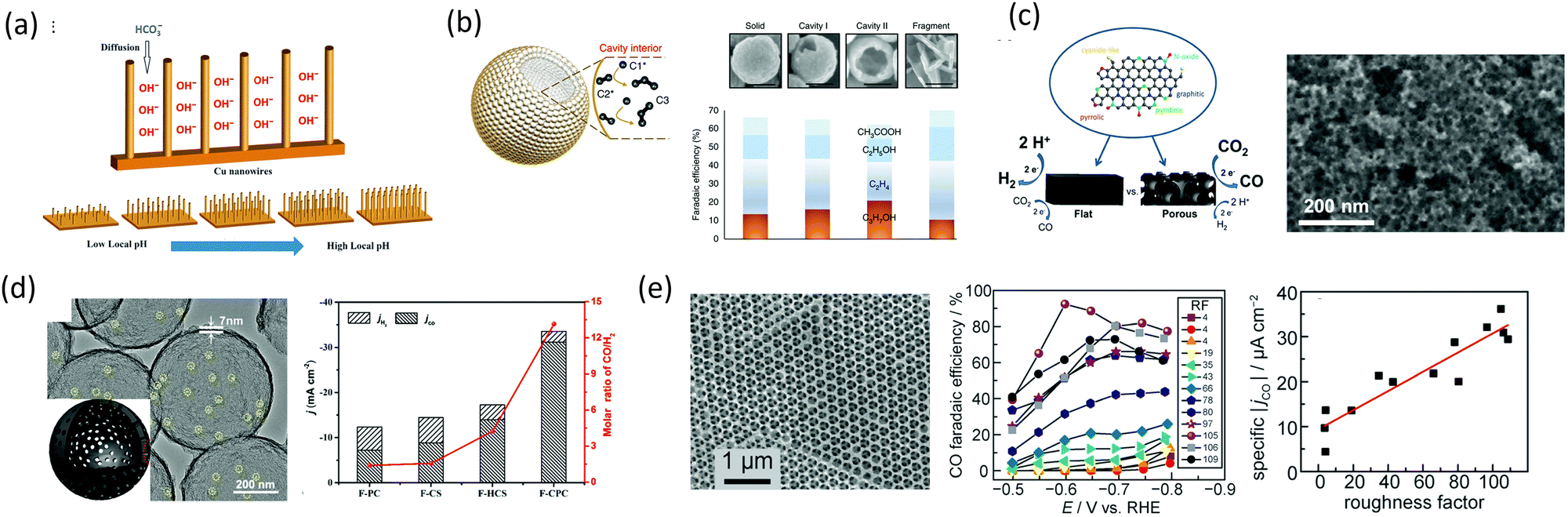 | ||
| Fig. 10 (a) Schematic of the diffusion of electrolytes into Cu nanowire arrays.77 Reproduced with permission from ref. 77, copyright 2016 Wiley (b) energy profile for the formation of the C3 intermediate. Red, grey, and orange balls represent oxygen, carbon, and copper, respectively and schematic showing that the cavity confinement effect boosts C2 species binding and further conversion to C3 and the corresponding faradaic efficiency.78 Reproduced with permission from ref. 78, copyright 2018 Nature (c) schematic showing the activity difference on flat and porous NC and TEM image of NC-27.84 Reproduced with permission from ref. 84, copyright 2019 Elsevier (d) TEM image of F-CPC. The insert shows the schematic structure and CO2RR performance of various catalysts at −1.0 V.87 Reproduced with permission from ref. 87, copyright 2020 American Chemical Society (e) SEM image of an Ag-IO film with CO FEs and ECSA-normalized CO current of Ag-IO films with varied RF.88 Reproduced with permission from ref. 88, copyright 2016 American Chemical Society. | ||
A few studies have been conducted while flowing CO2 in the presence of amines. Although they are not analogous to c-CO2RR conditions, they can help understand the role of amines in electrochemical reduction reactions. Chen et al. reported an increase in formate production in porous In catalyst and an increase in CO production in porous Ag and Zn catalyst in a 30 wt% MEA solution.89 Hossain et al. prepared nano-dendrites of Cu, Au, and Ag on a glassy carbon electrode and investigated CO2RR in a 0.05 M MEA solution, where they observed an enhancement in current densities with the nanostructured catalyst.90
The only study that reports the effect of porosity under c-CO2RR environment is by Kowalski et al. where they investigated the current densities and faradaic efficiencies between sputtered Ag electrodes and electrochemically roughened Ag electrodes (Fig. 11). They found that although c-CO2RR predominantly produces HER, increasing the porosity had an effect in slightly enhancing the CO production. They looked into amines, hydroxyls and methoxides as capture agents and in all of them the faradaic efficiency and jCO of CO increased with porosity. Maximum F.E. CO of ≈1.5% and ≈1% were reported for the electro reduction of methyl carbonate CH3OCOO− and bicarbonate HOCOO−, respectively (Fig. 11a and b). The F.E. CO for ammonium carbamate although increased, the maximum remained very low at ≈0.03% (Fig. 11c). They further normalized their jCO values to the mass transport limited jCO from the flux of dissolved CO2 and observed that the normalized jCO values crossed the limiting transport line for reduction of dissolved CO2 (Fig. 11d and e). They hypothesize that with porous electrodes the local pH within the pores becomes higher than at the entrance of the pores. This high pH suppresses HER resulting in more available sites for CO2 reduction to produce CO. Although the increase in jCO was higher than the calculated limiting value they still reported it as dissolved CO2 reduction. Furthermore, from the partial current density obtained from the different capture agents it was concluded that H2 was mostly produced from outside the pores as no changes in jH2 was observed with the as-sputtered electrodes and the electrochemically roughened electrodes. Whereas the increase in CO was the result of more CO2 being reduced within the pores. Thus internal mass transport and local reaction environment within and around the pores also play a role along with external mass transport in c-CO2RR systems.
 | ||
| Fig. 11 The faradaic efficiency of CO obtained from as-sputtered Ag electrode compared to roughened Ag electrode at different applied potentials (vs. Ag/AgCl) for (a) KCH2OCO2 (methyl carbonate). (b) KHOCO2 (bicarbonate) (c) NH4–NH2CO2 (ammonium carbamate). Comparison of (d) jCO and (e) jH2 between the sputtered Ag electrodes (solid dots) and the roughened Ag electrodes (hollow triangles). (f) The jCO is further normalized against the maximum flux of CO2 and it is observed that the rough electrodes are able to cross the transport-based limiting jCO which was rationalized from the local release of CO2 within the pores.12 Reproduced with permission from ref. 12, copyright 2024 American Chemical Society. | ||
In electrocatalysis, recent investigations have deployed the use of the Thiele modulus to understand the relative balance between kinetic and mass transport resistances inside catalyst pores. The Thiele modulus in catalysis is used to describe the relation between reaction rates and diffusion rates. For large values of Thiele modulus, the overall reaction rate is constrained by diffusion resistance, and the reaction predominantly occurs within the outer catalyst layers. Conversely, when Thiele modulus values are small, the overall reaction rate is restricted by the catalyst volume, specifically the total accessible pore surface area. Traditionally, this concept has been mostly used for fuel cell reactor development across multiple length scales, however its use in other electrochemical systems is somewhat limited. Recently, Thiele modulus analysis have been explored by a few researchers to understand the effectiveness factor of their catalyst. Kim et al. and Wang et al. used Thiele modulus modeling in gas diffusion electrodes for CO2 reduction and also developed analytical models to describe catalyst effectiveness.91,92 Similarly Wan et al., investigated the potential dependent Thiele modulus models to probe the utilization of pores as a function of catalyst particles and reactant properties. Using a 1D reaction diffusion model through the pores they developed the Thiele modulus model and delineated external and internal mass transport to effectively outline design principles for the electrocatalysts.93 The Thiele modulus can also be defined for c-CO2RR to understand and quantify the effectiveness of morphology and catalyst geometry. It will further be effective in understanding the relative timescales of reaction and diffusion within the pore of the catalyst to rationalize the increased CO2 reduction products observed within the pores under c-CO2RR conditions.
Since all studies point towards the free CO2 as the active species for reduction, the knowledge existing for CO2RR on the effect of porosity can further be used to understand c-CO2RR mechanisms. However, one study by Ma et al. reports the direct reduction of carbonate to formate which is discussed in detail in Section 5. The local OH− concentration dynamically influences the buffer reactions, and when imagined in the context of c-CO2RR there are additional buffer reactions that make these systems more complex. Suter et al. using a finite element model investigated different porosity and species concentration within the pores to predict CO and H2 current densities. The model predicts that OH− and CO32− concentration within the pores are higher than at the surface resulting in a high alkaline pH, which is more obvious in a thicker film, as can be seen from Fig. 12. Predicted CO FEs and current density increased with increasing RF factor which also aligned with experimental findings.94 Thus, a high alkaline pH is understood to suppress HER within the pores allowing more available sites for CO2 reduction. The same study also coupled the effect of pore diameter and film thickness to investigate the changes in CO FE and observed a linear dependency of roughness factor (RF) with film thickness and non-linear dependency of RF with pore diameter. They concluded that multiple combinations of pore diameter and film thickness can give similar faradaic efficiency. For instance, a FE CO of 90% can be achieved from a pore diameter of 200 nm and film thickness of 10 μm or a pore diameter of 100 nm and film thickness of 7 μm. Thus it was concluded that roughness factor alone is not enough to characterize a porous film.95 Experimentally, it is difficult to determine the local OH− or CO2 concentration inside the pores. A recent study by Bohme et al. developed a pOH sensor where a confocal laser scanning microscopy technique was used to map the pOH around the electrode during CO2RR. They were able to experimentally verify that the OH− concentration is high within the pores compared to outside the pores as suggested by many other studies mentioned before.96 When extending it to c-CO2RR, the high alkalinity of the pores can also suppress HER and promote CO2 reduction, as has been observed by Kowalski et al. through their experimental findings. Furthermore, the additional buffer reactions of amine deprotonation and carbamate consumption is also affected with changes in local pH that contribute to altering the selectivity of the products formed. Thus, it is important to be critical about all these factors while investigating different activity and stability descriptors for c-CO2RR.
 | ||
| Fig. 12 (a) Concentration profiles along the z-axis, where z = 0 is the innermost point inside the pore and z = 1 is the entrance of the pore, of OH−, CO32−, HCO3− and CO2versus their bulk concentration at a potential of −0.8 V vs. RHE. (b) Partial CO current density and (c) CO faradaic efficiency for Ag-IO with pore diameters of 150–400 nm and film thickness of 3, 6 and 15 μm.95 Reproduced with permission from ref. 95, copyright 2019 Royal Society of Chemistry. | ||
4.2 Tuning catalyst properties for c-CO2RR
Designing catalyst for c-CO2RR that can tune (i) electronic properties, (ii) binding properties, and (iii) intermediate concentration at the electrode–liquid interface will be necessary as the field evolves. Electronic properties in particular has been shown to play an important role in modifying the binding properties of key intermediates in CO2RR and can have the same effect on c-CO2RR. The upper shift of d-band energy level results in increased binding strength of the adsorption intermediates. This understanding has been used at different scales to tune the selectivity of reduction products, from single atom catalysts (SAC), to nanoparticles, and surfaces with different terminations.97Different SACs have been designed with different metal centers like Ni, Fe, Co, In, Bi, and Sn for producing CO and HCOO−.97–99 These dispersed metal sites along with their coordination environment create M–N4 moieties of superior performance over their metal counterparts. In this unique assembly the electronic structure of the metal center is modified by the adjacent coordination environment and this have been shown to modify the catalytic activity, selectivity and stability. Further optimization of electronic structure has been studied by looking into vacancy defects to generate the Ni–N3–V moiety or through heteroatom doping to generate Ni–N3P. Both of these approaches have been suggested to have superior performnace for c-CO2RR compared to the Ni–N4.100,101
For nanoparticles it is generally observed that the electrocatalytic performance is determined by the amount of undercoordinated sites where the CO2RR intermediates preferentially bind and is dependent on the ratio of surface to bulk atoms. Gu et al. observed that the Ni-based nanoparticles had lower faradaic efficiencies compared to Ni SAC.102 However, this behavior varies depending on the metal that is being used as the catalyst. Huang et al. investigated the production of HCOO− on In based catalysts and found that In2O3 nanocrystals which are 15 nm in diameter were more active than the nanocrystals which are 200 nm or In foil, which they attributed to the increased uncoordinated sites with lower particle size. On reducing the nanoparticle size to 5 nm they observed that the partial current densities for HCOO− decreased. This shows that smaller may not be always better in catalyst design and that a deep understanding of structure–property relationship is important when designing catalysts.103
Strategies like alloying have also been explored in numerous CO2RR studies in the past to tune the spatial distribution of reduction intermediates and enhance the selectivity towards certain products.104–106 For instance, Gao et al. designed a CuAg catalyst to improve the selectivity of CO2 reduction to ethylene. Using operando Raman spectroscopy they showed that the bimetallic catalyst produces more *CO binding sites that can then hydrogenate at neighboring sites and couple to make C2 products. This is often coupled with interface engineering strategies, as discussed in Section 5, to modify the local concentration of the active species and its intermediates.107
Although, these strategies are useful to design catalyst for improved activity and selectivity, however it is often argued for scalable technologies transport plays a more significant role than the catalyst itself. Hursan et al. in their study highlighted on this effect. Using N-doped carbon they designed electrodes of different porosity and depending on the pore size they observed different selectivity towards CO and H2 formation in CO2RR. They concluded that porosity modifies mass transport and consequently the CO2 adsorption properties that primarily governs CO2RR. Therefore, morphological attributes that determines transport affect the catalytic performance and needs to be given more importance while designing large scale electrolyzers.84
4.3 Engineering reactor design to tune mass transport and kinetics
The electrolyzer design and cell configuration are important considerations in c-CO2RR. The electrolyzer architecture and the operating conditions (e.g. temperature, pressure, potential, current) influence the transport of the active species and products between the bulk of the solvent and the electrode surface, as well as the temporal speciation of the capture agent and CO2 adducts. The reactor design will also determine the rate of equilibration between the species in the gas phase and those dissolved in the liquid. Recent studies on c-CO2RR have been carried out in different reactor designs which have different mass transport characteristics. The commonly used electrochemical reactors, the H-cell and the flow cell configuration, have also been used extensively for CO2RR in the past and recent work has shown that hydrodynamics in the cell indeed determine the product distribution obtained.63,108 To date, very few works have been reported for c-CO2RR but it can be anticipated that the type of reactors used will also impact the product distribution in this reaction. In some of the c-CO2RR systems discussed in this section, the membrane separating the cathode and anode compartments is used to control the generation and transport of protons that shift the equilibrium and release CO2 in the proximity of the catalyst (reverse direction of eqn (5)–(7)). Electrolyzer design is thus tightly tied to the performance of c-CO2RR systems and needs to be carefully studied.This section discusses four types of electrochemical cells (H-cell, compression, zero gap, and rotating electrode) that have been used for activity benchmarking in c-CO2RR, as shown in Fig. 13. Leverick et al., used an H-cell to investigate amines of varying pKa and sterics and reported the differences in faradaic efficiencies for producing CO as a function of pH and temperature.61 They observed that the partial current density of CO had a weak amine dependence but had a first order dependence with the partial pressure of CO2 in the system, which suggested that the active species undergoing reduction is the dissolved CO2 and not the amine. Safipour et al., investigated CO2RR in the presence of mono-ethanol amine (MEA) in a sandwich-type compression cell configuration (Fig. 13b) and observed a maximum partial current density for CO of 5 mA cm−2, which decreased with the increasing concentration of MEA. With the help of a 1D continumm model they report that the addition of MEA increases the concentration of MEAH+ which acts as a proton source for H2 and reduces the local concentration of CO2, which in turn reduces the partial current density of CO, with no evidence of any carbamate getting reduced.17 To increase the amount of reduced products from CO2 capture solutions, the Berlinguette group designed the bicarbonate electrolyzer as shown in Fig. 13c, where they used a bi-polar membrane or a proton exchange membrane in a membrane electrode assembly for supplying protons and locally release CO2 from bicarbonate in the solution and promote CO2RR.18,19 This increased the flux of CO2 to the surface of the electrode and high faradaic efficiencies for CO was achieved, but at a penalty of high input energy. The thermodynamic minimum energy for converting CO2 to CO is −1.33 V whereas the input voltage required to reach a current density of −100 mA cm−2 was −4.4 V in their electrolyzer, while lab-scale CO2RR electrolyzers have already demonstrated to achieve −200 mA cm−2 at cell voltages below −3 V.109,110 A similar setup was also used by Kim et al. to study the reduction of carbamates using single atom Ni catalyst and a high CO selectivity was achieved with FE of ≈69%.15 Shen et al., using the RCE cell investigated different mass transport regimes by changing the rotation speed and observed different faradaic efficiencies and partial current densities of CO, and further concluded that the produced CO is from the dissolved CO2 within the system. The RCE cell system is unique in that it measures the partial pressure of CO2 in the headspace of the cell, which is in a pseudo-equilibrium with the solution. The partial pressure of CO2 can be used to estimate the amount of free and dissolved CO2 in the bulk of the electrolyte and facilitates the discrimination between different carbon sources during c-CO2RR.16
 | ||
| Fig. 13 (a) Conventional H-type cell configuration used by Leverick et al. to study the jCO, jH2 and F.E. CO and FE H2 of c-CO2RR at different pH and applied potentials for amines of different pKa and sterics.61 Reproduced with permission from ref. 61, copyright 2023 American Chemical Society (b) a schematic of the compression cell type configuration used by Safipour et al. to investigate the species distribution near the surface of the electrode while studying CO2RR in the presence of MEA.17,111 Reproduced with permission from ref. 17 and 111, copyright 2023 American Chemical Society and 2012 Royal Society of Chemistry (c) the zero-gap membrane electrode assembly used as the bicarbonate electrolyzer by Li et al. and Lees et al. to release CO2 in the bulk of the solution by supplying protons to increase the FE CO.18,19 Reproduced with permission from ref. 18 and 19, copyright 2020 American Chemical Society and 2019 Elsevier (d) a schematic of the gas-tight rotating cylinder electrode cell used by Shen et al. to investigate mass transport limited conditions for c-CO2RR.16,63 Reproduced with permission from ref. 16 and 63, copyright 2023 Elsevier and 2022 Wiley. | ||
Transport of mass, heat, and charge affect the kinetics in c-CO2RR and therefore must be considered while designing electrolyzers for this reaction. A cell design with a thick hydrodynamic layer, like the H-cell, could slow down the local mass transfer of the species resulting in lower faradaic efficiency.112 Also, as the dominant product of c-CO2RR is H2, the design must include considerations for ways to suppress HER. For instance, a cell design with very good mass transport, like the RCE cell, promotes HER as along with CO2 and carbamate they also increase the flux of the bicarbonate and the ammonium cations to the electrode which act as a proton source.12 Furthermore, cell resistance and uniform current distribution are some other factors that can potentially affect product distributions in electrochemical systems, especially for organic solvents with lower conductivity.
The availability of the active species at the surface of the electrode determines the production efficiencies. Thus designing the catalyst geometry to efficiently increase the contact of the active species can enhance c-CO2RR. For instance, fundamental mass transport studies have mostly been carried out using the RDE/RRDE (rotating disk electrode/rotating ring disk electrode),113 however RDE setups present challenges of having low surface area, with poor hydrodynamics resulting in bubble formation at the tip of the electrode and concentration gradient along the radial direction of the disk.114 In comparison, the rotating cylinder electrode proposed by Jang et al. had a higher surface area, which leads to the availability of a higher concentration of CO2 near the surface of the electrode, which was eventually used for mass transport studies.63
CO2RR suffers from slow kinetics and is rapidly limited by mass transport of CO2 in most conventional cells used for fundamental studies. Similar difficulties can also be anticipated for c-CO2RR if species relevant to the catalytic process are present in small amounts, and thus it is important to maintain a high flux of the active species to the electrode. Changing the residence time of reactants and reaction intermediates can alter the product selectivity without altering the intrinsic kinetics of the electrode itself, and distinguishing the relative timescales for transport and reaction at the catalyst scale is increasingly necessary.63
5 Heterogeneous reaction mechanism
Although reaction mechanisms and reaction intermediates are known for CO2RR, little is known about c-CO2RR. It is unclear why hydrogen is the major product and what happens within the double layer. It is a common consensus that protonated amines act as the extra source of proton to facilitate hydrogen production, but how the carbamate is transported to the electrode–electrolyte interface is not known.61 Also, how the local pH or the buffer reactions play a role in such systems is not well-understood. To date c-CO2RR studies have mostly looked into the formation of CO and HCOO− as products and thus, delineating the different possible pathways at the surface of the electrode is important to gain deeper insights into the mechanisms relevant to c-CO2RR.Shen et al. proposed the competitive mechanism of electrochemical carbamate reduction on Ag as shown in Fig. 14. They proposed that cations present in the system help in bringing the carbamate to the electrode and can facilitate the electron transfer from the electrode to the carbamate for its further reduction. The carbamate can either decompose through a C–N bond cleavage to form CO2 and the amine RNH2 (blue pathway) or a direct proton–electron transfer on the carbamate to form the *RNHCOOH intermediate (orange pathway, *indicates a chemisorbed species on the Ag electrode). Once CO2 is formed, it can chemisorb on the catalyst surface as shown on the blue pathway and reduced subsequently to *CO via two proton–electron transfer steps, which is the typical CO2RR pathway on Ag.115 In contrast, the orange pathway directly reduces the carbamate, initially keeping the C–N bond formed. After forming *RNHCOOH, the C–N bond can break to generate *COOH on the surface (green path) or it can further reduce by proton–electron transfer to *RNHCO and water (orange path). Consequently, *RNHCO undergoes an additional proton–electron transfer on N and C–N bond cleaves to form *CO and restore the amine simultaneously. In short, along the pathway, C–N bond cleavage and further reduction of CO2 compete with the direct proton–electron transfer to O in the –CO2 group of the carbamate and C–N cleavage at a later step, thus creating three possible bifurcating pathways.16
 | ||
| Fig. 14 Proposed mechanism for the competition between electrochemical carbamate decomposition combined with CO2 electroreduction (blue pathway) and carbamate direct reduction (orange pathway). The C–N bond cleavage can occur at three possible steps in the mechanism. K is the alkali metal potassium.16 Reproduced with permission from ref. 16, copyright 2023 Elsevier. | ||
Kowalski et al. in their work describes a generic reaction network with different possible elementary pathways that could occur in c-CO2RR systems. They use KRCO2 as the captured CO2 adduct and study the different pathways that can occur when the R is CH3O (methyl carbonate), NH2 (carbamate) or OH (bicarbonate) as shown in Fig. 15.12 They show that after the adsorption of the KRCO2 on the Ag electrode four different pathways can occur. The first route is the cleavage of R–C bond by protonation from the proton source XH (XH is either the solvent or the capture agent) producing adsorbed CO2, RH and KX. This pathway then leads to the usual CO2RR pathway with the first proton-coupled electron transfer (PCET) producing *COOH and the second producing *CO and water. The second route modifies the order of PCET and protonation, starting from a PCET assisted cleavage of R–C bond followed by a second PCET to form *COOH. The final C–O bond cleavage in this pathway is chemically assisted by the proton transfer from XH. The third route starts with a PCET at an O atom forming KRCOOH from where it can either go through a PCET step to cleave C–OH bond to form RCO and then the R–C bond is cleaved in a final proton transfer step to make CO, water, and KX, or it can cleave the R–C bond first by PCET or proton transfer followed by C–O bond cleavage. The fourth route treats the first step as chemical protonation using the XH proton source, and the second and third steps can be electrochemical PCET. Thus, these different pathways provide an overview of the mechanisms that could exist for different capture agents in different c-CO2RR setups.
 | ||
| Fig. 15 (a) Reaction scheme for the reduction of the captured CO2 with a general capture agent (R–H, examples being NH2–H, CH3O–H, and HO–H). R represents the deprotonated capture agent (NH2 for NH3, OCH3 for methanol, or OH for H2O), XH represents a general proton source. In methanol solvent, the proton sources in our study include methanol and NH4+, written formally as NH4ClO4 to be in the XH form. In the water–amine solvent, this includes H2O, KHCO3, and NH4+. * represents the catalytic Ag site. All species containing * are absorbed on the site. Chemicals in red text represent the products evolving in the solvent at each step. The two most favored pathways from our calculations both initiate with a proton-coupled electron transfer and are highlighted in blue (initial R–C cleavage in the CO2 complex) or in red (final R–C cleavage). Reaction schemes for (b) CO2RR, and (c) HER are also recalled with similar notations.12 Reproduced with permission from ref. 12, copyright 2024 American Chemical Society. | ||
Recently Ma et al. reported the direct electroreduction of carbonate to formate using a Cu catalyst. Through DFT calculations they discussed four possible reaction pathways that can occur at the electrode. In Fig. 16, path 1 shows the adsorption of the CO2 through the O sites in the *OCO− bidentate form, in path 2 a molecular CO2 is attacked by a hydride to make formate, path 3 is a typical CO2RR pathway where the adsorbed CO2 undergoes a hydration to produce formate, and path 4 shows the direct carbonate reduction mechanism. They further discuss that the dominant pathway is a function of potential. At lower potentials between 0.0 and −0.4 V vs. RHE, the dominant species is *CO32− with only *H competiting for adsorption, while *CO2− and *OCO−* adsorb at potentials more negative than −0.4 and −0.8 V vs. RHE, respectively. At potentials more negative than −0.8 V vs. RHE, it is difficult to adsorb *OCO−*, and also for path 2 the *H− intermediate is unstable and mostly dominated by HER. Thus the only relevant path for formate production is path 3 in medium potential ranges. At larger potentials it will converge to CO, which can further undergo chemical hydration to make formate. But, at potentials more negative than −0.8 V vs. RHE the authors discuss that the *CO is more likely to reduce to C1 and C2+ products limiting the further reduction to formate. However, in their study formate production pathway corresponds to path 4, which is through the direct CO32− reduction. The first step in this pathway is to hydrogenate the adsorbed *CO32− into *HCO3−, which further reacts with H to give formate, while OH− is eliminated. In path 4, their proposed mechanism does not form CO which eliminates the possibility of forming any C2 products, as these products are primarily formed via CO–CO coupling. It provides a comprehensive insight into the heterogeneous mechanisms that can occur in c-CO2RR systems, especially for the formate producing pathway.116
 | ||
| Fig. 16 (a) Reaction network of eCO2RR and carbonate reduction mechanisms leading to HCOOH. The consensual eCO2RR mechanisms to formate start by ηO,O *OCO*− (path 1) or by *H− (path 2) adsorptions. Additionally, path 3 leads to *CO which can further evolve to HCOOH by hydration (path 3.1). *COOH can also evolve to HCOOH (path 3.2). Path 4 is the newly proposed carbonate mechanism that can have contributions from path 3 through the conversion of the *COOH intermediate (path 3.3). (b) Range of potential stability of different intermediates.116 Reproduced with permission from ref. 116, copyright 2023 American Chemical Society. | ||
The DFT studies can be further supported by operando analysis, including Raman, FTIR, UV-vis. For example, to experimentally verify the reduction of carbonate, Ma et al. mounted a carbonate complex, Cu2(CO3)(OH)2, on a glassy carbon working electrode and studied the reduction with in situ Raman spectra. They observed that copper hydroxide bands of Cu2(CO3)(OH)2 gradually disappear while the carbonate peaks become sharper. They concluded that the Cu2(CO3)(OH)2 was first converted to carbonate. After 900 s of electrolysis, they observed that the carbonate band disappeared, and they attributed this to the direct reduction of carbonate on Cu electrode. They also observed that after electrolysis the color of the electrode changed from green to brown which is the color of metallic Cu. They also used the same Cu2(CO3)(OH)2 mounted working electrode in a H-cell with a solution of 0.1 M KOH and they observed 1.01% of formate faradaic efficiency. Using this data, they solidify their claim of direct carbonate reduction. They describe the low faradaic efficiencies of formate as a result of the carbonate species detaching from the electrode driven by high HER activity. They also acknowledge that by introducing KOH and flowing N2 in their system they create conditions where the solution is free of dissolved CO2 which allows the study of carbonate reduction. In an analogous experiment with PbCO3 they observed a faradaic efficiency of 0.89% for formate. Their study concluded that PbCO3 can also be reduced in a similar mechanism to formate.116
Choi et al. investigated different transition metals, Ag, Au, Cu, and Sn, for direct reduction of ammonium carbamate. The binding energies of the protonated ammonium cation (ΔENH4+, solid lines) and CO2-captured carbamate anion (ΔEH2NCO2−, dotted lines) were plotted as a function of potential for Ag, Au, Cu, and Sn as shown in Fig. 17. They investigated the potential of zero charge (PZC) of all the metals, which influences the electrosorption properties in electrocatalysis, and found that all the PZCs are more positive than the experimental applied potentials (black triangles). Therefore, they hypothesize that the solvated NH4+ are preferentially accumulated on the surface of the electrode while the H2NCO2− are repelled due to its negative charge. They further discussed that the binding energies ΔENH4+ is stronger than ΔEH2NCO2− at the experimentally applied potentials, which limits the production of carbon based products.71 Thus, NH4+ will not only block the active sites but also serve as a proton source to promote HER, leading to the predominant production of hydrogen on all metals. Bringing the captured CO2 to the electrode surface while limiting HER activity from the capture agent is a major roadblock in c-CO2RR that needs to be overcome.
 | ||
| Fig. 17 The binding energies of ammonium cation NH4+ (ΔENH4+, solid lines) and carbamate anion H2NCO2− (ΔEH2NCO2−, dotted lines) as a function of potential for Ag, Au, Cu, and Sn. The potential of zero charge (PZC) for each metal is plotted with a vertical dash-dotted line. The applied potential to reach −12 mA cm−2 in the c-CO2RR experiments conducted by Choi et al. is marked with a triangle on the respective ΔENH4+ curve.71 Reproduced with permission from ref. 71, copyright 2024 American Chemical Society. | ||
Thus, tuning the local reaction environment becomes particularly important for c-CO2RR, which can provide a sufficient supply of the active species while suppressing the unwanted HER. Along with the bulk speciation, the local speciation can also vary with CO2 loading, temperature, pH, electrochemical operating conditions and the cations, as shown in Fig. 18. The common ways deployed until now to alter the reaction environment in the presence of amine capture agents is by (i) changing the types and concentration of the amines, (ii) addition of alkali cations, (iii) increasing the temperature, or (iv) incorporating surfactants to suppress HER.
 | ||
| Fig. 18 Conceptual local reaction environment for CO2 loaded amine solutions. Speciation at the gas–liquid interface, electrolyte bulk and liquid–electrode interface.16 Reproduced with permission from ref. 16, copyright 2023 Elsevier. | ||
Abdinejad et al. used MEA, ethylenediamine (EDA) and decylamine (DCA) with a Cu electrocatalyst in 0.1 M NaClO4 solution for CO2RR and observed that EDA gives the highest faradaic efficiency of CO which they attributed to the presence of two amine molecules that increases the capture efficiency of the system (Fig. 19a).117 Similarly Shen et al. studied the activity for c-CO2RR with ammonium carbamate (AC) and MEA using a electrodeposited Ag catalyst and observed similar faradaic efficiency of CO for both capture agents.16
 | ||
| Fig. 19 (a) Faradaic efficiency of CO and H2 over Cu catalyst in 0.1 M NaClO4 solution with MEA, ethylenediamine (EDA), and decylamine (DCA) at −0.78 V vs. RHE.117 Reproduced with permission from ref. 117, copyright 2020 American Chemical Society (b) comparison of equilibrium population and concentration of alkali and tetrabutylammonium (TBA+) cations in CO2-loaded 0.1 M EEA in DMSO.118 Reproduced with permission from ref. 118, copyright 2022 Royal Society of Chemistry (c) faradaic efficiency and applied potential during the electrochemical reduction of a 0.7 M MEA electrolyte with an Ag electrocatalyst at a fixed current density of 4 mA cm−2 at an electrode rotation speed of 800 rpm.16 Reproduced with permission from ref. 16, copyright 2023 Elsevier (d) product distributions of CO2 reduction over indium catalyst at −0.8 V vs. RHE in CO2-saturated 30 wt% MEA aqueous solution with different concentrations of CTAB surfactant.89 Reproduced with permission from ref. 89, copyright 2017 Wiley. | ||
Few studies have investigated the addition of alkali cations as a way to increase selectivity towards CO2 conversion. Monteiro et al. investigated CO2RR on Cu, Ag, and Au and showed that the presence of large alkali cations (K+ or Cs+) in particular can undergo weak hydration spheres that can be concentrated at the electrode surface and stabilise the CO2− intermediate via local electric field effect.119 In an amine containing solution it can further enhance charge transfer and destabilise the formation of carbamate to enhance the C–O bond cleavage. Khurram et al. reported a high CO current density with alkali cations in a solution of 2-ethoxyethylamine (EEA) in dimethylsulphoxide (DMSO) for CO2RR which depended on the size of the cations with K+ < Na+ < Li+, and attributed the behaviour to the unstable carbamate formation in the presence of large cations with easier desolvation and rapid pairing kinetics (Fig. 19b).118 As observed by Khurram et al. the size of the cations has an effect on the carbamate formation. With 1H NMR studies they showed that the proportion of carbamate formed in their solution was a function of the alkali cation present in their electrolyte. The Lewis acidity of the cations was found to dictate the amount of ammonium cation that will be formed. For alkali cations, the Lewis acidity decreases in the order Li+ > Na+ > K+, and thus it was found that the stronger Lewis acid, Li+, associates more strongly with the initial carbamic acid driving the reaction faster to the formation of carbamate. The interaction of the alkali cation with carbamate thus played a role in the binding of the cations to the carbamate anion. DFT studies also showed that the reaction of the carbamate anion with the alkali cations is spontaneous in decreasing order of Li+ > Na+ > K+. The type of anion present in the electrolyte did not play any significant role in the formation of carbamate. Larger cations like K+ and Cs+ have been reported to be able to increase the availability of CO2 near the surface of the electrode in CO2RR studies. The measurement of interfacial CO2 concentration for different alkali metal cations through in situ ATR-SEIRAS have also shown that Li+ has the highest CO2 concentration and K+ has the lowest CO2 concentration.120 This is primarily because the hydration sphere of cations inversely scales with the size of the cations where Li+ > Na+ > K+ > Cs+ which changes the local buffering capacity by changing the interfacial pKa.121 In contrast, for c-CO2RR larger cations can potentially help in reducing the local pH of the system near the electrode, enabling the local release of CO2 near the electrode from the captured-CO2 adduct. This can help in reducing HER and promote c-CO2RR activity. In current c-CO2RR studies the local pH is alkaline and thus tuning the cations to maintain pH close to the pKa of the capture agents can help prevent the deprotonation of the amines and the shifting of equilibrium reactions locally.
Temperature studies were also investigated for c-CO2RR and it was seen that higher temperatures increases the faradaic efficiency of CO formation. Shen et al. and Kim et al., investigated the temperature dependency and reached faradaic efficiency of ≈20–40% at higher temperatures which they attributed to the shift in equilibrium to have more free CO2 in the system (Fig. 19c). The effect of surfactants on CO2RR has also been studied in 30 wt% MEA by Chen et al., as they are known to reduce HER activity by blocking the active sites. Using CTAB they show that they can boost CO2 conversion to formate and CO, primarily through HER suppression. They further show that a high concentration of CTAB (>0.1 wt%) helps in producing CO while a low concentration (0.01 wt%) was sufficient to promote CO in indium electrodes. The highest formate faradaic efficiency of 45.4% and CO faradaic efficiency of 17.0% was achieved when combined with porous In electrodes (Fig. 19d).89
Although these techniques have been mostly investigated under CO2RR conditions, the same ideas can be translated to c-CO2RR conditions. The interactions at the liquid–electrode interface can be tuned to increase the selectivity towards CO. The trimethylammonium alkyl tail in surfactants like CTAB can align itself at the surface of the electrode creating hydrophobic interactions that suppresses HER in c-CO2RR studies.122,123 Similarly different sizes of alkali cations can also be combined with surfactants to alter the selectivity.124 Larger cations like Cs+ have been reported to be able to maintain a near neutral pH at the surface of the electrode increasing the availability of CO2 in CO2RR studies.125–127 In current c-CO2RR studies the local pH is alkaline and thus tuning the cations to maintain pH close to the pKa of the capture agents can help prevent the deprotonation of the amines and the shifting of equilibrium reactions locally. The use of ionomer coatings have also been studied to tailor microenvironments by controlling water and ion transport through the polymers. Ionomers like Nafion and Sustanion has shown that they can maintain a high local pH and CO2 concentration to increase C2+ products with Cu catalyst and CO with Ag catalyst.128–130 In addition, pulsed electrolysis can also be investigated to increase CO selectivity.131 The challenge in c-CO2RR systems is to bring the negatively charged carbamate species close to the cathode, which could be overcome by the pulsing technique. It can also reduce the local pH during the anodic cycle to shift the equilibrium and generate more carbamate species near the electrode that can potentially undergo reduction reactions.
6 Effect of different capture agents on c-CO2RR
Different capture agents behave differently under c-CO2RR conditions. So far only the changing binding energies and the CO2 loading capacity have been investigated,16,61 however it needs to be understood that the mass transport conditions, thermodynamics of the captured-CO2 adduct species and vapor–liquid equilibrium in different capture agents change at different conditions which needs due consideration. In theory, the best capture agent will be the one that has the minimum thermodynamic barrier to get reduced to products, but species with different equilibrium constants will have different rates of making CO2 in the bulk of the solution that will further influence the products that we see. For instance, as shown in Fig. 20, from a thermodynamic standpoint it can be observed that CO2 binding to the amines are the strongest which is around −60–80 kJ mol−1 followed by alcohol based capture agents −20 to −25 kJ mol−1, followed by bicarbonates −13 to −18 kJ mol−1.31,45 Thus when we talk about c-CO2RR, a larger thermodynamic barrier needs to be overcome while going from the carbamates to reduced products compared to the alkoxide based capture agents or the hydroxyl based capture agents. Furthermore, Appel et al. in their work showed that the partial pressure of CO2 also determines the free energy of the CO2-bound adduct and dictates the thermodynamic potential required to make reduced products.132 They further discuss that if the capture agent has the ability to bind CO2 stronger than what is required from a specific dilute stream then the process could lead to inefficiencies from the excess binding energy. Thus the influence of these different binding energies on c-CO2RR is important to understand for future research in c-CO2RR. | ||
| Fig. 20 Conceptual thermodynamic free energy change comparing c-CO2RR and CO2RR of amine, hydroxyl and alkoxide capture agents. The free energy of capture of a dilute stream of flue gas in amine is −60 to −80 kJ mol−1 (shown in red), in alkoxide is −20 to −25 kJ mol−1 (shown in green), and in alkali hydroxyls is −13 to −18 kJ mol−1 (shown in grey).31,45 The extra steps of release and compression in CO2RR (shown in blue lines) are responsible for the inefficiencies in the conventional CO2RR system. Although the barriers to go from captured-CO2 to reduced products are the same through the CO2RR and the c-CO2RR paths, the c-CO2RR process (shown in orange line) can facilitate the conversion in a one step process reducing the inefficiencies in the capture and conversion process. | ||
To investigate these changes, Kowalski et al. used methoxide as a capture agent to determine its activity for c-CO2RR and compared it to hydroxyls and amine capture agents using a silver electrocatalyst. Through DFT calculations they show that the nature of proton source and its pKa has a significant impact on the onset potentials for c-CO2RR. They hypothesized that a proton source with a low pKa and use of a solvent like methanol which has a low dielectric constant can improve c-CO2RR while penalizing the CO2RR. However, on Ag(111) surfaces they found that HER has a lower onset potential than CO2RR or c-CO2RR Fig. 21a–f, which is the dominant product also seen experimentally. As can be seen from (Fig. 21g and h), they measured the partial current density of CO and H2 as a function of potential for all the three capture agents. In all cases, the partial current density of CO observed was very low with H2 being the dominant product. For the ammonium carbamate, negiligible amounts of CO were observed as it was more selective for H2 evolution. The authors further normalized the partial current densities of CO observed to the limiting partial current density of CO to quantify the activity for c-CO2RR and determine the active species undergoing reduction. Using the method described in Section 3.3, they found that in all cases it was the free dissolved CO2 that is getting reduced (Fig. 21i). They also discussed that alcohol based capture agents are promising as the only proton source in such systems is the alcohol itself compared to more Bronsted acidic HCO3− or NH4+. Thus it can suppress HER which is the bottleneck for c-CO2RR systems (Fig. 21b).12 The work then needs to be combined with new catalyst design, moving away from Ag or other transition metals that are typically known for its good activity in CO2RR. Catalysts that are known to suppress HER to improve the activity for c-CO2RR are thus needed along with the ability to bring the negatively charged carbamate species to the surface of the electrode.
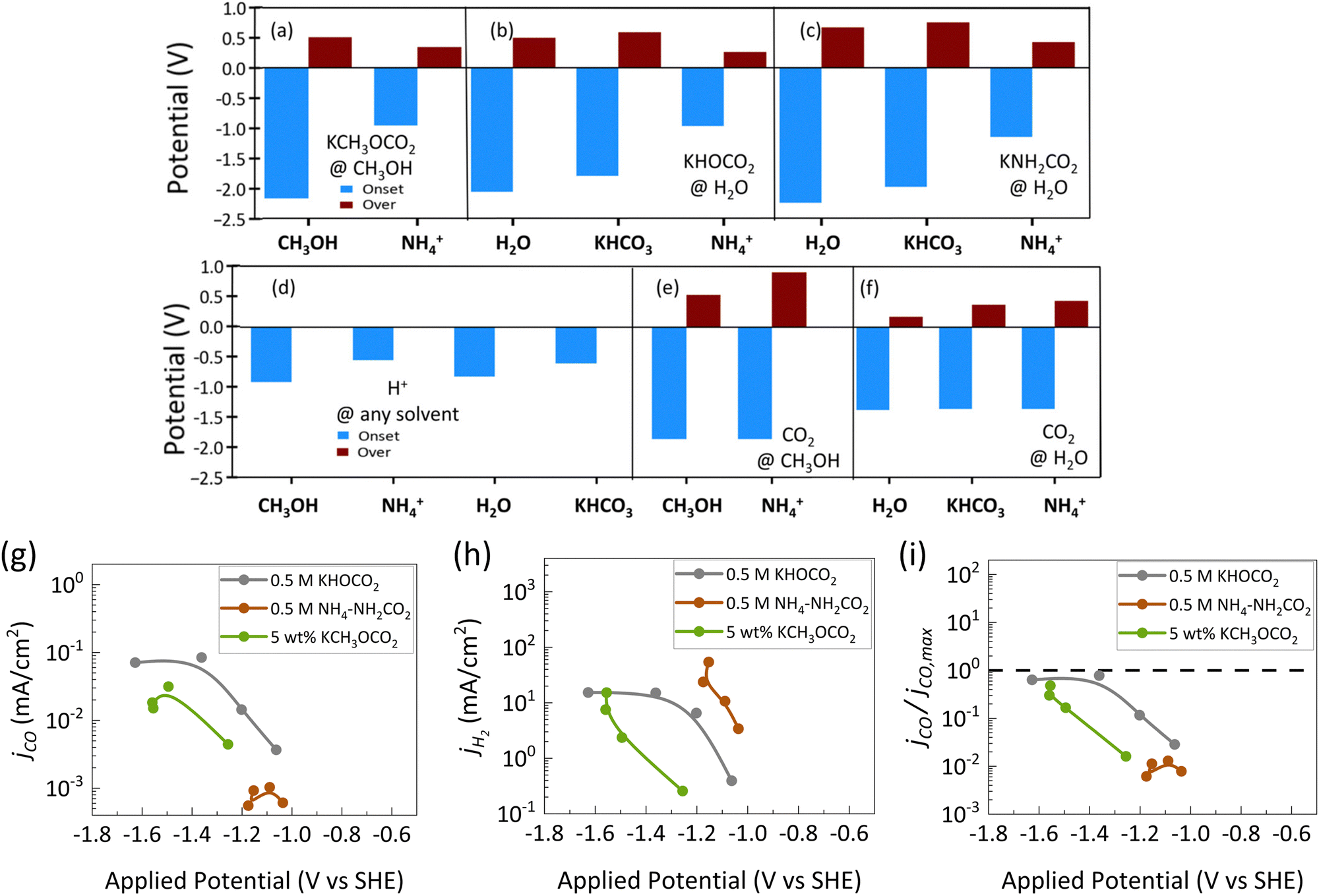 | ||
| Fig. 21 Calculated onset potential (blue, vs. SHE) and over-potential (brown) for the electroreduction of (a) methyl carbonate KCH3OCO2, (b) bicarbonate KHOCO2, (c) carbamate KNH2CO2 on Ag(111) using the various solvent-proton source combinations of CH3OH and H2O. The compound being electrochemically reduced is labeled in the top right of each panel, together with the solvent (notation: compound@solvent). The x axis denotes the proton source. Values for (d) HER, (e) CO2RR in CH3OH solvent, and (f) CO2RR in H2O solvent are also given for comparison. Onset potential is defined as the least negative potential lower than the equilibrium potential at which the ES becomes equal to 0.75 eV, while the overpotential is equal to the difference between the equilibrium potential and the onset potential. The pathway that has the smallest onset potential is displayed. Partial current densities of (g) CO and (h) H2 and (i) CO with respect to the maximum CO2 flux as a function of applied potential. Three CO2-captured complexes are considered: KHOCO2 (bicarbonate), NH4–NH2CO2 (ammonium carbamate) and KCH3OCO2 (methyl carbonate).12 Reproduced with permission from ref. 12, copyright 2024 American Chemical Society. | ||
Alkanolamine solutions for CO2 capture faces limitations with regard to high degradation rates, toxicity and high regeneration energy requirements. More recently amino acids have been proposed as an alternative to circumvent these challenges. Amino acids have advantages of low toxicity, low corrosivity, fast capture kinetics with CO2, and good resistance to oxidative and thermal degradation. Ramezani et al. compared different amino acid salts at different conditions to position their effectiveness with respect to alkanolamines, MEA in particular.133 In their comparison, they reported that amino acid salts like K-Lys (lysine), K-Pro (proline), and K-Sar (sarcosinate) have higher pKa and higher CO2 loading capacity than MEA. The high pKa of these salts makes the carbamate unstable and facilitates the formation of carbonates and bicarbonates which in turn reduces the heat of absorption in these systems. They further report that the CO2 absorption rate increased as the temperature and concentration of the amino acid salt increased. Thus, the advantages of amino acids makes them favorable for c-CO2RR. The challenge in c-CO2RR systems with amines is the strong binding of the CO2 in the form of stable carbamates that makes it hard to directly reduce the CO2-bound adduct. From the investigation of Ramezani et al., it was observed that the absorption rate and the desorption rate can be tuned by using smaller chains of amino acids that reduces the distance between the amino and the carboxyl groups. This along with sterically hindered amino acids can slow down the absorption rate and enhance the desorption rate due to the bulkier substituent group (Fig. 22a).133 For instance, K-Asp (asparagine), K-Tau (taurate), K-Ser (serine), K-Arg (arginine), K-Cys (cysteine), and K-Glu (glutamine) showed faster desorption rate than MEA and can be promising candidates for c-CO2RR (Fig. 22b). Thus for c-CO2RR it is important to achieve a balance between absorption and desorption kinetics to optimize the conversion.
 | ||
| Fig. 22 (a) The overall rate constant for CO2 absorption as a function of concentration for different amino acid salts at 298.15 K. (b) The desorption rate of 1 M potassium salts of different amino acid at 353.15 K. K-Lys: potassium lysine, K-Pro: potassium proline, K-Arg: potassium arginine, K-His: potassium histidine, K-Gly: potassium glycine, K-Ala: potassium alanine, K-Thr: potassium threonine, K-Tau: potassium taurate, K-Ser: potassium serine, K-Sar: potassium sarcosinate, and MEA: monoethanolamine.133 Reproduced with permission from ref. 133, copyright 2022 De Gruyter. | ||
Although amine capture agents are the state of the art for postcombustion capture processes due to its high capture efficiency, most c-CO2RR studies only show dissolved CO2 as the active species getting reduced. Selecting capture agents for c-CO2RR thus becomes more complicated. Amine capture agents binds to CO2via chemical absorption, however if dissolved CO2 is the main species getting reduced then other solvents, for instance the ones used in precombustion CO2 capture via physical absorption, can also be explored. Physical solvents exhibit characteristics such as low vapor pressure, high reactivity or absorptivity, facile regeneration, high thermal and mechanical resilience, minimal corrosiveness, and minimal environmental impact which proves ideal for CO2 capture.134,135 Such solvents also possess exceptional thermal stability, mitigating the risk of solvent loss and contamination while regenerating at elevated temperatures. Commercially available physical processes for CCS encompass selexol (dimethyl ether of polyethylene glycol (DMPEG)), rectisol (methanol), purisol (N-methyl-2-pyrolidone (NMP)), morphysorb, sulfolane (tetrahydrothiophenedioxide), and fluor (propylene carbonate) solvent.136,137 Selexol (a liquid glycol solvent) stands out among these processes, having been widely utilized in CO2 capture for decades. Its advantages over other physical solvents include lower vapor pressure, non-corrosive properties, reduced heat requirements, and inertness towards select gases.138
c-CO2RR can pose challenges on stability depending on the capture agent that is being used. CO2 capture solutions consist of several species that coexist in the solution which can trigger the interaction between the catalyst and the capture solution resulting into side reactions. There have been reports of morphological changes and corrosion of metal electrodes under c-CO2RR conditions.71 Thus, understanding the vapor–liquid equilibrium models and linking them with electrochemical c-CO2RR is important to determine the concentration of different species present in the capture solution and identify the species getting reduced or causing the instability of the catalyst. Recent studies have reported that direct reduction of CO2-capture solutions undergoes high HER.12,61,71 For instance, in amine capture solution the protonated amines contribute to the high HER. This reduces the generation of carbon products favoring the side reaction of HER. Therefore, according to the reported studies, the reduction of CO2-bound complex is not favored at the electrode surface. In addition, catalysts like Cu which is known to produce multi carbon products in CO2RR have been reported to undergo corrosion in the presence of amine capture agents, whereas catalysts like Au and Ag have been reported to be relatively stable and inert to corrosion.71 In the presence of ammonia, Cu was reported to be able to form ligands with the free amines in the solution to form copper–ammonia complex. This is possible even at reductive potential, making Cu not suitable for c-CO2RR with amine capture agents.139 Many other mechanisms have been proposed for cathodic corrosion, including cathodic etching in both aqueous and non-aqueous solutions, formation of (meta)stable metal hydrides at the cathode as possible intermediates that eventually are released from the cathode, and leaching due to the interaction of alkali metals with the cathode. These would also need to be investigated as new capture agents are developed to determine the stability of the catalysts (Chem. Rev., 2021, 121(17), 10241–10270). This requires a deep understanding of the degradation and restructuring mechanisms of the catalyst along with an understanding of the transport of different species during electrocatalysis to be able to develop strategies to mitigate these processes.
7 Effect of impurities on electrochemical c-CO2RR
Industrial flue gas contains contaminants like NOx, SOx and O2 that should be considered while developing c-CO2RR technologies. Several challenges have been identified by researchers on the impact of impurities for CO2RR that includes identifying catalysts that can maintain high selectivity and activity and designing suitable reactor configurations for viable operation. The presence of O2 which is 25 times less soluble in water than CO2 at STP conditions can result in significant loss of current density to oxygen reduction reaction (ORR). The lower standard reduction potential due to lower thermodynamic requirements and higher kinetic favorability promotes ORR compared to CO2RR. Furthermore, the higher diffusion rate of O2 compared to CO2 also plays a role in changes observed in selectivity. O2 can also oxidize the catalyst leading to changes in the oxidation state of the catalyst during catalytic turnover or lead to corrosion. To prevent catalyst oxidation and maintain CO2RR selectivity, strategies like deploying ionomer coatings which are hydrophilic in nature have been suggested.140 Hydrophilic nanopores have been argued to reduce the mass flux of O2 in to the electrode as the O2 needs to diffuse in the electrolyte-wet form, while the CO2 mass flux have been observed to not have any significant difference from different ionomer coatings. Hydrophilic support of TiO2 has also been proposed by Xu et al. that has shown to impede the effects of O2 an undergo stable CO2RR.141NOx and SOx impurities in flue gas have high solubility in water which dissolve to form acids in the solution as described in eqn (20)–(23). The formation of acids lowers the pH of the solution which then leads to increased HER, lowering the current densities for CO2RR. Both NO2 and SO2 also can adversely react with the catalyst to poison its surface and reduce the activity of the catalyst.142,143 NO2 concentrations above 1600 ppm have been shown to reduce the CO2RR faradaic efficiency on Cu catalyst. Similar results have been observed for SO2 on Cu catalysts. However, exception to this poisoning effect have been observed when using Ag or Sn catalysts. This has been attributed to the fact that they form Ag2S and SnS2 during CO2RR which are both thermodynamically unstable.144
SOx dissolution:
| SO2(g) + H2O(l) → SO32−(aq) + 2H+(aq) | (20) |
| SO3(g) + H2O(l) → SO42−(aq) + 2H+(aq) | (21) |
NOx dissolution:
| 2NO2(g) + H2O(l) → NO2−(aq) + NO3−(aq) + 2H+(aq) | (22) |
| NO2(g) + NO(g) + H2O(l) → 2NO2−(aq) + 2H+(aq) | (23) |
Therefore, similar effects could be observed in c-CO2RR. In the presence of O2, c-CO2RR which has a higher thermodynamic barrier than CO2RR, will result in even higher thermodynamic unfavorability while competing with ORR compared to CO2RR, unless the oxidized catalyst surface is a better catalyst for the absorption and activation of the CO2-bound adduct. Similarly, as alkaline environments are preferred for CO2 capture, unreacted NOx and SOx could accumulate in the capture agent and reduce the capture efficiency by lowering the pH of the capture solution resulting in lower CO2 loadings. Therefore, the system will have lower concentrations of CO2-bound adducts in the presence of SOx and NOx that can eventually be electrochemically reduced. However, there are instances where Ag and Sn have been reported to be stable in SO2 impurities. Therefore, for c-CO2RR catalysts which have stable activity and selectivity in the presence of impurities will be important to identify.
8 Energy comparison of CO2RR vs. c-CO2RR
The energy benefits of c-CO2RR can be extrapolated by comparing them with state-of-the-art CO2 electrolyzers. Most high-performance CO2 electrolyzer designs are inspired from the fuel cell community and are developed as zero-gap gas-fed electrolyzers to maximize the CO2 transport to the electrode and thus enable their operation at high current densities. These electrolyzers can typically operate at current densities higher than 100 mA cm−2 with a cell voltage of 3–3.5 V to make CO with a faradaic efficiency as high as 90%. Thus, these typically require 600–700 kJ mol−1 of energy to operate at standard temperature and pressure conditions. However, energy penalties lie at the anode where the bicarbonates or carbonate salts can be converted to CO2 which requires at least 254 kJ mol−1 and therefore the CO2 utilization rate is typically low in CO2 electrolyzers (Fig. 23).145 | ||
| Fig. 23 Energy comparison between the sequential route (conventional CO2RR route) vs. the integrated route (the c-CO2RR route).33 The optimistic scenario assumes that c-CO2RR performs at the same efficiency as the current state-of-the-art CO2 electrolyzers with a 90% F.E. CO at cell potential of 3 V, the baseline scenario uses Lee et al.'s report of 72% F.E. CO at −0.8 V vs. RHE,28 and the pessimistic scenario assumes a 40% F.E. CO at a large cell potential of 5 V. Reproduced with permission from ref. 33, copyright 2022 Nature. | ||
Compared to the CO2 electrolyzers, although the current c-CO2RR system requires significantly higher energy (800–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kJ mol−1), primarily because of the low CO production and high HER. Therefore, as discussed earlier, improving the CO production rates remains a challenge and addressing which can improve the energy requirements of c-CO2RR systems. This requires fundamental insights on reaction environments, catalysts activity and stability, and influence of different capture agents to optimize the c-CO2RR system. However, as reported by Li et al., considering that the c-CO2RR system can achieve similar faradaic efficiencies and current densities at similar cell voltage as the CO2 gas-fed electrolyzers, there is a potential of saving 44% of the total energy required (Fig. 23). This saving is due to the fact that c-CO2RR can save energy costs arising from capture agent regeneration, CO2 compression, and product purification needed in conventional CO2 reduction process.33
000 kJ mol−1), primarily because of the low CO production and high HER. Therefore, as discussed earlier, improving the CO production rates remains a challenge and addressing which can improve the energy requirements of c-CO2RR systems. This requires fundamental insights on reaction environments, catalysts activity and stability, and influence of different capture agents to optimize the c-CO2RR system. However, as reported by Li et al., considering that the c-CO2RR system can achieve similar faradaic efficiencies and current densities at similar cell voltage as the CO2 gas-fed electrolyzers, there is a potential of saving 44% of the total energy required (Fig. 23). This saving is due to the fact that c-CO2RR can save energy costs arising from capture agent regeneration, CO2 compression, and product purification needed in conventional CO2 reduction process.33
9 Conclusion and future outlook
The long term development of c-CO2RR technologies posseses many challenges that needs to be overcome to make this technology industrially viable. Along with the coupled effects of transport, kinetics, and thermodynamics that plays a role in c-CO2RR development, recent studies have reported corrosion and restructuring of the catalyst in the presence of amines as a crucial obstruction to further technology development.71 The activity and stability of the transition metal catalyst that were known for CO2RR thus do not apply to c-CO2RR anymore. Furthermore, alkoxide capture agents showed promise in suppressing HER, but the reaction when looked from the anodic half would probably oxidize the methyl carbonate to give back CO2, thus eliminating the purpose of c-CO2RR. Thus finding a combination of suitable capture agent, catalyst and operating condition is needed to advance the development of c-CO2RR. Although, the use of VLE models, effect of thermodynamic barriers, high HER, catalyst stability are discussed for selected capture agents, similar concepts and understanding are relevant for all capture agents that are used for c-CO2RR.To advance research in c-CO2RR the integration of capture agent, operando characterization, catalyst discovery, and reactor and catalyst design needs to be simultaneously investigated for achieving c-CO2RR and for potential scale-ups (Fig. 24). The selection of capture agents will determine the binding energy of CO2 to the capture agent and the thermodynamic energetic barriers relevant for direct reduction of the captured CO2-bound adduct. Catalyst discovery will involve investigating different catalysts for activity and stability under c-CO2RR by leveraging the use of both DFT calculations and experiments. These findings will further need to be linked with operando characterization to gain insights into the morphology, composition, crystallographic structural information to correlate experimental and theoretical results. Reactor and catalyst design also needs to be investigated in parallel to tune external and internal mass transport properties of the system and facilitate selectivity towards c-CO2RR. This screening loop of finding activity, stability and design descriptors for c-CO2RR is also beneficial to determine and address system-level challenges and accelerate the development of c-CO2RR processes (Fig. 24).
 | ||
| Fig. 24 Illustration showing the screening loop between capture agent discovery, catalyst discovery, reactor and catalyst design principles, and operando characterization that needs to be investigated for the eventual scale-up of c-CO2RR systems. | ||
Most literature on c-CO2RR reported C1 products, more specifically, CO. Table 1 summarizes the recent works on c-CO2RR. As the field evolves strategies for producing C2+ products should be explored. Currently, several challenges lie in c-CO2RR which should be investigated to gain more fundamental insight into the process. These include the higher thermodynamic barrier to reduce captured CO2-adduct, high HER, and identifying the carbon source getting reduced during electrolysis. Furthermore, catalysts like Cu which are known for producing multi carbon products undergo corrosion and restructuring when amines are used as capture agents.65 However, as reported by Choi et al., transition metals like Au and Ag do not undergo corrosion in the presence of amines.71 These metals are known for producing CO with faradaic efficiencies as high as 90% which perhaps explain why only C1 products have been observed. The understanding of corrosion mechanisms, which is one of the key bottlenecks in using catalysts like Cu in c-CO2RR needs to be addressed. Inhibiting cathodic corrosion can be achieved with the use of additives to tailor the electrical double layer or by alloying to alter the physico-chemical properties of the cathode. The addition of (poly)cationic ammonium salts can prevent the formation of parasitic hydrogen and corroding compounds in the double layer. Alloying can change the electronic band structure which can result in lower interaction between the cathode and the corrosive species or increase the energy barrier for reaction with the corrosion intermediates. The ternary alloy of CuSn7Pb15 has been shown to be physically and chemically stable towards the formation of formate in electroreduction of CO2 and can be employed in amine-based solvents in c-CO2RR.146 Therefore, a combination of design strategies is necessary for eventually extending the space of c-CO2RR towards C2+ products.
| Reactor | CO2 capture agent | Catalyst | Supporting electrolyte | Gas flow | c-CO2RR performance | Ref. |
|---|---|---|---|---|---|---|
| Zero-gap membrane electrode assembly | 5 M MEA | Ni–N/C | — | Ar | F.E. CO: 78.3% | Kim et al.15 |
| j CO: 2.6 mA cm−2 | ||||||
| 5 M MEA | Ag | — | Ar | F.E. CO: 38.3 | Kim et al.15 | |
| j CO: 0.79 mA cm−2 | ||||||
| Compression cell | 0.1 M MEA | Ag | 0.1 M KHCO3 | CO2 | j CO: 6 mA cm−2 | Safipour et al.17 |
| 0.2 M MEA | Ag | 0.1 M KHCO3 | CO2 | j CO: 3 mA cm−2 | ||
| H-cell | 2 M MEA, 2 M BAPN, 2 M n-BA, 2 M 2A1P, 2 M AMP, 2 M DMAE | Ag | 2 M KCl | N2 | F.E. CO: 1–20% | Leverick et al.61 |
| j CO: 0.01–1 mA cm−2 | ||||||
| RCE reactor | 0.2 M and 0.7 M AC, 0.2 and 0.7 M MEA, 0.2 M and 0.7 M KHCO3 | Ag | 0.099 M KClO4 + 0.001 M KOH | Ar | F.E. CO: 1–9% | Shen et al.16 |
| j CO: 0.01–0.4 mA cm−2 | ||||||
| RCE reactor | 0.5 M AC, 0.5 M KHCO3, 5 wt% methoxide | Ag | 0.099 M KClO4 + 0.001 M KOH | Ar | F.E. CO: 0.01–1.68% | Kowalski et al.12 |
| j CO: 0.001–0.28 mA cm−2 | ||||||
| RCE reactor | 0.2 M AC | Cu, Ag, Au, Sn, Ti | 0.099 M KClO4 + 0.001 M KOH | Ar | F.E. CO: ≈0.01% | Choi et al.71 |
| j CO: 12 mA cm−2 | ||||||
| Three electrode configuration | 2 M MEA | Ag | 2 M KCl | N2 | F.E. CO: 10% | Lee et al.28 |
| j CO: 0.6 mA cm−2 | ||||||
| Three electrode configuration | 2 M MEA | Ag | 2 M KCl | N2 | F.E. CO: 10% | Lee et al.28 |
| j CO: 0.6 mA cm−2 | ||||||
| H-cell | 0.05 M K2CO3 | Cu | — | N2 | F.E. HCOO−: 0.61% | Ma et al.116 |
In addition to the following, developing anodes for the captured CO2 media will guide the development of c-CO2RR technologies. Water oxidation reaction at the anode is kinetically sluggish and can lead to lower efficiency of the electrochemical reactor. Thus, choosing a catalyst that is thermodynamically favorable to water oxidation can improve the overall system efficiency. Furthermore, the loss of CO2 from the anode during c-CO2RR should be at similar rates as CO2RR considering a similar acidic microenvironment is generated at the anode during water oxidation. However, there could be added complexities to the development of catalyst materials for the anode for c-CO2RR. Cathodic corrosion that has been observed in c-CO2RR conditions, especially with Cu electrode in amine solutions, can occur at the anode too. Therefore, catalysts that are compatible with the CO2-captured complex are needed to be identified to prevent any anodic corrosion. Pt so far has been used for different c-CO2RR studies as the anode, but their stability in these capture solutions has not been investigated. Additionally, cheaper alternatives are required and needs to be tested for industrial scale electrolyzers that can operate at overpotentials similar to Pt.
Integrated CO2 capture and conversion posses benefits in terms of energy and capital cost, however there maybe trade-offs when trying to implement the capture and conversion in one unit. In current carbon capture and utilization processes, the CO2 is fed in a compressed form which increases the conversion efficiency of CO2 compared to if the whole process takes place at room temperature and atmospheric pressure. Thus the current technologies still outcompetes the conversion efficiency of c-CO2RR. Until now c-CO2RR technologies have mostly focused on batch mode of operation, and thus continuous mode of operation will simultaneously have to be investigated and compared with the current state-of-the-art conversion efficiency.147 Also, point sources of CO2 has a wide range of gas temperature, CO2 concentration, and contaminants which must also be explored during the development phase. A typical c-CO2RR system should be stable for thousand of hours at high current densities and operating in a continuous mode of operation.11
In addition to performance, the development of c-CO2RR can further be coupled with life-cycle and techno-economic analysis (TEA) analysis in parallel keeping in mind future viability. Li et al. in their work compared the TEA of a conventional CCU process to the integrated route of c-CO2RR. They found that at the current level of technology development there is no foreseen advantage of using the integrated route as the energy cost of converting captured CO2 to products is higher. This offsets the cost reduced from process intensification. Also due to higher cost of electricity than heat, the integrated route further uses up more costs. However in an optimistic scenario, where c-CO2RR reaches the same performance in terms of current density and faradaic efficiency, the overall energy can be reduced by 44% with a 22% savings in energy cost.33 Thus the development of c-CO2RR technologies is an attractive option for closing the carbon cycle using renewable electricity.
Acronym
| MEA | Monoethanolamine |
| AC | Ammonium carbamate |
| BAPN | β-Aminopropionitrile |
| n-BA | n-Butylamine |
| 2A1P | 2-Amino-1-propanol |
| AMP | Aminomethyl propanol |
| DMAE | Dimethylethanolamine |
| DEA | Diethanolamine |
| MDEA | Methyldiethanolamine |
| KHCO3 | Potassium bicarbonate |
| K2CO3 | Potassium carbonate |
| KClO4 | Potassium perchlorate |
Data availability
The review article consists of information and research findings which are already published in peer-reviewed journals. All the necessary citations are included in the main article and permission for reproducing the figures are also obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of California Office of the President and the National Laboratory Research Fees Program for supporting this work with award L22CR4468, which supports the Center for Direct Conversion of Captured CO2 into Chemicals and Fuels.Notes and references
- P. R. Shukla, J. Skea, E. Calvo Buendia, V. Masson-Delmotte, H. O. Pörtner, D. Roberts, P. Zhai, R. Slade, S. Connors and R. Van Diemenet al., Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems, IPCC, 2019 Search PubMed.
- R. Lindsey, Climate change: atmospheric carbon dioxide, 2020, https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide Search PubMed.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef PubMed.
- R. G. Grim, Z. Huang, M. T. Guarnieri, J. R. Ferrell, L. Tao and J. A. Schaidle, Energy Environ. Sci., 2020, 13, 472–494 RSC.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyeret al., Direct air capture of CO2with chemicals: a technology assessment for the APS Panel on Public Affairs, American Physical Society technical report, 2011.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- J. Valentine, A. Zoelle, S. Homsy, H. Mantripragada, M. Woods, N. Roy, A. Kilstofte, M. Sturdivan, M. Steutermann and T. Fout, Direct air capture case studies: sorbent system, National Energy Technology Laboratory (NETL), Pittsburgh, Pa, Morgantown, WV, technical report, 2022 Search PubMed.
- National Academies of Sciences, Division on Earth, Life Studies, Ocean Studies Board, Board on Chemical Sciences, Board on Earth Sciences, Board on Energy, Environmental Systems, Board on Atmospheric Sciences, Committee on Developing a Research Agenda for Carbon Dioxide Removal, Reliable Sequestration, Negative emissions technologies and reliable sequestration: a research agenda, National Academies Press, 2019 Search PubMed.
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- A. Aspelund and K. Jordal, Int. J. Greenhouse Gas Control, 2007, 1, 343–354 CrossRef CAS.
- M. C. Freyman, Z. Huang, D. Ravikumar, E. B. Duoss, Y. Li, S. E. Baker, S. H. Pang and J. A. Schaidle, Joule, 2023, 7, 631–651 CrossRef CAS.
- R. M. Kowalski, A. Banerjee, C. Yue, S. G. Gracia, D. Cheng, C. G. Morales-Guio and P. Sautet, J. Am. Chem. Soc., 2024, 146, 20728–20741 CrossRef CAS PubMed.
- Z. Zhang, A. L. Kummeth, J. Y. Yang and A. N. Alexandrova, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2123496119 CrossRef CAS PubMed.
- Y. C. Xiao, S. S. Sun, Y. Zhao, R. K. Miao, M. Fan, G. Lee, Y. Chen, C. M. Gabardo, Y. Yu and C. Qiu, et al. , Nat. Commun., 2024, 15, 7849 CrossRef CAS PubMed.
- J. H. Kim, H. Jang, G. Bak, W. Choi, H. Yun, E. Lee, D. Kim, J. Kim, S. Y. Lee and Y. J. Hwang, Energy Environ. Sci., 2022, 15, 4301–4312 RSC.
- K. Shen, D. Cheng, E. Reyes-Lopez, J. Jang, P. Sautet and C. G. Morales-Guio, Joule, 2023, 7, 1260–1276 CrossRef CAS.
- J. Safipour, A. Z. Weber and A. T. Bell, ACS Energy Lett., 2023, 8, 5012–5017 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. S. Prakash, J. Am. Chem. Soc., 2018, 140, 16873–16876 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- S. Kar, R. Sen, A. Goeppert and G. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS PubMed.
- R. Sen, A. Goeppert, S. Kar and G. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- Q. Fu, Y. Kuramochi, N. Fukushima, H. Maeda, K. Sato and H. Kobayashi, Environ. Sci. Technol., 2015, 49, 1225–1232 CrossRef CAS PubMed.
- R. Sleat, R. A. Mah and R. Robinson, Int. J. Syst. Evol. Microbiol., 1985, 35, 10–15 Search PubMed.
- S. Xu, B. Fu, L. Zhang and H. Liu, World J. Microbiol. Biotechnol., 2015, 31, 941–950 CrossRef CAS PubMed.
- N. Monnerie, P. G. Gan, M. Roeb and C. Sattler, Int. J. Hydrogen Energy, 2020, 45, 26117–26125 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip and Y.-C. Joo, et al. , Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2022, 13, 766–784 CrossRef.
- M. Li, K. Yang, M. Abdinejad, C. Zhao and T. Burdyny, Nanoscale, 2022, 14, 11892–11908 RSC.
- S. E. Jerng and B. M. Gallant, iScience, 2022, 25, 104558 CrossRef CAS PubMed.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- M. Li, E. Irtem, H.-P. Iglesias van Montfort, M. Abdinejad and T. Burdyny, Nat. Commun., 2022, 13, 5398 CrossRef CAS PubMed.
- D. J. Heldebrant, J. Kothandaraman, N. Mac Dowell and L. Brickett, Chem. Sci., 2022, 13, 6445–6456 RSC.
- S. H. Park, K. B. Lee, J. C. Hyun and S. H. Kim, Ind. Eng. Chem. Res., 2002, 41, 1658–1665 CrossRef CAS.
- R. L. Kent and B. Eisenberg, Hydrocarbon Processing, 1976, 55, 87–90 CAS.
- A. Aboudheir, P. Tontiwachwuthikul, A. Chakma and R. Idem, Chem. Eng. Sci., 2003, 58, 5195–5210 CrossRef CAS.
- K. Zhu, C. Yue, Z. Wei, J. Huang, M. Hu, Y. Ji, H. Liu, H. Zhu, W. Guo and F. Zhou, et al. , Adv. Mater. Sci. Eng., 2022, 2022, 6278342 Search PubMed.
- M. Haji-Sulaiman, M. K. Aroua and A. Benamor, Chem. Eng. Res. Des., 1998, 76, 961–968 CrossRef CAS.
- A. Bandi, M. Specht, T. Weimer and K. Schaber, Energy Convers. Manage., 1995, 36, 899–902 CrossRef CAS.
- F. Zeman, AIChE J., 2008, 54, 1396–1399 CrossRef CAS.
- J. K. Stolaroff, D. W. Keith and G. V. Lowry, Environ. Sci. Technol., 2008, 42, 2728–2735 CrossRef CAS PubMed.
- D. W. Keith, M. Ha-Duong and J. K. Stolaroff, Clim. Change, 2006, 74, 17–45 CrossRef CAS.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- P. C. De Carvalho Pinto, T. V. Batista, G. De Rezende Ferreira, G. P. Voga, L. C. Oliveira, H. S. Oliveira, L. A. De Souza and J. C. Belchior, ChemistrySelect, 2022, 7, e202202731 CrossRef.
- R. B. Said, J. M. Kolle, K. Essalah, B. Tangour and A. Sayari, ACS Omega, 2020, 5, 26125–26133 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, M. Paccagnini and H. Thomann, Energy Fuels, 2016, 30, 1223–1236 CAS.
- A. Banerjee, C. Yue, J. Choi and C. G. Morales-Guio, AIChE J., 2024, e18560 Search PubMed.
- H. Liu, M. Li, X. Luo, Z. Liang, R. Idem and P. Tontiwachwuthikul, AIChE J., 2018, 64, 2515–2525 CrossRef CAS.
- P. W. Derks, P. J. Huttenhuis, C. van Aken, J.-H. Marsman and G. F. Versteeg, Energy Procedia, 2011, 4, 599–605 CrossRef CAS.
- J. P. Jakobsen, J. Krane and H. F. Svendsen, Ind. Eng. Chem. Res., 2005, 44, 9894–9903 CrossRef.
- I. M. Bernhardsen and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 61, 27–48 CrossRef CAS.
- H. Seo, M. P. Nitzsche and T. A. Hatton, Acc. Chem. Res., 2023, 56, 3153–3164 CrossRef CAS PubMed.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, Energy Environ. Sci., 2020, 13, 3706–3722 RSC.
- S. Jin, M. Wu, Y. Jing, R. G. Gordon and M. J. Aziz, Nat. Commun., 2022, 13, 2140 CrossRef CAS PubMed.
- W. Jiang, W. Liu, Y. Wang, Z. Zhao, Q. Li, Y. Wu, T. Liu and H. Xie, Ind. Eng. Chem. Res., 2022, 61, 13578–13588 CrossRef CAS.
- A. E. Al-Rawajfeh and M. N. Al-Amaireh, Desalin. Water Treat., 2009, 7, 191–197 CrossRef CAS.
- J. I. Lee, F. D. Otto and A. E. Mather, J. Appl. Chem. Biotechnol., 1976, 26, 541–549 CrossRef CAS.
- W. Böttinger, M. Maiwald and H. Hasse, Fluid Phase Equilib., 2008, 263, 131–143 CrossRef.
- F. Y. Jou, A. E. Mather and F. D. Otto, Ind. Eng. Chem. Process Des. Dev., 1982, 21, 539–544 CrossRef CAS.
- G. Leverick, E. M. Bernhardt, A. I. Ismail, J. H. Law, A. Arifutzzaman, M. K. Aroua and B. M. Gallant, ACS Catal., 2023, 13, 12322–12337 CrossRef CAS.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084 CrossRef CAS PubMed.
- J. Jang, M. Rüscher, M. Winzely and C. G. Morales-Guio, AIChE J., 2022, 68, e17605 CrossRef CAS.
- C. Morales-Guio, J. Jang, M. Ruscher, M. Winzely, D. Rodriguez, E. Reyes-Lopez, S. Srivastava, P. Christofides and P. Sautet, Research Square, 2024 DOI:10.21203/rs.3.rs-4189647/v1.
- L. Yuan, S. Zeng, X. Zhang, X. Ji and S. Zhang, Mater. Rep.: Energy, 2023, 3, 100177 CAS.
- V. E. Nelson, C. P. O’Brien, J. P. Edwards, S. Liu, C. M. Gabardo, E. H. Sargent and D. Sinton, ACS Appl. Mater. Interfaces, 2024, 16, 50818–50825 Search PubMed.
- D. Segets, C. Andronescu and U.-P. Apfel, Nat. Commun., 2023, 14, 7950 CrossRef CAS PubMed.
- S. Appelhaus, L. Ritz, S.-V. Pape, F. Lohmann-Richters, M. R. Kraglund, J. O. Jensen, F. Massari, M. Boroomandnia, M. Romanò and J. Albers, et al. , Int. J. Hydrogen Energy, 2024, 95, 1004–1010 Search PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- J. Choi, S. Chiu, A. Banerjee, R. L. Sacci, G. M. Veith, C. Stieber, C. Hahn, A. N. Alexandrova and C. G. Morales-Guio, J. Phys. Chem. Lett., 2024, 15, 8007–8017 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Angew. Chem., 2016, 128, 9900–9904 CrossRef.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- F. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS PubMed.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna and P.-L. Hsieh, et al. , Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- F. Pan, A. Liang, Y. Duan, Q. Liu, J. Zhang and Y. Li, J. Mater. Chem. A, 2017, 5, 13104–13111 RSC.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed.
- S. Chen and S.-Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. Zhou, Angew. Chem., 2015, 127, 13905–13909 CrossRef.
- X. Cui, Z. Pan, L. Zhang, H. Peng and G. Zheng, Adv. Energy Mater., 2017, 7, 1701456 CrossRef.
- D. Hursán, A. A. Samu, L. Janovák, K. Artyushkova, T. Asset, P. Atanassov and C. Janáky, Joule, 2019, 3, 1719–1733 CrossRef PubMed.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- K. Shi, Z. Ren, Z. Meng and X. Feng, ChemCatChem, 2024, 16, e202301308 CrossRef CAS.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y.-M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- M. N. Hossain, S. Ahmad, I. S. da Silva and H.-B. Kraatz, Chem. – Eur. J., 2021, 27, 1346–1355 CrossRef CAS PubMed.
- J. H. Kim and T. J. Kang, ACS Appl. Mater. Interfaces, 2019, 11, 28894–28899 CrossRef CAS PubMed.
- J. Blake, J. Padding and J. Haverkort, Electrochim. Acta, 2021, 393, 138987 CrossRef CAS.
- C. T.-C. Wan, K. V. Greco, A. Alazmi, R. M. Darling, Y.-M. Chiang and F. R. Brushett, J. Electrochem. Soc., 2021, 168, 123503 CrossRef CAS.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed.
- S. Suter and S. Haussener, Energy Environ. Sci., 2019, 12, 1668–1678 RSC.
- A. Böhme, J. C. Bui, A. Q. Fenwick, R. Bhide, C. N. Feltenberger, A. J. Welch, A. J. King, A. T. Bell, A. Z. Weber and S. Ardo, et al. , Energy Environ. Sci., 2023, 16, 1783–1795 RSC.
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su and R. Crespo-Otero, et al. , Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- K. A. Gandionco, J. Kim, L. Bekaert, A. Hubin and J. Lim, Carbon Energy, 2024, 6, e410 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun, N. Liu and J. Zhou, Nanoscale, 2022, 14, 6846–6853 RSC.
- Z. Li, D. He, X. Yan, S. Dai, S. Younan, Z. Ke, X. Pan, X. Xiao, H. Wu and J. Gu, Angew. Chem., 2020, 132, 18731–18736 CrossRef.
- Y. Huang, X. Mao, G. Yuan, D. Zhang, B. Pan, J. Deng, Y. Shi, N. Han, C. Li and L. Zhang, et al. , Angew. Chem., 2021, 133, 15978–15982 CrossRef.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- Y. Xu, C. Li, Y. Xiao, C. Wu, Y. Li, Y. Li, J. Han, Q. Liu and J. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS PubMed.
- M. Ding, Z. Chen, C. Liu, Y. Wang, C. Li, X. Li, T. Zheng, Q. Jiang and C. Xia, Mater. Rep.: Energy, 2023, 3, 100175 CAS.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- N. B. Watkins, Z. J. Schiffer, Y. Lai, C. B. Musgrave III, H. A. Atwater, W. A. Goddard III, T. Agapie, J. C. Peters and J. M. Gregoire, ACS Energy Lett., 2023, 8, 2185–2192 CrossRef CAS.
- F. P. Garca de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, et al. , Science, 2020, 367, 661–666 CrossRef PubMed.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- A. Goyal, C. J. Bondue, M. Graf and M. T. Koper, Chem. Sci., 2022, 13, 3288–3298 RSC.
- J. Newman, J. Electrochem. Soc., 1966, 113, 1235 CrossRef CAS.
- E. L. Clark, S. Ringe, M. Tang, A. Walton, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, ACS Catal., 2019, 9, 4006–4014 CrossRef CAS.
- H. Ma, E. Ibáñez-Alé, R. Ganganahalli, J. Pérez-Ramrez, N. López and B. S. Yeo, J. Am. Chem. Soc., 2023, 145, 24707–24716 CAS.
- M. Abdinejad, Z. Mirza, X.-A. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- M. C. Monteiro, F. Dattila, B. Hagedoorn, R. Garca-Muelas, N. López and M. T. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- J. Wu, W. Li, K. Liu, A. Kucernak, H. Liu, L. Chai and M. Liu, Next Energy, 2023, 1, 100032 CrossRef.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- S. Banerjee, X. Han and V. S. Thoi, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- S. Sarkar, A. Maitra, S. Banerjee, V. S. Thoi and J. M. Dawlaty, J. Phys. Chem. B, 2020, 124, 1311–1321 CrossRef CAS PubMed.
- S. Banerjee, Z.-Q. Zhang, A. S. Hall and V. S. Thoi, ACS Catal., 2020, 10, 9907–9914 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- O. Ayemoba and A. Cuesta, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed.
- M. Sadeghpour, R. Yusoff and M. K. Aroua, Rev. Chem. Eng., 2017, 33, 183–200 CrossRef CAS.
- X. Ren, T. D. Myles, K. N. Grew and W. K. Chiu, J. Electrochem. Soc., 2015, 162, F1221 CrossRef CAS.
- E. W. Lees, B. A. Mowbray, D. A. Salvatore, G. L. Simpson, D. J. Dvorak, S. Ren, J. Chau, K. L. Milton and C. P. Berlinguette, J. Mater. Chem. A, 2020, 8, 19493–19501 RSC.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Acc. Chem. Res., 2022, 55, 484–494 CrossRef CAS PubMed.
- A. M. Appel and J. Y. Yang, ACS Energy Lett., 2024, 9, 768–770 CrossRef CAS.
- R. Ramezani, S. Mazinani and R. Di Felice, Rev. Chem. Eng., 2022, 38, 273–299 CrossRef CAS.
- C. Song, Q. Liu, S. Deng, H. Li and Y. Kitamura, Renewable Sustainable Energy Rev., 2019, 101, 265–278 CrossRef CAS.
- E. S. Rubin, H. Mantripragada, A. Marks, P. Versteeg and J. Kitchin, Prog. Energy Combust. Sci., 2012, 38, 630–671 CrossRef CAS.
- T. Wilberforce, A. Baroutaji, B. Soudan, A. H. Al-Alami and A. G. Olabi, Sci. Total Environ., 2019, 657, 56–72 CrossRef CAS PubMed.
- W. L. Theo, J. S. Lim, H. Hashim, A. A. Mustaffa and W. S. Ho, Appl. Energy, 2016, 183, 1633–1663 CrossRef CAS.
- A. Gautam and M. K. Mondal, Fuel, 2023, 334, 126616 CrossRef CAS.
- Y. Guan, J. Kümper, S. D. Mürtz, S. Kumari, P. J. Hausoul, R. Palkovits and P. Sautet, Chem. Sci., 2024, 15, 14485–14496 RSC.
- S. Mondal and S. C. Peter, Adv. Mater., 2024, 36, 2407124 Search PubMed.
- Y. Xu, J. P. Edwards, J. Zhong, C. P. O’Brien, C. M. Gabardo, C. McCallum, J. Li, C.-T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- D. J. Pimlott, A. Jewlal, B. A. Mowbray and C. P. Berlinguette, ACS Energy Lett., 2023, 8, 1779–1784 CrossRef CAS.
- M. Gautam, D. T. Hofsommer, S. S. Uttarwar, N. Theaker, W. F. Paxton, C. A. Grapperhaus and J. M. Spurgeon, Chem Catal., 2022, 2, 2364–2378 CrossRef CAS.
- Y. Kwon, B. Wu, N. Zhang, D. Hand, T. Mou, X. Han and Q. Chang, MRS Commun., 2024, 1–13 Search PubMed.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- M. D. J. Gálvez-Vázquez, P. Moreno-Garca, H. Guo, Y. Hou, A. Dutta, S. R. Waldvogel and P. Broekmann, ChemElectroChem, 2019, 6, 2324–2330 CrossRef.
- Y. Kim, E. W. Lees, C. Donde, A. M. Jewlal, C. E. Waizenegger, B. M. de Hepcée, G. L. Simpson, A. Valji and C. P. Berlinguette, Joule, 2024, 6, 3106–3125 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00285g |
| This journal is © The Royal Society of Chemistry 2025 |