DOI:
10.1039/D4RA06426G
(Paper)
RSC Adv., 2025,
15, 8471-8479
Effect of strontium on the performance of Ni/CBV20A catalyst in partial oxidation of methane for syngas and hydrogen production†
Received
5th September 2024
, Accepted 10th February 2025
First published on 19th March 2025
Abstract
Converting methane into syngas via partial oxidation of methane (POM) is a promising energy-efficient technology given its exothermic nature. Active nickel-based catalysts suffer from deactivation by carbon deposition and sintering. This study explores the novel use of mordenite zeolite (CBV20A) as a catalytic support for nickel (Ni) and using strontium (Sr) as a promoter. Ni5Srx/CBV20A samples with various Sr loadings were prepared and characterized using N2-sorption, X-ray diffraction, H2-temperature programmed reduction, temperature programmed desorption of CO2, and Transmission Electron Microscopy. Sr addition improved NiO reducibility at lower temperature and boosted basicity, enhancing CH4 conversion and H2 yield. The optimal catalyst, Ni5Sr2/CBV20A, exhibited the highest performance with 72% CH4 conversion, 47% H2 yield, and 2.6 H2/CO ratio at 700 °C and 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 mL g−1 h−1. Results show that at a high gas hourly space velocity (GHSV) of 72000 mL g−1 h−1, a combustion and reforming reaction mechanism is preferred, while at a low GHSV of 14400 mL g−1 h−1, a direct partial oxidation mechanism predominates.
400 mL g−1 h−1. Results show that at a high gas hourly space velocity (GHSV) of 72000 mL g−1 h−1, a combustion and reforming reaction mechanism is preferred, while at a low GHSV of 14400 mL g−1 h−1, a direct partial oxidation mechanism predominates.
Introduction
Nowadays, the production of hydrogen (H2) and syngas, a mixture composed mainly of hydrogen and carbon monoxide (CO), is primordial to meet the increasing demand of the chemical industry and the growing environmental awareness. In this regard, hydrogen and syngas represent valuable feedstocks for the production of ammonia,1 liquid hydrocarbons and chemicals such as methanol and ethanol, among other high value products.2 While most of the hydrogen and syngas are currently produced from fossil fuels,3 the depletion of these reserves calls for the urgent need for alternative production methods.
Methane (CH4) is the major constituent of natural gas and shale gas reserves.4 It is also a major product from the anaerobic digestion of biomass and municipal solid waste, disposed of worldwide in large quantities.5 Furthermore, methane is the second most potent greenhouse gas, which is also emitted from anthropogenic activities worldwide. The abundance and availability of methane makes it an attractive feedstock for fuels and chemical synthesis. In this regard, the conversion of methane into valuable syngas is highly beneficial. Hydrogen and syngas can be produced from methane using steam reforming of methane (SRM), dry reforming of methane (DRM), or partial oxidation of methane (POM).6,7 The first two processes (SRM and DRM) are highly endothermic and require high energy demand. The high temperatures needed for these reactions cause sintering and heavy carbon deposition, leading to catalyst deactivation.7 POM is an exothermic reaction that does not require any heat supply; hence it is less energy intensive. In addition, it generates H2-rich syngas (H2/CO ratio of 2), ideal to produce methanol and synthetic fuels through Fischer–Tropsch Synthesis (FTS).8 This reaction is characterized by high methane conversion, high selectivity to syngas,9 and a rapid reaction rate. It also requires smaller reactor, compared to conventional reforming.10
In such applications, nickel is an interesting active metal because of its availability and more economical cost compared to noble metals. Indeed, Ni-based catalysts are highly active in POM;11,12 however, they are susceptible to deactivation by carbon deposition and metal sintering at high temperatures.13 In this regard, studies have shown that the support plays a major role in the stability of Ni-based catalysts in POM.14,15 For example, the strong interaction between Ni and La2O3 support resulted in high catalytic activity, reaching around 80% CH4 conversion with more than 90% H2 selectivity at 800 °C and atmospheric pressure.14 In contrast, the weak Ni–ZrO2 interaction led to catalyst deactivation because of Ni particles sintering.14 Moreover, high syngas yield was achieved in POM using fibrous Ni/Al2O3 catalysts, owing to better Ni dispersion, higher metal–support interaction and improved mass transfer, compared to monolithic spherical catalysts.15 In the same context, the confinement of Ni nanoparticles inside well-structured porous supports such as MCM-4110 or SBA-1616 can enhance the metal–support interaction and improve the durability of the catalyst by avoiding sintering and carbon deposition. Among porous supports, zeolites are interesting candidates for being available, cheap, and characterized with well-defined structure, high surface area, high thermal stability, and tunable active sites. Hence, they are expected to improve the stability and durability of Ni-based catalysts.17–19 The use of mordenite as a support for Ni-based catalysts resulted in high ethanol conversion and high hydrogen selectivity in steam reforming of bioethanol.20 In partial oxidation of butane, the use of mordenite as a support for Ni-based catalysts showed better activity than ZSM-5 where butane conversion and hydrogen selectivity reached 80% and 56%, respectively on Ni/mordenite compared to only 73% and 48%, respectively on Ni/ZSM-5 after 5 hours on stream at 700 °C.21 Mordenite-type zeolites were found to be characterized with large pore size which can promote product desorption in direct partial oxidation of methane to methanol.22 Rhenium supported on mordenite showed better performance than other types of supports such as Al2O3, SiO2 and MgO in partial oxidation of methane to syngas, due to the easier reduction of Re species on this support and to the stabilization of the resulting low-valent Re species.23 Furthermore, mordenite CBV20A proved to be efficient for applications including benzene conversion to benzyl alcohol,24 benzene alkylation with propylene,25 and as support for gold nanoparticles in CO oxidation.26 Yet, its use in partial oxidation of methane for syngas production remains limited. Only one paper17 evaluated the use of mordenite CBV20A as a support for Rh-based catalysts for POM, and the resulting Rh/MOR-IE sample maintained around 84% methane conversion and 91% CO selectivity for 50 h at 600 °C and 1.2 × 106 mL g−1 h−1.
In addition to the importance of selecting proper support, the choice of promoter is a vital factor in achieving high catalytic performance.9,27,28 Among the options available, alkaline earth elements are gaining attention given their abundancy and low cost. These group 2 elements include beryllium (Be), magnesium (Mg), calcium (Ca), strontium (Sr), barium (Ba), and radium (Ra) which is a radioactive element. Strontium has the second largest atomic radius after barium and is thus characterized by low charge/size ratio or charge density. The large size of Sr stabilized CO2 as bidentate species.29 Sr2+-mediated CO2 was reported to be a good oxidizing agent.30 In indirect pathways of POM, greater interaction of CO2 with the catalyst surface facilitated the oxidation of CH4 by CO2 towards the formation of syngas. Sr addition over Ni/La2O3 catalyst was found to induce the generation of high amount of lattice oxygen surface species which promoted C–H activation.29 The promotional addition of Sr over Ni-based catalyst supported over titania–zirconia,31 zirconia–alumina,32 or tungsten–zirconia30 were found to induce enhanced reducibility. The metallic Ni formed after reduction of NiO is the active center of C–H dissociation. Overall, the use of Sr seems to be advantageous in the mean of C–H activation/dissociation as well as CO2 stabilization/activation during partial oxidation of methane. Furthermore, strontium was found to be effective in enhancing metal dispersion,27,33 increasing metal–support interaction,28,34 improving catalyst basicity,35 and boosting catalytic activity and stability by limiting carbon deposition36,37 in several applications such as dry reforming of methane,27,34,37,38 steam reforming of methane,39 hydrogenation,33 cracking,40 and CO2 methanation.28,35,41,42 In partial oxidation of methane, the use of up to 1% Sr in Co/Al2O3 enhanced the initial activity but the catalyst quickly deactivated afterwards.36 An amount of 2 wt% Sr was needed to maintain the stability of the catalyst and achieve 82% CH4 conversion, 89% H2 selectivity, and a H2/CO ratio close to 2. Moreover, the addition of 1 wt% Sr to Ni supported on SAPO-5 boosted methane conversion and hydrogen yield to 47 and 42%, respectively, in comparison to 40 and 30%, respectively, on the non-promoted sample at 600 °C.9 Similarly, the 2 wt% Sr-promoted Ni/TiZr sample reached 46% CH4 conversion and 42% H2 yield and decreased the H2/CO molar ratio from 4.25 on Ni/TiZr to 3.75.31 These two recent studies show the promotional effect of Sr on Ni-based catalysts in POM for syngas generation, in comparison to other types of promoters. Yet, in the two studies, the Sr loading was fixed at either 1 or 2 wt%. From this perspective, it would be interesting to further increase Sr loading beyond these values, in an attempt to achieve even higher performance in POM.
In this work, we try to combine the beneficial effects of mordenite CBV20A support for improved catalytic stability and durability with the advantages of Sr promoter in terms of enhanced methane conversion and hydrogen yield. To date, the use of Sr as a promoter for Ni catalysts supported on mordenite zeolite support for POM was never addressed before. For this purpose, Ni5Srx/CBV20A samples are prepared with 1, 2 and 3 wt% Sr loading and evaluated in partial oxidation of methane at 700 °C. The properties of the fresh and used catalysts are evaluated using N2 physisorption, TPR, XRD, CO2-TPD, FTIR, TGA, Raman, TEM, and NH3-TPD, to determine the optimum catalyst.
Experimental
Materials
Ni(NO3)2·6H2O (Purity 98%, Alfa Aesar) and Sr(NO3)2 (Aldrich) and commercial MOR (CBV20A, Zeolyst) were used in this work.
Catalyst preparation
The catalysts were prepared with 5 wt% Ni and varying amounts of Sr (1, 2, and 3 wt%) supported on CBV20A zeolite using the impregnation method. The appropriate amount of nickel nitrate (Ni(NO3)2·6H2O) and strontium nitrate (Sr(NO3)2) were dissolved in 20 mL of distilled water. The CBV20A zeolite support was gradually added to each solution while stirring continuously at 80 °C for sufficient time to allow complete impregnation. After impregnation, the samples were dried at 120 °C overnight for 12 hours, and then calcined at 600 °C for 4 hours. The resulting samples with 5 wt% Ni and 1, 2, or 3 wt% Sr supported on CBV20A zeolite were labelled Ni5Srx/CBV20A (x = 1, 2, 3).
Catalyst characterization
The X-ray diffraction (XRD) analysis of fresh samples was conducted by means of a Bruker D8-Discover diffractometer using Cu Kα1 radiation (λ = 0.15406 nm) operated at 40 mA and 40 kV. The N2 adsorption–desorption isotherms were obtained on a Micromeritics Tristar II 3020 surface area analyzer using 0.2–0.3 g of sample degassed and analyzed using the Barrett, Joyner & Halenda (BJH) method. Temperature-programmed hydrogen reduction (H2-TPR) and temperature-programmed carbon dioxide desorption (CO2-TPD) measurements were performed on a Micromeritics AutoChem II chemisorption device using a thermal conductivity detector over 70 mg sample. In H2-TPR, H2 absorption is monitored up to 1000 °C under 10% H2/He gas, whereas in CO2-TPD, CO2 desorption is monitored upon raising the temperature to 800 °C under 10% CO2/He gas. The Transmission Electron Microscopy (TEM) was conducted at 200 kV using an aberration-corrected JEM-ARM200F (JEOL) with a CEOS corrector. The particle size distribution was estimated by statistical counting of at least 175 spherical particles using the software “Comptage des Particules, Laboratoire de Réactivité de Surface, France”. The spent catalysts underwent Raman analysis within the 1250–3000 cm−1 range using a Laser Raman Spectrometer (JASCO, Japan) with a 532 nm beam excitation and 1.6 mW laser intensity. The exposure time was set to 10 seconds with 3 accumulations. The spectra were processed using Spectra Manager Ver.2 software (JASCO, Japan). The carbon deposition on the spent catalysts is measured using thermogravimetric analysis (TGA-51 by Shimadzu, Kyoto, Japan). In this analysis, 10–20 mg of the spent catalyst is heated in the presence of nitrogen at a rate of 20 °C min−1. The FTIR measurements were collected using a Nicolet Is-10 model (USA) Infrared spectrophotometer adopting the KBr technique. The samples were measured as KBr disks by mixing the sample with KBr (spectroscopic grade), where the solid samples were transferred to the cell after melting using an infrared lamp. The spectra of all the studied samples were measured under ambient conditions between 400 and 4000 cm−1 with a suitable scan resolution of 4 cm−1 and a scan rate of 16 cm min−1.
Catalyst activity test
0.1 g of catalyst sample was packed in a tubular stainless steel fixed-bed reactor (PID Eng & Tech, 9 mm I. D.) equipped with a K-type thermocouple fitted axially at the center of the catalyst bed for temperature control. Before the reaction, each catalyst was reduced by flowing hydrogen at a 30 mL min−1 flow rate for 1 h at 800 °C. Afterward, the reactor was purged with N2 to remove the hydrogen gas from the system. Then the temperature was stabilized at 700 °C. The packed catalyst was exposed to a mixture of CH4 and O2 gases in a 2:1 ratio, with a total feed rate of CH4, O2, and N2 set to 24 mL min−1 and the resulting space velocity held at 14400 mL g−1 h−1. An additional test was also performed at 72000 mL g−1 h−1. The product stream is analyzed by a gas chromatograph equipped with a Porapak Q column, molecular sieve columns, and a thermal conductivity detector. The composition of effluent gases was calculated by the normalization method, and the equations for the determination of CH4 conversion, H2 yield, and H2/CO ratio used are as follows:| |
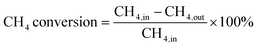 | (1) |
| |
 | (2) |
| |
 | (3) |
| |
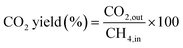 | (4) |
| |
 | (5) |
For comparison, thermodynamic equilibrium data were calculated using the HSC 10 Chemistry software, by selecting CH4, O2, and N2 as gaseous input at a molar ratio of CH4:O2:N2 = 2:1:1, identical to the reaction conditions. The resulting products included CH4, O2, H2, CO, CO2, N2, and H2O, in gaseous phase. The equilibrium composition was obtained at 700 °C and 1 atm, with and without considering solid carbon deposition C(s) in the exiting stream.
Results and discussion
The N2 sorption isotherms of all calcined samples (Fig. 1) exhibit type IV isotherm, characteristic of mesoporous materials.43 The presence of H4 hysteresis loop is typical of mesoporous zeolites.43 Upon addition of Sr, the BET surface area decreases from 373 m2 g−1 for Ni5/CBV20A to 331 and 333 m2 g−1 for Ni5Sr1/CBV20A and Ni5Sr2/CBV20A, respectively, and further decreases to 315 m2 g−1 when 3 wt% Sr is added to Ni5/CBV20A (Table 1). Similarly, the pore volume slightly decreases from 0.14 cm3 g−1 for Ni5/CBV20A to 0.12 and 0.13 cm3 g−1 for Ni5Sr1/CBV20A and Ni5Sr2/CBV20A, respectively. This is also accompanied by a negligible reduction in the pore size from 8.0 nm for Ni5/CBV20A to 7.8 and 7.9 nm when 1 and 2 wt% Sr are added to Ni5/CBV20A, respectively. The slight decrease in pore volume and pore size upon Sr addition might be indicative of the presence of Ni and Sr species outside the zeolitic framework. Compared to other types of zeolites, the samples supported on mordenite CBV20A presents lower pore size (Table 1) than those supported on mordenite CBV10A (15.7 nm) and ZSM-11 or CP810B (13.7 nm),44 yet higher than comparable Ni-based samples supported on ZSM-8 (CBV3024E) having a pore size of 5.55 nm44 or on ZSM-5 with a pore size of 3.4 nm.45
 |
| | Fig. 1 (a) N2 sorption isotherms and (b) adsorption and (c) desorption porosity distribution for fresh Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples. | |
Table 1 Textural properties, total hydrogen consumption and desorbed CO2 amounts of fresh Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples
| Sample |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
H2 consumption (cm3 g−1) |
CO2 desorbed (cm3 g−1) |
| Ni5/CBV20A |
373 |
0.14 |
8.0 |
14.97 |
5.58 |
| Ni5Sr1/CBV20A |
331 |
0.12 |
7.8 |
17.75 |
3.91 |
| Ni5Sr2/CBV20A |
333 |
0.13 |
7.9 |
20.07 |
4.26 |
| Ni5Sr3/CBV20A |
315 |
0.12 |
8.1 |
18.01 |
4.89 |
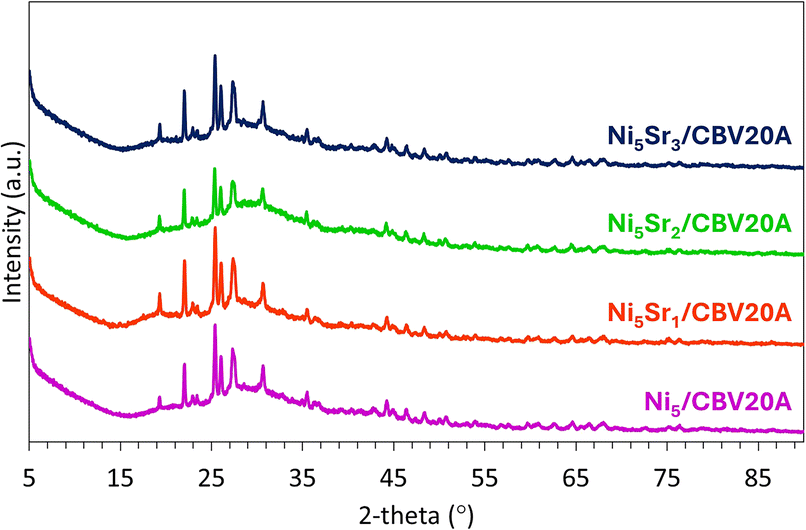
The XRD patterns of fresh Ni5/CBV20A and Ni5Srx/CBV20A samples (Fig. 2) show characteristic peaks of the mordenite structure, principally between 20 and 35°.46 In detail, the peaks observed at 19.6, 22.2, 23.1, 25.6, 26.3, 27.7, 30.8, and 35.6°, indexed as (330), (150), (241), (202), (350), (511), (332), and (442), respectively, are typical of mordenite (JCPDS 00-006-0239) with an orthorhombic crystal system and a space group of Cmc21. The representative peaks of NiO at 37.2, 43.2, and 62.7°47 and SrO2 at 26.9, 28.4, 35.6, 45.2, 48.5, and 51.2°, indexed as (002), (101), (110), (112), (103), and (200), respectively, are barely distinguishable from the support. Interestingly, this indicates the formation of very small NiO and SrO2 nanoparticles highly dispersed inside the mordenite zeolitic structure.
 |
| | Fig. 2 XRD patterns of fresh Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples. | |
During H2-TPR, NiO and SrO2 nanoparticles are reduced into metallic Ni and SrO, respectively. The reduction takes place in two major steps (Fig. 3), the first between 300 and 370 °C, attributed to the reduction of free NiO species48 or NiO in weak interaction with the support,47 and the second between 370 and 600 °C, for NiO species in strong interaction with the support.47 The addition of Sr causes an increase in the area of the first peak, implying an easier reduction of NiO into metallic Ni at lower temperature. This suggests lower metal–support interaction and improved NiO accessibility and reducibility upon the addition of Sr, unlike previous reports showing a higher metal–support interaction achieved upon Sr addition to Ni/Al2O3.28,34 Indeed, the reduction of Sr2+ to metallic Sr is not easy because it is a highly electropositive element (doubly charged positive ion). Yet, Sr2+ has a larger ionic radius than Ni2+, which may cause lattice distortions and introduces oxygen vacancies or defects. Such oxygen vacancies can facilitate the reduction of Ni2+ to Ni0 by increasing the mobility of surface oxygen species, allowing their reduction at lower temperature, as reported on CeO249 and Y2O350 supports and on Rh/Ce-SBA-15 catalysts.51 Such behavior has been also described on Sr-doped Ni-La2O3 catalysts.29 A similar enhancement of reducibility at lower temperature was observed upon the addition of 2 wt% Sr to Ni/TiZr31 and up to 3 wt% Sr to Ni/WO3 + ZrO2 catalytic systems.52 It is worth noting that the addition of Sr increases the hydrogen consumption from 14.97 cm3 g−1 for the Sr-free Ni5/CBV20A sample to 17.75 and 20.07 cm3 g−1 as the Sr content increases to 1 and 2 wt%, respectively (Table 1). These values are relatively close to the theoretical values of 18.99, 21.55 and 24.10 cm3 g−1 hydrogen consumption upon the reduction of either 5 wt% Ni or 5 wt% Ni promoted with 1 or 2 wt% Sr, respectively. These numbers suggest that more than 80% reduction is achieved on these samples, under the employed experimental conditions, and further validate the higher mobility of surface oxygen species.51
 |
| | Fig. 3 H2-TPR profiles of Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples. | |
The basicity of the samples is evaluated using CO2-TPD (Fig. 4). It is commonly recognized that peaks at low temperatures, between 50 and 200 °C, are attributed to low basic sites, between 200 and 400 °C to moderate basic sites, and above 400 °C to strong basic sites.41 For all samples, the small peak observed at around 100 °C can be assigned to the presence of low strength basic sites, as also observable on Sr-promoted Ni/Al2O3 catalysts.34 On the Ni5/CBV20A sample, most of the basic sites fall between 200 and 400 °C, characteristic of moderate strength. The increase in Sr content from 1 to 3 wt% increases the amount of CO2 adsorbed from 3.91 to 4.89 cm3 g−1, respectively, implying an improvement of the surface basicity and an increase in the concentration of basic sites. In addition, as the Sr content exceeds 2 wt%, the peak at around 470 °C becomes more intense, suggesting an improvement in the strength of the basic sites. The presence of an additional peak at around 620 °C for Ni5Sr2/CBV20A and Ni5Sr3/CBV20A and an extra one at around 750 °C Ni5Sr3/CBV20A is indicative of the presence of very strong basic sites on the surfaces of these two samples. The beneficial effect of Sr in boosting the basicity of the catalyst was also reported elsewhere on Ni/Al2O3 catalysts for dry reforming of methane34 and Ni/MCM-41 for dry and steam reforming of methane.39
 |
| | Fig. 4 CO2-TPD profiles of Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples. | |
The FTIR spectra of Ni5/CBV20A and Ni5Srx/CBV20A samples in the 400–4000 cm−1 range are reported in Fig. 5. The bands observed at about 3400 cm−1 and 1640 cm−1 are attributed to the stretching and bending vibrations of OH hydroxyl groups in water, respectively.37 The bands between 450 and 1200 cm−1 confirm the presence of zeolite structures.53 In detail, the band in the range of 350–470 cm−1 corresponds to pore opening mode and the one around 550–650 cm−1 is assigned to the vibration of double five-membered rings. The bands at around 806 and 1050 cm−1 are attributed to symmetric and asymmetric stretching of Si–O–Si bridges, respectively.9,10
 |
| | Fig. 5 FTIR spectra of Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) samples. | |
The partial oxidation of methane (eqn (6)) generates H2-rich syngas, while the complete oxidation (eqn (7)) produces CO2 and H2O. The production of syngas can be enhanced through dry reforming (eqn (8)) and steam reforming (eqn (9)). Additionally, methane decomposition (eqn (10)) may occur, causing unintended carbon deposition on the catalyst surface. The hydrogenation of CO (eqn (11)) or CO2 (eqn (12)) is part of the methanation process.
| |
 | (6) |
| | |
CH4(g) + 2O2(g) = CO2(g) + 2H2O(g)
| (7) |
| | |
CO2(g) + CH4(g) = 2CO(g) + 2H2(g)
| (8) |
| | |
H2O(g) + CH4(g) = CO(g) + 3H2(g)
| (9) |
| | |
CH4(g) = C(s) + 2H2(g)
| (10) |
| | |
CO(g) + 3H2(g) = CH4(g) + H2O(g)
| (11) |
| | |
CO2(g) + 4H2(g) = CH4(g) + 2H2O(g)
| (12) |
In literature, two mechanisms have been proposed for the reaction: the Direct Partial Oxidation (DPO) or the Combustion and Reforming Reaction (CRR).11,54 In direct partial oxidation, the CH4 and O2 reactants dissociate on the active sites and the adsorbed species directly react and combine towards syngas formation. In combustion and reforming reaction, methane undergoes total combustion to CO2 and H2O (eqn (7)), and the dry and steam reforming reactions between CH4 and either CO2 (eqn (8)) or H2O (eqn (9)) generate the final syngas product.
The catalytic activity of Ni5/CBV20A and Ni5Srx/CBV20A samples was compared in partial oxidation of methane using a CH4/O2 ratio of 2 at 700 °C and a GHSV equivalent to 14400 mL g−1 h−1 (Fig. 6). At equilibrium, under uniform reaction conditions (CH4:O2:N2 = 2:1:1 at 700 °C), the CH4 conversion, H2 yield, and H2/CO molar ratio achieve 86%, 86% and 2.4, respectively without accounting for carbon deposition, and 88%, 88%, and 2.5, respectively, when considering carbon deposition. The Sr-free Ni5/CBV20A sample shows the lowest CH4 conversion of about 63% after 240 min on stream, with an H2/CO ratio of 2.62. On this sample, the H2 and CO yields reached 40 and 19%, respectively, while the CO2 yield was around 21%. All the promoted samples showed higher CH4 conversion and better H2 and CO yields. The best performance was observed on the Ni5Sr2/CBV20A sample that exhibited the highest CH4 conversion of about 72% with an H2/CO ratio of 2.60. On this sample, a maximum H2 yield of 47% was achieved with the minimum CO2 yield of 19%. These values are the closest to the thermodynamic equilibrium values calculated using the HSC Chemistry software, as indicated previously. This indicates the beneficial effect of Sr addition to Ni-based catalysts in promoting methane conversion and boosting hydrogen production during the partial oxidation of methane. On the Sr-free sample, the decrease in methane conversion and hydrogen yield with time suggests the occurrence of the methanation reactions (eqn (11) and (12)) where hydrogen is consumed to produce more methane. The addition of Sr on the promoted samples limits the occurrence of these undesirable side reactions, in favor of the main POM reaction for syngas production. At the reaction temperature of 700 °C, Al-Fatesh et al. have found that the direct POM pathway, which generates syngas in a single step, is dominant over Sr-promoted Ni-based samples.31
 |
| | Fig. 6 Variation of (a) CH4 conversion, (b) H2/CO molar ratio, (c) CO yield, (d) H2 yield, and (e) CO2 yield with time on stream for Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) catalysts at 700 °C and 1 atm (GHSV = 14400 mL g−1 h−1). | |
When the samples are tested at higher gas hourly space velocity (72000 mL g−1 h−1), the promoted catalyst maintained a higher catalytic performance compared to the unpromoted sample (Fig. 7). The Ni5Sr3/CBV20A catalyst reached 39% CH4 conversion, 37% H2 yield, and 28% CO2 yield compared to only 31%, 29%, and 22%, respectively on Ni5/CBV20A at 700 °C. This means that the promotional effect of Sr remains effective, even when the samples are tested under more severe conditions. Nevertheless, these values are much lower than those achieved at low GHSV. Chang-lin et al.36 observed similar behavior on Sr-promoted Co-based catalysts and clarified that a higher GHSV does not allow sufficient time for the reactants on the surface of the catalyst. In more detail, the CO2 resulting from the total oxidation reaction (eqn (7)) does not have enough time to react further with residual methane through the reforming reactions (eqn (8) and (9)) to generate syngas. This explains the higher CO2 yield achieved at high GHSV. The competition between steam and dry reforming of methane can also be a reason behind the high CO2 yield.31 In such cases, steam reforming would be more efficient than dry reforming, which results in high hydrogen production and high H2/CO molar ratio in the range of 3.48–4.27.31 However, since the H2/CO molar ratio is much lower in the current case (2.3–2.4), this means that the reforming reactions are not taking place, and the presence of CO2 is majorly coming from the total oxidation reaction.
 |
| | Fig. 7 Variation of (a) CH4 conversion, (b) H2/CO molar ratio, (c) H2 yield, and (d) CO2 yield with time on stream for Ni5/CBV20A and Ni5Sr3/CBV20A catalysts at 700 °C and 1 atm (GHSV = 72000 mL g−1 h−1). | |
The catalytic results obtained in this work suggest that, at low GHSV, the direct pathway of POM is preferred on the Sr-promoted sample (Fig. 10). In this case, the CH4 and O2 reactants dissociate on the nickel active sites and the adsorbed species directly react and combine towards syngas formation. The production of H2 and CO thus occurs in a single step. This can be confirmed by the H2/CO molar ratio close to 2.31 At high GHSV, the increase in CO2 yield on the Sr-promoted sample implies the occurrence of the indirect pathway, known as the Combustion and Reforming Reaction (CRR). In this case, methane undergoes total combustion to CO2 and H2O (eqn (7)), and the reforming reactions between CH4 and CO2 (eqn (8)) or H2O (eqn (9)) generate the final syngas product (Fig. 10). This indirect pathway was found to be also preferential at high GHSV on Sr-promoted Co-based catalysts.36
At low GHSV, the Ni5Sr2/CBV20A sample showed a deactivation of only 1% in CH4 conversion and 8% in H2 yield, compared to 6% and 15%, respectively on the non-promoted Ni5/CBV20A sample. Similarly, at high GHSV, the Ni5Sr3/CBV20A sample showed a deactivation of less than 7% in CH4 conversion with a stable H2 yield, while the deactivation reached 16% and 17% in CH4 conversion and H2 yield, respectively on Ni5/CBV20A. These values show a much higher catalytic stability of the Sr-promoted samples compared to the non-promoted one. Indeed, the addition of Sr to Ni5/CBV20A improved the basicity of the samples by creating strong and very strong basic sites on the surface of the catalyst. The presence of these strong basic sites favors the adsorption and dissociation of CO2, resulting from possible side reactions, thus creating more oxygen-free spaces on the surface and contributing to the elimination of carbon deposits.34,39 The lower carbon formation extends the lifetime of the catalyst and limits its deactivation over time. This explains the higher performance and better catalytic stability of Ni5Sr2/CBV20A at 14400 mL g−1 h−1 and Ni5Sr3/CBV20A at 72000 mL g−1 h−1, compared to the non-promoted sample.
While comparisons with other catalytic systems might not be evident due to either large differences in catalyst compositions or variations in the catalytic test conditions, it can be noted that Ni5Sr1/CBV20A is slightly more performing than 5Ni + 1Sr/SAPO-5 sample9 and Ni5Sr2/CBV20A is less performing than Ni/TiZr promoted with 2 wt% Sr under similar test conditions.31 In numbers, the Ni5Sr1/CBV20A sample shows around 67% CH4 conversion compared to 64% on 5Ni + 1Sr/SAPO-5 and the Ni5Sr2/CBV20A shows around 72% CH4 conversion and 47% H2 yield compared to 77% and 82%, respectively on NiSr/TiZr at 700 °C and 14400 mL g−1 h−1. This can be correlated to the pore size of CBV20A (around 7.8–7.9 nm), which is larger than that of SAPO-5 (7.0 nm) yet lower than that of TiZr support (8.7 nm). Indeed, the presence of larger pore diameters facilitates the transport of reactants in and products out from the support structure resulting in higher catalytic performance.
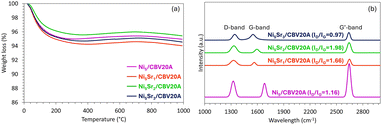
After catalytic testing, carbon deposition was first quantified by TGA (Fig. 8a). On the TGA curves, the major weight loss detected at temperatures lower than 200 °C is assigned to the removal of physisorbed water.36 On all the spent catalysts, the total weight loss does not exceed 6%, suggesting limited carbon deposits. This is in agreement with the previous discussion about the effect of Sr addition in favoring POM and limiting undesirable side reactions. It is interesting to mention that the Ni5Sr2/CBV20A sample presents the lowest weight loss of only 4.5%, indicative of its high resistance to carbon deposition. The subsequent increase in weight after 400 °C can be attributed to Ni re-oxidation on the surface.55
 |
| | Fig. 8 (a) TGA profiles and (b) Raman spectra of the used Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1–3 wt%) catalysts. | |
Raman spectroscopy was then employed to evaluate the quality of carbon deposition (Fig. 8b). The first peak observed at around 1350 cm−1 is the D-band representative of amorphous carbon or any defective or disordered carbon,9 the second one at around 1580 cm−1 is the G-band characteristic of sp2-bonded planar graphene sheets or crystalline graphite,55 and the third peak at around 2690 cm−1 is known as the G′-band, attributable to a two-phonon, second order scattering that describes long-range order of crystalline carbon.56 The intensity ratio of the D-band with respect to the G-band (ID/IG) can be used to assess the degree of graphitization of the deposited carbon. On the Ni5Sr2/CBV20A sample, the D-band is larger and more intense than the G-band, suggesting that the carbon deposited on this promoted sample is amorphous carbon. The highest ID/IG ratio, close to 2 (Fig. 8b), obtained in this case indicates the formation of the minimum graphitic carbon over Ni5Sr2/CBV20A catalyst.57 Consequently, carbon deposited on Ni5Sr2/CBV20A can be easily gasified during the reaction, resulting in a superior performance in POM.
The TEM images of Ni5/CBV20A and Ni5Sr2/CBV20A before and after catalytic testing in POM (Fig. 9) confirm the absence of carbon deposition on the used samples, in agreement with the limited weight loss observed on the TGA curves. The results show an increase in particle size from 17.7 to about 19.9 nm on the spent Ni5/CBV20A catalyst and from 13.9 to about 21.8 nm on the spent Ni5Sr2/CBV20A catalyst. Thus, the dispersion of metallic nanoparticles is quite maintained, despite the sintering observed on the promoted sample. The nanoparticles on Ni5/CBV20A and Ni5Sr2/CBV20A samples seem to be larger than the catalysts pore sizes of about 8 nm evaluated by physisorption, implying the presence of Ni and Sr species outside the zeolite. Furthermore, the NiO nanoparticles observed on CBV20A are larger than those observed on ZSM-5 catalyst reported in another study for DRM.45 This can be attributed to the larger pore size of CBV20A compared to ZSM-5, as described earlier.
 |
| | Fig. 9 TEM images and average particle size of (a) fresh Ni5/CBV20A, (b) used Ni5/CBV20A, (a′) fresh Ni5Sr2/CBV20A and (b′) used Ni5Sr2/CBV20A catalysts. | |
 |
| | Fig. 10 Proposed reaction mechanism at low and high gas hourly space velocity. | |
The samples presented in this work show superior performance of the Sr-promoted Ni-based catalysts. The use of CBV20A support was beneficial in maintaining the stability of the catalysts for the tested duration. The addition of 2 wt% Sr was sufficient to achieve higher CH4 conversion, lower H2/CO ratio, closer to 2, higher H2 and CO yields, as well as lower CO2 yield compared to the non-promoted sample.
The catalytic evaluation of Ni5/CBV20A and Ni5Sr2/CBV20A samples for longer test duration under the same operating conditions at 700 °C, 1 atm, and GHSV = 14400 mL g−1 h−1 show a preservation of high and constant CH4 conversion with a steady hydrogen yield of the Sr-promoted sample after more than 20 hours on stream (Fig. S1a†). In numbers, the CH4 conversion and H2 yield decrease from 74% and 50% after 1 hour on stream to only 66% and 45%, respectively, after 21 hours on stream. However, upon testing the Ni5/CBV20A sample, the CH4 conversion decreases from 66% to 61% while the H2 yield declines from 48% to 38% after 21 hours on stream. Consequently, it appears the hydrogen yield is reduced by 21% for Ni5/CBV20A, compared to only 7.5% for Ni5Sr2/CBV20A. After catalytic testing, the TGA and Raman results (Fig. S1b and c†) show limited weight loss that does not exceed 6%, confirming the limited carbon deposition, that can be easily gasified, on both samples. These results confirm, once again, the promotional effect of Sr that leads to higher methane conversion and hydrogen yield and provides enhanced stability, particularly in terms of hydrogen yield, due to enhanced NiO reducibility.
Conclusions
The partial oxidation of methane to syngas using Ni-based catalysts supported on mordenite (CBV20A) and promoted with strontium (Sr) was investigated in this work. The catalytic performance of Ni5/CBV20A and Ni5Srx/CBV20A (where x = 1, 2, and 3 wt%) catalysts was evaluated at 700 °C and different GHSV. The addition of Sr favored the reduction of NiO at lower temperatures, enhancing NiO reducibility and accessibility. It also increased the amount and strength of basic sites on the surface of the catalysts. All Sr-promoted samples exhibited higher CH4 conversion, and H2 and CO yields compared to the Sr-free Ni5/CBV20A catalyst, highlighting the promotional effect of Sr in POM. Among the tested samples, Ni5Sr2/CBV20A showed the best catalytic performance with a CH4 conversion of about 72%, a H2 yield around 47%, and an H2/CO ratio of 2.60 at 700 °C and 14400 mL g−1 h−1. The superior activity of the Sr-promoted samples compared to the unpromoted sample was maintained at higher GHSV, indicating the effectiveness of Sr addition even under more severe conditions. Moreover, it was shown that the direct pathway of POM was preferred on the Sr-promoted samples at low GHSV, given the H2/CO molar ratio close to 2. At high GHSV (72000 mL g−1 h−1), the indirect pathway, involving the total combustion of methane to CO2 and H2O followed by reforming reactions to syngas, was more prominent, as indicated by the increased CO2 yield. These findings highlight the potential of Sr-promoted Ni/mordenite catalysts for efficient syngas production via POM.
Data availability
Data for this article are available at King Saud University, Chemical Engineering Department.
Author contributions
Abdulaziz Al-Anazi: data curation; methodology; writing – original draft. Marie-Nour Kaydouh: data curation; formal analysis; investigation; software; visualization; writing – original draft. Omer Bellahwel: data curation; methodology; formal analysis. Ahmed A. Ibrahim: software; writing – review & editing. Abdulaziz A. M. Abahussain: validation; visualization. Vijay Kumar Srivastava: investigation; writing – review & editing. Anis H. Fakeeha: supervision; writing – review & editing. Naif S. Almuqati: data curation; methodology; formal analysis. Raja Alotaibi: visualization; methodology; conceptualization. Ahmed S. Al-Fatesh: funding acquisition; formal analysis; validation; methodology; writing – review & editing. Nissrine El Hassan: funding acquisition; investigation; supervision; validation; visualization; writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to extend their sincere appreciation to Researchers Supporting Project Number (RSPD2025R612), King Saud University, Riyadh, Saudi Arabia. This work was also supported by the Lebanese American University President's Intramural Research Fund PIRF I0046.
Notes and references
- Z. Abdin, A. Zafaranloo, A. Rafiee, W. Merida, W. Lipinski and K. R. Khalilpour, Renewable Sustainable Energy Rev., 2020, 120, 109620 Search PubMed
 .
. - J. Holladay, J. Hu, D. King and Y. Wang, Catal. Today, 2009, 139, 244 Search PubMed .
- H. Villafán-Vidales, C. Arancibia-Bulnes, D. Riveros-Rosas, H. Romero-Paredes and C. Estrada, Renewable Sustainable Energy Rev., 2017, 75, 894 Search PubMed .
- Q. Wang, X. Chen, A. N. Jha and H. Rogers, Renewable Sustainable Energy Rev., 2014, 30, 1 Search PubMed .
- M. F. Abushammala, N. E. A. Basri, H. Basri, A. H. El-Shafie and A. A. H. Kadhum, Waste Manage. Res., 2011, 29, 863 Search PubMed .
- M. Boscherini, A. Storione, M. Minelli, F. Miccio and F. Doghieri, Energies, 2023, 16, 6375 Search PubMed .
- K. Ahmad, K. Polychronopoulou and M. Abi Jaoude, Fuel, 2022, 320, 123877 Search PubMed .
- M. M. Ghouri, S. Afzal, R. Hussain, J. Blank, D. B. Bukur and N. O. Elbashir, Comput. Chem. Eng., 2016, 91, 38 CrossRef CAS .
- A. Al-Anazi, O. Bellahwel, C. Kavitha, J. Abu-Dahrieh, A. A. Ibrahim, S. Santhosh, A. E. Abasaeed, A. H. Fakeeha and A. S. Al-Fatesh, Catalysts, 2024, 14, 316 CrossRef CAS .
- Y. Li, J. Wang, C. Ding, L. Ma, Y. Xue, J. Guo, S. Wang, Y. Meng, K. Zhang and P. Liu, RSC Adv., 2019, 9, 25508 Search PubMed .
- A. I. Osman, Chem. Eng. Technol., 2020, 43, 641 CrossRef CAS .
- A. H. Fakeeha, Y. Arafat, A. A. Ibrahim, H. Shaikh, H. Atia, A. E. Abasaeed, U. Armbruster and A. S. Al-Fatesh, Processes, 2019, 7, 141 CrossRef CAS .
- I. A. Makaryan, E. A. Salgansky, V. S. Arutyunov and I. V. Sedov, Energies, 2023, 16, 2916 CrossRef CAS .
- J. Barbero, M. Peña, J. Campos-Martin, J. Fierro and P. Arias, Catal. Lett., 2003, 87, 211 CrossRef CAS .
- Y. Ma, Y. Ma, Z. Zhao, X. Hu, Z. Ye, J. Yao, C. Buckley and D. Dong, Renewable Energy, 2019, 138, 1010 CrossRef CAS .
- Z. S. Shooli, A. Izadbakhsh and A. M. Sanati, React. Kinet., Mech. Catal., 2018, 124, 873 CrossRef .
- Y. Hou, S. Ogasawara, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2017, 7, 6132 RSC .
- S. Yasuda, R. Osuga, Y. Kunitake, K. Kato, A. Fukuoka, H. Kobayashi, M. Gao, J.-y. Hasegawa, R. Manabe, H. Shima, S. Tsutsuminai and T. Yokoi, Commun. Chem., 2020, 3, 129 CrossRef CAS PubMed .
- K. A. Chalupka, W. K. Jozwiak, J. Rynkowski, W. Maniukiewicz, S. Casale and S. Dzwigaj, Appl. Catal., B, 2014, 146, 227 CrossRef CAS .
- J. Da Costa-Serra, M. Navarro, F. Rey and A. Chica, Int. J. Hydrogen Energy, 2012, 37, 7101 CrossRef CAS .
- A. Mosayebi and R. Abedini, J. Ind. Eng. Chem., 2014, 20, 1542 CrossRef CAS .
- M. Álvarez, P. Marín and S. Ordóñez, Ind. Eng. Chem. Res., 2021, 60, 9409 CrossRef PubMed .
- L. Li, A. Shrotri, K. Kato, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2023, 13, 5190 RSC .
- N. Candu, M. Florea, S. Coman and V. Parvulescu, Appl. Catal., A, 2011, 393, 206 Search PubMed .
- A. N. van laak, R. W. Gosselink, S. L. Sagala, J. D. Meeldijk, P. E. de Jongh and K. P. de Jong, Appl. Catal., A, 2010, 382, 65 CrossRef CAS .
- R. Camposeco, N. A. Sánchez-Flores and R. Zanella, Front. Nanotechnol., 2024, 6, 1359629 Search PubMed .
- E.-h. Yang, Y.-s. Noh, S. Ramesh, S. S. Lim and D. J. Moon, Fuel Process. Technol., 2015, 134, 404 Search PubMed .
- C. Liang, X. Hu, T. Wei, P. Jia, Z. Zhang, D. Dong, S. Zhang, Q. Liu and G. Hu, Int. J. Hydrogen Energy, 2019, 44, 8197 CrossRef CAS .
- K. Sutthiumporn and S. Kawi, Int. J. Hydrogen Energy, 2011, 36, 14435 CrossRef CAS .
- A. Y. Elnour, A. H. Fakeeha, A. A. Ibrahim, A. I. Osman, A. E. Abasaeed, S. F. Adil, R. Kumar and A. S. Al-Fatesh, Res. Chem. Intermed., 2024, 50, 1211 Search PubMed .
- A. S. Al-Fatesh, D. M. Vadodariya, K. M. Banabdwin, A. A. Ibrahim, A. H. Fakeeha, S. F. Adil, R. Kumar and A. A. M. Abahussain, Catal. Lett., 2024, 154, 4625 Search PubMed .
- A. S. Al-Fatesh, A. A. Ibrahim, A. I. Osman, A. E. Abasaeed, M. F. Alotibi, S. A. Alfatesh, D. W. Rooney, A. H. Fakeeha and C.-Y. Yin, Energy Sci. Eng., 2023, 11, 3780 Search PubMed .
- U. R. Pillai and E. Sahle-Demessie, Appl. Catal., A, 2005, 281, 31 CrossRef CAS .
- A. A. Ibrahim, A. H. Fakeeha and A. S. Al-Fatesh, Int. J. Hydrogen Energy, 2014, 39, 1680 CrossRef CAS .
- M. Mikhail, P. Da Costa, J. Amouroux, S. Cavadias, M. Tatoulian, M. E. Galvez and S. Ognier, Appl. Catal., B, 2021, 294, 120233 CrossRef CAS .
- Y. Chang-lin, Z. Xiao-chun, W. Wei-zheng, H. Jiu-biao, C. Xi-rong and W. Long-fu, J. Fuel Chem. Technol., 2012, 40, 1222 Search PubMed .
- A. Owgi, A. Jalil, M. Aziz, M. Alhassan, H. Hambali, W. Nabgan, R. Saravanan and A. Hatta, Fuel, 2023, 340, 127592 Search PubMed .
- D. S. José-Alonso, M. Illán-Gómez and M. Román-Martínez, Catal. Today, 2011, 176, 187 CrossRef .
- M. H. Estalkhi, M. Yousefpour, H. Koohestan and Z. Taherian, Int. J. Hydrogen Energy, 2024, 68, 1344 Search PubMed .
- S. Li, Z. Wang, H. Zhang, Z. Liu, J. Wang, Q. Zhu, X. Li and Y. Chen, J. Anal. Appl. Pyrolysis, 2017, 123, 269 Search PubMed .
- E. H. Cho, Y.-K. Park, K. Y. Park, D. Song, K. Y. Koo, U. Jung, W. R. Yoon and C. H. Ko, Chem. Eng. J., 2022, 428, 131393 Search PubMed .
- F. Namvar, M. Hajizadeh-Oghaz, M. A. Mahdi, S. H. Ganduh, F. Meshkani and M. Salavati-Niasari, Int. J. Hydrogen Energy, 2023, 48, 3862 CrossRef CAS .
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051 Search PubMed .
- A. S. Al-Fatesh, A. A. Ibrahim, A. H. Fakeeha, A. I. Osman, Y. M. Alanazi, F. S. Almubaddel and A. E. Abasaeed, Nanomaterials, 2024, 14, 1320 CrossRef CAS PubMed .
- U. Ashik, S. Asano, S. Kudo, D. Pham Minh, S. Appari, E. Hisahiro and J. Hayashi, Catalysts, 2020, 10, 21 CrossRef CAS .
- K. Nakamoto, M. Ohshiro and T. Kobayashi, J. Environ. Chem. Eng., 2017, 5, 513 CrossRef CAS .
- M. N. Kaydouh, N. El Hassan, A. Davidson, S. Casale, H. El Zakhem and P. Massiani, Microporous Mesoporous Mater., 2016, 220, 99 CrossRef CAS .
- L. Karam, M. C. Bacariza, J. M. Lopes, C. Henriques, P. Massiani and N. El Hassan, Int. J. Hydrogen Energy, 2020, 45, 28626 Search PubMed .
- X. Wu and S. Kawi, Cryst. Growth Des., 2010, 10, 1833 Search PubMed .
- G. B. Sun, K. Hidajat, X. S. Wu and S. Kawi, Appl. Catal., B, 2008, 81, 303 Search PubMed .
- X. Wu and S. Kawi, Catal. Today, 2009, 148, 251 CrossRef CAS .
- K. Acharya, A. S. Al-Fatesh, G. Almutairi, A. H. Fakeeha, A. A. Ibrahim, A. E. Abasaeed, M. R. H. Siddiqui and R. Kumar, Catal. Lett., 2024, 154, 2023 CrossRef CAS .
- H. U. Hambali, A. Abdul Jalil, A. A. Abdulrasheed, T. J. Siang and D.-V. N. Vo, J. Energy Inst., 2020, 93, 1535 CrossRef .
- R. Ma, B. Xu and X. Zhang, Catal. Today, 2019, 338, 18 Search PubMed .
- C. Miao, S. Chen, K. Shang, L. Liang and J. Ouyang, ACS Appl. Mater. Interfaces, 2022, 14, 47616 Search PubMed .
- M. S. Ferrandon, C. Byron, G. Celik, Y. Zhang, C. Ni, J. Sloppy, R. A. McCormick, K. Booksh, A. V. Teplyakov and M. Delferro, Appl. Catal., A, 2022, 629, 118379 CrossRef CAS .
- S. Dama, S. R. Ghodke, R. Bobade, H. R. Gurav and S. Chilukuri, Appl. Catal., B, 2018, 224, 146 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Vijay Kumar Srivastavac,
Anis H. Fakeehaa,
Naif S. Almuqatid,
Raja Alotaibid,
Ahmed S. Al-Fatesh
a,
Vijay Kumar Srivastavac,
Anis H. Fakeehaa,
Naif S. Almuqatid,
Raja Alotaibid,
Ahmed S. Al-Fatesh