
Recent progress in emerging molecular targeted therapies for intrahepatic cholangiocarcinoma
Younghoon
Kim
 abc,
Jaewon
Song
d,
Namkyoung
Kim
b and
Taebo
Sim
*abcde
abc,
Jaewon
Song
d,
Namkyoung
Kim
b and
Taebo
Sim
*abcde
aKU-KIST Graduate School of Converging Science and Technology, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Korea
bDepartment of Biomedical Sciences, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea. E-mail: TBSIM@yuhs.ac; Tel: +822 2228 0797
cClinical Candidate Discovery & Development Institute, Yonsei University College of Medicine, Seoul, Korea
dGraduate School of Clinical Drug Discovery & Development, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea
eGraduate School of Medical Science, Brain Korea 21 Project, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul, 03722, Republic of Korea
First published on 6th February 2025
Abstract
Cholangiocarcinoma (CCA) is a diverse group of epithelial malignant tumors arising from the biliary tract, characterized by high molecular heterogeneity. It is classified into intrahepatic (iCCA) and extrahepatic CCA (eCCA) based on the location of the primary tumor. CCA accounts for approximately 15% of all primary liver cancers, with iCCA comprising 10–20% of all CCAs. iCCA is especially known for its characteristic aggressiveness and refractoriness, leading to poor prognosis. Despite the increasing global incidence and mortality rates, surgery remains the only available standard treatment approach for a subset (25%) of patients with early-stage, resectable iCCA. The paucity of effective systemic medical therapies restricts therapeutic options for patients with advanced or metastatic iCCA. In the past decade, advances in the understanding of the molecular complexity of these tumors have provided fruitful insights for the identification of promising new druggable targets and the development of feasible therapeutic strategies that may improve treatment outcomes for patients with iCCA. In this review, we aim to highlight critical up-to-date studies and medicinal chemistry aspects, focusing on novel targeted approaches utilizing promising candidates for molecular targeted therapy in iCCA. These candidates include aberrations in isocitrate dehydrogenase (IDH) 1/2, fibroblast growth factor receptor (FGFR), B-Raf proto-oncogene (BRAF), neurotrophic tyrosine receptor kinase (NTRK), human epidermal growth factor receptor 2 (HER2), and programmed cell death protein 1 (PD-1)/programmed cell death-ligand 1 (PD-L1). Furthermore, this review provides an overview of potential inhibitors aimed at overcoming acquired drug resistance in these actionable targets for iCCA.
1. Introduction
Cholangiocarcinoma (CCA) is a heterogeneous group of malignancies emerging in the epithelium of the biliary ducts. Although it is a rare type of tumor, comprising only 3% of all gastrointestinal malignancies, it remains one of the most common primary hepatic cancers, accounting for approximately 15% of all hepatobiliary malignancies.1 CCA is classified into two subgroups based on the anatomical site of the primary tumor, namely, intrahepatic (iCCA) and extrahepatic CCA (eCCA), which is further categorized as perihilar (pCCA or Klatskin tumor) and distal CCA (dCCA).2 iCCA arises from the small biliary tree within the periphery of the liver, pCCA arises at the level or junction of the right and/or left hepatic ducts, and dCCA usually develops at the common bile duct.3According to the latest studies on epidemiology, iCCA comprises roughly 10–20% of all CCAs, and its incidence and mortality rate have been steadily increasing over the past decades, regardless of geographical factors. In contrast, a decrease has been noted in the case of eCCA.4,5 High incidence rates of iCCA are reported in South Korea, China, Thailand, and other Southeast Asian countries (nearly 10 per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people/year), where parasite infections were or still are prevalent.6,7 On the other hand, reports show that the incidence rate of iCCA is relatively lower in countries without endemic parasite infections (<1 per 100000 people/year).1,8 Despite the numerous risk factors associated with the development of CCA, chronic inflammation and irritation of the biliary epithelium, including cirrhosis, liver flukes, type II diabetes, and chronic pancreatitis, are considered the most significant risk factors for both iCCA and eCCA.9,10 However, despite the well-documented risk factors mentioned above, more than 50% of patients are reported to have sporadically developed CCAs, with no identifiable risk factors.1,11
000 people/year), where parasite infections were or still are prevalent.6,7 On the other hand, reports show that the incidence rate of iCCA is relatively lower in countries without endemic parasite infections (<1 per 100000 people/year).1,8 Despite the numerous risk factors associated with the development of CCA, chronic inflammation and irritation of the biliary epithelium, including cirrhosis, liver flukes, type II diabetes, and chronic pancreatitis, are considered the most significant risk factors for both iCCA and eCCA.9,10 However, despite the well-documented risk factors mentioned above, more than 50% of patients are reported to have sporadically developed CCAs, with no identifiable risk factors.1,11
iCCA is marked by a characteristically unsatisfactory treatment outcome with a 5 year survival rate of less than 10–15%.12,13 Although surgical resection remains the best curative treatment option, most patients are diagnosed when the tumor is already at an advanced and non-operable stage due to asymptomatic nature and molecular heterogeneity of the disease, hampering its diagnosis in time. Moreover, the median overall survival (mOS) of patients with advanced iCCA drops to less than 1 year.4 However, only one subset (25%) of patients is diagnosed in the early stages with surgically resectable tumor.14
In addition, these patients often need to undergo adjuvant therapy with capecitabine owing to the high recurrence rate after surgical resection (∼70%).15–17 Unfortunately, the treatment options for patients with advanced or metastasized disease are limited. In fact, systemic chemotherapy with a combination of gemcitabine and cisplatin has been used as the standard first-line treatment for patients with advanced or metastasized CCA in the palliative setting. In a previous study, the use of gemcitabine and cisplatin together was proven to be more effective than gemcitabine alone by improving the mOS from 8.1 to 11.7 months.18 Furthermore, recent clinical investigation (ABC-06, phase III) revealed that FOLFOX, a combination of 5-fluorouracil and oxaliplatin, could provide a new option as a standard treatment regimen in the second-line setting. However, patients who have already progressed from gemcitabine and cisplatin only marginally benefited from FOLFOX and the vast majority (69%) of patients was reported to experience adverse effects related to FOLFOX such as neutropenia, fatigue, and infection.19 Therefore, effective novel therapeutic approaches for the treatment of iCCA are still a great unmet medical need.
Considering the current limitations of traditional chemotherapies, the high molecular heterogeneity of iCCA presents a new opportunity for innovative treatment approaches.20 Targeted therapies against various actionable molecular alterations employing either small molecules or monoclonal antibodies have increased in popularity owing to their advantages, such as high efficiency, enhanced safety profile, and high specificity, compared to traditional chemotherapy.21–25 During the last decade, comprehensive molecular profiling enabled by evolving genetic sequencing technologies shed light on the rapid improvement in understanding the pathogenesis of iCCA, which has led to the identification of multiple promising targetable molecular alterations. In addition, these molecular targets have been reported to be more frequently observed in iCCA, comprising approximately 50% of cases, than in eCCA.26,27 Based on previous studies, several promising targets have been elucidated for the treatment of iCCA including IDH1/2 (15–25%),28–31 FGFR (10–20%),32–34 BRAF (<5%),29,35 NTRK (<5%),30,36 HER2 (4–6%),37,38 and PD-1/PD-L1 (20%). These advancements in the molecular profiling of iCCA offer a promising avenue for the development of personalized and targeted treatments, which have the potential to greatly improve patient outcomes and overall survival rates. Herein, we summarize the currently identified promising molecular targets of iCCA and review the potential anti-cancer agents mainly focusing on those targeting emerging molecular alterations as well as their medicinal chemistry aspects, aiming to provide insights into drug discovery campaigns for iCCA.
2. Pathogenesis and targetable molecular alterations of iCCA
The pathogenesis of iCCA is most commonly associated with chronic biliary inflammation and stasis caused by a variety of disease processes. These conditions can bring about biliary obstruction and abnormal bile acid signaling, which eventually lead to the proliferation of cholangiocytes through growth factor activation. This can be further facilitated with the release of pro-inflammatory cytokines, resulting in the accumulation of molecular aberrations.39,40 Moreover, these aberrations are generally caused by impaired DNA damage repair and tumor suppressor genes, as well as activation of proto-oncogenes, which collectively promote the malignant transformation of cholangiocytes and the subsequent development of CCA. A combination of genetic and epigenetic changes together with the dysregulation of key signaling pathways, such as elevated and aberrant cyclooxygenease-2 (COX2), HER2, IL-6, and RAS/RAF/mitogen-activated protein kinase (MAPK) signaling transduction, contribute to the initiation and progression of CCA, leading to the heterogeneous nature of this disease.41,42The advancement of next-generation DNA sequencing (NGS) technologies has significantly enhanced our comprehension of the molecular landscape of iCCA, paving a new way for the implementation of targeted therapies, which can provide a paradigm shift in therapeutic strategies for advanced and metastasized CCAs.43,44 Although molecular alterations in other types of cancer often differ between early and advanced stages, the molecular profile of iCCA remains relatively consistent regardless of the disease stage, allowing the more accurate identification of targetable molecular alterations and providing patients with better treatment opportunities.45 Based on previous studies, up to 70% of iCCA patients are reported to harbor at least one gene alteration actionable by targeted therapy, including aberrations in IDH1/2, FGFR, BRAF, HER2, and NTRK, and overexpression of PD-1/PD-L1. Among them, mutations in IDH1/2 and gene fusions of FGFR2 (25% and 10–15% in iCCA patients, respectively) were identified as predominant and specific genetic alterations exclusively occurring in iCCA.28,46–49 To visually summarize these findings, we included a figure that integrates the molecular targets and signaling pathways implicated in iCCA (Fig. 1). This figure highlights the roles of key mutations and aberrations (e.g., IDH1/2 mutations and FGFR2 fusions) and their downstream signaling pathways, such as RAS/MAPK and PI3K/AKT, which contribute to tumorigenesis. It also demonstrates the actionable therapeutic strategies, including IDH1/2 inhibitors (ivosidenib (1) and enasidenib (6)), FGFR2 inhibitors (pemigatinib (25) and futibatinib (26)), BRAF/MEK inhibitors (dabrafenib (51) plus trametinib), NTRK inhibitors (entrectinib (58) and larotrectinib (60)), HER2-targeted therapy (trastuzumab), and immune checkpoint inhibitors (pembrolizumab or nivolumab targeting PD-1/PD-L1).
 | ||
| Fig. 1 Molecular targets and signaling pathways in intrahepatic cholangiocarcinoma (iCCA) with corresponding therapeutic strategies. Key receptors and targets, including IDH1/2, FGFR2, HER2, NTRK, and PD-1/PD-L1, are shown at the cell membrane or intracellular locations. Downstream signaling pathways, such as RAS/MAPK and PI3K/AKT, are highlighted to depict their roles in promoting cell proliferation, survival, and differentiation. Specific therapeutic agents targeting these pathways are indicated, including IDH1/2 inhibitors (ivosidenib (1) and enasidenib (6)), FGFR2 inhibitors (pemigatinib (25) and futibatinib (26)), BRAF/MEK inhibitors (dabrafenib (51) plus trametinib), and NTRK inhibitors (entrectinib (58) and larotrectinib (60)). Additional therapeutic agents include the KRAS inhibitor (sotorasib), CDK4/6 inhibitor (palbociclib), and antibody-based therapies targeting HER2 (trastuzumab) and PD-1/PD-L1 (pembrolizumab and nivolumab), demonstrating their clinical relevance in targeted therapies for iCCA. Small-molecule inhibitors and their targets are labeled with blue text and blue boxes, while antibody-based therapies and their targets are labeled with pink text and pink boxes to distinguish the therapeutic classes. | ||
Although strong evidence for the prognostic value of IDH1/2 is yet to be established, several studies suggest that mutations in IDH1/2 can serve as a favorable prognostic predictor of iCCA.50,51 Specifically, AG-120 (ivosidenib, 1), a small molecule inhibitor for mtIDH1, received approval from the U.S. Food and Drug Administration (FDA) in 2021 for the treatment of patients with advanced, metastatic, and chemotherapy-resistant CCA harboring IDH1 mutation.52 The molecular aberration of FGFR2 frequently occurring in iCCA is usually gene fusion of FGFR2 with the partner protein BICC1 producing the FGFR2–BICC1 fusion protein, which is responsible for the constitutive activation of several signaling pathways related to carcinogenesis.53 Furthermore, among the FGFR isoforms, gene fusions and rearrangements of FGFR2 are most commonly observed in iCCA.29 The results from the phase II clinical trial (FIGHT-202) suggest that 10–16% of patients with advanced or metastasized CCA possessing altered FGFR2 benefitted from treatment with an FGFR1–3 inhibitor, pemigatinib (25). Encouraged by these promising results, pemigatinib (25) received accelerated FDA approval in April 2020 as the first targeted therapy for advanced CCA.54,55
In addition, BRAF mutations play a significant role in the constitutive activation of the key signaling element in the MAPK cascade, which is the primary signaling cascade in the RAS-RAF-MEK-ERK pathway.56,57 In the study conducted by Goeppert et al. using the immunohistochemical screening approach, they found out that BRAF mutation, although less often encountered (<5%), is restricted to iCCA, which provides a positive diagnostic implication by narrowing the screening effort.35 NTRK is also one of the promising therapeutic targets in various malignancies. It is responsible for carcinogenesis by gene fusion or translocation. Zou et al. examined NTRK gene expression by immunohistochemistry (IHC) assay from surgical samples of 160 patients with iCCA and their study showed that translocation of the NTRK1 gene occurred in 5.6% (9/160) of patients. This suggests that NTRK inhibitors can potentially be effective in treating the subset of iCCA patients with NTRK gene alteration.58 HER2 is another promising therapeutic target for iCCA. It is highly involved in signal transduction via the MAPK and phosphoinositide 3-kinase (PI3K) signaling pathways.59 Furthermore, the overexpression and amplification of HER2 often indicate poor prognosis due to their high correlation with the invasiveness and proliferation of CCA cells.60
Besides these molecular targets, the immune checkpoint pathway has also been investigated as a potential therapeutic target in iCCA. PD-1 and PD-L1, which are ligands of PD-1, play an important role in inhibiting the activation of T cells, and their upregulation has been observed in various cancers, including iCCA.61–63 Several clinical trials have evaluated the efficacy of PD-1/PD-L1 inhibitors in advanced or metastatic solid tumors, with promising results.64,65 In fact, pembrolizumab, a PD-1 inhibitor, has been granted accelerated FDA approval for addressing solid tumors that are unresectable or have metastasized.66 However, further studies are needed to identify optimized mono- and combination therapeutic strategies for enhancing the antitumor immune responses in iCCA.
3. Molecular targeted therapies
Recent advancements in molecular profiling of iCCA have identified several promising molecular alterations for targeted therapy, including IDH1/2, FGFR, BRAF, NTRK, HER2, and PD-1/PD-L1. This article is distinguished from other reviews on iCCA by providing a comprehensive overview of these promising targets, specifically focusing on their current status, molecular basis, medicinal chemistry aspects, and the potential underlying causes of acquired resistance. The aim is to provide distinct perspectives that can guide drug discovery programs for iCCA.3.1. IDH 1/2
Isocitrate dehydrogenase (IDH) plays a crucial role as a metabolic enzyme in the tricarboxylic acid (TCA) cycle. It consists of three isoforms, IDH1, IDH2, and IDH3, where IDH1 is found in the cytosol and peroxisomes, whereas IDH2 and IDH3 are located in the mitochondria. IDHs originally work by catalyzing the oxidative decarboxylation of isocitrate to produce 2-oxoglutarate (2-OG) using NADP+ and Mg2+. Catalysis of this reaction by IDH1 and IDH2 is reversible, while IDH3 catalyzes the same reaction in an irreversible manner. Owing to their significance in homeostasis by regulating the energy and REDOX balance, IDHs have been comprehensively studied as a part of metabolism research in the last few decades.67 However, given that the reports on the involvement of mutated IDH1 and IDH2 in various human diseases have been widespread in recent years, molecular aberrations in IDH 1/2 have attracted tremendous attention in drug discovery.68,69 The pathogenic mechanisms of mutated IDHs associated with cancer have been the most extensively studied.70,71 IDHs, when mutated, lose their original enzymatic role and acquire a gain-of-function activity with neomorphic character, resulting in the accumulation of D-2-hydroxyglutarate (2-HG), which is also referred to as ‘oncometabolite’ (Fig. 2A).72 2-HG, being structurally similar to 2-OG, inhibits 2-OG-oxygenases from participating in histone and DNA demethylation in a competitive manner. Consequently, high levels of 2-HG promote epigenetic dysregulation, abnormal cellular differentiation, and oncogenesis.71 | ||
| Fig. 2 (A) Reactions catalyzed by wtIDH1/2 and mtIDH1/2. (B) Dimeric interface of IDH1 and inhibitors binding to this pocket. Figure modified from ref. 73. Copyright© 2023 (licensed under CC-BY 4.0), JMC. | ||
Oncogenic mutations in IDH1 and IDH2 are observed in several human cancers, including acute myeloid leukemia (AML), glioma, chondrosarcoma, and iCCA.74 Among 10–25% of IDH1/2 mutations are observed in iCCA cases, where IDH1, being more frequently mutated than IDH2, shows up to 50% of mutation rate in iCCA, while the contribution of IDH2 is minimal compared to IDH1 with a mutation rate of ∼5%.75–77 After discovering that gain-of-function mutations in IDHs are highly associated with a wide range of cancer types, both solid and hematologic, this has caused a significant shift in drug discovery approaches, leading to the exploration of targeted therapies that focus on the development of synthetic molecules, aiming to effectively block the aberrant activities of mutated IDHs (mtIDHs). Several small molecule inhibitors against mtIDHs are currently under clinical investigation for patients with iCCA or advanced CCA including AG-120 (ivosidenib, 1), IDH305 (2), FT-2102 (olutasidenib, 3), GSK864 (4), BAY-1436032 (5), AG-221 (enasidenib, 6), and AG-881 (vorasidenib, 7).78–82 Notably, based on the results from a clinical study (ClarIDHy, AG120-C-005), AG-120 (ivosidenib, 2) received FDA approval in August 2021 for the treatment of patients with unresectable advanced and metastatic CCA harboring mtIDH1 (Fig. 3).
 | ||
| Fig. 3 Structures of mtIDH1/2 inhibitors. | ||
The frequently occurring mutations take place in arginine position 132 in IDH1 and 140 or 172 in IDH2.83 These mutations result in the conversion of arginine to histidine, cysteine, serine, lysine, glycine, and valine, which have a shorter length and less basic property. In most cases of mtIDH inhibitors, crystallographic studies suggest that they do not show direct interaction with the active substrate site of mtIDH, but they bind to an allosteric pocket on each monomer located at the dimeric interface of mtIDH involving a bundle of α-helices.84–86 In wild-type IDH (wtIDH), this pocket is usually closed due to the ionic interaction of R132 with D275 of the α10 regulatory segment. Alternatively, this ionic interaction is lost in mtIDH, for example in IDHR132H, because of the inappropriate positioning of the mutated residue destabilizing the interaction, which allows mtIDH variants to exhibit an accessible allosteric pocket to mtIDH inhibitors. This endows them with remarkable selectivity to mutated IDH variants over wtIDH (Fig. 2B).87 Moreover, Liu et al. suggested that the relatively weak and separate binding of Mg2+ and 2-OG in mtIDH1 is related to the selective inhibition observed in the allosteric inhibitors, presumably by disrupting the binding of Mg2+ to the active site.88 Understanding the structural and molecular basis of binding features manifested by mtIDH inhibitors is an important factor in the design of improved inhibitors for the mutated variants of IDHs, showing promise as targeted therapies for iCCA harboring these mutations. A brief history, biological activities, status of clinical trials, binding modes, and medicinal chemistry aspects of promising mtIDH1/2 inhibitors for the treatment of iCCA are summarized in this section (Table 1).
| Target protein | Compound | Discovery/development | Scaffold | Key interacting residues | PDB | Biological activities/DMPK/clinical outcome | Clinical trial/status/patents | References |
|---|---|---|---|---|---|---|---|---|
| mtIDH1 | AG-120 (Ivosidenib, 1) | Agios | Phenylglycine | Ile128, Leu120 | — | IDH1R132H IC50 = 12 nM | Phase III (NCT02989857)/FDA-approved/WO2021/026436 WO2013107291 | 31, 89 |
| U87-MG (IDH1R132H) GI50 = 19 nM | ||||||||
| E h = 0.15 | ||||||||
| Efflux ratio in Caco2 cell = 2 | ||||||||
| mOS = 10.8 mos. | ||||||||
| mtIDH1 | IDH305 (2) | Novartis | Pyrimidinyl-oxazolidinone | Leu120, Ile128, Ser278 | 6B0Z | IDH1R132H IC50 = 18 nM | Phase I (NCT02381886)/on-going/WO2013046136 A1 | 78 |
| HCT116 (IDH1R132H) GI50 = 24 nM | ||||||||
| Clint in rat liver microsome = 45 μL min−1 mg−1 | ||||||||
| PPB (rat) = 88% | ||||||||
| Brain/plasma ratio in rat (total/unbound) = 0.61/0.18 | ||||||||
| Solubility = 130 μM at pH 6.8 | ||||||||
| mtIDH1 | GSK321 (14) | GlaxoSmithKline (GSK) | Tetrahydropyrazolopyridine | Ile128, Pro127, Trp124, Arg119, Leu120 | 5DE1 | Inhibition of intracellular 2-HG production: EC50 in HT-1080 (IDH1R132C) = 85 nM | — | 83 |
| mtIDH1 | BAY-1436032 (5) | Bayer | Aminobenzimidazole | Ser280, His132 | 5LGE (IDH1R132H, docking result) | IDH1R132H IC50 = 15 nM | Phase I (NCT02746081)/on-going/WO/2015121210 A1, US10442772B2 | 90 |
| HT1080 (IDH1R132C) GI50 = 135 nM | ||||||||
| ORR = 11% | ||||||||
| mtIDH2 | AG-221 (Enasidenib, 6) | Agios/Celgene | Diaminotriazine | Gln316 | 5I96 | IDH2R140Q/WT IC50 = 30 nM (at 16 h) | Phase I & II (NCT01915498)/FDA-approved | 91 |
| U87MG (IDH2R140Q) GI50 = 10 nM | ||||||||
| PCT/US2016/064615 | ||||||||
| TF-1 (IDH2R140Q) GI50 = 20 nM | ||||||||
| mtIDH1/2 | AG-881 (Vorasidenib, 7) | Agios/Servier | Diaminotriazine | Gln277 | 6VEI | IDH1R132H IC50 = 6 nM | Phase III (NCT04164901)/FDA-approved | 82 |
| IDH2R140Q IC50 = 12 nM | ||||||||
| Brain/plasma ratio in rat = 0.65 | ||||||||
| PFS = 27.7 mos. |
AG-120 (ivosidenib, 1) and phenylglycine derivatives. Agios Pharmaceuticals has made pioneering achievements in the discovery of novel mtIDH inhibitors. The starting point for phenylglycine-based mtIDH inhibitors was compound 8, having an IC50 of 0.09 μM against IDHR132H, which was identified via high-throughput screening (HTS) reported by Popovici-Muller and colleagues (Fig. 4). The phenylglycine scaffold was prepared through Ugi reaction and multicomponent synthesis. It is also noteworthy that the enantioselectivity for this reaction could be improved with the use of chiral phosphoric acid as a catalyst.92 Although the X-ray cocrystal data of the series of derivatives is not resolved to date, several studies suggest that these compounds also selectively bind to the allosteric pocket formed by mtIDH1, similar to other inhibitors.84,93 Initially, the effect of the α-carbon stereocenter was analyzed by chiral synthesis, showing that (s)-enantiomer is responsible for the activity observed in the racemate. Further optimization was performed with the racemate, enabling rapid structure–activity relationship (SAR) studies by changing the position with a cyclopentyl group to cyclohexyl group, which increased the activity by roughly two-fold (IDHR132H IC50 = 0.05 μM). Alternatively, changing this position to smaller groups (cyclobutyl, tert-butyl, cyclopropyl), aromatic rings, or rings with heteroatoms (morpholine and piperidine) did not give favorable IC50 values (IDHR132H IC50 = 0.20–53.1 μM). In addition, the thiophene position was investigated with the aim to improve the physicochemical properties and decrease the overall lipophilicity. In the case of aromatic amines and fused nitrogen heterocycles, they maintained the biochemical activity; however, clog
P was also increased, retaining the high hydrophobicity. AGI-5198 (9) having thiophene replaced with 2-imidazole exhibited the lowest clogP value and retained its biochemical activity against IDHR132H (IC50 = 0.07 μM). AGI-5198 (9) was also equipotent in U87 (glioblastoma cells) cellular assays (IC50 = 0.07 μM) and showed reasonable pharmacokinetic (PK) results with AUC of 20800 h ng mL−1 at 50 mg kg−1. Moreover, AGI-5198 (9) displayed effective tumor 2-HG inhibition (∼90%) in vivo in a U87 R132H tumor xenograft mouse.94 Subsequently, extensive optimization efforts of AGI-5198 (9) by the same team from Agios led to the discovery of the first mtIDH1 inhibitor to have entered human clinical trials, AG-120 (1). The in vitro drug metabolism and pharmacokinetic (DMPK) profiling of AGI-5198 (9) revealed that this compound displays unfavorable metabolic stability (hepatic excretion ratio (Eh = 0.93) with cyclohexyl and imidazole rings being possible metabolic soft spots. To increase its metabolic stability, the o-methyl group was modified to o-chloride and a fluorinated cycloalkyl group was introduced to avoid unwanted oxidative metabolism. Then, by scanning heterocycles at the 2-methylimidazole position, pyridines bearing a cyanide group were found to be effective in achieving the targeted activity and DMPK properties. In addition, the incorporation of a difluorinated phenyl ring identified that AGI-14100 (10) possesses single-digit nanomolar enzymatic and cellular activities including favorable metabolic stability (Eh = 0.23). However, further assessment of AGI-14100 (10) with the human pregnane X receptor (hPXR) assay showed that it potentially induces the cytochrome P450 (CYP) 3A4 isotype. Given that the CYP activity is highly proportional to the hydrophobicity of the compound, they attempted to alleviate CYP induction by increasing the polarity of the molecule. Although modifying pyrrolidinone and cyanopyridine did not bring about the desired results, replacing one of the fluorine atoms with nitrogen incorporated in the ring dramatically reduced hPXR activation, which led to the discovery of AG-120 (1). The putative binding mode of AG-120 (1) shows that two terminal carbonyl groups provide key interactions by forming hydrogen bonds with Tyr272 and Ala111, in addition to hydrogen bonds formed by the cyanopyridine group with Ile128 and Leu120.89 AG-120 (1) demonstrated potent antiproliferative activities in multiple cancers cells harboring endogenous or overexpressed mtIDH such as U87-MG (R132H), HT1080 (R132C), COR-L105 (R132C), and HCCC-9810 (R132S) with GI50 values in the range of 8 to 20 nM. Moreover, AG-120 (1) is capable of reducing the 2-HG level by 96% at a concentration as low as 0.5 μM. In a randomized phase 3 clinical trial with patients with iCCA, the mOS of patients given AG-120 (1) was 10.8 months, whereas it was 9.7 months in the placebo group. In addition, the 12 month overall survival rate for the AG-120 (1) group increased by 10% compared to the placebo group (48% and 38%, respectively) (NCT02989857).31,95 Based on these results, AG-120 (1) was granted FDA-approval for the treatment of patients with metastatic or advanced unresectable chemotherapy refractory CCA harboring an mtIDH1 on August 25, 2021.
 | ||
| Fig. 4 Optimization process of AG-120 (ivosidenib, 1). | ||
Pyrimidinyloxazolidinone derivatives. Pyrimidinyloxazolidinone derivatives have been extensively investigated by Cho and colleagues from Novartis. IDH125 (11) derived from an HTS hit gave significant clues on the SAR preference for this series of mtIDH1 inhibitors (IDHR132H IC50 = 0.22 μM). HTS was performed by biochemical assay measuring 2-HG levels with LC/MS using the IDHR132H homodimer.96 The (S)-amine center was found to be preferred for achieving binding affinity against mtIDH1, although the effects of chirality at oxazolidinone sometimes varied. IDH125 (11) was anticipated to inhibit other kinases due to its amino-pyrimidine core, which is also one of the most widely used scaffold in kinase inhibitors with structural features resembling the purine motif of ATP (Fig. 5A). However, IDH125 (11) did not show kinase activity across the internal and external panel tested. X-ray cocrystal determination of IDH125 (11) revealed that it binds to the allosteric pocket near the substrate binding site, which is formed by the destabilized ionic interaction between Arg132 and Asn271.85 Nitrogen at the 3-position and 2-amino group participated in hydrogen bonds with the protein backbone residue Leu120 and the methyl group at the chiral center occupied the small hydrophobic pocket formed by Arg109, Glu110, and Ile128.96 Consequently, substituting these positions was quite intolerable, while substituents other than isopropyl were tolerable for the oxazolidinone part. Given that modifying oxazolidinone or pyrimidine did not provide additional potent analogues, the researchers moved on to the exploration of the phenyl group. Although large substituents such as naphthyl and biphenyl were also tolerable (IDHR132H IC50 = 0.10–0.25 μM), IDH662 (12) bearing a p-phenoxyphenyl ring showed a significant increase in selectivity towards wtIDH (IC50 = 1.03 μM) over IDHR132H (IC50 = 0.010 μM), which suggests that the allosteric pocket is preferably accessible in the mtIDHs. IDH662 (12) also potently inhibited 2-HG production in HCT116 cells harboring IDHR132H (IC50 = 0.022 μM). However, IDH662 (12) did not exhibit desired efficacy in an in vivo mouse pharmacodynamic (PD) study with an HCT116-IDHR132H xenograft model presumably due to its high plasma protein binding (PPB, 99%).96 In addition, metabolic studies predicted that benzylamine was identified to be labile for oxidative metabolism. The researchers expected to replace this position with heterocycles to mitigate the potential oxidation at the benzylic carbon. It is also noteworthy that reactions with less nucleophilic benzylamines only proceeded with 2-fluoro-4-oxazoldiinonyl pyrimidine instead of 2-chloro-4-oxazolidinonyl pyrimidine. Given that the phenyl-pyrimidine analogue displayed enhanced intrinsic clearance in the rat microsome (Clint = 240 μL min−1 mg−1), retaining the pyrimidine and biaryl system in the subsequent SAR led to the identification of the optimized hit compound IDH889 (13, IDHR132H IC50 = 0.020 μM). IDH889 (13) was shown to effectively inhibit new 2-HG production without eliminating 2-HG from mtIDH1 tissue in an HCT-116-IDH1R132H xenograft model. Moreover, it showed favorable brain distribution (1.4 brain/blood ratio, 30 mg kg−1 po) with excellent permeability, and also efflux was not seen in Caco-2 and MDR1-MDCK cells.96 The Novartis group attempted to further optimize the DMPK profile of IDH889 (13) owing to its high in vitro intrinsic clearance (Clint), plasma protein binding (PPB), and poor solubility, which may require a high efficacious dose in humans (>10 g BID).78 To balance adequate polarity and lipophilicity, the researchers maintained the optimized biaryl system of IDH889 (13). Also, based on the crystal structure of IDH889 (13), one ortho nitrogen in the first ring participating in a hydrogen bond with Ser278 was also retained for the SAR study. Throughout the SAR, the meta-substituent at the terminal pyridine ring was found to be important in achieving potency. The bulky tert-butyl was tolerable at this position; however, it showed an increased Clint in rats. The trifluoromethyl group provided the most balanced potency and in vitro clearance (IDHR132H IC50 = 0.077 μM; Clint = 101 μL min−1 mg−1). In addition, incorporating a methyl group at the 2-position of the first pyridine ring further enhanced the potency (IDHR132H IC50 = 0.012 μM; Clint = 101 μL min−1 mg−1). Subsequently, the substituent effects of the oxazolidinone part were explored. Modification of the isopropyl group to methyl and ethyl groups effectively reduced the in vitro clearance at the expense of decreased cellular activity (Clint = 36 and 58 μL min−1 mg−1, respectively). Installing a fluorine and the chirality preference analysis led to the identification of the optimized lead compound IDH305 (2), which exhibited an improved balance between DMPK and mtIDH1 inhibitory activity compared to IDH889 (13). Moreover, IDH305 (2) also binds to IDH1R132H in a similar manner to IDH889 (13) (Fig. 5B). In a PK/PD study with an HCT116-IDH1R132H/+ xenograft model, IDH135 effectively reduced the tumor 2-HG concentration (∼87% 2-HG maximal inhibition). Moreover, the intravenous administration of IDH305 (2) at 5 mg kg−1 in rats displayed a brain/plasma ratio of 0.61, indicating that IDH305 (2) is a blood–brain barrier (BBB) penetrant. Encouraged by these results, IDH305 (2) was selected as a clinical candidate for the treatment of AML, chondrosarcoma, and CCA (NCT02381886).78,97,98
 | ||
| Fig. 5 (A) Structures of pyrimidinyloxazolidinone-based mtIDH1 inhibitors. (B) Binding mode of IDH305 (2) in IDH1R132H. | ||
Tetrahydropyrazolopyridine derivatives. Researchers from GlaxoSmithKline Pharmaceuticals (GSK) investigated mtIDH1 inhibitors based on the tetrahydropyrazolopyridine scaffold (Fig. 6A).83 GSK321 (4) derived from an HTS hit potently inhibited mtIDH1 enzymes (IC50 = 4.6 nM (R132H), 3.6 nM (R132C), and 2.9 nM (R132G)). Although GSK321 (4) modestly inhibited wtIDH as well, it was not capable of inducing inhibition in IDH2 enzymes. GSK990 (14) is a structurally related analogue of GSK321 (4) and does not inhibit wtIDH and mtIDH1/2, making it an effective negative control. In a mechanistic evaluation in HT-1080 cells harboring IDH1R132H mutation, GSK321 (4) effectively inhibited the intracellular production of 2-HG (EC50 = 85 nM), whereas GSK990 (14) did not cause any changes in the intracellular 2-HG level. Moreover, chemical proteomic analysis of GSK321 (4) demonstrated that it is a highly selective inhibitor of IDH1. This team also studied the crystal structure of GSK321 (4) to IDH1R132H homodimer, indicating that it is an allosteric inhibitor of mtIDH1 with the 4-fluorophenyl group providing a favorable lock in a lipophilic pocket (Fig. 6B). Alternatively, the poor inhibitory activity of GSK990 (14) could be due to the presence of the polar imidazole group, possibly causing steric clash with the residues aligned in this lipophilic region.83 Further optimization of GSK321 (4) led to the identification of GSK864 (15) with enhanced PK profile to be used for in vivo evaluation. In the preclinical in vivo studies, a reduced 2-HG level in IDH1 was observed in an IDH1 mutant AML mouse xenograft model as well as a decrease in leukemic blasts upon treatment with GSK864 (15).83
 | ||
| Fig. 6 (A) Structures of tetrahydropyrazolopyridine-based mtIDH1 inhibitors. (B) Binding mode of GSK321 (4) in IDH1R132H. | ||
Aminobenzimidazole derivatives. According to the screening of a library containing over 3 million compounds, researchers from Bayer identified a potent lead compound (16) based on the aminobenzimidazole scaffold (IC50 IDHR132H, 30 nM), which further led to the identification of BAY-1436032 (5) upon subsequent optimization processes (Fig. 7A).90 BAY-1436032 (5) displayed potent inhibitory activity against IDHR132H (IC50 = 15 nM), whilst not inhibiting wtIDH1 and IDH2 with IC50 values of 20 μM and >100 μM, respectively. Besides its allosteric binding mode, providing the selectivity for mtIDH1 over wtIDH1, the structural analysis based on the cocrystal structure of homodimeric IDH1R132H and structurally similar compound 17 (due to inability to obtain cocrystalized BAY-1436032 (5) and IDH1R132H complex) revealed that the carboxylic acid group directly interacts with His132 by forming a salt bridge. Additionally, the aminobenzimidazole scaffold participates in a hydrogen bond with the protein backbone residue Ser280, while the cyclohexyl group and isopropylphenyl group are completely buried in hydrophobic sub-pockets adjacent to the binding site (Fig. 7B). BAY-1436032 (5) effectively inhibited 2-HG production in cells bearing the following IDH1 mutations: R132H, R132C, R132G, R132S and R132L (∼>70% inhibited, 500 nM). Moreover, it also potently inhibited HT1080 cells (IDH1R132C) having high intracellular 2-HG levels with an IC50 value of 135 nM.90 The effects of potential PK/PD and safety upon dose escalation of BAY-1436032 (5) were investigated first-in-human in a phase I clinical trial (NCT02746081) with a subset of 81 non-randomized participants possessing eligible mtIDH1 solid tumors including iCCA, which demonstrated that BAY-1436032 (5) is orally well-tolerated when patients are treated with 1500 mg twice daily. Furthermore, oral administration of BAY-1436032 (5) resulted in rapid and robust suppression of plasma 2-HG (median maximal reduction = 76%) to levels associated with many healthy iCCA subjects. Although effects on other solid tumors were modest, only a subset of patients with lower grade glioma (LGG) showed clinical benefit with an objective response rate (ORR) of 11%.80,99
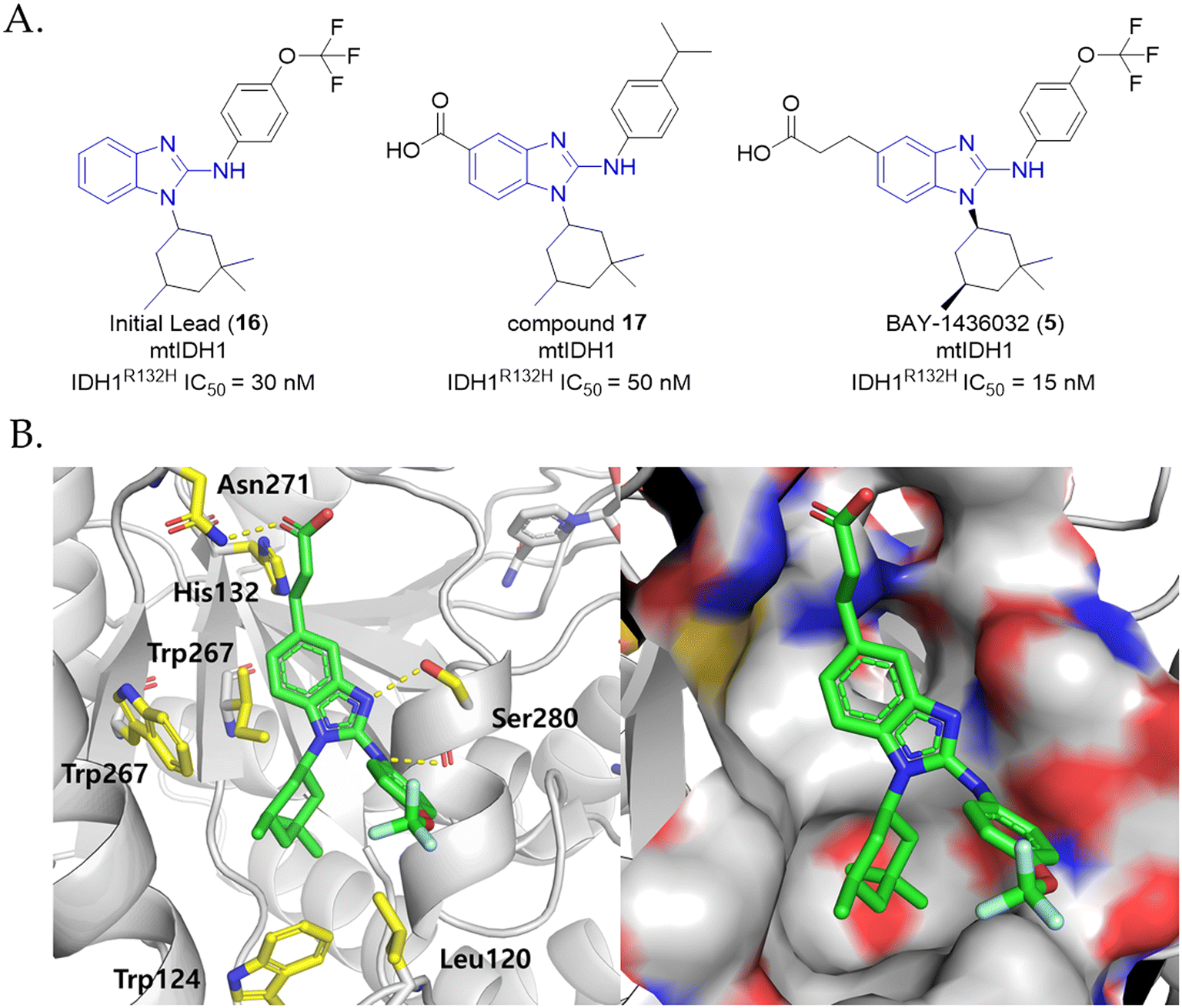 | ||
| Fig. 7 (A) Structures of aminobenzimidazole-based mtIDH1 inhibitors. (B) Putative binding mode of BAY-1436032 (5) in IDH1R132H. | ||
AG-221 (enasidenib, 6) and AG-881 (vorasidenib, 7), and diaminotriazine derivatives. The initial hit-to-lead optimization of a diaminotriazine-based hit compound provided an improved analogue (18) showing a sub-100 nanomolar IC50 against the IDH2R140Q homodimer (Fig. 8A).100 However, despite its potent enzymatic and cellular activities, further development of this compound was limited due to its high lipophilicity, leading to poor solubility and in vitro microsomal stability. Subsequent optimization was performed by installing mildly polar substituents around the triazine core to increase the overall physicochemical properties, which led to the identification of an orally available and potent mtIDH2 selective diaminotriazine analogue with improved solubility and metabolic stability (solubility at pH 7.4 = 23 μM; HLM Eh = 0.16). AG-221 (6) is a selective binder of either homo- or heterodimers of mtIDH2 over wtIDH1 and IDH1R132H variants. Among the IDH2 mutants, AG-221 (6) more potently inhibited cancer cells harboring R140Q mutation (IC50 = 0.1–0.2 μM) than those harboring R172K mutation (IC50 = 0.5–1.6 μM). The cocrystal structure of AG-221 (6) complexed with IDH2R140Q revealed that the nitrogen from triazine and amine of the 2-methyl-2-propanol group form a pair of hydrogen bonds with Gln316 (Fig. 8B). In addition, the trifluoromethyl moiety forms a halogen bond with the Asp312 residue of mtIDH2. The high hydrophobic nature of the pockets in the homodimeric interface is responsible for the steric clash or desolvation penalty, providing unfavorable interactions when larger polar substituents are installed. The PK/PD study of AG-221 (6) with a U87MG-IDH2R140Q mouse xenograft model demonstrated that dose-dependent PK was seen upon the administration of AG-221 (6) with a half-life of 6–7 h.100 Owing to the abundance of mtIDH2 in AML patients, AG-221 (6) is extensively investigated in patients with advanced AML. In this regard, the phase 1/2 clinical trial data for AG-221 (6) shows that its administration is well-tolerated and brings about clinical benefits in patients with relapsed/refractory AML.101 The clinical effects of AG-221 (6) on advanced solid tumors such as glioma, chondrosarcoma, and iCCA are still being investigated.81
 | ||
| Fig. 8 (A) Structures of diaminotriazine-based mtIDH2 inhibitors. (B) Binding mode of AG-221 (enasidenib, 6) in mtIDHR140Q. | ||
Encouraged by the consecutive FDA approval of the mtIDH inhibitors AG-120 (ivosidenib, 1) and AG-221 (enasidenib, 6) for patients with mtIDH cancers, Agios Pharmaceuticals sought to fix the low brain drug exposure exhibited by AG-120 (ivosidenib, 1) and AG-221 (enasidenib, 6) to expand their use in the treatment of glioma harboring mtIDHs (Fig. 9A).82 These researchers also focused on the observation that targeting co-occurring mtIDH1 and mtIDH2 in the same tumor can have potential therapeutic use as in the case of long-term treatment of LGG. Initially, it was assumed that lowering the topological polar surface area (tPSA) and number of hydrogen bond donors/acceptors would increase its blood–brain barrier (BBB) penetration. AG-12026 (19), which was identified during the discovery programs of AG-221 (ivosidenib, 1), served as an adequate starting point for subsequent optimization. AG-12026 (19) is a good BBB penetrant and exhibits potent inhibitory activities against both mtIDH1 and mtIDH2, which is unusual considering the selective nature manifested by most allosteric mtIDH inhibitors.82 The rapid evaluation of SAR studies with several triazine compounds led to another analogue (AG-15056, 20) with a brain to plasma ratio of 1.5, whilst also inhibiting both mtIDH1 and mtIDH2 enzymes. Given that other compounds showing dual mtIDH1 and mtIDH2 inhibition shared the same diaminotriazine core, further optimization was focused on this series of compounds. Moreover, the evaluation of two stereocenters of AG-15056 (20) revealed that the (R,R)-configuration is optimal for achieving superior potency. The cocrystal structure of AG-15056 (20) bound to IDH2R140Q shows that the inhibitor exhibits a significant feature in its binding mode to the protein, which is the symmetrical interaction of the trifluoromethyl moiety with Asp273 on both sides of the pocket. Due to the conserved residues present in most parts of the binding site, AG-15056 (20) is capable of possessing similar affinities to both mtIDH1 and mtIDH2.82 This effect is further supported by the SAR investigation of the triazine 6-position. When trifluoromethyl was removed or changed with various polar substituents, a great loss in potency was observed. The introduction of the 2-chloropyridyl group at this position showed inhibitory activity comparable to 2-trifluoromethylpyridiyl (IC50 = 32 nM and 6 nM, respectively). Subsequently, upon exploration of symmetric substituents, the absence of a methyl group resulted in a decrease in enzymatic potency. Analogues with symmetrical difluorocyclobutyl and difluorocyclopentyl substituents showed single-digit nanomolar activities (IC50 = 9 nM and 6 nM, respectively). In addition, the best symmetric substituents from the 2-trifluoromethyl pyridyl system were installed in the 2-chloropyridyl system. Although these two substituents did not further increase the potency, changing them to trifluoro-2-methylpropane resulted in sub-nanomolar potency, leading to the identification of AG-881 (vorasidenib, 7; IDH1R132H/wtIDH1 IC50 = 0.6 nM). The cocrystal structure of AG-881 (vorasidenib, 7) in complex with the IDH1R132H and IDH2R140Q homodimers revealed that AG-881 (7) symmetrically binds to the same allosteric pocket present in both proteins, which is also consistent with previous findings in AG-15056 (20) (Fig. 9B).82 The phase I clinical trial of AG-881 (vorasidenib, 7) in patients with advanced solid tumors including glioma demonstrated that it shows antitumor activity in non-enhancing glioma with an ORR and PFS of 18% and 36.8%, respectively, while antitumor activity was not observed in enhancing glioma (no ORR; PFS = 3.6 months) (NCT02481154).102 Based on the favorable safety and antitumor activity of AG-881 (7) in the phase I clinical trial, the effects of AG-881 (vorasidenib, 7) versus the placebo group were evaluated in a randomized phase III INDIGO study (NCT04164901).103
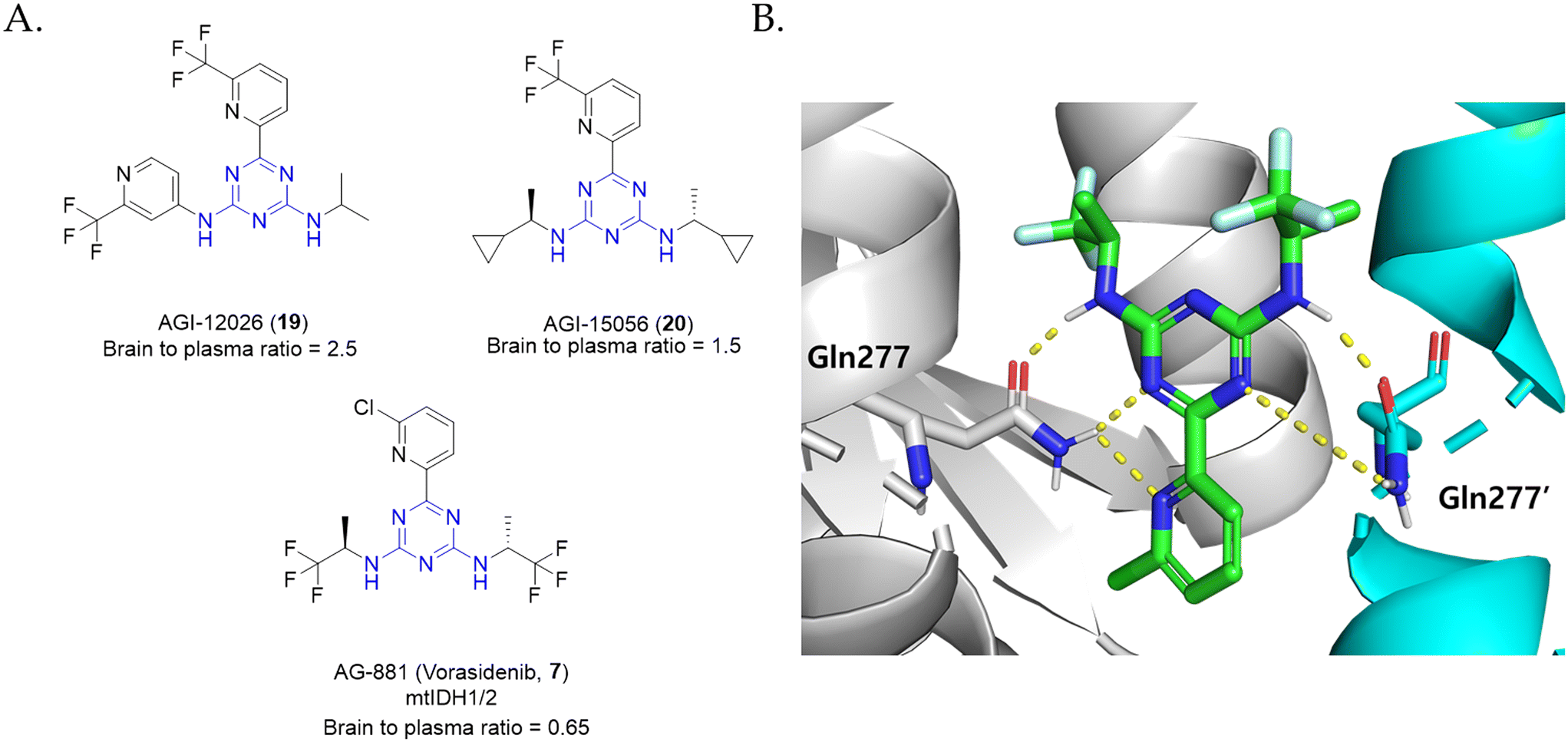 | ||
| Fig. 9 (A) Structures of diaminotriazine-based mtIDH1/2 inhibitors. (B) Binding mode of AG-881 (vorasidenib, 7) in mtIDHR132H. | ||
Based on the reports of two clinical cases by Cleary et al., one patient with iCCA acquired a newly formed IDH2R172K mutation after 17 month treatment with ivosidenib (1) and the other patient developed a novel IDH1D279N mutation after one year treatment with ivosidenib (1).104 Additionally, in patients with AML, where post-therapy acquired mtIDH is more extensively studied, IDH1S280F is reported to cause drug resistance to ivosidenib (1) presumably due to the replacement of the relatively small serine to bulky phenylalanine residue, causing steric hindrance in the dimeric interface, which contributes to the reduced binding affinity of ivosidenib (1).105,106 This is further supported by the study conducted by Reinbold et al.107 In their evaluation, ivosidenib (1) did not inhibit R132C/S280F and R132H/S280H, while it showed modest inhibition against R132C/S280A (IC50 = 992 nM), implying steric hindrance. Also, possibly the loss of the hydrogen bond with the serine hydroxyl group may contribute to the reduced binding affinity of ivosidenib (1). Moreover, according to their study, other mtIDH1 inhibitors such as GSK864, IDH224, DS1001B, and FT2102 (olutasidenib, 3) are capable of suppressing the 2-HG levels in R132C/S280F and R132H/S280F overexpressed cells. These inhibitors differ from ivosidenib (1) in that 2 molecules of these inhibitors stoichiometrically bind to each mtIDH1 dimer, whereas 1 molecule of ivosidenib (1) binds to the dimer.107 Among them, FT2102 (olutasidenib, 3), which is a quinolinone-based allosteric inhibitor of IDH1R132H, most effectively suppressed 2-HG production in LN-18 cells (glioma) overexpressed with either R132C/S280F or R132H/S280F (Fig. 10).107 In the reports by Cleary et al., an in silico evaluation was performed to determine the binding feature of ivosidenib (1) to the R132H/D279N double mutant.104 The orientation of asparagine residue and the reshaped pocket around it result in steric clash with the chlorophenyl group, conferring resistance.
 | ||
| Fig. 10 Structures of potential IDH 1/2 inhibitors for overcoming drug-resistance. | ||
Moreover, the researchers identified that a structurally unrelated covalent mtIDH1 inhibitor, LY3410738 (21), effectively suppressed the production of 2-HG in TF-1 cells expressing the IDH1R132H/D279N double mutation.104 LY3410738 (21) forms more solid interactions including a covalent bond with Cys269 and additional hydrogen bonds with Ile128 and Leu120 buried deeply in the cavity. Moreover, the binding mode of LY3410738 (21) is retained similarly in both IDHR132C and IDHR132C/D279N being minimally affected by the conformation changes brought by double mutation, which contributes to the maintained binding affinity of LY3410738 (21).104 LY3410738 (21) is being evaluated in a clinical trial for the treatment of iCCA and other advanced solid tumors harboring mtIDH1 (NCT04521686).108
Alternatively, a selective mtIDH2 inhibitor shows weak inhibitory activities against IDH2R172K, which is a point mutation often encountered as an acquired mutation in patients treated with AG-221 (6), or also as an isoform switching mutation in patients treated with ivosidenib (1). TQ05310 (22) is a close analogue of AG-221 (6), which does not only exhibit superior inhibition of IDH2R140Q compared to AG-221 (6), but also potently inhibits the IDH2R172K mutation (IC50 of TQ05310 (21) = 38 nM vs. IC50 of AG-221 (6) = 625 nM). Although the binding features of TQ0310 and AG-221 (6) could not be compared in detail due to the absence of a cocrystalized IDHR172K structure, the researchers proposed that Gln316 is the critical residue contributing to the binding of TQ0310 and AG-221 (6) to IDH2R140Q.109
3.2. FGFR
Fibroblast growth factor receptors (FGFRs) are a subfamily of receptor tyrosine kinases (RTKs) consisting of four main family members (FGFR1–4) and an additional receptor also known as fibroblast growth factor like 1 (FGFRL1 or FGFR5).110 The components of FGFR1–4 include three immunoglobulin-like domains (IgI–IgIII), a transmembrane (TM) domain, an intracellular tyrosine kinase domain composed of two parts (kinase 1 and kinase 2), and an acidic box.110 Usually, fibroblast growth factors (FGFs) bind to the receptor in the ligand-binding pocket formed by the IgII and IgIII domains.111 Among the 22 reported FGFs, there are only 18 of these known to interact with FGFR1–4, leading to stimulation of kinase activity upon the induction of dimerization, while FGFR5 acting as a decoy receptor for FGFs lacks kinase activity due to the absence of the kinase domain and is reported to regulate FGFR signaling via a negative feedback mechanism.112–114 The acidic box plays a crucial role in the regulation of FGFR activities by interacting with the heparan–sulfate-binding site.115,116 Dimerized FGFRs induce a conformational change in the receptor structure, providing dramatically increased (∼100-fold) kinase activities and resulting in a cascade of intracellular phosphorylation of receptor kinase domains. This activates downstream signal transduction pathways such as phospho-inositide-3kinase (PI3K)/Akt, extracellular signal-regulated kinase (ERK)/mitogen activated protein kinase (MAPK), and signal transducer and activator of transcription (STAT), promoting cell differentiation, proliferation, development, angiogenesis, and survival.112,117 In addition, alternative splicing of FGFR into various isoforms and tissue-specific expression of ligands and receptors contribute to the regulation of specific FGF-FGFR interactions.118 Accordingly, the alteration of FGFRs causing aberrant signaling is highly associated with a broad range of human cancers.119–121In fact, alteration of FGFR1–4 is detected in approximately 10% of all human cancers, which is mostly attributed to the activation of single-nucleotide variants (SNVs), gene fusion, and chromosomal translocation, causing gain-of-function mutation of FGFRs.119,120 Helsten and colleagues extensively investigated the molecular landscape of FGFR in cancer by analyzing the frequencies of aberrant FGFRs in 4853 solid tumors. This analysis demonstrated that FGFR aberrations were present in 7.1% of the cases examined, with FGFR1 being the most frequently encountered alteration among the FGFR aberrations.120 In addition, they found that gene amplification of FGFR was observed in the most cases (66%), followed by gene mutation (26%) and gene rearrangement (8%).120 Especially, in some types of cancer such as iCCA and urothelial cancer, the alteration in FGFR increased to 10–30% and this increased frequency is also reported to be FGFR isotype-specific.119 For example, 15–20% of alterations in FGFR1 are observed in head and neck cancer and non-small cell lung cancer (NSCLC) and 15% of alterations in FGFR3 is seen in urothelial cancers.120,122–124 In patients with iCCA, major aberrations are observed in FGFR2 (10–15%), while although rare, aberrant FGFR1 and 3 are detected in some cases.29,47,50 Overexpression of FGFR4 has been also reported to be related to poor prognosis in CCA.125 However, it is worth noting that FGFR2, besides being the major aberrant FGFR responsible for the disease, it is almost exclusively detected in iCCA.47,126 The significant impact of FGFR2 compared to other FGFR isotypes can be attributed to its tissue-specific expression in cholangiocytes and its frequent gene fusions (e.g., FGFR2-BICC1 and FGFR2-TACC3) in iCCA.127,128 These fusions enable ligand-independent dimerization and activation of pathways such as MAPK and PI3K/AKT, promoting tumorigenesis.129 These unique features of FGFR2 underline its specificity and importance in iCCA and highlight its potential as a key therapeutic and diagnostic target in this disease.
Furthermore, it is also important to understand several oncogenic drivers of FGFRs. For instance, activating mutations of FGFRs make the receptor more prone to dimerization, leading to increased kinase activity and affinity to FGF ligands. Somatic activation of SNVs of FGFRs occurs in the intracellular kinase domain and the transmembrane domain, making it distinct from SNVs of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) usually occurring in the kinase domain.130 SNVs in FGFR are mostly observed in FGFR2 compared to FGFR1 and 3, which predominantly occur in the transmembrane (Y375C) and extracellular domain (Y375C, C382Y/R). In addition, FGFRs readily undergo chromosomal rearrangements with several partner proteins, resulting in gene fusion, which also constitutively activates FGFR.110,120 Gene fusions of FGFR2 are frequently detected in CCA and gene fusions of FGFR3 are observed in glioblastoma and urothelial cancers, whereas gene fusions of FGFR1 and 4 are rarely seen in solid tumors.120,131,132 The commonly encountered gene fusions of FGFR2 include FGFR2–BICC1 (bicaudal family RNA binding protein 1), FGFR2–PPHLN1 (periphilin 1), and FGFR2–TACC3 (transforming acidic coiled-coil containing protein 3).53,133 FGFR2–BICC1 gene fusion is frequently observed in iCCA, which makes the receptor constitutively active and leads to the uncontrolled activation of the MAPK and PI3K pathways.132 Overall, gene fusions of FGFRs result in increased dimerization capability, acting as a strong oncogenic driver.
Given the high relevance of FGFR alterations in specific types of cancer, such as iCCA and urothelial cancer, targeting aberrant FGFR signaling has emerged as a promising therapeutic approach.110,120,121 In particular, FGFR2 is the major aberrant FGFR responsible for iCCA, and therefore has become an important druggable target for the treatment and diagnosis of this disease.29,126,132 Boosted by the development of selective inhibitors against FGFRs, there are several FGFR inhibitors being evaluated in pre-clinical or clinical trials, which include derazantinib (23) and infigratinib (24) (Fig. 11). In 2020, pemigatinib was approved by the FDA as the first FGFR inhibitor for the treatment of advanced or metastatic CCA, which was based on the findings from its clinical trial (FIGHT-202, NCT02924376).134 Moreover, futibatinib (26) was granted accelerated approval from the FDA for the treatment of iCCA based on the results of a multi-center, single-arm clinical trial (NCT02052778), where futibatinib (26) displayed an ORR of 42%.135–137 In just less than a decade, approaches for FGFR inhibition have provided a successful avenue for the treatment of iCCA, which was considered an undruggable disease until recently. A brief history, biological activities, status of clinical trials, binding modes, and medicinal chemistry aspects of promising FGFR inhibitors for the treatment of iCCA are summarized in this section (Table 2).
 | ||
| Fig. 11 Structures of FGFR inhibitors for the treatment of iCCA. | ||
| Target protein | Compound | Discovery/development | Scaffold | Key interacting residues | PDB | Biological activities/DMPK/clinical outcome | Clinical trial/status/patents | References |
|---|---|---|---|---|---|---|---|---|
| Pan-FGFR | Derazantinib (ARQ-087, 23) | ArQule | 5,6-Dihydro-benzo[h]quinazolin-2-amine | Ala567 | 3RI1 (FGFR2, docking result) | FGFR2 IC50 = 1.8 nM | Phase II (NCT03230318)/terminated/WO2010/078421 A1 | 138, 139 |
| FGFR1 IC50 = 4.5 nM | ||||||||
| FGFR3 IC50 = 4.5 nM | ||||||||
| FGFR4 IC50 = 34 nM | ||||||||
| NCI-H716 (colon cancer) GI50 = 100 nM | ||||||||
| SNU16 (gastric cancer) GI50 = 100 nM | ||||||||
| Pan-FGFR | Infigratinib (BGJ398, 24) | Novartis/QED therapeutics | N-Pyrimidin-4-yl-urea | Ala564, Asp641 | 3TT0 | FGFR1 IC50 = 0.9 nM | Phase II (NCT02150967)/FDA-approval withdrawn | 140, 141 |
| FGFR2 IC50 = 1.4 nM | ||||||||
| FGFR3 IC50 = 1.0 nM | ||||||||
| FGFR4 IC50 = 60 nM | ||||||||
| RT112 (bladder cancer) GI50 = 5 nM | ||||||||
| Bioavailability (mouse) = 32% | ||||||||
| Bioavailability (rat) = 51% | ||||||||
| ORR = 23% | ||||||||
| mPFS = 7 mos. | ||||||||
| FGFR1–3 | Pemigatinib (INCB045828, 25) | Incyte | 1-Ethyl-1,3,4,7-tetrahydro-2H-pyrrolo[3′,2′:5,6]-pyrido[4,3-d]pyrimidin-2-one | Ala564 | 2FGI | FGFR1 IC50 = 0.4 nM | Phase II (NCT02924376)/FDA-approved | 142, 143 |
| FGFR2 IC50 = 0.5 nM | ||||||||
| FGFR3 IC50 = 1.2 nM | ||||||||
| WB IC50 KatoIII = 11 nM | ||||||||
| Solubility in FaSSIF = 11 mM | ||||||||
| Rat PK AUC = 7670 nM h | ||||||||
| Cyno PK AUC = 6190 nM h | ||||||||
| ORR = 36% | ||||||||
| mDoR = 9.1 mos. | ||||||||
| mOS = 17.5 mos. | ||||||||
| Pan-FGFR | Futibatinib (TAS-120, 26) | Taiho pharmaceuticals | Pyrrolopyrimidine | Ala567, Cys491 | 7OZY (FGFR2, docking result) | FGFR2 IC50 = 1.0 nM | Phase I/II (NCT02052778)/FDA-approved | 144–146 |
| EGFRdel19/T790M IC50 = >5000 nM | ||||||||
| SNU-16 (gastric cancer) GI50 = 3.7 nM | ||||||||
| ORR = 42% | ||||||||
| mDoR = 9.7 mos. |
Non-kinase selective FGFR inhibitor, ARQ-087 (derazantinib, 23). Derazantinib (23) is a pan-FGFR inhibitor with a novel tricyclic 5,6-dihydro-benzo[h]quinazolin-2-amine scaffold discovered by the biopharmaceutical company ArQule. It is a non-kinase selective inhibitor, which is also capable of potently inhibiting RET, DDR2, and PDGFRβ having IC50 values of 3–4 nM.138 The discovery of derazantinib (23) was initially facilitated by in silico virtual screening of compounds, leading to the identification of ARQ-523 (27) as an important chemical motif for this series of inhibitors, which can bind to the hydrophobic pocket exclusively present in inactive kinase structures (Fig. 12A). ARQ-523 (27) displays a unique binding mode, in which it forms strong hydrophobic interactions with unphosphorylated (inactive) FGFR2, thus inhibiting autophosphorylation of the protein with an IC50 value of 18 μM.139 Moreover, the incorporation of an additional phenyl group to the 6-position of the scaffold identified the analogue ARQ-069 (S-enantiomer, 29) with enhanced binding affinity to unphosphorylated FGFR1 and 2 (IC50s = 0.84 and 1.23 μM, respectively) in an enantiospecific manner. In contrast, its counterpart ARQ-068 (R-enantiomer, 28) only poorly bound to FGFR1 and 2 (IC50 > 30 μM).139
 | ||
| Fig. 12 (A) Structures of 5,6-dihydro-benzo[h]quinazolin-2-amine analogues and derazantinib (ARQ-087, 23). (B) Binding mode of ARQ-069 (29) in FGFR2. (C) Putative binding mode of derazantinib (ARQ-087, 23) in FGFR2. | ||
Through kinome-wide profiling of ARQ-069 (29) using a panel of 96 kinases, it was determined that this compound displays inhibition of 9 kinases by more than >30%. Notably, among the four kinases were FGFR1, FGFR2, FGFR2 (N549H), and FGFR3, suggesting its potential as an FGFR inhibitor. In addition, ARQ-069 (29) also inhibited Aurora A, Aurora C, c-Raf-1, VEGFR2, and CK1δ. Moreover, further mechanistic studies revealed that ARQ-069 (29) differs from other type II kinase inhibitors in its binding mode. It binds to the autoinhibited conformation of the kinase and stabilizes the inactive conformation of FGFR1 and 2 by accessing the hydrophobic allosteric back pocket, without requiring interactions with gatekeeper or lysine/glutamic acid residues (Fig. 12B).139
The subsequent endeavors focused on the potency optimization described in the patent resulted in the identification of ARQ-087 (derazantinib, 23), which is an ATP-competitive analogue of ARQ-069 (29) with significantly enhanced biochemical inhibitory activities against FGFR2 (IC50 = 1.8 nM), FGFR1, and FGFR3 (IC50 = 4.5 nM for both) (WO 2010/078421 A1).147 In three cell lines with highly expressed FGFR2 levels (NCI-H716, SNU-16, and KATO-III), derazantinib (23) was capable of inhibiting the phosphorylation of FGFR2 in all three cell lines and its downstream molecules including MEK, ERK, and AKT, indicating that derazantinib (23) targets FGFR2. Furthermore, derazantinib (23) was well-tolerated in in vivo xenograft models up to 75 mg kg−1 and induced partial or complete regression in FGFR2-dependent tumor models (NCI-H716, SNU-16, and Ba/F3-FGFR2), while this compound could not inhibit tumor growth in a xenograft model without FGFR2-dependency (Ba/F3-INSR). Based on these results, derazantinib (23) was investigated in a phase I clinical trial in humans, where it showed favorable tolerability at a dose of 300 mg daily and manageable toxicities (NCT01752920). Derazantinib (23) is under a pivotal clinical trial studies in patients with metastasized or inoperable iCCA having FGFR2 alterations (NCT03230318).148
Selective FGFR inhibitors.
BGJ398 (infigratinib, 24). Selective kinase inhibitors can serve as useful tool compounds for target validation and superior therapeutic agents compared to non-selective multi-kinase inhibitors due to their improved toxicity profile. However, the discovery of highly selective kinase inhibitors still remains a challenging task for medicinal chemists. Researchers from Novartis applied a nonconventional strategy to design a new series of kinase inhibitors with an N-pyrimidin-4-yl-urea scaffold derived from a pyrido[2,3-d]pyrimidin-7-one motif (31) (Fig. 13A). Through this approach, they obtained a prototype compound (32) featuring a pseudo-ring structure that showed promising activity against FGFR1, with an IC50 value of 570 nM.140
 | ||
| Fig. 13 (A) Summary of optimization strategies for BGJ398 (infigratinib, 24). (B) Binding mode of BGJ398 (infigratinib, 24) in FGFR1. | ||
During the lead optimization process with the prototype compound, the researchers introduced a 3,5-dimethoxyphenyl moiety in the pyrimidinyl urea scaffold. The 3,5-dimethoxyphenyl moiety, which is now a widely used functionality in FGFR inhibitors, is responsible for providing high affinity and selectivity for FGFRs by optimally filling a complementary hydrophobic pocket. Moreover, it should be noted that the 3,5-dimethoxyphenyl group should adopt a perpendicular orientation to optimally bind to the ATP binding site of FGFR.140 However, compound 32 exhibits a significantly high deconjugation energy barrier (1.5 kcal mol−1) to be overcome, as determined by ab initio calculation. Thus, to mitigate this penalty, researchers attempted to introduce chlorine atoms in the ortho positions of the dimethoxyphenyl moiety, which hypothetically stabilize the perpendicular conformation by inducing intramolecular steric hindrance. In agreement with the hypothesis, mono-chloride compound 33 exhibited a 4-fold improvement in biochemical activity (IC50 = 125 nM). The addition of a second chlorine atom afforded the most potent derivative BGJ398 (24) with a single digit nanomolar biochemical activity (IC50 = 5 nM).
It is also noteworthy that BGJ398 (24) did not inhibit any of five CYP450 isoenzymes (3A4, 2C9, 2C19, 2D6, and 3A4) up to a concentration of 10 μM and its evaluation in a panel of 76 kinases revealed that BGJ398 (24) is a highly selective kinase inhibitor, showing nanomolar biochemical activities against only 8 kinases including FGFR1–4 (IC50 = 0.9–60 nM).140 The pharmacokinetic study of BGJ398 (24) with an RT112 mouse xenograft model (bladder cancer) showed that its bioavailability was 32%. The in vivo efficacy of this compound was also measured with an RT112 rat xenograft model. Tumor regression was observed at doses of 10 and 15 mpk (66% and 73% regression, respectively). In addition, the pharmacodynamic analysis of BGJ398 (24) revealed significant reductions in the levels of pFRS2 and pMAPK in a dose-dependent manner. This observation supports the mechanism of action of BGJ398 (24), indicating that it exerts anti-tumor effects by inhibiting FGFRs. The docking model of BGJ398 (24) in FGFR1 indicates that in addition the critical hydrogen bond involving the aniline NH and the pyrimidine nitrogen with alanine 558, the perpendicular orientation of the dichlorodimethoxyphenyl group to the ATP back pocket plays a crucial role in inducing the optimal conformation upon binding to the ATP pocket (Fig. 13B).140
In 2018, the intellectual property rights of BGJ398 (24) were licensed to QED Therapeutics, a subsidiary of BridgeBio Pharmaceuticals, from Novartis. As a result, BGJ398 (24) became the lead candidate of QED Therapeutics.149 To further evaluate the clinical activity of BGJ398 (24), a multicenter, open-label, phase II trial was conducted, which involved previously treated patients with iCCA or eCCA carrying FGFR2 aberrations (NCT02150967). When taken orally at a dose of 125 mg per day for 21 consecutive days, the results showed that the ORR was 23%, the median duration of response (DoR) was 5 months, and the median PFS was 7 months with hyperphosphatemia being the most-common adverse event, which is reported in 77% of patients.141,150 Thus, based on the promising clinical activity of BGJ398 (24), the FDA granted the first approval of infigratinib (24) for previously treated patients with metastatic or advanced CCA on May 28th, 2021.149 However, in May 2024, the FDA announced the withdrawal of this approval.
Pemigatinib (INCB045828, 25). Pemigatinib (25) is a potent and selective inhibitor of FGFR1–3 with a novel tricyclic urea scaffold, which was developed by researchers from Incyte Corporation. An HTS screening of 20
000 compounds developed in-house identified a screening hit (34) displaying potent inhibitory activities against both FGFR1 and JAK2 with IC50 values of 6 nM and 1 nM, respectively (Fig. 14A).142 To improve the kinase selectivity, the researchers proposed incorporating a 3,5-dimethoxybenzene ring based on the structural superimposition of the screening hit compound (34) and PD173074 (35). The resulting hybrid scaffold (36) demonstrated significantly enhanced selectivity for FGFR1 with a slight decrease in FGFR1 potency. To restore FGFR activity, the researchers decided to rigidify the ethylene linker through cyclization to afford compound 37, which would supposedly reduce the flexibility of the structure, thus enabling the 3,5-dimethoxybenzene moiety to properly extend towards the complementary hydrophobic pocket of FGFR. As anticipated, the synthesized compound 37 having a tricyclic urea scaffold successfully regained FGFR1 activity, while preserving its selectivity over JAK2.142
 | ||
| Fig. 14 (A) Identification of initial hit compound 37. (B) Structural modification of hit compound 38 leading to the discovery of pemigatinib (25). | ||
Accordingly, tricyclic compound 37 was selected as a starting point for subsequent optimization using a series of in vitro assays, including biochemical assays, cellular assays with H1581 and KatoIII cells, and a whole blood (WB) assay to predict the in vivo activity of compounds. To increase the potency of FGFR, halide groups were introduced at the ortho positions of the 3,5-dimethoxybenzene ring, following a similar strategy employed in the development of infigratinib (24).142 As a result, compound 38 bearing a bis-ortho-fluoride group exhibited dramatically inhibitory activities in both enzymatic and cellular assays, with an IC50 value of 0.1 nM for FGFR1 and WB GI50 value of 6 nM for KatoIII cells. However, although compound 38 demonstrated favorable in vitro ADME properties and in vivo PK profile in rats with an AUC of 10600 nM·h, the exposure and bioavailability were found to be rather limited in cynomolgus monkey (cyno) PK (AUC = 1690 nM·h; BA = 15%). The researchers hypothesized that the inadequate cyno PK profile can be attributed to the low aqueous solubility of compound 38, which was measured to be 0.3 μg mL−1. Thus, to address this issue, the team focused on increasing its solubility with an aim to improve its cyno PK profile in their subsequent SAR studies.142
Three strategies were initially implemented in this effort including installing polar groups, reducing planarity, and scaffold morphing of pyrrole to lactam ring. Unfortunately, none of these options could achieve the desired goal (Fig. 14B). Upon revisiting the pyrrole series, the researchers proposed that introducing substituents at the 2-position of the pyrrole ring would place them within the solvent-exposed region, allowing various structural modifications for ADME and PK improvement.142 As anticipated, it was found that an analogue bearing a morpholine with a methylene linker displayed significantly enhanced PK profiles in rats and monkeys with AUC values of 8193 and 11804 nM·h, respectively. However, this compound suffered from slight inhibition of CYP3A4 (4% inhibition at 10 μM), which could be relieved by modifying the N-methyl group of cyclic urea to an N-ethyl group, resulting in the identification of pemigatinib (25). The detailed preclinical study of pemigatinib (25) revealed that it did not inhibit CYP isotypes and exhibited favorable DMPK profiles such as high permeability (11 × 10−6 cm s−1 in Caco2 cells), favorable metabolic stability within an intrinsic clearance of 0.8 L h−1 kg−1 when evaluated in human liver microsomes, and excellent in vivo dog PK profiles (AUC = 22100 μM·h; BA = 98%).151
According to the binding mode proposed by the researchers, as determined from the docking study with the X-ray cocrystal structure, compound 34, PD173074 (35), and pemigatinib (25) reside in the ATP pocket, the pyrrolopyridine motif of pemigatinib (25) forms hydrogen bonds with the Ala564 residue located in the hinge region of the protein backbone and the tricyclic urea part is situated within a hydrophobic pocket near the Leu630 residue, while the ethyl group is placed in the hydrophobic ribose pocket (Fig. 15A and B). It is worth noting that the 2,6-difluoro-3,5-dimethoxybenzene ring of pemigatinib (25) stands at a right angle to the scaffold, effectively occupying the hydrophobic pocket adjacent to the Phe642 residue. Additionally, the morpholine moiety is extended towards the solvent region.142
 | ||
| Fig. 15 (A) Binding mode for superimposition of compounds 34 (magenta) and 35 (cyan) in FGFR1. (B) Binding mode of pemigatinib (25, green) in FGFR1. | ||
In a recent study, Lin et al. discovered that pemigatinib (25) exhibits retained excellent potency as in FGFR wild-type against the FGFR2V564I gatekeeper mutation (IC50 = 7 nM).152 This study also investigated the effect of mutations, where the valine residue is replaced by a slightly bulkier methionine residue (FGFR1V561M and FGFR3V555M) or a phenylalanine residue (FGFR2V564F). The results demonstrated that pemigatinib (25) retained tolerable activity against the FGFR1V561M and FGFR3V555M mutations, with IC50 values of 149 nM and 107 nM, respectively. However, a significant reduction in activity was observed in the FGFR2V564F mutation (IC50 = 263 nM), where the valine is substituted with a much bulkier phenylalanine residue.152 The authors of this study suggested that this decrease in activity is likely due to the steric clash between the 3,5-dimethoxybenzene ring of pemigatinib (25) and the Met/Phe residue, thus introducing a smaller group in place of the 3,5-dimethoxybenzene ring, and increasing the inhibitor flexibility would help mitigate the bulky gatekeeper residues.
Based on these preclinical results, the efficacy of pemigatinib (25) was also evaluated in a clinical setting. In FIGHT-202 (NCT02924376), a multicenter, international, open-label phase II clinical trial, enrolled with 107 CCA patients harboring FGFR alteration, the ORR was observed to be sufficient (ORR = 36%) to represent efficacy with a median DoR of 9.1 months.134 Although the administration of pemigatinib (25) was found to be commonly associated with hyperphosphatemia as an adverse event, the promising outcomes from the FIGHT-202 trial resulted in the accelerated approval of pemigatinib (25) by the FDA on April 17, 2020 for the treatment of patients with advanced or metastatic CCA harboring FGFR alteration.134
An irreversible FGFR inhibitor, futibatinib (TAS-120, 26). Futibatinib (TAS-120, 26) is an irreversible and selective inhibitor of FGFR1–4 developed by Taiho Pharmaceuticals, which was derived from a hit compound capable of inhibiting both EGFR and FGFR. Covalent inhibitors that irreversibly bind to the adenosine triphosphate (ATP) binding site of kinases have attracted significant attention in the field of cancer drug discovery, which stems from their superior potency, selectivity, and prolonged duration compared to conventional reversible inhibitors. Moreover, the tight binding mode of the covalent inhibitors makes them less susceptible to kinase mutations that may occur in the drug-binding sites. This characteristic feature provides an effective approach to counteract the common acquired drug resistance mechanisms observed in kinases.135,144
Through screening of an in-house compound library, a mutant EGFR/FGFR dual inhibitor (39) featuring a pyrrolopyrimidine scaffold was identified. To assess the impact of stereoisomerism, enantiomers of compound 39 together with their pyrazolopyrimidine analogues were synthesized (Fig. 16A). The findings revealed that R-analogues exhibited more potent inhibitory activities against mutant EGFR than FGFR2, whereas S-analogues demonstrated enhanced potency and selectivity towards FGFR2.144 Considering the need for further optimization, the S-pyrazolopyrimidine analogue (40) was chosen as the starting hit compound for the swift exploration of its structure–activity relationship (SAR), primarily due to its ease of synthetic accessibility. To enhance potency and selectivity for FGFR2, a 3,5-dimethoxybenzene ring was introduced, given that the quinoline group is anticipated to reach into the hydrophobic pocket located near the gatekeeper residue adjacent to the ATP binding site. This pocket is typically occupied by the 3,5-dimethoxybenzene moiety found in most FGFR inhibitors. Interestingly, only compound 26 with a 3,5-dimethoxybenzene ring attached to the scaffold via an alkyne linker showed single digit nanomolar activity against FGFR2 (3 nM), while others (direct-bond, alkane, and amide linkages) demonstrated reduced inhibitory activities.144 More importantly, a substantial decrease in FGFR2 inhibitory activity was observed upon replacing the acrylamide with a propionamide moiety. This result suggests that the acrylamide moiety is involved in the formation of a covalent bond with a target cysteine residue (C491) located within a glycine-rich loop of FGFR.144 The potency and selectivity of the lead compound (futibatinib, 26) was well-translated in cell-based assays using two human gastric cancer cells, SNU-16 (with FGFR alteration) and SNU-1 (without FGFR alteration). The results indicate that the lead compound exhibited strong inhibitory activity against FGFR-dependent SNU-16 cells with a GI50 value of 3.7 nM. In addition, it did not show significant inhibitory effects on SNU-1 (GI50 > 5000 nM), highlighting the selective inhibitory activity of the lead compound.144 The selective kinase inhibition of futibatinib (26) was further supported by kinome profiling utilizing a panel of 387 kinases. At a concentration of 100 nM, futibatinib (26) exhibited significant inhibition of ligand binding for FGFR1–4 by 94.8% to 99.1% at 100 nM. Notably, only two kinases exhibited inhibition greater than 50%.144
 | ||
| Fig. 16 (A) Summary of optimization strategies for futibatinib (TAS-120, 26). (B) Putative binding mode of futibatinib (26) in FGFR2. | ||
In addition, the researchers performed a peptide mapping analysis of the modified protein with Glu-C endopeptidase. This analysis revealed a distinct modification solely at the C491 residue of FGFR2, indicating the formation of a covalent bond between futibatinib (26) and C491 of FGFR2.144 Although Kalyukina et al. disclosed that the binding mode of futibatinib (26) features a covalent bond with Cys488 in the X-ray cocrystal structure of futibatinib (26) bound to FGFR1, the docking results of futibatinib (26) with FGFR2 indicate that it forms a hydrogen bond with Ala567 and a covalent bond with Cys491, and the corresponding cocrystal structure of futibatinib (26) complexed with FGFR2 remains unresolved (Fig. 16B). This underscores the importance of conducting further structural studies to gain a more comprehensive understanding of the binding mechanism between futibatinib (26) and FGFR2.153
Futibatinib (26) displayed moderate in vitro microsomal stability in humans and mice with % residual activity of 66% and 69%, respectively. In an in vivo mouse PK study, futibatinib (26) exhibited an area under curve (AUC) level of 23.9 μM·h, which is likely attributed to its covalent nature.144 Moreover, futibatinib (26) achieved a significant antitumor efficacy at the lowest administered dose without signs of any toxicity in in vivo mouse models. These models included xenografts with AN3CA cells harboring FGFR2K310R and FGFR2N549K point mutations and SNU-16 cells with FGFR2 amplification.144 In preclinical studies, it is noteworthy that futibatinib (26) showed stronger inhibitory activity against a wider range of acquired mutations in FGFR2, including gatekeeper and molecular brake mutations, than other FGFR inhibitors. This enhanced activity is expectedly due to the covalent nature of futibatinib (26), which renders it less susceptible to on-target mutations.135
Building upon the potent therapeutic potential of futibatinib (26) in targeting tumors with FGFR alterations, subsequently futibatinib (26) was assessed in a clinical setting. Specifically, it was assessed in individuals diagnosed with advanced iCCA exhibiting FGFR aberration who had received one or more previous treatment regimens. In a Phase II clinical trial, the ORR of futibatinib (26) was 42% and its median DoR was 9.7 months with hyperphosphatemia being the most commonly observed adverse effect.135 Furthermore, it is important to note that futibatinib (26) received accelerated approval from the FDA on September 30, 2022 for patients with unresectable or metastatic iCCA harboring rearrangements involving FGFR2 or other fusion alterations.136,137,145 This FDA approval serves as a significant milestone in the field of oncology given that it provides another targeted treatment option for patients with iCCA harboring FGFR2 alterations, which is also a crucial event addressing the medical unmet need for this specific patient population.
LY2874455 (41). LY2874455 (41), which was developed by Eli Lilly and Co., is a selective pan-FGFR inhibitor based on an indazole scaffold (Fig. 17A). This compound displays sub-nanomolar to single nanomolar inhibitory activities against wild-type FGFR1–4 (IC50 = 0.3–5.2 nM). Wu and colleagues investigated the effects of LY2874455 (41) on FGFR gatekeeper mutants. Unexpectedly, it was initially found out that LY2874455 (41) retained excellent potency against FGFR4V555L and FGFR4V550M mutants with IC50 values of 6.2 and 6.0 nM, respectively.158 Considering that ponatinib, which also binds to FGFRs in an ATP-competitive manner, is totally ineffective on the same mutants (IC50 > 1000 nM), it was surprising that LY2874455 (41) could mitigate the steric hindrance induced by point mutations at the gatekeeper residues. Afterward, this team also tested the impact of LY2874455 (41) on gatekeeper mutations of FGFR1–3. Their findings demonstrated that this compound also effectively inhibits FGFR1–3 gatekeeper mutants, displaying IC50 values of 0.57, 0.26, and 2.11 nM, respectively.158 The docking results of LY2874455 (41) with FGFR1V561M suggest that this compound forms a pair of hydrogen bonds with the backbone amino acids Ala564 and Glu562 (Fig. 17B). In addition, upon careful analysis of the crystal structures of LY2874455 (41) in complex with FGFR4V550L/M, it was observed that LY287445 (41), by adopting a chair-like conformation, is situated within the ATP-binding pocket with significant distances from the mutated gatekeeper residues Leu550 and Met550 (4.4 Å and 4.8 Å, respectively).158,159 This spatial arrangement allows LY287445 (41) to evade the potential steric hindrance caused by the mutated gatekeeper residues, thereby maintaining its exceptional potency against those mutations. These findings were further supported by the MD (molecular dynamics) studies performed by Dehghanian et al. Their study demonstrated that LY2874455 (41), which binds to the active conformation of the FGFR4 induced by the gatekeeper V550 mutation, increases the rigidity around the hinge and αD helix regions, resulting in the stabilization of LY2874455 (41) in complex with the FGFR4V550L mutation.160 Although the effects of LY2874455 (41) on individuals with FGFR gatekeeper mutations have not been extensively investigated, the results from the phase I clinical study suggest that LY2874455 (41), which demonstrates a linear PK and an effective half-life of approximately 12 h, is well-tolerable among patients with advanced solid tumors.161 Considering its significant potency against FGFR gatekeeper mutations, LY2874455 (41) has the potential to serve as a promising lead compound for overcoming drug resistance caused by FGFR gatekeeper mutations.
 | ||
| Fig. 17 (A) Structure of LY2874455 (41). (B) Binding mode of LY2874455 (41) in FGFR1V561M. | ||
Pyridinyltriazine derivatives. Ryu et al. from our laboratory recently reported pyridinyltriazine analogues derived from structures of infigratinib (24), which exhibit enhanced inhibitory activities against FGFR-acquired mutations, including the gatekeeper mutants.162 Considering that the FDA-approved panFGFR inhibitor for the treatment of CCA, infigratinib (24), is resistant to various acquired FGFR2 mutations such as gatekeeper mutations (V564F) and molecular brake mutations (N549H/K, E565A, K641R, and K659M), we aimed to develop FGFR inhibitors with enhanced activities against FGFR acquired mutations.
Accordingly, we employed the scaffold morphing strategy, wherein an N-methylurea moiety was cyclized into a pyridine ring and the pyrimidine scaffold was replaced with a triazine ring (Fig. 18A). It was speculated from the quantum mechanics calculations that the pyridinyltriazine scaffold would have a smaller dihedral angle (0.4°) than that of pyrimidinylurea moiety in infigratinib (24) (5.2°), enabling more favorable alignment of the pseudo six-membered ring with the binding site.162 The preliminary results from the SAR study demonstrated that compound 42, which has an identical (4-(4-ethylpiperazin-1-yl)phenyl) group as infigratinib (24), exhibited more enhanced inhibitory activities on the FGFR2V564F gatekeeper mutation compared to infigratinib (24) and pemigatinib (25).
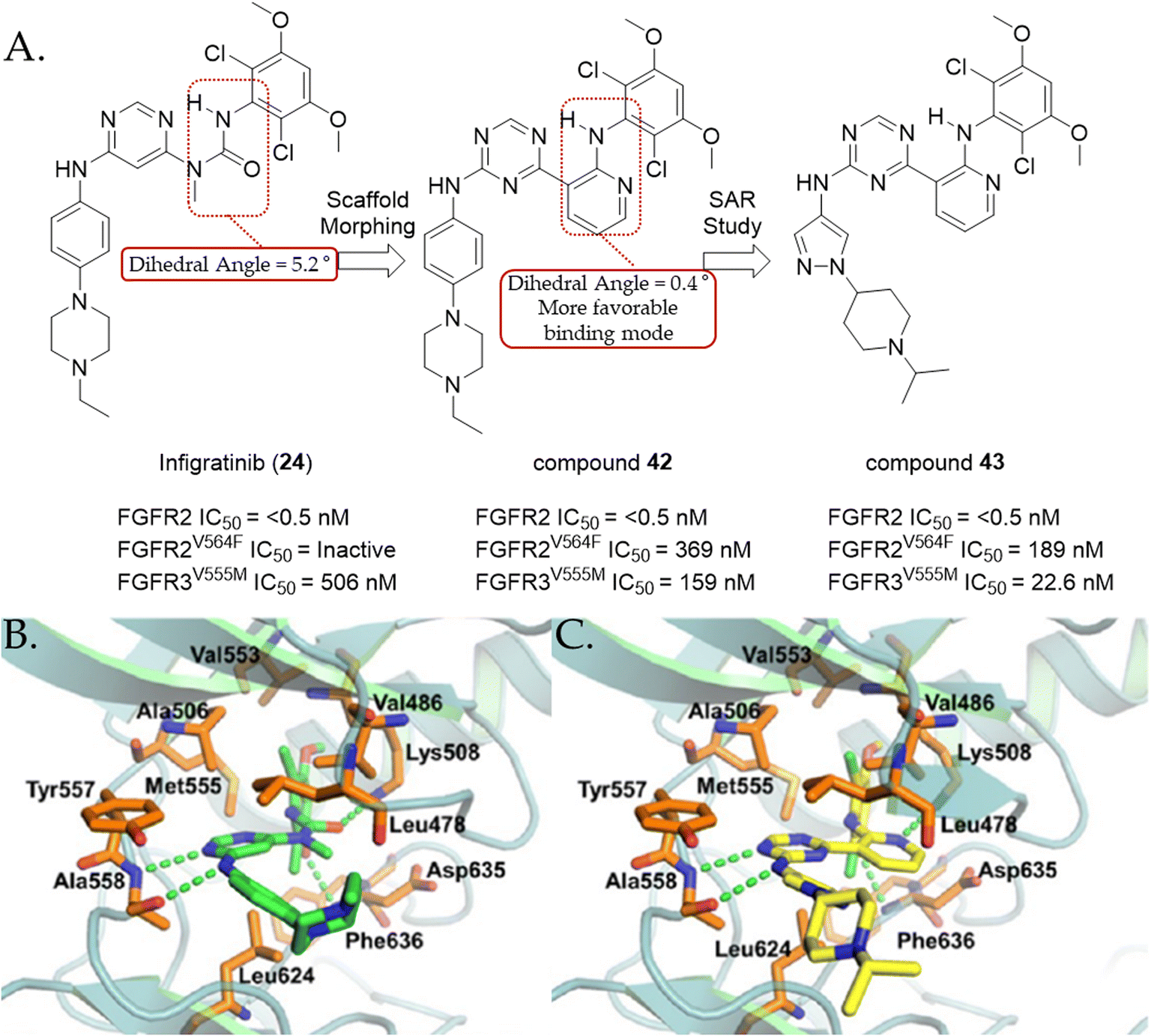 | ||
| Fig. 18 (A) Structure of infigratinib (24) and pyridinyltriazine derivatives (42 and 43). Binding modes of (B) infigratinib (24) and (C) compound 43 in FGFR3V555M. Reprinted from J. Med. Chem. 2022, 65(8), 6017–6038. Copyright 2022, the American Chemical Society.162 | ||
Further efforts on SAR studies have identified that compound 43 possessing a pyrazolyl moiety exhibits 2- and 7-fold increased activities compared to compound 42 in FGFR2V564F (IC50 = 189.1 nM) and FGFR3V555M (IC50 = 22.3 nM) and sub-nanomolar inhibitory activities against both FGFR2N549H and FGFR3 (IC50s = <0.5 nM), respectively. It is also noteworthy that compound 43 displayed comparable or higher activities to infigratinib (24) in cancers cells harboring various FGFR mutations such as AN3CA (FGFR2K310R/N549K), KMS-11 (FGFR3Y373C), and J82 (FGFR3K652E).162 The results from the docking studies using an FGFR3V550M homology model indicated that compound 43 participates in a 4.8-fold enhanced π-cation interaction with the Lys508 residue relative to infigratinib (24) (Fig. 18B and C). Moreover, the triazine ring participating in hydrophobic interactions with Leu478 and Leu624 would reinforce the binding affinity of compound 43 to acquired FGFR mutation. In addition, kinome-wide screening of compound 43 against a panel of 359 kinases demonstrated that compound 43 is a selective panFGFR inhibitor, which inhibited only 11 other kinases by >90% besides FGFR1–4.
Encouraged by these findings, we evaluated the in vivo efficacy of compound 43 using FGFR3V555M Ba/F3 and AN3CA xenograft mouse models. The results demonstrated that compound 43 exhibited a substantial decrease in tumor mass with an absence of weight loss upon treatment with it.162 This research provides fruitful insight into the application of the scaffold morphing strategy to overcome FGFR acquired mutations.
KIN-3248 (44). KIN-3248 (44), a next-generation irreversible selective panFGFR inhibitor, which is currently under development by Kinnate Biopharma Inc (Fig. 19A and B). The aim is to utilize it for the treatment of patients with iCCA, urothelial carcinoma (UC), and other solid tumors that manifest primary alterations of FGFR2 and FGFR3. Although limited data is available, there have been reports stating that KIN-3248 (44) maintains strong inhibitory activities in drug-resistant secondary mutations of FGFR2 and FGFR3 at nanomolar levels in biochemical assays.163,164 Recently reported data in a patent (WO2023107979 A1) revealed that KIN-3248 (44) selectively inhibits FGFR isotypes, achieving over 90% inhibition, including TNK1. In addition to nanomolar biochemical activities against FGFR isotypes, this compound effectively inhibited FGFR2 auto-phosphorylation in FGFR2-amplified cancer cells such as SNU-16 (pFGFR2 EC50 = 3.01 nM) and KATO-III (pFGFR2 EC50 = 5.53 nM). The putative binding mode of KIN-3248 (44) in FGFR2 suggests that KIN-3248 (44) forms hydrogen bond interactions with the backbone residues Ala567 and Glu565, together with irreversible covalent interaction with Cys491, providing high selectivity towards FGFR isotypes.
 | ||
| Fig. 19 (A) Structure of KIN-3248 (44). (B) Putative binding mode of KIN-3248 (44) in FGFR2. | ||
Furthermore, this compound demonstrated effectiveness in CCA cell lines with engineered FGFR2 fusion (CCLP-1 cells) and secondary mutation expression. The potential of KIN-3248 (44) is currently being assessed in a phase I clinical trial (KN4802, NCT05242822).165 It is anticipated that KIN-3248 (44) can provide a groundbreaking approach to address the medical needs unmet by existing therapies in iCCA patients with FGFR2 and/or FGFR3 alterations. This includes individuals who have developed drug resistance due to gatekeeper, molecular brake, and activation loop mutations.
3.3. BRAF
The v-raf murine sarcoma viral oncogene homolog B1, also known as the B-Raf proto-oncogene (BRAF), is a serine/threonine kinase belonging to the rapidly accelerated fibrosarcoma (RAF) family. The RAF family consists of three isoforms, ARAF, BRAF, and CRAF, among which BRAF has the highest mutation frequency in all cancers (7–10%).166 One of the most well-known BRAF mutations is the V600E mutation, which involves a substitution of valine (V) with glutamic acid (E) at position 600 in the protein amino acid sequence. This mutation leads to a constitutively activated BRAF protein that signals cells to grow and divide uncontrollably. The V600E mutation is particularly associated with melanoma, wherein nearly half of the tumors harbor an activating BRAF mutant,167 but their role in iCCA is less common compared to other cancers. However, there have been reports of BRAF mutations being present in a subset of iCCA cases. For instance, in a cohort study recently conducted by Xin et al., they identified that iCCA patients harboring the BRAFV600E variant is closely associated with poor prognosis and increased rate of recurrence.168 Vemurafenib (49) and dabrafenib (52) are FDA-approved drugs for the treatment of melanoma patients harboring the BRAFV600E variant. A brief history, biological activities, status of clinical trials, binding modes, and medicinal chemistry aspects of promising BRAF inhibitors for the treatment of iCCA are summarized in this section (Table 3).| Target protein | Compound | Discovery/development | Scaffold | Key interacting residues | PDB | Biological activities/DMPK/clinical outcome | Clinical trial/status/patents | References |
|---|---|---|---|---|---|---|---|---|
| BRAF | Vemurafenib (48) | Plexxikon/Roche | Azaindole | Gln530, Cys532 | 3OG7 | BRAFV600E IC50 = 31 nMORR = 33% | Phase II (NCT01524978)/terminated | 169–171 |
| BRAF | Dabrafenib (51) | GlaxoSmithKline (GSK)/Novartis | Biarylthiazole | Cys532 | 4XV2 | BRAFV600E IC50 = 0.7 nM | Phase II (NCT02684058)/FDA-approved (in combination with trametinib) | 172, 173 |
| Rat po DNAUC = 729 ng·h mL−1 mg−1 kg−1 | ||||||||
| Dog po DNAUC = 3754 ng h mL−1 mg−1 kg−1 | ||||||||
| ORR = 53% | ||||||||
| mPFS = 9.0 mos. | ||||||||
| BRAF | PLX8394 (54) | Plexxikon/Fore Biotherapeutics | Azaindole | Gln530, Cys532 | 4XV1 | BRAFV600E IC50 = 3.8 nM | Phase I/IIa (NCT02428712)/FDA fast-track designation | 174, 175 |
| HT29 (colon cancer) GI50 = 320 nM | ||||||||
| Colo-205 (colon cancer) GI50 = 40 nM |
Vemurafenib (48). Vemurafenib (48) is an azaindole-based BRAF inhibitor discovered by Plexxikon and developed by Roche. Researchers at Plexxikon adopted a fragment-based drug discovery approach to expand the chemical space and unique binding modalities.169 The initial step of this strategy started with screening of a library comprised of 20
000 scaffold-like compounds with enriched chemical diversity. Among the screened library, 238 compounds initially obtained from the screening were cocrystalized with PIM-1 and FGFR1, while BRAF crystallization was optimized. This effort led to the identification of a 7-azaindole scaffold (45) as a promising starting point (Fig. 20A). Subsequent validation was achieved by synthesizing a more potent derivative, a 3-substituted azaindole (compound 46), guided by insights from the cocrystal structure. The iterative process of refinement, utilizing both compound Y and the BRAF cocrystal structure, culminated in the identification of PLX4720 (47) and vemurafenib (48) as the ultimate outcomes of this optimization journey (Fig. 20B and C, respectively).169
 | ||
| Fig. 20 (A) Fragment-based drug discovery of vemurafenib (49). Binding modes of (B) compound 47 (green) and (C) vemurafenib (48, cyan) in BRAFV600E. | ||
PLX4720 (47) is a sister compound of vemurafenib (48) exhibiting a slightly enhanced PK profile in rodents and it has been extensively studied by this team. It is noteworthy that PLX4720 (47) is highly selective to the BRAFV600E variant. This heightened selectivity is attributed to its novel binding mode. PLX4720 (47) specifically binds to the active state (DFG-in conformation) of BRAF and its selectivity over other kinases is attributed to its propyl group directed towards the unique small pocket of the RAF family, aptly labeled as the RAF-selective pocket.176 This characteristic binding mode is highly likely to also be mirrored in the case of vemurafenib (48).
Vemurafenib (48) received FDA approval in 2011 for the treatment of patients with advanced or metastatic melanoma harboring the BRAFV600E variant and in 2017 for patients with Erdheim–Chester disease (ECD) with the BRAFV600E mutant.177 Moreover, it is currently also being evaluated in other non-melanoma solid tumors harboring BRAFV600 mutations. Subbiah and colleagues undertook a multi-cohort study (NCT01524978) involving 172 patients with 26 distinct forms of cancer, including CCA. This study demonstrated that administering vemurafenib (48) to these patients resulted in an ORR of 33% and a median response duration of 13 months.178 Furthermore, recent findings suggest that combining vemurafenib (48) with other chemical agents does not significantly enhance OS or PFS in patients with BRAFV600E solid tumors. This highlights the need to explore the rapidly evolving molecular landscape of BRAF activation to develop effective therapeutic strategies for addressing this challenge.170
Dabrafenib (51). Another FDA-approved drug specifically indicated for cancers harboring the BRAFV600E variant is a thiazole-based inhibitor dabrafenib (51) developed by the global pharmaceutical company GlaxoSmithKline (GSK). The efforts in identifying this promising BRAFV600E inhibitor started with initial hit compound 49 (Fig. 21). However, compound 49 suffered from rapid metabolism in higher animal species, guiding the direction of the medicinal chemistry optimization towards improving its pharmacokinetic profile in dogs. Given that the sulfonamide moiety was identified as a crucial part for achieving high enzymatic potency, the SAR studies were focused on modifying the aminopyridine moieties mostly by introducing simple truncated alkyl chains. Dabrafenib (51) binds to BRAFV600E in a similar manner with vemurafenib (48), forming hydrogen bond interactions with residues Asp594, Phe 595, and Cys532 (Fig. 22).179,180 Installing a polar alkylsulfonyl chain (compound 50) has led to increased oral dose-normalized AUC (DNAUC) in rats, while maintaining potent BRAFV600E activity. However, in the metabolite analysis, the isopropyl group and alkylsulfonyl group were identified as metabolic soft spots. The former at the thiazole core was found to be susceptible to oxidation, while the latter was prone to N-dealkylation. Further optimization of these functionalities resulted in the discovery of dabrafenib (51), which exhibited a significantly improved PK profile in dogs (dog po DNAUC = 3754 ng h mL−1 mg−1 kg−1).172
 | ||
| Fig. 21 Optimization process of dabrafenib (51). | ||
 | ||
| Fig. 22 Binding mode of vemurafenib (48, cyan), dabrafenib (51, green), and overlay of the two in BRAFV600E. | ||
It is also noteworthy that dabrafenib (51) showed enhanced activities in a clinical setting when it was used in combination with the MEK inhibitor trametinib. Subbiah et al. recently reported the results of a phase 2 clinical trial evaluating the combination of dabrafenib (51) and trametinib in patients with advanced rare cancers harboring BRAFV600E mutation (NCT02684058). Within the entire participant pool, a specific subset comprised of 43 patients afflicted with BTCs, including CCA cases, exhibited an ORR of 53%, accompanied by an mPFS of 9.0 months. These compelling clinical outcomes resulted in the accelerated approval of the dabrafenib (51)-trametinib combination therapy by the FDA for the treatment of patients with rare cancers carrying the BRAFV600E variant, a milestone achieved on June 22nd, 2022.181
PLX7904 (53) and PLX8394 (54). Despite the promising clinical effectiveness of vemurafenib (48) and dabrafenib (51), paradoxical activation of the MAPK/ERK signaling pathway confers acquired drug-resistance to these BRAF inhibitors.182 Thus, to mitigate this, Zhang et al. screened various vemurafenib (48) derivatives possessing different terminal sulfonamide functionalities to identify next-generation BRAF inhibitors, also called ‘paradox breakers’, which are potent BRAFV600E inhibitors capable of preventing paradoxical activation of the MAPK signaling pathway. Each compound was assessed using the ERK pathway inhibition index (EPII), calculated as the ratio between the activation of pERK by the compound and its inhibition of pERK. The initial hit compound PLX7683 (52, EPII > 100) containing an N-ethylmethyl-sulfamide group instead of the propylsulfonamide moiety in vemurafenib (48) displayed potent pERK inhibition (in A375 and COLO829 cells) without showing activation of pERK in mutant RAS cells tested (B9, IPC-298, and HCT116), wherein vemurafenib (48) and dabrafenib (51) exhibited EPIIs of 11 and 4, respectively (Fig. 23). Introducing a 2-cyclopropylpyrimidine moiety at the 5-position on the azaindole scaffold resulted in PLX7904 (53), which showed slightly increased BRAFV600E activity. It should also be noted that this compound, unlike vemurafenib (48), did not induce elevation of the pERK levels in cancer cells known to activate the MAPK pathway through upstream signal feedback into RAS (A431 and SKBR3 cells). Further optimization led to the identification of PLX8394 (54), exhibiting a 5-fold enhanced EPII (>50
000) compared to PLX7904 (53).179 PLX7904 (53) and its further optimized counterpart, PLX8394 (54), share a highly similar chemical structure with vemurafenib (48). The researchers were prompted to investigate how this minor chemical alteration can bring about significant changes in their biological impact as paradox breakers.
 | ||
| Fig. 23 Optimization process of PLX8394 (54). | ||
To gain insights into this, the X-ray cocrystal structure of PLX7904 (53) bound to BRAFV600E was obtained. Although the overall binding mode of PLX7904 (53) closely resembles that of vemurafenib (48), with both compounds occupying the same binding pocket, the close contact between the methyl group of the N-ethylmethyl sulfonamide functionality and Leu505 within this pocket is the notable difference observed as compared to vemurafenib (48) (Fig. 24A). This close contact of PLX7904 (53) and Leu505 led to an outward shift in the α-C helix, ultimately disrupting the RAF dimer interface (Fig. 24B).179 Moreover, it is worth noting that PLX7904 (53) and PLX8394 (54) remained sensitive against the vemurafenib (48)-resistant BRAFL505H mutant cell (YUMAC-BRAFL505H GI50s = 0.389 and 0.095 μM, respectively). This further reinforces the critical role played by the Leu505 residue in differentiating the paradox breaker from first-generation RAF inhibitors.183
 | ||
| Fig. 24 (A) Binding mode showing the superimposition of vemurafenib (48, cyan) and PLX7904 (53, green) in BRAFV600E. (B) X-ray cocrystal structure illustrating the binding mode of PLX7904 (53, green; PDB: 4XV1) in BRAFV600E. Leu505 residue is shown in sphere, with the red circle highlighting the close contact between the N-methyl group from PLX7904 (53) and the Leu505 residue. | ||
In addition, PLX8394 (54) was licensed to Fore Biotherapeutics and underwent clinical investigation, also recognized by the designation FORE8394. The preclinical efficacy of PLX8394 (54) was evaluated in a BRAF-mutant lung adenocarcinoma (LA) model. The results demonstrated that PLX8394 (54) is capable of potently suppressing the growth of vemurafenib (48)-resistant LA cells (HCC364VR1) as well as the MAPK signaling pathway in vitro. This efficacy was well-translated in an in vivo evaluation using HCC364 cell xenograft mouse models upon oral administration at a dose of 150 mg kg−1 d−1.182 The clinical efficacy of PLX8394 (54) is being investigated in patients with advanced unresectable solid tumors, including CCA (NCT02428712) as a single agent or combined with a CYP3A4 inhibitor, cobicistat.184 Furthermore, PLX8394 (54) received a fast-track orphan drug designation from the U. S. FDA for the treatment of patients with advanced solid tumors and central nervous system (CNS) malignancies harboring class I/II BRAF alterations. This designation was granted based on the findings from the phase I/IIa clinical trial (NCT02428712).174
SIJ1227 (56) and SIJ1777 (57). BRAF mutations can be classified into class I, II, and III. Class I BRAF mutants, represented by V600 variants, are characterized by their RAS-independent status and function as constitutively activated RAF monomers in terms of kinase activity. BRAF mutations of class II, including BRAFG464 and BRAFG469 mutants, function independently of RAS and exist as constitutive dimers with kinase activities ranging from intermediate to high. In contrast, class III BRAF mutations, which include BRAFG466 and BRAFD594 mutants, require RAS for activation and function as heterodimers with notably lower or negligible kinase activities. Vemurafenib (48) is capable of inhibiting class I BRAFV600 mutants, which are RAF monomers. However, it is insensitive to BRAF mutations categorized as class II and class III.185,186 Moreover, the paradox breaker, PLX8394 (55), demonstrates the limited efficacy on BRAF mutations classified as class II and class III as well, increasing the demand for the development of pan-class BRAF inhibitors.187
Our experience on extensive studies using a multi-targeted type II kinase inhibitor, GNF-7 (55), and its derivatives for application in kinase inhibition led to the identification of the first small molecules possessing strong anti-proliferative activities against cancer cells with BRAF class I, II, and III mutants.188 We found that GNF-7 (55) exhibits potent inhibitory activities in biochemical assays against BRAFV600E (class I) and BRAFG469A (class II) with IC50 values of 6.3 nM and 17.3 nM, respectively (Fig. 25). The anti-proliferative activities of GNF-7 (55) on cancer cells harboring BRAF class I, II, and III mutations were also evaluated. The results showed that GNF-7 (55) could significantly suppress the growth of C8161 (melanoma; BRAFG464E; class II mutant), H1775 (NSCLC; BRAFG469A; class II), and H1666 (NSCLC; BRAFG466V; class III mutant) cells with GI50 values ranging from 2–54 nM, while vemurafenib (48) and PLX8394 (54) showed sub-micromolar to micromolar activities in those cells. With the aim to enhance the potency shown by GNF-7 (55), we designed SIJ1227 (56) as an analogue of GNF-7 (55) with a substituted pyrazole functionality on the left-side of the 3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one scaffold. Although SIJ1227 (56) is as potent as GNF-7 (55) with respect to its anti-proliferative activities against C8161, H1775, and H1666 cells (GI50s = 0.019, 0.007, and 0.020 μM, respectively), it should be noted that SIJ1227 (56) more effectively inhibited the growth of SK-MEL-2 cells (BRAFWT) than GNF-7 (55, GI50 = 20 nM).
 | ||
| Fig. 25 Structures of GNF-7 (55) and its derivatives, SIJ1227 (56) and SIJ1777 (57). | ||
Encouraged by these results, we wanted to further investigate the effects of GNF-7 (55) derivatives on inhibition of BRAF class I, II, and III mutants. We prepared six GNF-7 (55) derivatives having substituted pyrazoles and evaluated their activities against cancer cells with wild-type or mutated BRAF. Although all six derivatives potently inhibited the cancer cells, SIJ1777 (57) having a simple methylated pyrazole-3-amine exhibited two-digit nanomolar anti-proliferative activities in melanoma cells as well as 3–5-fold enhanced activities as compared to GNF-7 (55) against WM3670 and WM3629 cells harboring BRAF class III mutations (GI50s = 0.040 μM for both). It should be noted that both vemurafenib (48) and PLX8394 (54) are insensitive to the tested cell lines. Collectively, GNF-7 (55) analogues, specifically SIJ1227 (56) and SIJ1777 (57), demonstrate significantly superior potency compared to vemurafenib (48) and PLX8394 (54) in suppressing BRAF class II and class III, including class I mutations. Our research offers valuable insights into the development of innovative BRAF inhibitors designed to effectively target all classes of BRAF mutants.189
3.4. NTRK
The neurotrophin receptor tyrosine kinase (NTRK) genes 1–3 encode tropomyosin-receptor kinase (TRK) A, TRKB, and TRKC, respectively. All TRKs share highly homologous protein structures. TRK proteins originally play a critical role in neuronal development; however, somatic mutations and overexpression can lead to various types of cancer, including colorectal, biliary tract, lung cancer, and leukemia.190,191 Gene fusions of NTRK, such as ETV6-NTRK3, TPM3-NTRK1, and TPR-NTRK1, represent the most frequent mechanism of oncogenic activation for TRKs.192 Though rare, it has been reported that approximately 3.5% of patients with iCCA carry NTRK fusions.30,190,193,194 This suggests that targeting NTRK genes can serve as a useful strategy for the treatment of patients with diverse cancer types, including iCCA. There are currently two FDA-approved NTRK inhibitors, namely entrectinib (58) and larotrectinib (59), which are first-generation NTRK inhibitors (Fig. 26). Entrectinib (58), developed by Roche, is an indazole-based orally available small molecule inhibitor with blood–brain barrier (BBB) permeability. Due to positive outcomes from clinical trials based on its potent inhibitory activities against TRKA-C (IC50s = 1–5 nM), ALK (IC50 = 12 nM), and ROS1 (IC50 = 7 nM), entrectinib (58) is approved by FDA and specifically indicated for the treatment of ROS1-positive NSCLC and solid tumors with NTRK gene fusions.195–197 Larotrectinib (60) is a highly selective pan-TRK inhibitor having a novel pyrazolopyrimidine scaffold. It was initially discovered by Array Biopharma, and then licensed to Loxo Oncology. | ||
| Fig. 26 Structures of NTRK inhibitors (58–60). | ||
Clinical trials investigating the efficacy of entrectinib (58) and larotrectinib (59) in iCCA patients with NTRK gene fusions are still relatively limited. However, data from broader studies in solid tumors with NTRK gene fusions have demonstrated remarkable responses to these targeted therapies. The STARTRK-2 trial (NCT02568267), which evaluated entrectinib (58) in patients with various solid tumors harboring NTRK, ROS1, or ALK gene fusions, showed substantial antitumor activity against solid tumors harboring NTRK gene fusion with an ORR of 67.1%. These findings indicate the potential benefits of entrectinib (58) for iCCA patients with NTRK gene fusions.192,198 Similarly, larotrectinib (59) has shown impressive results in clinical trials targeting NTRK gene fusions across various cancer types.199 The phase II NAVIGATE trial (NCT02576431) demonstrated robust and durable responses in patients with NTRK gene fusion-positive tumors, and one of the two CCA patients responded positively to larotrectinib (59) treatment.199,200 These results further support the efficacy of larotrectinib (59) as a promising treatment option for patients with NTRK gene fusions, which may also be present in iCCA.
The first-generation NTRK inhibitors were reported to exhibit reduced activities against acquired secondary mutations occurring at the solvent-front region and xDFG motif such as TRKAG595R, TRKAG667R, and TRKCG623R.201–203 Moreover, mutations at the solvent-front are frequently observed in clinical cases of acquired resistance to TKIs including ALK and ROS1 inhibitors. The substitution of the small glycine residue for the bulky arginine residue in this region induces steric hindrance to TKIs binding to the ATP site. LOXO-195 (selitrectinib, 60), which was identified through in silico evaluation, is a highly potent and selective NTRK inhibitor with a novel macrocyclic structure capable of mitigating this acquired resistance. Structural modeling supports that LOXO-195 (60) does not induce steric hindrance to the bulky and positively charged arginine residue.203
The first two cases of patients developing acquired resistance to larotrectinib (59) were treated with LOXO-195 (selitrectinib, 60). For instance, an adult patient having colorectal cancer harboring an LMNA-NTRK1 fusion and a G595R acquired mutation experienced a rapid clinical response to LOXO-195 (60), leading to a 38% reduction in tumor burden. Similarly, a pediatric patient with infantile fibrosarcoma harboring an ETV6-NTRK3 fusion also showed at least 30% regression in tumor burden.203 LOXO-195 (60) is currently being investigated in a clinical setting, showing an ORR of 50% in patients with NTRK solvent-front mutation.204 These results strongly suggest that LOXO-195 (60) holds potential as a valuable additional treatment option for patients with iCCA carrying NTRK gene fusions.
3.5. HER2
The human epidermal growth factor receptor (HER) family plays a crucial role in the tumorigenesis of several human cancers.205 Its members regulate multiple signaling pathways to modulate cell functions, including proliferation, differentiation, and survival. This family consists of four members, including HER1, HER2, HER3, and HER4. The first three can be activated by ligands such as EGF, EGF-like molecules, or neuregulin ligands, triggering downstream signaling pathways such as MAPK, PI3K/AKT, and JAK/STAT pathways, whereas HER2 follows a different activation mechanism.206,207 There is no known direct HER2 activating ligand thus far, but it is considered to be activated upon heterodimerization with its family members such as HER1 and HER3.208 Moreover, insulin-like growth factor receptor 1 (IGF1) is also reported to activate HER2 by heterodimerization.209An elevated level of HER2, either through amplification or overexpression, has been reported in multiple solid cancers, including breast cancer, gastrointestinal tumors (gastric cancer and gastroesophageal cancer) and CCA.210 In CCA cells, HER2 aberrations are linked to more aggressive tumor behavior, characterized by heightened invasiveness, motility, and proliferation compared to CCA cells expressing lower levels of HER2.211
Several studies suggest that the HER2 expression status is correlated with the pathogenesis of CCA. The HER2 level was upregulated in 27.8% (5/18),212 52.6% (20/38),213 55% (45/81)214 iCCA patients. Moreover, Ata et al. revealed that there was a correlation between the lower cytoplasmic HER2 scores and longer survival of tfap, including CCA (p = 0.052).215 Preclinical studies of anti-HER2 inhibitors indicate that the HER2 targeting strategy can be effective for targeting CCA. A previous study showed that targeting EGFR and or HER2 suppressed CCA cell growth in vitro and in vivo.216
Zanidatamab (ZW25). Zanidatamab is a bispecific antibody targeting both the juxtamembrane extracellular domain and the dimerization domain of HER2.217 It was designated for FDA breakthrough therapy in 2020 for metastatic biliary tract cancers (BTCs), which express HER2 and encompass iCCA, eCCA, and gallbladder cancer. In a phase 1 clinical trial, unresectable, locally advanced or metastatic BTC patients having HER2 overexpression (5 iCCA, 4 eCCA, and 11 GBC) were treated 20 mg kg−1 for every 2 weeks.218 Among the seventeen patients for whom response assessments were conducted, eight of them achieved an ORR of 47%.218 In the phase 2b study (HERIZON-BTC-01), higher response rates were observed with HER IHC 3+ patients than 2+, with a manageable safety profile.219
Trastuzumab (herceptin). Trastuzumab is a monoclonal antibody that targets HER2 and has been approved for the treatment of HER2-positive breast cancer.220 Several clinical trials investigated the use of trastuzumab in HER2-positive CCA. Some of these trials evaluated trastuzumab as a single agent (NCT03613168), while others studied its combination with chemotherapy or other targeted therapies.221,222 The promising results from these studies underscore the significance of HER2 inhibition, either alone or in combination, as a viable strategy for targeting CCA.
Antibody–drug conjugates (ADCs). ADCs are monoclonal antibodies with chemotherapeutic conjugation. ADCs have the benefit of reduced off-target cytotoxicity due to their target-specific binding. Clinical trials are ongoing with HER2 antibody-based ADCs such as RC48-ADC or MRG002-ADC.223,224 In addition to zanidatamab, trastuzumab, and ADCs, there are ongoing pre-clinical/clinical studies exploring other HER2 inhibitors for CCA treatment.225,226 These inhibitors may target HER2 through various mechanisms, blocking its signaling pathways and providing potential alternative treatments for HER2-positive CCA patients.
3.6. Immunotherapy and combination therapies for the treatment of iCCA
| Combination therapy | Clinical trial ID | Study objective | Phase | Key findings/outcomes |
|---|---|---|---|---|
| Pembrolizumab + chemotherapy | KEYNOTE-966 (NCT04003636) | Evaluate efficacy of pembrolizumab with gemcitabine and cisplatin in advanced CCA | Phase 3 | mOS: 12.7 months (95% CI 11.5–13.6) in combination group vs. 10.9 months (95% CI 9.9–11.6) in chemotherapy alone group234 |
| Nivolumab + ipilimumab (anti-CTLA4) | NCT02834013 | Assess combined effect of nivolumab (anti-PD-1) and ipilimumab (anti-CTLA4) | Phase 2 | Combination did not improve 6 month PFS but showed higher OS rates in a small cohort at 2 years235 |
| Nivolumab + chemotherapy | NCT02923934 | Evaluate nivolumab with gemcitabine and cisplatin | Phase 2 | Combination treatment showed limited benefit with no significant improvement in 6 month PFS but a small increase in OS in specific cohorts235 |
| Atezolizumab + molecular targeted therapy | NCT03201458 | Explore combination of PD-L1 inhibitor (atezolizumab) with MEK inhibitor (cobimetinib) | Phase 2 | Prolonged PFS: 3.65 months in combination group vs. 1.87 months in atezolizumab monotherapy group. Adverse events reported (skin rash, GI issues)236 |
3.7. Other potential molecular targets for iCCA
Although IDH1 and FGFR2 mutations account for 10–25% of patients with iCCA, which is more frequent than other subtypes of CCAs, it is important to note that a significant portion of iCCA patients still may not benefit therapeutically from the current targeted therapies. Therefore, finding alternative actionable molecular targets is crucial in drug discovery efforts focusing on iCCA patients and advancing the development of targeted therapies. This section provides a brief overview of additional potential targets, highlighting the novel approaches for iCCA targeted therapy, in addition to the well-established targets.| Target | Compound | Structure | Mechanism | References |
|---|---|---|---|---|
| CDKN2A/B loss | Palbociclib |
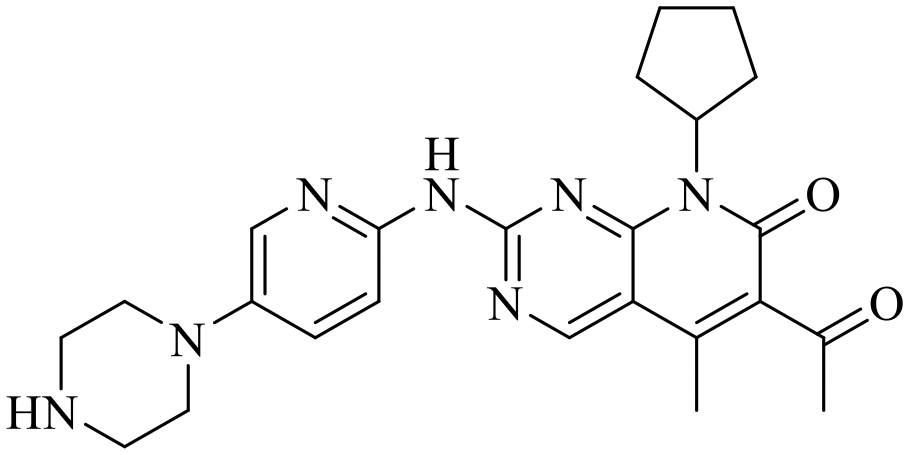
|
CDK4/6 inhibition | 246, 247 |
| KRAS | Sotorasib |

|
KRASG12C inhibition | 249, 252 |
| Chromatin remodeling genes | Vorinostat |

|
HDAC inhibition | 50 |
| Chromatin remodeling genes | Azacitidine |

|
DMNT inhibition | 50 |
| Chromatin remodeling genes | Tazemetostat |

|
EZH2 inhibition | 50 |
| BRCA1/2 | Olaparib |

|
PARP inhibition | 256 |
4. Summary and future prospects
Recent advancements in understanding the molecular complexity of iCCA have paved the way for significant therapeutic breakthroughs. Thus, to provide a clearer understanding of the currently available therapeutic options, a concise summary of FDA-approved drugs for iCCA and metastatic or unresectable CCA is presented in Table 6. One of the most common alterations in iCCA is mutations in IDH1/2, which drive tumorigenesis through the production of the oncometabolite 2-HG. This metabolite disrupts cellular metabolism and epigenetic regulation, contributing to cancer progression. The approval of ivosidenib (1), a first-in-class IDH1 inhibitor, highlights the therapeutic potential of targeting IDH1 in iCCA. Similarly, IDH2 inhibitors such as enasidenib (6) have shown promise in related malignancies and are being evaluated for iCCA treatment. Future efforts will likely focus on expanding the use of IDH inhibitors to earlier disease stages, identifying predictive biomarkers for patient selection, and exploring their synergy with therapies targeting the tumor microenvironment or metabolic pathways.| Drug name | Target | Mechanism of action | Indication | Treatment type | Approval year |
|---|---|---|---|---|---|
| Ivosidenib (1) | IDH1 | IDH1 inhibitor | IDH1-mutant iCCA | Single | 2021 |
| Pemigatinib (25) | FGFR2 | FGFR2 inhibitor | FGFR2 fusion-positive or arranged iCCA | Single | 2020 |
| Futibatinib (26) | FGFR2 | FGFR2 inhibitor | FGFR2 fusion-positive or arranged iCCA | Single | 2022 |
| Dabrafenib (51) | BRAF | BRAF inhibitor | BRAFV600E-mutant metastatic or unresectable CCA | Combined with trametinib | 2022 |
| Pembrolizumab | PD-1 | Immune checkpoint inhibitor (PD-1) | PD-L1-positive metastatic or unresectable CCA | Combined with gemcitabine and cisplatin | 2023 |
FGFR2 fusions and rearrangements are highly specific to iCCA and serve as key oncogenic drivers by enabling ligand-independent activation of signaling pathways such as MAPK and PI3K/AKT. FGFR inhibitors, including infigratinib (24), pemigatinib (25), and futibatinib (26), have transformed the treatment landscape, with pemigatinib (25) receiving FDA approval as the first targeted therapy for FGFR2-altered iCCA. However, acquired resistance to FGFR inhibitors, which is often driven by gatekeeper mutations or secondary alterations, remains a significant challenge. Research efforts will likely focus on developing next-generation FGFR inhibitors, expanding combination strategies with immunotherapies or chemotherapy, and exploring pan-FGFR inhibitors with improved efficacy and tolerability.
Although BRAF mutations are relatively rare in iCCA (<5%), they play a critical role in activating the MAPK pathway, contributing to tumor progression. Targeting BRAF with dabrafenib (51), in combination with the MEK inhibitor trametinib, has shown efficacy in BRAF-mutant iCCA. These combination regimens have been adapted from their success in other cancers, such as melanoma. Future research will aim to overcome resistance mechanisms to BRAF inhibitors and identify biomarkers to better stratify patients for treatment.
HER2 overexpression or amplification is observed in a subset of iCCA cases and is associated with aggressive tumor behavior and poor prognosis, making it an actionable target for therapy. HER2-targeting monoclonal antibodies, such as trastuzumab, have shown promise in HER2-positive iCCA. Additionally, ADCs such as trastuzumab deruxtecan are being evaluated for their enhanced efficacy. Combination therapies involving HER2 inhibitors and chemotherapy are also under investigation to improve outcomes. Future strategies will focus on developing next-generation HER2-targeted therapies, including bispecific antibodies and HER2-selective tyrosine kinase inhibitors (TKIs), and refining patient selection based on HER2 amplification and mutation profiles.
NTRK fusions, though rare in iCCA, are highly actionable targets that result in the constitutive activation of tyrosine kinase signaling pathways. Pan-TRK inhibitors such as entrectinib (58) and larotrectinib (59) have shown remarkable efficacy in patients with NTRK fusions, including those with iCCA. However, given that research on NTRK-targeted therapies in iCCA is still in its early stages, key areas of focus will include developing next-generation TRK inhibitors to address resistance mutations and implementing broader genomic screening for NTRK fusions in iCCA patients.
The PD-1/PD-L1 immune checkpoint pathway plays a critical role in immune evasion, with PD-L1 overexpression observed in a subset of iCCA cases. Immune checkpoint inhibitors such as pembrolizumab and nivolumab have shown durable responses in PD-L1-positive iCCA patients, and clinical trials are ongoing to further evaluate their efficacy. Efforts are also being made to explore combination therapies involving PD-1/PD-L1 inhibitors and other agents, such as chemotherapy, FGFR inhibitors, and other immune checkpoint inhibitors such as CTLA-4 inhibitors. Additionally, in future efforts, targeting novel immune checkpoint pathways beyond PD-1/PD-L1, such as LAG-3 and TIGIT, represents a promising direction for future immunotherapy in iCCA.
These advancements in targeted therapy and immunotherapy represent significant progress in addressing the heterogeneity of iCCA, though challenges such as resistance mechanisms and limited patient stratification remain. Continued research and innovation in combination strategies, next-generation inhibitors, and novel therapeutic targets will shape the future of iCCA treatment and improve the outcomes for patients.
5. Conclusion and perspectives
The molecular complexity and aggressive nature of intrahepatic cholangiocarcinoma (iCCA) pose significant challenges in its diagnosis and treatment, particularly given its poor prognosis in advanced stages. Although surgery remains the standard approach for early-stage resectable iCCA, the majority of patients with advanced or metastatic iCCA have limited therapeutic options due to the lack of effective systemic medical therapies. However, recent advances in the understanding of the molecular complexity of these tumors have provided new opportunities for targeted therapy development. The identification of promising druggable targets such as IDH1/2 mutations, FGFR and NTRK gene fusions and rearrangements, HER2 amplification, BRAF aberrations, and PD-1/PD-L1 overexpression, together with FDA-approved drugs such as ivosidenib (1), infigratinib (24), pemigatinib (25), and futibatinib (26) offer potential for the development of novel therapeutic strategies for patients with iCCA.In addition, despite decades of experience with molecular targeted therapies, cancer has shown the ability to develop drug resistance through the induction of point mutations. As a result, there is a pressing need to identify new molecules that can overcome these resistance mechanisms. An important strategy in targeted therapy is to analyze potential secondary mutations and design molecules that can effectively counteract them. This approach is crucial for staying ahead of the adaptive responses of cancer cells and developing novel treatments that can address drug resistance. In addition, given that IDH1 and FGFR2 mutations are found in 10–25% of iCCA cases, it is likely that most patients will not benefit therapeutically from currently targeted therapies using IDH1 and FGFR2 as actionable targets. Thus, to maximize the number of patients who can benefit from the effective treatment options, it is still in necessary to conduct further research to investigate new molecular targets in iCCA. Exploring CDKN2A/B loss, KRAS mutations, chromatin remodeling genes (ARID1A, BAP1, PBRM1), BRCA1/2 mutations, and NTRK fusions reveals promising avenues for the development of personalized targeted therapies for iCCA. As previously noted, diversifying therapeutic approaches beyond well-established targets is crucial for advancing iCCA treatment options. Ongoing investigations on drugs such as entrectinib, larotrectinib, palbociclib, sotorasib, olaparib, and entrectinib offer possible breakthroughs in alternate treatment choices for iCCA.
Hence, additional research and clinical trials are required to comprehensively assess the effectiveness and safety of these targeted approaches, ultimately enhancing the prognosis and survival of patients grappling with this devastating disease. Moreover, it is envisioned that the application of new platform technologies such as PROTAC (PROteolysis-Targeting Chimera) or incorporation of various modalities as forms of drug–drug conjugates (chaperone binders) and antibody–drug conjugates can increase opportunities for the treatment of patients with iCCA harboring various actionable molecular targets.
6. Materials and methods
A comprehensive search was conducted utilizing the following databases to acquire recent and pertinent references in regard to actionable targets of iCCA: PubMed, Science-Direct, Springer, ACS, NIH, Google Scholar, MEDLINE, EBSCO, Web of Science, http://ClinicalTrials.gov, and Sci-Finder from 2010 to 2024. The keywords used included ‘cholangiocarcinoma’, ‘small molecule inhibitors’, ‘molecular targets of iCCA’, ‘IDH inhibitors for iCCA’, ‘FGFR inhibitors for iCCA’, ‘BRAF inhibitors for iCCA’, ‘NTRK inhibitors for iCCA’, ‘HER2 inhibitors for iCCA’, and ‘immunotherapy for iCCA’ alone or combined with the keywords ‘derivatives’, ‘anticancer’, ‘signaling pathway’, ‘apoptosis’, ‘cell cycle’, ‘anti-proliferative activities’, ‘drug-resistance’, ‘angiogenesis’, ‘metastasis’, ‘kinase inhibition’, ‘point mutation’, ‘acquired mutation’, ‘overcoming drug-resistance’, and ‘drug discovery’.English-language references were exclusively considered for inclusion in this review due to language barriers. Compound entries and references were chosen based on the following criteria: availability of research focused on a) actionable targets of iCCA and corresponding inhibitors, b) structural optimization process, c) X-ray co-crystal structure and binding mode of the inhibitors, d) in vitro and in vivo studies, and e) patents and clinical studies. Conversely, compound entries and references were excluded based on the following criteria: a) research not focused on actionable targets of iCCA and advanced CCA, b) research without sufficient information on structure–activity relationship and optimization process, and c) research without sufficient information on biological evaluation.
Molecular modeling
The co-crystal structures of IDH1R132H (PDB: 5LGE) and FGFR2 (PDB:3RI1 and 7OZY) were obtained from the Protein Data Bank. Molecular docking studies were conducted using previously described methods.259List of abbreviations
| 2-HG | D-2-Hydroxyglutarate |
| ADCs | Antibody–drug conjugates |
| AML | Acute myeloid leukemia |
| BA | Bioavailability |
| BRAF | V-raf murine sarcoma viral oncogenes homolog B1 |
| BRCA 1/2 | Breast cancer type genes 1/2 |
| BTC | Biliary tract cancer |
| CDKN2A/B | Cyclin-dependent kinase inhibitor 2A/B |
| CTLA4 | Cytotoxic T lymphocyte antigen 4 |
| CYP | Cytochrome P450 |
| DMPK | Drug metabolism and pharmacokinetics |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| FGFR | Fibroblast growth factor receptors |
| HER2 | Human epidermal growth factor receptor 2 |
| hPXR | Human pregnane X receptor |
| ICIs | Immune checkpoint inhibitors |
| IDH1 | Isocitrate dehydrogenase 1 |
| iCCA | Intrahepatic cholangiocarcinoma |
| KRAS | Kirsten rat sarcoma viral oncogene |
| LA | Lung adenocarcinoma |
| MAPK | Mitogen activated protein kinase |
| mDOR | Median duration of response |
| MEK | Mitogen-activated protein kinase |
| mOS | Median overall survival |
| mPFS | Median progression-free survival |
| NTRK | Neurotropic tyrosine kinase receptor |
| NSCLC | Non-small cell lung cancer |
| ORR | Objective response rate |
| PARP | Poly(ADP-ribose) polymerase |
| PD-1/PD-L1 | Programmed cell death protein 1/programmed cell death-ligand 1 |
| PI3K | Phospho-inositide-3 kinase |
| PK/PD | Pharmacokinetics/pharmacodynamics |
| PPB | Plasma protein binding |
| RTK | Receptor tyrosine kinase |
| SNV | Single-nucleotide variant |
| STAT | Signal transducer and activator of transcription |
| VEGFR | Vascular endothelial growth factor receptor |
Data availability
No primary research results were included, and no original experimental data were generated or analyzed in this review. However, molecular docking studies were conducted as part of the analysis, and the datasets generated and/or analyzed from these studies are available from the authors upon reasonable request.Author contributions
Younghoon Kim: conceptualization, visualization, writing – original draft, and writing – review & editing. Jaewon Song: formal analysis, visualization, and writing – review & editing. Namkyoung Kim: writing – original draft. Taebo Sim: writing – review & editing and supervision. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Taebo Sim is a shareholder of Magicbullettherapeutics Inc.Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant number: RS-2024-00404555), the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (NRF-2021R1A2C3011992), Brain Korea 21 Project, and the KU-KIST Graduate School of Converging Science and Technology Program. Graphical abstract and Fig. 1 were created with http://BioRender.com (license numbers: HI27VEBRXU and TN27QZQC5Z, respectively).References
- S. A. Khan, S. Tavolari and G. Brandi, Cholangiocarcinoma: Epidemiology and risk factors, Liver Int., 2019, 39, 19–31 CrossRef PubMed.
- C. J. O'Rourke, P. Munoz-Garrido and J. B. Andersen, Molecular targets in cholangiocarcinoma, Hepatology, 2021, 73, 62–74 CrossRef PubMed.
- L. Izquierdo-Sanchez, A. Lamarca, A. La Casta, S. Buettner, K. Utpatel, H.-J. Klümpen, J. Adeva, A. Vogel, A. Lleo and L. Fabris, Cholangiocarcinoma landscape in Europe: Diagnostic, prognostic and therapeutic insights from the ENSCCA Registry, J. Hepatol., 2022, 76, 1109–1121 CrossRef CAS PubMed.
- J. M. Banales, J. J. Marin, A. Lamarca, P. M. Rodrigues, S. A. Khan, L. R. Roberts, V. Cardinale, G. Carpino, J. B. Andersen and C. Braconi, Cholangiocarcinoma 2020: the next horizon in mechanisms and management, Nat. Rev. Gastroenterol. Hepatol., 2020, 17, 557–588 CrossRef PubMed.
- I. Endo, M. Gonen, A. C. Yopp, K. M. Dalal, Q. Zhou, D. Klimstra, M. D'Angelica, R. P. DeMatteo, Y. Fong and L. Schwartz, Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection, Ann. Surg., 2008, 248, 84–96 CrossRef PubMed.
- A. A. Florio, J. Ferlay, A. Znaor, D. Ruggieri, C. S. Alvarez, M. Laversanne, F. Bray, K. A. McGlynn and J. L. Petrick, Global trends in intrahepatic and extrahepatic cholangiocarcinoma incidence from 1993 to 2012, Cancer, 2020, 126, 2666–2678 CrossRef PubMed.
- R. Kitphati, O. Watanawong, T. Wongsaroj and C. Nithikathkul, National Program of Opisthorchiasis in Thailand; Situation and Policy Strategy, Int. J. Geoinformatics, 2021, 17(2), 61–68 Search PubMed.
- T. Chung and Y. N. Park, Up-to-date pathologic classification and molecular characteristics of intrahepatic cholangiocarcinoma, Front. Med., 2022, 817 Search PubMed.
- A. Gupta and E. Dixon, Epidemiology and risk factors: intrahepatic cholangiocarcinoma, Hepatobiliary Surg. Nutr., 2017, 6, 101 CrossRef PubMed.
- N. Guedj, Pathology of Cholangiocarcinomas, Curr. Oncol., 2022, 30, 370–380 CrossRef PubMed.
- B. Blechacz, Cholangiocarcinoma: current knowledge and new developments, Gut Liver, 2017, 11, 13 CrossRef PubMed.
- C. Tawarungruang, N. Khuntikeo, N. Chamadol, V. Laopaiboon, J. Thuanman, K. Thinkhamrop, M. Kelly and B. Thinkhamrop, Survival after surgery among patients with cholangiocarcinoma in Northeast Thailand according to anatomical and morphological classification, BMC Cancer, 2021, 21, 1–12 CrossRef PubMed.
- H. Kayhanian, E. C. Smyth and C. Braconi, Emerging molecular targets and therapy for cholangiocarcinoma, World J. Gastrointest. Oncol., 2017, 9, 268 CrossRef PubMed.
- R. J. Coelen, E. Roos, J. K. Wiggers, M. G. Besselink, C. I. Buis, O. R. Busch, C. H. Dejong, O. M. van Delden, C. H. van Eijck and P. Fockens, Endoscopic versus percutaneous biliary drainage in patients with resectable perihilar cholangiocarcinoma: a multicentre, randomised controlled trial, Lancet Gastroenterol. Hepatol., 2018, 3, 681–690 CrossRef PubMed.
- B. N. Reames, F. Bagante, A. Ejaz, G. Spolverato, A. Ruzzenente, M. Weiss, S. Alexandrescu, H. P. Marques, L. Aldrighetti and S. K. Maithel, Impact of adjuvant chemotherapy on survival in patients with intrahepatic cholangiocarcinoma: a multi-institutional analysis, HPB, 2017, 19, 901–909 CrossRef PubMed.
- Y. Sota, T. Einama, K. Kobayashi, I. Fujinuma, T. Tsunenari, Y. Takihata, T. Iwasaki, Y. Miyata, K. Okamoto and Y. Kajiwara, Recurrent cholangiocarcinoma with long-term survival by multimodal treatment: A case report, Mol. Clin. Oncol., 2021, 14, 1–4, DOI:10.3892/mco.2021.2288.
- J. H. Kim, Y. K. Cheon, T. Y. Lee, S. H. Lee and H. Chung, Effect of age on the prognosis of intrahepatic cholangiocarcinoma, Korean J. Intern. Med., 2022, 38, 39–47 CrossRef PubMed.
- J. Valle, H. Wasan, D. H. Palmer, D. Cunningham, A. Anthoney, A. Maraveyas, S. Madhusudan, T. Iveson, S. Hughes and S. P. Pereira, Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer, N. Engl. J. Med., 2010, 362, 1273–1281 CrossRef CAS PubMed.
- A. Lamarca, D. H. Palmer, H. S. Wasan, P. J. Ross, Y. T. Ma, A. Arora, S. Falk, R. Gillmore, J. Wadsley and K. Patel, Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial, Lancet Oncol., 2021, 22, 690–701 CrossRef CAS PubMed.
- J. W. Valle, A. Lamarca, L. Goyal, J. Barriuso and A. X. Zhu, New Horizons for Precision Medicine in Biliary Tract CancersPrecision Medicine in BTC, Cancer Discovery, 2017, 7, 943–962 CrossRef CAS PubMed.
- H.-Y. Min and H.-Y. Lee, Molecular targeted therapy for anticancer treatment, Exp. Mol. Med., 2022, 54, 1670–1694 CrossRef CAS PubMed.
- Y. Yang, S. Li, Y. Wang, Y. Zhao and Q. Li, Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective, Signal Transduction Targeted Ther., 2022, 7, 329 CrossRef CAS PubMed.
- T. F. Greten, R. Schwabe, N. Bardeesy, L. Ma, L. Goyal, R. K. Kelley and X. W. Wang, Immunology and immunotherapy of cholangiocarcinoma, Nat. Rev. Gastroenterol. Hepatol., 2023, 1–17 Search PubMed.
- Y. R. Murciano-Goroff, A. B. Warner and J. D. Wolchok, The future of cancer immunotherapy: microenvironment-targeting combinations, Cell Res., 2020, 30, 507–519 CrossRef PubMed.
- C. Robert, A decade of immune-checkpoint inhibitors in cancer therapy, Nat. Commun., 2020, 11, 3801 CrossRef CAS PubMed.
- L. Verlingue, D. Malka, A. Allorant, C. Massard, C. Ferté, L. Lacroix, E. Rouleau, N. Auger, M. Ngo and C. Nicotra, Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial, Eur. J. Cancer, 2017, 87, 122–130 CrossRef PubMed.
- K. Mody, P. Jain, S. M. El-Refai, N. S. Azad, D. J. Zabransky, M. Baretti, R. T. Shroff, R. K. Kelley, A. B. El-Khouiery and A. J. Hockenberry, Clinical, genomic, and transcriptomic data profiling of biliary tract cancer reveals subtype-specific immune signatures, JCO Precis. Oncol., 2022, 6, e2100510 CrossRef PubMed.
- D. Moris, M. Palta, C. Kim, P. J. Allen, M. A. Morse and M. E. Lidsky, Advances in the treatment of intrahepatic cholangiocarcinoma: an overview of the current and future therapeutic landscape for clinicians, Ca-Cancer J. Clin., 2023, 73, 198–222 CrossRef PubMed.
- M. A. Lowery, R. Ptashkin, E. Jordan, M. F. Berger, A. Zehir, M. Capanu, N. E. Kemeny, E. M. O'Reilly, I. El-Dika and W. R. Jarnagin, Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for InterventionMolecular Profiling of Biliary Tract Cancer, Clin. Cancer Res., 2018, 24, 4154–4161 CrossRef CAS PubMed.
- J. S. Ross, K. Wang, L. Gay, R. Al-Rohil, J. V. Rand, D. M. Jones, H. J. Lee, C. E. Sheehan, G. A. Otto and G. Palmer, New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing, Onco Targets Ther, 2014, 19, 235–242 CAS.
- G. K. Abou-Alfa, T. Macarulla, M. M. Javle, R. K. Kelley, S. J. Lubner, J. Adeva, J. M. Cleary, D. V. Catenacci, M. J. Borad and J. Bridgewater, Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study, Lancet Oncol., 2020, 21, 796–807 CrossRef CAS PubMed.
- M. Javle, T. Bekaii-Saab, A. Jain, Y. Wang, R. K. Kelley, K. Wang, H. C. Kang, D. Catenacci, S. Ali and S. Krishnan, Biliary cancer: utility of next-generation sequencing for clinical management, Cancer, 2016, 122, 3838–3847 CrossRef CAS PubMed.
- M. Javle, M. Lowery, R. T. Shroff, K. H. Weiss, C. Springfeld, M. J. Borad, R. K. Ramanathan, L. Goyal, S. Sadeghi and T. Macarulla, Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma, J. Clin. Oncol., 2018, 36, 276 CrossRef CAS PubMed.
- A. Jain, M. J. Borad, R. K. Kelley, Y. Wang, R. Abdel-Wahab, F. Meric-Bernstam, K. A. Baggerly, A. O. Kaseb, H. O. Al-Shamsi and D. H. Ahn, Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype, JCO Precis. Oncol., 2018, 2, 1–12 Search PubMed.
- B. Goeppert, L. Frauenschuh, M. Renner, S. Roessler, A. Stenzinger, F. Klauschen, A. Warth, M. N. Vogel, A. Mehrabi and M. Hafezi, BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma, Mod. Pathol., 2014, 27, 1028–1034 CrossRef CAS PubMed.
- A. Lamarca, J. Barriuso, M. G. McNamara and J. W. Valle, Molecular targeted therapies: ready for “prime time” in biliary tract cancer, J. Hepatol., 2020, 73, 170–185 CrossRef CAS PubMed.
- H. Kim, R. Kim, H. R. Kim, H. Jo, H. Kim, S. Y. Ha, J. O. Park, Y. S. Park and S. T. Kim, HER2 Aberrations as a novel marker in advanced biliary tract cancer, Front. Oncol., 2022, 12 CAS.
- S. Galdy, A. Lamarca, M. G. McNamara, R. A. Hubner, C. A. Cella, N. Fazio and J. W. Valle, HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: a potential therapeutic target?, Cancer Metastasis Rev., 2017, 36, 141–157 CrossRef CAS PubMed.
- S. Rizvi and G. J. Gores, Pathogenesis, diagnosis, and management of cholangiocarcinoma, Gastroenterology, 2013, 145, 1215–1229 CrossRef CAS PubMed.
- J. B. Andersen and S. S. Thorgeirsson, Genomic decoding of intrahepatic cholangiocarcinoma reveals therapeutic opportunities, Gastroenterology, 2013, 144, 687–690 CrossRef PubMed.
- D. Sia, V. Tovar, A. Moeini and J. Llovet, Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies, Oncogene, 2013, 32, 4861–4870 CrossRef CAS PubMed.
- K. Maemura, S. Natsugoe and S. Takao, Molecular mechanism of cholangiocarcinoma carcinogenesis, J. Hepatobiliary Pancreat. Sci., 2014, 21, 754–760 CrossRef PubMed.
- R. Xue, L. Chen, C. Zhang, M. Fujita, R. Li, S.-M. Yan, C. K. Ong, X. Liao, Q. Gao and S. Sasagawa, Genomic and transcriptomic profiling of combined hepatocellular and intrahepatic cholangiocarcinoma reveals distinct molecular subtypes, Cancer Cell, 2019, 35(932–947), e938 Search PubMed.
- M. Rimini, E. Loi, C. Fabregat-Franco, V. Burgio, S. Lonardi, M. Niger, M. Scartozzi, I. G. Raposelli, G. Aprile and F. Ratti, Next-generation sequencing analysis of cholangiocarcinoma identifies distinct IDH1-mutated clusters, Eur. J. Cancer, 2022, 175, 299–310 CrossRef CAS PubMed.
- T. Boerner, E. Drill, L. M. Pak, B. Nguyen, C. S. Sigel, A. Doussot, P. Shin, D. A. Goldman, M. Gonen and P. J. Allen, Genetic determinants of outcome in intrahepatic cholangiocarcinoma, Hepatology, 2021, 74, 1429–1444 CrossRef CAS PubMed.
- G. Aitcheson, A. Mahipal and B. V. John, Targeting FGFR in intrahepatic cholangiocarcinoma [iCCA]: leading the way for precision medicine in biliary tract cancer [BTC]?, Expert Opin. Invest. Drugs, 2021, 30, 463–477 CrossRef CAS PubMed.
- H. Nakamura, Y. Arai, Y. Totoki, T. Shirota, A. Elzawahry, M. Kato, N. Hama, F. Hosoda, T. Urushidate and S. Ohashi, Genomic spectra of biliary tract cancer, Nat. Genet., 2015, 47, 1003–1010 CrossRef CAS PubMed.
- J. M. Cleary, S. Raghavan, Q. Wu, Y. Y. Li, L. F. Spurr, H. V. Gupta, D. A. Rubinson, I. J. Fetter, J. L. Hornick and J. A. Nowak, FGFR2 Extracellular Domain In-Frame Deletions Are Therapeutically Targetable Genomic Alterations That Function as Oncogenic Drivers in CholangiocarcinomaFGFR2 Extracellular Domain In-Frame Deletions, Cancer Discovery, 2021, 11, 2488–2505 CrossRef CAS PubMed.
- S. Rizvi, S. A. Khan, C. L. Hallemeier, R. K. Kelley and G. J. Gores, Cholangiocarcinoma—evolving concepts and therapeutic strategies, Nat. Rev. Clin. Oncol., 2018, 15, 95–111 CrossRef CAS PubMed.
- Y. Jiao, T. M. Pawlik, R. A. Anders, F. M. Selaru, M. M. Streppel, D. J. Lucas, N. Niknafs, V. B. Guthrie, A. Maitra and P. Argani, Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas, Nat. Genet., 2013, 45, 1470–1473 CrossRef CAS PubMed.
- P. Wang, Q. Dong, C. Zhang, P.-F. Kuan, Y. Liu, W. R. Jeck, J. B. Andersen, W. Jiang, G. L. Savich and T.-X. Tan, Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas, Oncogene, 2013, 32, 3091–3100 CrossRef CAS PubMed.
- S. J. Casak, S. Pradhan, L. A. Fashoyin-Aje, Y. Ren, Y.-L. Shen, Y. Xu, E. C. Y. Chow, Y. Xiong, J. F. Zirklelbach and J. Liu, FDA Approval Summary: Ivosidenib for the treatment of patients with advanced unresectable or metastatic, chemotherapy refractory cholangiocarcinoma with an IDH1 mutation, Clin. Cancer Res., 2022, 28, 2733–2737 CrossRef CAS PubMed.
- F. Li, M. N. Peiris and D. J. Donoghue, Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma, Cytokine Growth Factor Rev., 2020, 52, 56–67 CrossRef CAS PubMed.
- D. Romero, Benefit from pemigatinib in cholangiocarcinoma, Nat. Rev. Clin. Oncol., 2020, 17, 337–337 CrossRef PubMed.
- S. M. Hoy, Pemigatinib: first approval, Drugs, 2020, 80, 923–929 CrossRef PubMed.
- D. Planchard, B. Besse, H. J. Groen, P.-J. Souquet, E. Quoix, C. S. Baik, F. Barlesi, T. M. Kim, J. Mazieres and S. Novello, Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial, Lancet Oncol., 2016, 17, 984–993 CrossRef CAS PubMed.
- Z. Yao, N. M. Torres, A. Tao, Y. Gao, L. Luo, Q. Li, E. de Stanchina, O. Abdel-Wahab, D. B. Solit and P. I. Poulikakos, BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition, Cancer Cell, 2015, 28, 370–383 CrossRef CAS PubMed.
- Y. Zou and Y. Ji, The translocations of FGFR2 and NTRK1 in intrahepatic cholangiocarcinoma and their discordance in status by FISH and IHC, Cancer Res., 2021, 81, 353–353 CrossRef.
- M. Javle, C. Churi, H. C. Kang, R. Shroff, F. Janku, R. Surapaneni, M. Zuo, C. Barrera, H. Alshamsi and S. Krishnan, HER2/neu-directed therapy for biliary tract cancer, J. Hematol. Oncol., 2015, 8, 1–9 CrossRef CAS PubMed.
- C. Vivaldi, L. Fornaro, C. Ugolini, C. Niccoli, G. Musettini, I. Pecora, A. Cacciato Insilla, F. Salani, G. Pasquini and S. Catanese, HER2 overexpression as a poor prognostic determinant in resected biliary tract cancer, Onco Targets Ther, 2020, 25, 886–893 CAS.
- J. M. Taube, A. Klein, J. R. Brahmer, H. Xu, X. Pan, J. H. Kim, L. Chen, D. M. Pardoll, S. L. Topalian and R. A. Anders, Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti–PD-1 TherapyAssociation of PD-1 and Ligands with Response to Anti–PD-1, Clin. Cancer Res., 2014, 20, 5064–5074 CrossRef CAS PubMed.
- Y. Sato, M. Kinoshita, S. Takemura, S. Tanaka, G. Hamano, S. Nakamori, M. Fujikawa, Y. Sugawara, T. Yamamoto and A. Arimoto, The PD-1/PD-L1 axis may be aberrantly activated in occupational cholangiocarcinoma, Pathol. Int., 2017, 67, 163–170 CrossRef CAS PubMed.
- F. Sabbatino, V. Villani, J. H. Yearley, V. Deshpande, L. Cai, I. T. Konstantinidis, C. Moon, S. Nota, Y. Wang and A. Al-Sukaini, PD-L1 and HLA Class I Antigen Expression and Clinical Course of the Disease in Intrahepatic CholangiocarcinomaPD-L1 and HLA Class I in Intrahepatic Cholangiocarcinoma, Clin. Cancer Res., 2016, 22, 470–478 CrossRef CAS PubMed.
- S. Izreig, F. Alzahrani, J. Earles, S. Mehra, B. L. Judson, Z. Pan and R. W. Rahmati, Hyperprogression of a Sinonasal Squamous Cell Carcinoma Following Programmed Cell Death Protein-1 Checkpoint Blockade, JAMA Otolaryngol. Head Neck Surg., 2020, 146, 1176–1178 CrossRef PubMed.
- Y. Bang, T. Doi, F. De Braud, S. Piha-Paul, A. Hollebecque, A. A. Razak, C. Lin, P. Ott, A. He and S. Yuan, 525 Safety and efficacy of pembrolizumab (MK-3475) in patients (pts) with advanced biliary tract cancer: Interim results of KEYNOTE-028, Eur. J. Cancer, 2015, 3, S112 CrossRef.
- V. Subbiah, D. Solit, T. Chan and R. Kurzrock, The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB)≥ 10: a decision centered on empowering patients and their physicians, Ann. Oncol., 2020, 31, 1115–1118 CrossRef CAS PubMed.
- S. Tommasini-Ghelfi, K. Murnan, F. M. Kouri, A. S. Mahajan, J. L. May and A. H. Stegh, Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease, Sci. Adv., 2019, 5, eaaw4543 CrossRef PubMed.
- M. S. Waitkus, B. H. Diplas and H. Yan, Biological role and therapeutic potential of IDH mutations in cancer, Cancer Cell, 2018, 34, 186–195 CrossRef CAS PubMed.
- T. Fujii, M. R. Khawaja, C. D. DiNardo, J. T. Atkins and F. Janku, Targeting isocitrate dehydrogenase (IDH) in cancer, Discov. Med., 2016, 21, 373–380 Search PubMed.
- L. Dang, K. Yen and E. Attar, IDH mutations in cancer and progress toward development of targeted therapeutics, Ann. Oncol., 2016, 27, 599–608 CrossRef CAS PubMed.
- C. J. Pirozzi and H. Yan, The implications of IDH mutations for cancer development and therapy, Nat. Rev. Clin. Oncol., 2021, 18, 645–661 CrossRef CAS PubMed.
- S. Liu, M. Abboud, V. Mikhailov, X. Liu, R. Reinbold and C. J. Schofield, Differentiating Inhibition Selectivity and Binding Affinity of Isocitrate Dehydrogenase 1 Variant Inhibitors, J. Med. Chem., 2023, 66, 5279–5288 CrossRef CAS PubMed.
- P. S. Ward, J. Patel, D. R. Wise, O. Abdel-Wahab, B. D. Bennett, H. A. Coller, J. R. Cross, V. R. Fantin, C. V. Hedvat and A. E. Perl, The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate, Cancer Cell, 2010, 17, 225–234 CrossRef CAS PubMed.
- T. Ma, F. Zou, S. Pusch, Y. Xu, A. Von Deimling and X. Zha, Inhibitors of mutant isocitrate dehydrogenases 1 and 2 (mIDH1/2): an update and perspective, J. Med. Chem., 2018, 61, 8981–9003 CrossRef CAS PubMed.
- A. N. Boscoe, C. Rolland and R. K. Kelley, Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: a systematic literature review, J. Gastrointest. Oncol., 2019, 10, 751 CrossRef PubMed.
- W. Chan-On, M.-L. Nairismägi, C. K. Ong, W. K. Lim, S. Dima, C. Pairojkul, K. H. Lim, J. R. McPherson, I. Cutcutache and H. L. Heng, Exome sequencing identifies distinct mutational patterns in liver fluke–related and non-infection-related bile duct cancers, Nat. Genet., 2013, 45, 1474–1478 CrossRef CAS PubMed.
- J. Mondesir, C. Willekens, M. Touat and S. de Botton, IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives, J. Blood Med., 2016, 171–180 CAS.
- Y. S. Cho, J. R. Levell, G. Liu, T. Caferro, J. Sutton, C. M. Shafer, A. Costales, J. R. Manning, Q. Zhao and M. Sendzik, Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor, ACS Med. Chem. Lett., 2017, 8, 1116–1121 CrossRef CAS PubMed.
- J. A. Caravella, J. Lin, R. B. Diebold, A.-M. Campbell, A. Ericsson, G. Gustafson, Z. Wang, J. Castro, A. Clarke and D. Gotur, Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor, J. Med. Chem., 2020, 63, 1612–1623 CrossRef CAS PubMed.
- A. Wick, O. Bähr, M. Schuler, K. Rohrberg, S. P. Chawla, F. Janku, D. Schiff, V. Heinemann, Y. Narita and H.-J. Lenz, Phase I Assessment of Safety and Therapeutic Activity of BAY1436032 in Patients with IDH1-Mutant Solid TumorsmIDH1i BAY1436032 Ph I in Solid Tumors, Clin. Cancer Res., 2021, 27, 2723–2733 CrossRef CAS PubMed.
- Celgene Corporation, Study of Orally Administered Enasidenib (AG-221) in Adults With Advanced Solid Tumors, Including Glioma, or Angioimmunoblastic T-cell Lymphoma, With an IDH2 Mutation, 2014 Search PubMed.
- Z. Konteatis, E. Artin, B. Nicolay, K. Straley, A. K. Padyana, L. Jin, Y. Chen, R. Narayaraswamy, S. Tong and F. Wang, Vorasidenib (AG-881): a first-in-class, brain-penetrant dual inhibitor of mutant IDH1 and 2 for treatment of glioma, ACS Med. Chem. Lett., 2020, 11, 101–107 CrossRef CAS PubMed.
- U. C. Okoye-Okafor, B. Bartholdy, J. Cartier, E. N. Gao, B. Pietrak, A. R. Rendina, C. Rominger, C. Quinn, A. Smallwood and K. J. Wiggall, New IDH1 mutant inhibitors for treatment of acute myeloid leukemia, Nat. Chem. Biol., 2015, 11, 878–886 CrossRef CAS PubMed.
- S. Liu, M. I. Abboud, T. John, V. Mikhailov, I. Hvinden, J. Walsby-Tickle, X. Liu, I. Pettinati, T. Cadoux-Hudson and J. S. McCullagh, Roles of metal ions in the selective inhibition of oncogenic variants of isocitrate dehydrogenase 1, Commun. Biol., 2021, 4, 1243 CrossRef CAS PubMed.
- X. Xie, D. Baird, K. Bowen, V. Capka, J. Chen, G. Chenail, Y. Cho, J. Dooley, A. Farsidjani and P. Fortin, Allosteric mutant IDH1 inhibitors reveal mechanisms for IDH1 mutant and isoform selectivity, Structure, 2017, 25, 506–513 CrossRef CAS PubMed.
- E. I. Juritz, J. P. Bascur, D. E. Almonacid and F. D. González-Nilo, Novel Insights for Inhibiting Mutant Heterodimer IDH1 wt-R132H in Cancer: An In-Silico Approach, Mol. Diagn. Ther., 2018, 22, 369–380 CrossRef CAS PubMed.
- B. Yang, C. Zhong, Y. Peng, Z. Lai and J. Ding, Molecular mechanisms of “off-on switch” of activities of human IDH1 by tumor-associated mutation R132H, Cell Res., 2010, 20, 1188–1200 CrossRef CAS PubMed.
- S. Liu, M. Abboud, V. Mikhailov, X. Liu, R. Reinbold and C. J. Schofield, Differentiating Inhibition Selectivity and Binding Affinity of Isocitrate Dehydrogenase 1 Variant Inhibitors, J. Med. Chem., 2023, 66(7), 5279–5288 CrossRef CAS PubMed.
- Q. Zheng, S. Tang, X. Fu, Z. Chen, Y. Ye, X. Lan, L. Jiang, Y. Huang, J. Ding and M. Geng, Discovery and structure-activity-relationship study of novel conformationally restricted indane analogues for mutant isocitric dehydrogenase 1 (IDH1) inhibitors, Bioorg. Med. Chem. Lett., 2017, 27, 5262–5266 CrossRef CAS PubMed.
- S. Pusch, S. Krausert, V. Fischer, J. Balss, M. Ott, D. Schrimpf, D. Capper, F. Sahm, J. Eisel and A.-C. Beck, Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo, Acta Neuropathol., 2017, 133, 629–644 CrossRef CAS PubMed.
- K. Yen, J. Travins, F. Wang, M. D. David, E. Artin, K. Straley, A. Padyana, S. Gross, B. DeLaBarre and E. Tobin, AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations, Cancer Discovery, 2017, 7, 478–493 CrossRef CAS PubMed.
- M. J. Buskes, A. Coffin, D. M. Troast, R. Stein and M.-J. Blanco, Accelerating Drug Discovery: Synthesis of Complex Chemotypes via Multicomponent Reactions, ACS Med. Chem. Lett., 2023, 14(4), 376–385 CrossRef CAS PubMed.
- Y. Wang, S. Tang, H. Lai, R. Jin, X. Long, N. Li, Y. Tang, H. Guo, X. Yao and E. L.-H. Leung, Discovery of novel IDH1 inhibitor through comparative structure-based virtual screening, Front. Pharmacol., 2020, 11, 579768 CrossRef CAS PubMed.
- J. Popovici-Muller, J. O. Saunders, F. G. Salituro, J. M. Travins, S. Yan, F. Zhao, S. Gross, L. Dang, K. E. Yen and H. Yang, Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo, ACS Med. Chem. Lett., 2012, 3, 850–855 CrossRef CAS PubMed.
- A. X. Zhu, T. Macarulla, M. M. Javle, R. K. Kelley, S. J. Lubner, J. Adeva, J. M. Cleary, D. V. Catenacci, M. J. Borad and J. A. Bridgewater, Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical ClarIDHy trial, JAMA Oncol., 2021, 7, 1669–1677 CrossRef PubMed.
- J. R. Levell, T. Caferro, G. Chenail, I. Dix, J. Dooley, B. Firestone, P. D. Fortin, J. Giraldes, T. Gould and J. D. Growney, Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1, ACS Med. Chem. Lett., 2017, 8, 151–156 CrossRef CAS PubMed.
- C. D. DiNardo, A. D. Schimmer, K. W. Yee, A. Hochhaus, A. Kraemer, R. D. Carvajal, F. Janku, P. Bedard, C. Carpio and A. Wick, A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations, Blood, 2016, 128, 1073 CrossRef.
- Novartis Pharmaceuticals, A Study of IDH305 in Patients With Advanced Malignancies That Harbor IDH1R132 Mutations, 2015, NCT02381886, https://clinicaltrials.gov/study/NCT02381886 Search PubMed.
- Bayer, Phase I Study of BAY1436032 in IDH1-mutant Advanced Solid Tumors, 2016, NCT02746081, https://clinicaltrials.gov/study/NCT02746081 Search PubMed.
- K. Yen, J. Travins, F. Wang, M. D. David, E. Artin, K. Straley, A. Padyana, S. Gross, B. DeLaBarre and E. Tobin, AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 MutationsAG-221 Therapy for IDH2-Mutant AML, Cancer Discovery, 2017, 7, 478–493 CrossRef CAS PubMed.
- E. M. Stein, C. DiNardo, J. K. Altman, R. Collins, D. J. DeAngelo, H. M. Kantarjian, M. A. Sekeres, A. T. Fathi, I. W. Flinn and A. E. Frankel, Safety and efficacy of AG-221, a potent inhibitor of mutant IDH2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: results of a phase 1/2 trial, Blood, 2015, 126, 323 CrossRef.
- I. K. Mellinghoff, M. Penas-Prado, K. B. Peters, H. A. Burris III, E. A. Maher, F. Janku, G. M. Cote, M. I. de la Fuente, J. L. Clarke and B. M. Ellingson, Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial, Clin. Cancer Res., 2021, 27, 4491–4499 CrossRef CAS PubMed.
- Servier, Study of Vorasidenib (AG-881) in Participants With Residual or Recurrent Grade 2 Glioma With an IDH1 or IDH2 Mutation, (INDIGO), 2020, NCT04164901, https://clinicaltrials.gov/study/NCT04164901 Search PubMed.
- J. M. Cleary, B. Rouaisnel, A. Daina, S. Raghavan, L. A. Roller, B. M. Huffman, H. Singh, P. Y. Wen, N. Bardeesy and V. Zoete, Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma, NPJ Precis. Oncol., 2022, 6, 61 CrossRef CAS PubMed.
- J. Lyu, Y. Liu, H. Cao, M. Chen, Y. F. Madanat, Y. Zhang, Z. Gu, P. Kaphle, K. E. Dickerson and P. Yenerall, Elucidating Mechanisms of Acquired Resistance to IDH Inhibition By Saturation Variant Screening of Base-Edited Leukemia Cells, Blood, 2020, 136, 3 CrossRef.
- S. Choe, H. Wang, C. D. DiNardo, E. M. Stein, S. de Botton, G. J. Roboz, J. K. Altman, A. S. Mims, J. M. Watts and D. A. Pollyea, Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML, Blood Adv., 2020, 4, 1894–1905 CrossRef CAS PubMed.
- R. Reinbold, I. C. Hvinden, P. Rabe, R. A. Herold, A. Finch, J. Wood, M. Morgan, M. Staudt, I. J. Clifton and F. A. Armstrong, Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors, Nat. Commun., 2022, 13, 4785 CrossRef CAS PubMed.
- J. M. Pauff, K. P. Papadopoulos, F. Janku, A. A. Turk, L. Goyal, R. T. Shroff, T. Shimizu, M. Ikeda, N. S. Azad and J. M. Cleary, A phase I study of LY3410738, a first-in-class covalent inhibitor of mutant IDH1 in cholangiocarcinoma and other advanced solid tumors, 2021, NCT04521686, https://clinicaltrials.gov/study/NCT04521686 Search PubMed.
- M. Gao, H. Zhu, L. Fu, Y. Li, X. Bao, H. Fu, H. Quan, L. Wang and L. Lou, Pharmacological characterization of TQ 05310, a potent inhibitor of isocitrate dehydrogenase 2 R140Q and R172K mutants, Cancer Sci., 2019, 110, 3306–3314 CrossRef CAS PubMed.
- I. S. Babina and N. C. Turner, Advances and challenges in targeting FGFR signalling in cancer, Nat. Rev. Cancer, 2017, 17, 318–332 CrossRef CAS PubMed.
- K. H. Tiong, L. Y. Mah and C.-O. Leong, Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers, Apoptosis, 2013, 18, 1447–1468 CrossRef CAS PubMed.
- D. M. Ornitz and N. Itoh, The fibroblast growth factor signaling pathway, Wiley Interdiscip. Rev.:Dev. Biol., 2015, 4, 215–266 CrossRef CAS PubMed.
- M. Katoh and H. Nakagama, FGF receptors: cancer biology and therapeutics, Med. Res. Rev., 2014, 34, 280–300 CrossRef CAS PubMed.
- B. Trueb, Biology of FGFRL1, the fifth fibroblast growth factor receptor, Cell. Mol. Life Sci., 2011, 68, 951–964 CrossRef CAS PubMed.
- J. Kalinina, K. Dutta, D. Ilghari, A. Beenken, R. Goetz, A. V. Eliseenkova, D. Cowburn and M. Mohammadi, The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition, Structure, 2012, 20, 77–88 CrossRef CAS PubMed.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Heparan sulfate proteoglycans Cold Spring Harb, Perspect. Biol., 2011, 3, a004952 Search PubMed.
- N. Turner and R. Grose, Fibroblast growth factor signalling: from development to cancer, Nat. Rev. Cancer, 2010, 10, 116–129 CrossRef CAS PubMed.
- A. A. Belov and M. Mohammadi, Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology, Cold Spring Harbor Perspect. Biol., 2013, 5, a015958 Search PubMed.
- M. A. Krook, J. W. Reeser, G. Ernst, H. Barker, M. Wilberding, G. Li, H.-Z. Chen and S. Roychowdhury, Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance, Br. J. Cancer, 2021, 124, 880–892 CrossRef CAS PubMed.
- T. Helsten, S. Elkin, E. Arthur, B. N. Tomson, J. Carter and R. Kurzrock, The FGFR Landscape in Cancer: Analysis of 4, 853 Tumors by Next-Generation SequencingFGFR Aberrations in Cancer, Clin. Cancer Res., 2016, 22, 259–267 CrossRef CAS PubMed.
- R. Dienstmann, J. Rodon, A. Prat, J. Perez-Garcia, B. Adamo, E. Felip, J. Cortes, A. Iafrate, P. Nuciforo and J. Tabernero, Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors, Ann. Oncol., 2014, 25, 552–563 CrossRef CAS PubMed.
- R. S. Heist, M. Mino-Kenudson, L. V. Sequist, S. Tammireddy, L. Morrissey, D. C. Christiani, J. A. Engelman and A. J. Iafrate, FGFR1 amplification in squamous cell carcinoma of the lung, J. Thorac. Oncol., 2012, 7, 1775–1780 CrossRef CAS PubMed.
- J. Zhang, L. Zhang, X. Su, M. Li, L. Xie, F. Malchers, S. Fan, X. Yin, Y. Xu and K. Liu, Translating the Therapeutic Potential of AZD4547 in FGFR1-Amplified Non–Small Cell Lung Cancer through the Use of Patient-Derived Tumor Xenograft ModelsAZD4547 Is Active in FGFR1-Amplified Squamous NSCLC PDTX Models, Clin. Cancer Res., 2012, 18, 6658–6667 CrossRef CAS PubMed.
- T. L. Rose, W. H. Weir, G. M. Mayhew, Y. Shibata, P. Eulitt, J. M. Uronis, M. Zhou, M. Nielsen, A. B. Smith and M. Woods, Fibroblast growth factor receptor 3 alterations and response to immune checkpoint inhibition in metastatic urothelial cancer: a real world experience, Br. J. Cancer, 2021, 125, 1251–1260 CrossRef CAS PubMed.
- Y.-M. Wu, F. Su, S. Kalyana-Sundaram, N. Khazanov, B. Ateeq, X. Cao, R. J. Lonigro, P. Vats, R. Wang and S.-F. Lin, Identification of targetable FGFR gene fusions in diverse cancers, Cancer Discovery, 2013, 3, 636–647 CrossRef CAS PubMed.
- O. Neumann, T. C. Burn, M. Allgäuer, M. Ball, M. Kirchner, T. Albrecht, A.-L. Volckmar, S. Beck, V. Endris and H. Goldschmid, Genomic architecture of FGFR2 fusions in cholangiocarcinoma and its implication for molecular testing, Br. J. Cancer, 2022, 127, 1540–1549 CrossRef CAS PubMed.
- P. L. S. Uson Junior, T. T. DeLeon, J. M. Bogenberger, R. K. Pai, H. E. Kosiorek, J. Yin, D. H. Ahn, M. B. Sonbol, T. Bekaii-Saab and A. S. Mansfield, FGFR2-IIIb expression by immunohistochemistry has high specificity in cholangiocarcinoma with FGFR2 genomic alterations, Dig. Dis. Sci., 2022, 1–9 Search PubMed.
- A. Vogel, O. Segatto, A. Stenzinger and A. Saborowski, FGFR2 inhibition in cholangiocarcinoma, Annu. Rev. Med., 2023, 74, 293–306 CrossRef CAS PubMed.
- X. Pu, L. Qi, J. W. Yan, Z. Ai, P. Wu, F. Yang, Y. Fu, X. Li, M. Zhang and B. Sun, Oncogenic activation revealed by FGFR2 genetic alterations in intrahepatic cholangiocarcinomas, Cell Biosci., 2023, 13, 208 CrossRef CAS PubMed.
- M. A. Krook, I. M. Silverman, K. Murugesan, G. Ernst, J. Reeser, M. Wing, G. M. Frampton, R. C. Newton, J. R. Jackson and L. A. Albacker, Pan-cancer analysis of FGFR1-3 genomic alterations to reveal a complex molecular landscape, J. Clin. Oncol., 2020, 38, 3620 Search PubMed.
- D. Singh, J. M. Chan, P. Zoppoli, F. Niola, R. Sullivan, A. Castano, E. M. Liu, J. Reichel, P. Porrati and S. Pellegatta, Transforming fusions of FGFR and TACC genes in human glioblastoma, Science, 2012, 337, 1231–1235 CrossRef CAS PubMed.
- Y. Arai, Y. Totoki, F. Hosoda, T. Shirota, N. Hama, H. Nakamura, H. Ojima, K. Furuta, K. Shimada and T. Okusaka, Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma, Hepatology, 2014, 59, 1427–1434 CrossRef CAS PubMed.
- L. Chen, Y. Zhang, L. Yin, B. Cai, P. Huang, X. Li and G. Liang, Fibroblast growth factor receptor fusions in cancer: opportunities and challenges, J. Exp. Clin. Cancer Res., 2021, 40, 1–15 CrossRef PubMed.
- T. H. Patel, L. Marcus, M. N. Horiba, M. Donoghue, S. Chatterjee, P. S. Mishra-Kalyani, R. N. Schuck, Y. Li, X. Zhang and J. Fourie Zirkelbach, FDA approval summary: pemigatinib for previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with FGFR2 fusion or other rearrangement, Clin. Cancer Res., 2023, 29, 838–842 CrossRef CAS PubMed.
- L. Goyal, F. Meric-Bernstam, A. Hollebecque, J. W. Valle, C. Morizane, T. B. Karasic, T. A. Abrams, J. Furuse, R. K. Kelley and P. A. Cassier, Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma, N. Engl. J. Med., 2023, 388, 228–239 CrossRef CAS PubMed.
- R. Roskoski, Futibatinib (Lytgobi) for cholangiocarcinoma, Trends Pharmacol. Sci., 2023, 44(3), 190–191 CrossRef CAS PubMed.
- Y. Y. Syed, Futibatinib: First Approval, Drugs, 2022, 1–7 Search PubMed.
- T. G. Hall, Y. Yu, S. Eathiraj, Y. Wang, R. E. Savage, J.-M. Lapierre, B. Schwartz and G. Abbadessa, Preclinical activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation, PLoS One, 2016, 11, e0162594 CrossRef PubMed.
- S. Eathiraj, R. Palma, M. Hirschi, E. Volckova, E. Nakuci, J. Castro, C.-R. Chen, T. C. Chan, D. S. France and M. A. Ashwell, A novel mode of protein kinase inhibition exploiting hydrophobic motifs of autoinhibited kinases: discovery of ATP-independent inhibitors of fibroblast growth factor receptor, J. Biol. Chem., 2011, 286, 20677–20687 CrossRef CAS PubMed.
- V. Guagnano, P. Furet, C. Spanka, V. Bordas, M. Le Douget, C. Stamm, J. Brueggen, M. R. Jensen, C. Schnell and H. Schmid, Discovery of 3-(2, 6-dichloro-3, 5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase, J. Med. Chem., 2011, 54, 7066–7083 CrossRef CAS PubMed.
- M. M. Javle, S. Roychowdhury, R. K. Kelley, S. Sadeghi, T. Macarulla, D. T. Waldschmidt, L. Goyal, I. Borbath, A. B. El-Khoueiry and W.-P. Yong, Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement, 2021, NCT02150967, https://www.clinicaltrials.gov/study/NCT02150967 Search PubMed.
- L. Wu, C. Zhang, C. He, D. Qian, L. Lu, Y. Sun, M. Xu, J. Zhuo, P. C. Liu and R. Klabe, Discovery of pemigatinib: a potent and selective fibroblast growth factor receptor (FGFR) inhibitor, J. Med. Chem., 2021, 64, 10666–10679 CrossRef CAS PubMed.
- G. K. Abou-Alfa, V. Sahai, A. Hollebecque, G. M. Vaccaro, D. Melisi, R. M. d. T. Al-Rajabi, A. S. Paulson, M. J. Borad, D. H. Gallinson and A. G. Murphy, Pemigatinib for previously treated locally advanced/metastatic cholangiocarcinoma (CCA): Update of FIGHT-202, 2021, NCT02924376, https://clinicaltrials.gov/study/NCT02924376 Search PubMed.
- S. Ito, S. Otsuki, H. Ohsawa, A. Hirano, H. Kazuno, S. Yamashita, K. Egami, Y. Shibata, I. Yamamiya and F. Yamashita, Discovery of Futibatinib: The First Covalent FGFR Kinase Inhibitor in Clinical Use, ACS Med. Chem. Lett., 2023, 14, 396–404 CrossRef CAS PubMed.
- S. U. Gandhy, S. J. Casak, S. L. Mushti, J. Cheng, S. Subramaniam, H. Zhao, M. Zhao, Y. Bi, G. Liu and J. Fan, FDA Approval Summary: Futibatinib for Unresectable Advanced or Metastatic, Chemotherapy Refractory Intrahepatic Cholangiocarcinoma with FGFR2 Fusions or Other Rearrangements, Clin. Cancer Res., 2023, CCR-23-1042 Search PubMed.
- L. Goyal, F. Meric-Bernstam, A. Hollebecque, J. W. Valle, C. Morizane, T. B. Karasic, T. A. Abrams, J. Furuse, Y. He and N. Soni, FOENIX-CCA2: A phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements, 2020, NCT02052778, https://clinicaltrials.gov/study/NCT02052778 Search PubMed.
- S. Ali, M. Ashwell, M. Tandon, N. Namdev, J.-M. Lapierre, N. Westlund, R. Palma, R.-Y. Yang, Y. Liu and D. Vensel, Substituted 5, 6-dihydro-6-phenylbenzo [F] isoquinolin-2-amine compounds, WO2010078421A1, 2013.
- V. Mazzaferro, B. F. El-Rayes, M. Droz dit Busset, C. Cotsoglou, W. P. Harris, N. Damjanov, G. Masi, L. Rimassa, N. Personeni and F. Braiteh, Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma, Br. J. Cancer, 2019, 120, 165–171 CrossRef CAS PubMed.
- C. Kang, Infigratinib: first approval, Drugs, 2021, 81, 1355–1360 CrossRef CAS PubMed.
- M. Javle, S. Roychowdhury, R. K. Kelley, S. Sadeghi, T. Macarulla, K. H. Weiss, D.-T. Waldschmidt, L. Goyal, I. Borbath and A. El-Khoueiry, Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study, Lancet Gastroenterol. Oncol., 2021, 6, 803–815 Search PubMed.
- P. C. Liu, H. Koblish, L. Wu, K. Bowman, S. Diamond, D. DiMatteo, Y. Zhang, M. Hansbury, M. Rupar and X. Wen, INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models, PLoS One, 2020, 15, e0231877 CrossRef CAS PubMed.
- Q. Lin, X. Chen, L. Qu, M. Guo, H. Wei, S. Dai, L. Jiang and Y. Chen, Characterization of the cholangiocarcinoma drug pemigatinib against FGFR gatekeeper mutants, Commun. Chem., 2022, 5, 100 CrossRef CAS PubMed.
- M. Kalyukina, Y. Yosaatmadja, M. J. Middleditch, A. V. Patterson, J. B. Smaill and C. J. Squire, TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure, ChemMedChem, 2019, 14, 494–500 CrossRef CAS PubMed.
- M. A. Krook, R. Bonneville, H.-Z. Chen, J. W. Reeser, M. R. Wing, D. M. Martin, A. M. Smith, T. Dao, E. Samorodnitsky and A. Paruchuri, Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy, Cold Spring Harb. Mol. Case Stud., 2019, 5, a004002 CrossRef CAS PubMed.
- S. Yue, Y. Li, X. Chen, J. Wang, M. Li, Y. Chen and D. Wu, FGFR-TKI resistance in cancer: current status and perspectives, J. Hematol. Oncol., 2021, 14, 1–14 CrossRef PubMed.
- L. Goyal, S. K. Saha, L. Y. Liu, G. Siravegna, I. Leshchiner, L. G. Ahronian, J. K. Lennerz, P. Vu, V. Deshpande and A. Kambadakone, Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion–positive cholangiocarcinoma, Cancer Discovery, 2017, 7, 252–263 CrossRef CAS PubMed.
- L. Qu, X. Chen, H. Wei, M. Guo, S. Dai, L. Jiang, J. Li, S. Yue, Z. Chen and Y. Chen, Structural insights into the potency and selectivity of covalent pan-FGFR inhibitors, Commun. Chem., 2022, 5, 5 CrossRef CAS PubMed.
- D. Wu, M. Guo, X. Min, S. Dai, M. Li, S. Tan, G. Li, X. Chen, Y. Ma and J. Li, LY2874455 potently inhibits FGFR gatekeeper mutants and overcomes mutation-based resistance, Chem. Commun., 2018, 54, 12089–12092 RSC.
- D. Wu, M. Guo, M. A. Philips, L. Qu, L. Jiang, J. Li, X. Chen, Z. Chen, L. Chen and Y. Chen, Crystal structure of the FGFR4/LY2874455 complex reveals insights into the Pan-FGFR selectivity of LY2874455, PLoS One, 2016, 11, e0162491 CrossRef PubMed.
- F. Dehghanian and S. Alavi, Molecular mechanisms of the anti-cancer drug, LY2874455, in overcoming the FGFR4 mutation-based resistance, Sci. Rep., 2021, 11, 16593 CrossRef CAS PubMed.
- M. Michael, Y.-J. Bang, Y. S. Park, Y.-K. Kang, T. M. Kim, O. Hamid, D. Thornton, S. C. Tate, E. Raddad and J. Tie, A phase 1 study of LY2874455, an oral selective pan-FGFR inhibitor, in patients with advanced cancer, Target Oncol., 2017, 12, 463–474 CrossRef PubMed.
- S. Ryu, Y. Nam, N. Kim, I. Shin, E. Jeon, Y. Kim, N. D. Kim and T. Sim, Identification of pyridinyltriazine derivatives as potent panFGFR inhibitors against gatekeeper mutants for overcoming drug resistance, J. Med. Chem., 2022, 65, 6017–6038 CrossRef CAS PubMed.
- R. Williams, An Important Source to Expand Targeted Therapy Access, J. Precis. Med., 2022, 8 Search PubMed.
- A. Franovic, A. Mohan, S. Uryu, Q. Wu, P. Jiang, N. Miller, J. Tyhonas, N. Timple, P. Severson and R. Kania, Activity of KIN-3248, a next-generation pan-FGFR inhibitor, against acquired FGFR-gatekeeper and molecular-brake drug resistance mutations, J. Clin. Oncol., 2022, 40, 461 Search PubMed.
- L. Goyal, C. A. Perez, S. Kato, M. Sharma, B. Garmezy, K. Kobayashi, A. Franovic, B. Tam and C. Voong, Design and rationale of a first-in-human (FIH) phase 1/1b study evaluating KIN-3248, a next-generation, irreversible (irrev), pan-FGFR inhibitor (FGFRi), in adult patients with solid tumors harboring FGFR2 and/or FGFR3 gene alterations, 2022, NCT05242822, https://clinicaltrials.gov/study/NCT05242822 Search PubMed.
- R. Yaeger and R. B. Corcoran, Targeting alterations in the RAF–MEK pathway, Cancer Discovery, 2019, 9, 329–341 CrossRef CAS PubMed.
- F. Sanchez-Vega, M. Mina, J. Armenia, W. K. Chatila, A. Luna, K. C. La, S. Dimitriadoy, D. L. Liu, H. S. Kantheti and S. Saghafinia, Oncogenic signaling pathways in the cancer genome atlas, Cell, 2018, 173, 321–337 CrossRef CAS PubMed , e310.
- H.-Y. Xin, R.-Q. Sun, J.-X. Zou, P.-C. Wang, J.-Y. Wang, Y.-H. Ye, K.-X. Liu, Z.-Q. Hu, Z.-J. Zhou and J. Fan, Association of BRAF Variants With Disease Characteristics, Prognosis, and Targeted Therapy Response in Intrahepatic Cholangiocarcinoma, JAMA Netw. Open, 2023, 6, e231476–e231476 CrossRef PubMed.
- G. Bollag, J. Tsai, J. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Vemurafenib: the first drug approved for BRAF-mutant cancer, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- B. E. Nelson, J. Roszik, F. Janku, D. S. Hong, S. Kato, A. Naing, S. Piha-Paul, S. Fu, A. Tsimberidou and M. Cabanillas, BRAF v600E–mutant cancers treated with vemurafenib alone or in combination with everolimus, sorafenib, or crizotinib or with paclitaxel and carboplatin (VEM-PLUS) study, NPJ Precis. Oncol., 2023, 7, 19 CrossRef CAS PubMed.
- D. M. Hyman, I. Puzanov, V. Subbiah, J. E. Faris, I. Chau, J.-Y. Blay, J. Wolf, N. S. Raje, E. L. Diamond and A. Hollebecque, Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations, N. Engl. J. Med., 2015, 373, 726–736 CrossRef CAS PubMed.
- T. R. Rheault, J. C. Stellwagen, G. M. Adjabeng, K. R. Hornberger, K. G. Petrov, A. G. Waterson, S. H. Dickerson, R. A. Mook Jr., S. G. Laquerre and A. J. King, Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors, ACS Med. Chem. Lett., 2013, 4, 358–362 CrossRef CAS PubMed.
- R. F. van Golen and T. J. Dekker, Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer, Lancet Oncol., 2020, 21, e515 CrossRef PubMed.
- M. I. De La Fuente, J. Rodon Ahnert, R. Yaeger, F. Y.-C. Tsai, F. Janku, N. A. Butowski, C. E. Allen, N. R. Ammakkanavar, J. W. Taylor and G. Michelson, Safety and efficacy of the novel BRAF inhibitor FORE8394 in patients with advanced solid and CNS tumors: Results from a phase 1/2a study, 2023, NCT02428712, https://clinicaltrials.gov/study/NCT02428712 Search PubMed.
- K. Koumaki, G. Kontogianni, V. Kosmidou, F. Pahitsa, E. Kritsi, M. Zervou, A. Chatziioannou, V. L. Souliotis, O. Papadodima and A. Pintzas, BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600E CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis, Biochim. Biophys. Acta, 2021, 1867, 166061 CrossRef CAS PubMed.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong and N. K. Haass, Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity, Proc. Nat. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS PubMed.
- P. A. Oneal, V. Kwitkowski, L. Luo, Y. L. Shen, S. Subramaniam, S. Shord, K. B. Goldberg, A. E. McKee, E. Kaminskas and A. Farrell, FDA Approval Summary: Vemurafenib for the Treatment of Patients with Erdheim-Chester Disease with the BRAF V600 Mutation, Onco Targets Ther, 2018, 23, 1520–1524 CAS.
- V. Subbiah, I. Puzanov, J.-Y. Blay, I. Chau, A. C. Lockhart, N. S. Raje, J. Wolf, J. Baselga, F. Meric-Bernstam and J. Roszik, Pan-cancer efficacy of vemurafenib in BRAF V600-mutant non-melanoma cancers, Cancer Discovery, 2020, 10, 657–663 CrossRef CAS PubMed.
- C. Zhang, W. Spevak, Y. Zhang, E. A. Burton, Y. Ma, G. Habets, J. Zhang, J. Lin, T. Ewing and B. Matusow, RAF inhibitors that evade paradoxical MAPK pathway activation, Nature, 2015, 526, 583–586 CrossRef CAS PubMed.
- J. C. Stellwagen, G. M. Adjabeng, M. R. Arnone, S. H. Dickerson, C. Han, K. R. Hornberger, A. J. King, R. A. Mook Jr., K. G. Petrov and T. R. Rheault, Development of potent B-RafV600E inhibitors containing an arylsulfonamide headgroup, Bioorg. Med. Chem. Lett., 2011, 21, 4436–4440 CrossRef CAS PubMed.
- V. Subbiah, R. J. Kreitman, Z. A. Wainberg, A. Gazzah, U. Lassen, A. Stein, P. Y. Wen, S. Dietrich, M. J. de Jonge and J.-Y. Blay, Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: the phase 2 ROAR trial, Nat. Med., 2023, 1–10 Search PubMed.
- R. A. Okimoto, L. Lin, V. Olivas, E. Chan, E. Markegard, A. Rymar, D. Neel, X. Chen, G. Hemmati and G. Bollag, Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer, Proc. Nat. Acad. Sci. U. S. A., 2016, 113, 13456–13461 CrossRef CAS PubMed.
- J. Choi, S. F. Landrette, T. Wang, P. Evans, A. Bacchiocchi, R. Bjornson, E. Cheng, A. L. Stiegler, S. Gathiaka and O. Acevedo, Identification of PLX 4032-resistance mechanisms and implications for novel RAF inhibitors, Pigment Cell Melanoma Res., 2014, 27, 253–262 CrossRef CAS PubMed.
- F. Janku, U. N. Vaishampayan, V. Khemka, M. Bhatty, E. J. Sherman, J. Tao, J. R. Whisenant, D. S. Hong, N. Bui and S. Kummar, Phase 1/2 precision medicine study of the next-generation BRAF inhibitor PLX8394, 2018, NCT02428712, https://clinicaltrials.gov/study/NCT02428712 Search PubMed.
- Z. Yao, R. Yaeger, V. S. Rodrik-Outmezguine, A. Tao, N. M. Torres, M. T. Chang, M. Drosten, H. Zhao, F. Cecchi and T. Hembrough, Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS, Nature, 2017, 548, 234–238 CrossRef CAS PubMed.
- Z. Karoulia, Y. Wu, T. A. Ahmed, Q. Xin, J. Bollard, C. Krepler, X. Wu, C. Zhang, G. Bollag and M. Herlyn, An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling, Cancer Cell, 2016, 30, 485–498 CrossRef CAS PubMed.
- Z. Yao, Y. Gao, W. Su, R. Yaeger, J. Tao, N. Na, Y. Zhang, C. Zhang, A. Rymar and A. Tao, RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling, Nat. Med., 2019, 25, 284–291 CrossRef CAS PubMed.
- S.-H. Choi, I. Shin, N. Kim, Y. Nam and T. Sim, The first small molecules capable of strongly suppressing proliferation of cancer cells harboring BRAF class I/II/III mutations, Biochem. Biophys. Res. Commun., 2020, 532, 315–320 CrossRef CAS PubMed.
- N. Kim, I. Shin, J. Lee, E. Jeon, Y. Kim, S. Ryu, E. Ju, W. Cho and T. Sim, Novel and potent small molecules against melanoma harboring BRAF class I/II/III mutants for overcoming drug resistance, Int. J. Mol. Sci., 2021, 22, 3783 CrossRef CAS PubMed.
- E. Cocco, M. Scaltriti and A. Drilon, NTRK fusion-positive cancers and TRK inhibitor therapy, Nat. Rev. Clin. Oncol., 2018, 15, 731–747 CrossRef CAS PubMed.
- J. F. Hechtman, NTRK insights: best practices for pathologists, Mod. Pathol., 2022, 35, 298–305 CrossRef PubMed.
- R. C. Doebele, A. Drilon, L. Paz-Ares, S. Siena, A. T. Shaw, A. F. Farago, C. M. Blakely, T. Seto, B. C. Cho and D. Tosi, Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials, Lancet Oncol., 2020, 21, 271–282 CrossRef CAS PubMed.
- J. Du, X. Lv, Z. Zhang, Z. Huang and E. Zhang, Revisiting targeted therapy and immunotherapy for advanced cholangiocarcinoma, Front. Immunol., 2023, 14, 1142690 CrossRef CAS PubMed.
- A. Demols, L. Rocq, M. Charry, N. De Nève, A. Verrellen, A. Ramadhan, C. Van Campenhout, S. De Clercq, I. Salmon and N. D'Haene, NTRK gene fusions in biliary tract cancers, Ann. Oncol., 2020, 31, S233 CrossRef.
- L. Marcus, M. Donoghue, S. Aungst, C. E. Myers, W. S. Helms, G. Shen, H. Zhao, O. Stephens, P. Keegan and R. Pazdur, FDA approval summary: entrectinib for the treatment of NTRK gene fusion solid tumors, Clin. Cancer Res., 2021, 27, 928–932 CrossRef CAS PubMed.
- Food and Drug Administration, FDA approves entrectinib for NTRK solid tumors and ROS-1 NSCLC, 2019, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc Search PubMed.
- M. Menichincheri, E. Ardini, P. Magnaghi, N. Avanzi, P. Banfi, R. Bossi, L. Buffa, G. Canevari, L. Ceriani and M. Colombo, Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros oncogene 1 kinase (ROS1), and pan-tropomyosin receptor kinases (Pan-TRKs) inhibitor, J. Med. Chem., 2016, 59, 3392–3408 CrossRef CAS PubMed.
- L. Paz-Ares, F. Barlesi, S. Siena, M.-J. Ahn, A. Drilon, A. Conley, C. Rolfo, J. Wolf, T. Seto and R. Doebele, Patient-reported outcomes from STARTRK-2: A global phase II basket study of entrectinib for ROS1 fusion-positive non-small-cell lung cancer and NTRK fusion-positive solid tumours, ESMO Open, 2021, 6, 100113 Search PubMed.
- A. Drilon, T. W. Laetsch, S. Kummar, S. G. DuBois, U. N. Lassen, G. D. Demetri, M. Nathenson, R. C. Doebele, A. F. Farago and A. S. Pappo, Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children, N. Engl. J. Med., 2018, 378, 731–739 CrossRef CAS PubMed.
- D. S. Hong, S. G. DuBois, S. Kummar, A. F. Farago, C. M. Albert, K. S. Rohrberg, C. M. van Tilburg, R. Nagasubramanian, J. D. Berlin and N. Federman, Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials, Lancet Oncol., 2020, 21, 531–540 Search PubMed.
- G. Harada, N. J. Choudhury, A. M. Schram, E. Rosen, Y. R. Murciano-Goroff, C. J. Falcon, C. Wilhelm, L. A. Kaplanis, D. Liu and J. C. Chang, Mechanisms of acquired resistance to TRK inhibitors, J. Clin. Oncol., 2022, 40, 3104 Search PubMed.
- E. Cocco, A. M. Schram, A. Kulick, S. Misale, H. H. Won, R. Yaeger, P. Razavi, R. Ptashkin, J. F. Hechtman and E. Toska, Resistance to TRK inhibition mediated by convergent MAPK pathway activation, Nat. Med., 2019, 25, 1422–1427 CrossRef CAS PubMed.
- A. Drilon, R. Nagasubramanian, J. F. Blake, N. Ku, B. B. Tuch, K. Ebata, S. Smith, V. Lauriault, G. R. Kolakowski and B. J. Brandhuber, A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion–positive solid tumors, Cancer Discovery, 2017, 7, 963–972 CrossRef CAS PubMed.
- D. Hyman, S. Kummar, A. Farago, B. Geoerger, M. Mau-Sorensen, M. Taylor, E. Garralda, R. Nagasubramanian, M. Natheson and L. Song, Abstract CT127: Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi), Cancer Res., 2019, 79, CT127–CT127 CrossRef.
- P. van der Geer, T. Hunter and R. A. Lindberg, Receptor protein-tyrosine kinases and their signal transduction pathways, Annu. Rev. Cell Biol., 1994, 10, 251–337 CrossRef CAS PubMed.
- S. Grant, L. Qiao and P. Dent, Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival, Front. Biosci., 2002, 7, d376–d389 CrossRef CAS PubMed.
- L. Geng, Z. Wang, X. Yang, D. Li, W. Lian, Z. Xiang, W. Wang, X. Bu, W. Lai and Z. Hu, et al., Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors, Theranostics, 2015, 5, 1154–1165, DOI:10.7150/thno.12398.
- N. Iqbal and N. Iqbal, Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications, Mol. Biol. Int., 2014, 852748, DOI:10.1155/2014/852748.
- R. Nahta, L. X. Yuan, B. Zhang, R. Kobayashi and F. J. Esteva, Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells, Cancer Res., 2005, 65, 11118–11128, DOI:10.1158/0008-5472.CAN-04-3841.
- C. M. Connell and G. J. Doherty, Activating HER2 mutations as emerging targets in multiple solid cancers, ESMO Open, 2017, 2, e000279, DOI:10.1136/esmoopen-2017-000279.
- W. Treekitkarnmongkol and T. Suthiphongchai, High expression of ErbB2 contributes to cholangiocarcinoma cell invasion and proliferation through AKT/p70S6K, World J. Gastroenterol., 2010, 16, 4047–4054, DOI:10.3748/wjg.v16.i32.4047.
- N. H. Chow, S. M. Huang, S. H. Chan, L. R. Mo, M. H. Hwang and W. C. Su, Significance of c-erbB-2 expression in normal and neoplastic epithelium of biliary tract, Anticancer Res., 1995, 15, 1055–1059 CAS.
- Y. Ito, T. Takeda, Y. Sasaki, M. Sakon, T. Yamada, S. Ishiguro, S. Imaoka, M. Tsujimoto, S. Higashiyama and M. Monden, et al., Expression and clinical significance of the erbB family in intrahepatic cholangiocellular carcinoma, Pathol., Res. Pract., 2001, 197, 95–100, DOI:10.1078/0344-0338-00016.
- W. Jin, ErBb Family Proteins in Cholangiocarcinoma and Clinical Implications, J. Clin. Med., 2020, 9(7), 2255, DOI:10.3390/jcm9072255.
- A. Ata, A. Polat, E. Serinsoz, M. A. Sungur and A. Arican, Prognostic value of increased HER2 expression in cancers of pancreas and biliary tree, Pathol. Oncol. Res., 2015, 21, 831–838, DOI:10.1007/s12253-014-9847-x.
- Z. Zhang, R. A. Oyesanya, D. J. Campbell, J. A. Almenara, J. L. Dewitt and A. E. Sirica, Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy, Hepatology, 2010, 52, 975–986, DOI:10.1002/hep.23773.
- N. E. Weisser, G. Wickman, L. Abraham, J. O'Toole, B. Harbourne, J. Guedia, C. W. Cheng, P. Chan, D. Browman and M. R. Gold, The bispecific antibody zanidatamab's (ZW25's) unique mechanisms of action and durable anti-tumor activity in HER2-expressing cancers, Cancer Res., 2021, 81, 1005–1005 CrossRef.
- F. Meric-Bernstam, D. L. Hanna, A. B. El-Khoueiry, Y.-K. Kang, D.-Y. Oh, J. M. Chaves, S. Y. Rha, E. P. Hamilton, S. Pant and M. M. Javle, Zanidatamab (ZW25) in HER2-positive biliary tract cancers (BTCs): Results from a phase I study, 2021, NCT02892123, https://clinicaltrials.gov/study/NCT02892123 Search PubMed.
- J. J. Harding, J. Fan, D. Y. Oh, H. J. Choi, J. W. Kim, H. M. Chang, L. Bao, H. C. Sun, T. Macarulla and F. Xie, et al., Zanidatamab for HER2-amplified, unresectable, locally advanced or metastatic biliary tract cancer (HERIZON-BTC-01): a multicentre, single-arm, phase 2b study, Lancet Oncol., 2023, 24, 772–782, DOI:10.1016/S1470-2045(23)00242-5.
- A. C. Pinto, F. Ades, E. de Azambuja and M. Piccart-Gebhart, Trastuzumab for patients with HER2 positive breast cancer: delivery, duration and combination therapies, Breast, 2013, 22(Suppl 2), S152–S155, DOI:10.1016/j.breast.2013.07.029.
- C. K. Lee, H. J. Chon, J. Cheon, M. A. Lee, H. S. Im, J. S. Jang, M. H. Kim, S. Park, B. Kang and M. Hong, et al., Trastuzumab plus FOLFOX for HER2-positive biliary tract cancer refractory to gemcitabine and cisplatin: a multi-institutional phase 2 trial of the Korean Cancer Study Group (KCSG-HB19-14), Lancet Gastroenterol. Hepatol., 2023, 8, 56–65, DOI:10.1016/S2468-1253(22)00335-1.
- M. Javle, M. J. Borad, N. S. Azad, R. Kurzrock, G. K. Abou-Alfa, B. George, J. Hainsworth, F. Meric-Bernstam, C. Swanton and C. J. Sweeney, et al., Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): a multicentre, open-label, phase 2a, multiple basket study, Lancet Gastroenterol. Hepatol., 2023, 22, 1290–1300, DOI:10.1016/S1470-2045(21)00336-3.
- J. Wang, Y. Liu, Q. Zhang, J. Feng, J. Fang, X. Chen, Y. Han, Q. Li, P. Zhang and P. Yuan, RC48-ADC, a HER2-targeting antibody-drug conjugate, in patients with HER2-positive and HER2-low expressing advanced or metastatic breast cancer: a pooled analysis of two studies, J. Clin. Oncol., 2021, 39, 1022 Search PubMed.
- J. Li, Y. Guo, J. Xue, W. Peng, X. Ge, W. Zhao, C. Dai, L. Xue, W. Tang and C. Hu, First-in-human phase I study of anti-HER2 ADC MRG002 in patients with relapsed/refractory solid tumors, J. Clin. Oncol., 2020, 38, TPS1101 Search PubMed.
- J. J. Harding, S. Piha-Paul, R. H. Shah, J. M. Cleary, D. Quinn, I. Braña, V. Moreno, M. Borad, S. Loi and I. Spanggaard, Targeting HER2 mutation-positive advanced biliary tract cancers with neratinib: Final results from the phase 2 SUMMIT ‘basket'trial, J. Clin. Oncol., 2022, 40, 4079 CrossRef.
- Z. Bai, Z. Guo, J. Liu, Y.-A. Chen, Q. Lu, P. Zhang, L. Hong, Y. Wang and J. Dong, Lapatinib suppresses HER2-overexpressed cholangiocarcinoma and overcomes ABCB1–mediated gemcitabine chemoresistance, Front. Oncol., 2022, 12, 860339 CrossRef CAS PubMed.
- V. Velcheti and K. Schalper, Basic Overview of Current Immunotherapy Approaches in Cancer, Am. Soc. Clin. Oncol. Educ. Book, 2016, 35, 298–308, DOI:10.1200/EDBK_156572.
- H. Nakamura, Y. Arai, Y. Totoki, T. Shirota, A. Elzawahry, M. Kato, N. Hama, F. Hosoda, T. Urushidate and S. Ohashi, et al., Genomic spectra of biliary tract cancer, Nat. Genet., 2015, 47, 1003–1010, DOI:10.1038/ng.3375.
- M. Gutierrez-Larranaga, E. Gonzalez-Lopez, A. Roa-Bautista, P. M. Rodrigues, A. Diaz-Gonzalez, J. M. Banales, M. Lopez-Hoyos, A. Santos-Laso and J. Crespo, Immune Checkpoint Inhibitors: The Emerging Cornerstone in Cholangiocarcinoma Therapy?, Liver Cancer, 2021, 10, 545–560, DOI:10.1159/000518104.
- Y. Ye, L. Zhou, X. Xie, G. Jiang, H. Xie and S. Zheng, Interaction of B7-H1 on intrahepatic cholangiocarcinoma cells with PD-1 on tumor-infiltrating T cells as a mechanism of immune evasion, J. Surg. Oncol., 2009, 100, 500–504, DOI:10.1002/jso.21376.
- B. A. Weinberg, J. Xiu, M. R. Lindberg, A. F. Shields, J. J. Hwang, K. Poorman, M. E. Salem, M. J. Pishvaian, R. F. Holcombe and J. L. Marshall, et al., Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets, J. Gastrointest. Oncol., 2019, 10, 652–662, DOI:10.21037/jgo.2018.08.18.
- A. Rizzo, A. D. Ricci and G. Brandi, Recent advances of immunotherapy for biliary tract cancer, Expert Rev. Gastroenterol. Hepatol., 2021, 15, 527–536, DOI:10.1080/17474124.2021.1853527.
- S. Ahn, J. C. Lee, D. W. Shin, J. Kim and J. H. Hwang, High PD-L1 expression is associated with therapeutic response to pembrolizumab in patients with advanced biliary tract cancer, Sci. Rep., 2020, 10, 12348, DOI:10.1038/s41598-020-69366-4.
- R. K. Kelley, M. Ueno, C. Yoo, R. S. Finn, J. Furuse, Z. Ren, T. Yau, H. J. Klumpen, S. L. Chan and M. Ozaka, et al., Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial, Lancet, 2023, 401, 1853–1865, DOI:10.1016/S0140-6736(23)00727-4.
- V. Sahai, K. A. Griffith, M. S. Beg, W. L. Shaib, D. Mahalingam, D. B. Zhen, D. A. Deming and M. M. Zalupski, A randomized phase 2 trial of nivolumab, gemcitabine, and cisplatin or nivolumab and ipilimumab in previously untreated advanced biliary cancer: BilT-01, Cancer, 2022, 128, 3523–3530, DOI:10.1002/cncr.34394.
- M. Yarchoan, L. Cope, A. N. Ruggieri, R. A. Anders, A. M. Noonan, L. W. Goff, L. Goyal, J. Lacy, D. Li and A. K. Patel, et al., Multicenter randomized phase II trial of atezolizumab with or without cobimetinib in biliary tract cancers, J. Clin. Invest., 2021, 131(24), e152670, DOI:10.1172/JCI152670.
- G. Zhou, D. Sprengers, S. Mancham, R. Erkens, P. P. C. Boor, A. A. van Beek, M. Doukas, L. Noordam, L. Campos Carrascosa and V. de Ruiter, et al., Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules, J. Hepatol., 2019, 71, 753–762, DOI:10.1016/j.jhep.2019.05.026.
- X. J. Guo, J. C. Lu, H. Y. Zeng, R. Zhou, Q. M. Sun, G. H. Yang, Y. Z. Pei, X. L. Meng, Y. H. Shen and P. F. Zhang, et al., CTLA-4 Synergizes With PD1/PD-L1 in the Inhibitory Tumor Microenvironment of Intrahepatic Cholangiocarcinoma, Front. Immunol., 2021, 12, 705378, DOI:10.3389/fimmu.2021.705378.
- M. Luo and L. Fu, The effect of chemotherapy on programmed cell death 1/programmed cell death 1 ligand axis: some chemotherapeutical drugs may finally work through immune response, Onco Targets Ther, 2016, 7, 29794–29803, DOI:10.18632/oncotarget.7631.
- T. Bekaii-Saab, M. A. Phelps, X. Li, M. Saji, L. Goff, J. S. Kauh, B. H. O'Neil, S. Balsom, C. Balint and R. Liersemann, et al., Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers, J. Clin. Oncol., 2011, 29, 2357–2363, DOI:10.1200/JCO.2010.33.9473.
- R. T. Shroff, M. Yarchoan, A. O'Connor, D. Gallagher, M. L. Zahurak, G. Rosner, C. Ohaji, S. Sartorius-Mergenthaler, R. Parkinson and V. Subbiah, et al., The oral VEGF receptor tyrosine kinase inhibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma, Br. J. Cancer, 2017, 116, 1402–1407, DOI:10.1038/bjc.2017.119.
- M. A. Lowery, M. Bradley, J. F. Chou, M. Capanu, S. Gerst, J. J. Harding, I. E. Dika, M. Berger, A. Zehir and R. Ptashkin, et al., Binimetinib plus Gemcitabine and Cisplatin Phase I/II Trial in Patients with Advanced Biliary Cancers, Clin. Cancer Res., 2019, 25, 937–945, DOI:10.1158/1078-0432.CCR-18-1927.
- S. Dushyanthen, Z. L. Teo, F. Caramia, P. Savas, C. P. Mintoff, B. Virassamy, M. A. Henderson, S. J. Luen, M. Mansour and M. H. Kershaw, et al., Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer, Nat. Commun., 2017, 8, 606, DOI:10.1038/s41467-017-00728-9.
- P. J. R. Ebert, J. Cheung, Y. Yang, E. McNamara, R. Hong, M. Moskalenko, S. E. Gould, H. Maecker, B. A. Irving and J. M. Kim, et al., MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade, Immunity, 2016, 44, 609–621, DOI:10.1016/j.immuni.2016.01.024.
- T. Bekaii-Saab, J. Bridgewater and N. Normanno, Practical considerations in screening for genetic alterations in cholangiocarcinoma, Ann. Oncol., 2021, 32, 1111–1126 CrossRef CAS PubMed.
- X. Song, X. Liu, H. Wang, J. Wang, Y. Qiao, A. Cigliano, K. Utpatel, S. Ribback, M. G. Pilo and M. Serra, Combined CDK4/6 and pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma, Clin. Cancer Res., 2019, 25, 403–413 CrossRef CAS PubMed.
- W. Fan, C. Wang, X. Zhong, Y. Zheng, T. Chen, M. Huang and S. Su, A refractory case of CDKN2A/B loss metastatic intrahepatic cholangiocarcinoma achieving a partial response after first-line treatment with palbociclib, OncoTargets Ther., 2023, 23–29 CrossRef PubMed.
- L. Huang, Z. Guo, F. Wang and L. Fu, KRAS mutation: from undruggable to druggable in cancer, Signal Transduction Targeted Ther., 2021, 6, 386 CrossRef CAS PubMed.
- M. Tanaka, A. Kunita, M. Yamagishi, H. Katoh, S. Ishikawa, H. Yamamoto, J. Abe, J. Arita, K. Hasegawa and T. Shibata, KRAS mutation in intrahepatic cholangiocarcinoma: Linkage with metastasis-free survival and reduced E-cadherin expression, Liver Int., 2022, 42, 2329–2340 CrossRef CAS PubMed.
- S.-L. Zhou, H.-Y. Xin, R.-Q. Sun, Z.-J. Zhou, Z.-Q. Hu, C.-B. Luo, P.-C. Wang, J. Li, J. Fan and J. Zhou, Association of KRAS variant subtypes with survival and recurrence in patients with surgically treated intrahepatic cholangiocarcinoma, JAMA Surg., 2022, 157, 59–65 CrossRef PubMed.
- J. H. Strickler, H. Satake, T. J. George, R. Yaeger, A. Hollebecque, I. Garrido-Laguna, M. Schuler, T. F. Burns, A. L. Coveler and G. S. Falchook, Sotorasib in KRAS p. G12C–mutated advanced pancreatic cancer, N. Engl. J. Med., 2023, 388, 33–43 CrossRef CAS PubMed.
- D. S. Hong, M. G. Fakih, J. H. Strickler, J. Desai, G. A. Durm, G. I. Shapiro, G. S. Falchook, T. J. Price, A. Sacher and C. S. Denlinger, KRASG12C inhibition with sotorasib in advanced solid tumors, N. Engl. J. Med., 2020, 383, 1207–1217 CrossRef CAS PubMed.
- J. Mandal, P. Mandal, T.-L. Wang and I.-M. Shih, Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response, J. Biomed. Sci., 2022, 29, 71 CrossRef CAS PubMed.
- A. Kalyan and R. T. Shroff, Cholangiocarcinoma: The Present and Future of Targeted Therapies, touchREVIEWS in Oncology & Haematology, 2022, 18(1), 9–15 Search PubMed.
- G. Spizzo, A. Puccini, J. Xiu, R. M. Goldberg, A. Grothey, A. F. Shields, S. P. Arora, M. E. Salem, F. Battaglin and Y. Baca, Molecular profile of BRCA-mutated biliary tract cancers, ESMO Open, 2020, 5, e000682 CrossRef PubMed.
- F. Xiong, J. Gong and Q. Wang, Olaparib and pembrolizumab treatment for BRCA1-mutated and PD-L1-positive intrahepatic cholangiocarcinoma recurrence and metastasis: a case report, OncoTargets Ther., 2020, 6385–6391 CrossRef CAS PubMed.
- P. LoRusso, Olaparib in Treating Patients With Advanced Glioma, Cholangiocarcinoma, or Solid Tumors With IDH1 or IDH2 Mutations, 2023, NCT03212274, https://clinicaltrials.gov/study/NCT03212274?tab=table Search PubMed.
- E. Chen, Study of Olaparib and Durvalumab in IDH-Mutated Solid Tumors, 2024, NCT03991832, https://clinicaltrials.gov/study/NCT03991832#collaborators-and-investigators Search PubMed.
- M. Bahcall, T. Sim, C. P. Paweletz, J. D. Patel, R. S. Alden, Y. Kuang, A. G. Sacher, N. D. Kim, C. A. Lydon and M. M. Awad, Acquired MET D1228V mutation and resistance to MET inhibition in lung cancer, Cancer Discovery, 2016, 6, 1334–1341 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |