DOI:
10.1039/D5RA00208G
(Paper)
RSC Adv., 2025,
15, 4702-4707
Insights into the structural diversity of cembranoids from Sarcophyton glaucum and their bioactivity†
Received
9th January 2025
, Accepted 29th January 2025
First published on 12th February 2025
Abstract
Three capnosane-based cembranoids (1–3), four normal cembranoids (4–7), and one sarsolenane-based cembranoid (8) were isolated from the soft coral Sarcophyton glaucum, including three new compounds: glacunoids A (1), B (4), and C (7). The chemical structures were elucidated using IR, MS, and NMR spectroscopy, with the absolute configurations of 1, 2, and 6 confirmed by single-crystal X-ray diffraction analysis. The bioactivity of the isolated compounds was evaluated for their effects on alkaline phosphatase (ALP) activity. All compounds demonstrated the ability to enhance ALP activity, with compound 8 exhibiting the most potent effect, achieving an ALP activity of 139.41 ± 8.06% without detectable cytotoxicity. This study represents the first report of three distinct structural types of cembranoids from a single soft coral species.
1 Introduction
Soft corals of the genus Sarcophyton (family Alcyoniidae) are prolific sources of diverse secondary metabolites, particularly diterpenes of the cembrane type. These metabolites have garnered significant scientific interest due to their structural diversity and broad spectrum of biological activities, including antitumor, antifouling, and anti-inflammatory effects.1,2 Among these, cembranoids represent a prominent subclass, characterized by their unique structural frameworks and functional group diversity, which contribute to their pharmacological potential.1,2 Continuing our exploration of bioactive natural products from marine organisms, we focused on the soft coral Sarcophyton glaucum (Quoy & Gaimard, 1833) (Fig. 1),3–5 a widely distributed species in tropical and subtropical marine ecosystems. Previous investigations into S. glaucum revealed its capacity to produce an array of cembrane-type diterpenoids, which exhibited notable bioactivities ranging from cytotoxicity to ecological defense mechanisms.1,2 In this study, we report the isolation and characterization of eight cembranoids from S. glaucum, classified into three structural subtypes: capnosane-based cembranoids (compounds 1–3),6,7 normal cembranoids (compounds 4–7),8 and a sarsolenane-based cembranoid (compound 8)7 (Fig. 2). Among these, three compounds—glacunoid A (1), a capnosane-based cembranoid, and two normal cembranoids, glacunoids B (4) and C (7)—were identified as new natural products. The chemical structures of these new compounds were elucidated using a combination of spectroscopic techniques, including infrared (IR), mass spectrometry (MS), one-dimensional (1D), and two-dimensional (2D) nuclear magnetic resonance (NMR) spectroscopy. Additionally, the absolute configurations of compounds 1, 2, and 6 were determined through single-crystal X-ray crystallography (Fig. 3), providing further insight into their stereochemistry. This study not only expands the chemical diversity of cembranoids from S. glaucum but also contributes to the understanding of their structural and biological significance.
 |
| | Fig. 1 Soft coral Sarcophyton glaucum. | |
 |
| | Fig. 2 Structures of metabolites 1–8. | |
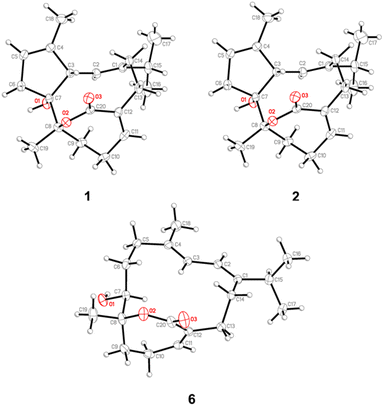 |
| | Fig. 3 A computer-generated ORTEP plot revealing the absolute structures of 1, 2, and 6. | |
2 Results and discussion
Our current results reported the isolation and structure elucidation of a novel capnosane-based cembranoids, glacunoid A (1) and two normal cembranoids, glacunoids B (4) and C (7). The biosynthesis of capnosane from cembranoids involves the enzymatic transformation of a 14-membered macrocyclic cembrane skeleton derived from geranylgeranyl diphosphate (GGPP). Key steps include cyclization of GGPP to form the cembrane ring, followed by intricate rearrangements that generate the distinctive bicyclo[7.2.1] ring system characteristic of capnosane. Oxidative tailoring, mediated by cytochrome P450 enzymes, introduces functional groups such as hydroxyls, carbonyls, and epoxides, enhancing chemical complexity and bioactivity. Final modifications, such as esterification, complete the structure, showcasing the sophisticated enzymatic processes that diversify cembranoids into bioactive natural products like capnosane.9
Glacunoid A (1) was isolated and crystalized as colorless needles. Its infrared (IR) absorptions showed the presence of both hydroxy (3448 cm−1) and lactone (1669 cm−1) functional groups. The molecular formula of 1 was determined to be C20H28O3 by HRESIMS (m/z 339.1928 [M + Na]+) and 13C NMR data (Table 1), suggesting seven degrees of unsaturation. Resonances of one ester carbonyl carbon (δ 170.1) and six olefinic carbons (δ 146.0, C; 144.3, CH; 141.5, C; 133.8, C; 120.5, CH; 120.0, CH) in the 13C NMR and DEPT spectral data accounted for four double-bond equivalents, which indicated that 1 was a tricyclic compound. The 1H NMR spectrum of 1 (Table 1) revealed the presence of resonances of three olefinic methine protons [δ 6.33 dd (8.4, 6.0); 5.47d (12.0); 5.36 brs] and four methyls [δ 1.48 s; 1.45 s; 1.08 d (6.6); 1.05 d (6.6)]. The spectroscopic data of 1 (1H and 13C NMR) were similar to those of 2, except for the absence of the 4,18-double bond and 4,5-single bond signals, both were replaced by signals of a 4,18- single bond and 4,5-double bond in 1. Moreover, single-crystal X-ray crystallographic analysis using Cu Kα (λ = 1.54178 Å) radiation source [Flack parameter x = 0.16(5)] (CCDC 2401636) allowed the assignment of the absolute configuration of 1. An Oak Ridge Thermal–Ellipsoid Plot (ORTEP) diagram (Fig. 3) showed the absolute configuration of the stereogenic carbons of 1 to be (3S,7R,8R).
Table 1 1H and 13C NMR data for 1
| C/H |
δHa (J in Hz) |
δC,b multc |
C/H |
δHa (J in Hz) |
δC,bmultc |
| Spectra obtained in CDCl3 at 600 MHz. Spectra obtained in CDCl3 at 150 MHz. Attached protons were determined by DEPT experiments. |
| 1 |
|
146.0, C |
11 |
6.33 dd (8.4, 6.0) |
144.3, CH |
| 2 |
5.47 d (12.0) |
120.5, CH |
12 |
|
133.8, C |
| 3 |
3.33 d (11.4) |
50.9, CH |
13 |
2.95 dd (11.4, 5.4) |
31.9, CH2 |
| 4 |
|
141.5, C |
|
2.00 m |
|
| 5 |
5.36 brs |
120.0, CH |
14 |
2.65 m |
26.8, CH2 |
| |
2.01 m |
|
| 15 |
2.25 m |
34.4, CH |
| 6 |
2.86 d (15.6,3.0) |
42.4, CH2 |
16 |
1.05 d (6.6) |
23.0, CH3 |
| 2.18 d (15.6) |
| 7 |
|
90.0, C |
17 |
1.08 d (6.6) |
21.6, CH3 |
| 8 |
|
86.5, C |
18 |
1.48 s |
15.5, CH3 |
| 9 |
2.56 t (13.2) |
38.6, CH2 |
19 |
1.45 s |
27.6, CH3 |
| 1.80 dd (13.2, 6.0) |
| 10 |
2.35 m |
22.2 CH2 |
20 |
|
170.1, C |
| 2.07 m |
Glacunoid B (4) was isolated as a colorless oil. Its molecular formula C20H28O3 was established by HRESIMS (m/z 339.1928, [M + Na]+) and 13C NMR data, implying, seven degrees of unsaturation. By the analysis of 13C NMR and DEPT spectra of 4, the carbons signals were assigned into four methyls, six sp3 methylenes, one sp3 methine, three sp2 methines, and six quaternary carbons (including one oxygenated δ 86.2). The remaining two signals appearing in the lower field region of the spectrum were due to the quaternary carbons of one lactone carbon (δ 170.5) and one ketone carbonyl (δ 208.4). Careful analysis of 1H–1H COSY correlations led to the establishment of five-part structures in Fig. 3. The molecular framework of 4 was further established by an HMBC experiment (Fig. 4).
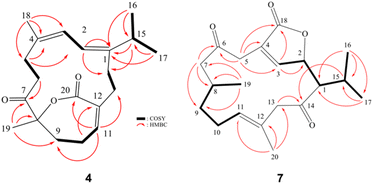 |
| | Fig. 4 Key HMBC and COSY correlations of 4 and 7. | |
The two rings and their connectivity were elucidated based on the following key HMBC correlations, both methyls H3-16 and H3-17 to C-1 and C-15, H3-18 to C-3, C-4 and C-5, H3-19 to C-7, C-8 and C-9, H-2 to C-1 and C-14, H2-5 to C-3 and C-4, H2-6 to C-7, H2-10 to C-12, H-11 to C-13 and C-20. Thus, 4 was found to possess three double bonds at C-1/C-2, C-3/C-4, and C-11/C-12, one ketone group at C-7, and one α,β-unsaturated ε-lactone at C-8/C-12. Linking all the above functional groups to the cembrane skeleton yielded a gross structure of 4. The 13C NMR data of 4 were found to be quite similar to those of 5. We then carefully examined the NOESY correlations of 4. The NOESY spectrum (Fig. 4) showed a correlation of H3-18 with H-3, but not with H-2, H-3 with H3-16 revealing the Z configuration of the C-1/C-2 and C-3/C-4 double bonds. The comparison of its 1H and 13C NMR with those of 5 indicated that 4 is the 1Z and 3Z isomer of 5. Based on the well-established 8S configuration of 4 and the fact that both 4–6 could be the precursors of each other by dehydration, the structure of 4 was then established.
Compound 7 was isolated as a colorless oil. The HRESIMS (m/z 359.1928, [M + Na]+) and 13C NMR data of 7 indicated the molecular formula C20H28O3. IR absorptions were observed at 1761 and 1723 cm−1, suggesting the presence of carbonyl groups in 7. The 13C NMR and DEPT spectroscopic data (Table 2), showed signals of four methyls, five sp3 methylenes, three sp3 methines, one sp3 oxymethine, two sp2 methines, and five sp2 quaternary carbons (including two ketones and one ester carbonyl). From the 1H NMR spectrum of 7, the resonances of two olefinic protons [δH 7.03 dd (3.0, 1.2) and δH 5.24 dd (9.0, 5.4)], one oxygenated methine [δH 5.21 dd (9.6, 1.8)] and four methyls [δH 1.69 s, 1.14 d (7.2); 1.06 d (7.2); 0.95 d (6.6)] were observed. The planar structure and all the 1H and 13C chemical shifts of 7 were elucidated by 2D NMR spectroscopic analysis, in particular 1H–1H COSY and HMBC experiments (Fig. 4). Thus, 7 was found to possess two double bonds at C-3/C-4 and C-11/C-12, two ketone groups at C-6 and C-14, and an α,β-unsaturated γ-lactone moiety at C-4, C-3, C-2, and C-18. The relative configuration of 7 was elucidated from the NOESY spectrum, which was compatible with that of 7 obtained by the MM2 force field calculations suggesting the most stable conformations as shown in Fig. 5. In the NOESY spectrum, it was found that H3-19 (δ 1.44, s) showed NOE interactions with one of the methylene protons at C-10 (δH 1.95, m). Therefore, H3-19 and one of proton H-10 (δH 1.95, m) were located on the α-face, while the other (δH 2.20, m) was oriented as β H-10. The NOE correlations observed between H3-20 with H-10β and H-1 reflected the β-orientations of H-1. Furthermore, the NOE correlations between the H-1 and H-2 suggested the β-orientation of H-2. From the above findings and further analysis of other NOE interactions (Fig. 6), the structure of 7 was fully established.
Table 2 1H and 13C NMR data for 4 and 7
| C/H |
4 |
7 |
| δHa (J in Hz) |
δC,b multc |
δHa (J in Hz) |
δC,b multc |
| Spectra obtained in CDCl3 at 600 MHz. Spectra obtained in CDCl3 at 150 MHz. Attached protons were determined by DEPT experiments. |
| 1 |
|
140.7, C |
2.85 m |
58.6, CH |
| 2 |
5.21 d (11.4) |
118.8, CH |
5.21 dd (9.6, 1.8) |
80.3, CH |
| 3 |
6.05 d (11.4) |
120.2, CH |
7.03 dd (3.0, 1.2) |
150.4, CH |
| 4 |
|
138.8, C |
|
127.7, C |
| 5 |
3.15 dt (15.0, 4.8) |
23.5, CH2 |
3.42 d (16.2) |
40.2, CH2 |
| 1.81 m |
|
3.16 d (16.2) |
|
| 6 |
2.95 m |
39.8, CH2 |
|
205.6, C |
| 2.33 t (4.8) |
|
|
|
| 7 |
|
208.4, C |
2.32 d (7.2) |
49.8, CH2 |
| 8 |
|
86.2, C |
1.69 m |
28.2, CH |
| 9 |
2.60 m; 2.24 m |
37.4, CH2 |
1.42 m |
35.7, CH2 |
| 10 |
2.37 m; 2.16 m |
22.6, CH2 |
2.20 m; 1.95 m |
24.3, CH2 |
| 11 |
5.78 dt (7.8, 2.0) |
131.4, CH |
5.24 dd (9.0, 5.4) |
131.0, CH |
| 12 |
|
136.6, C |
|
128.1, C |
| 13 |
2.63 m; 2.39 m |
26.3, CH2 |
3.27 d (12.6) |
56.2, CH2 |
| |
|
2.84 d (13.2) |
|
| 14 |
2.37 m; 2.18 m |
25.5, CH2 |
|
208.1, C |
| 15 |
3.01 m |
28.9, CH |
2.30 m |
28.9, CH2 |
| 16 |
1.04 d (7.2) |
21.1, CH3 |
1.14 d (7.2) |
20.7, CH3 |
| 17 |
1.02 d (7.2) |
20.7, CH3 |
1.06 d (7.2) |
19.3, CH3 |
| 18 |
1.86 s |
26.5, CH3 |
|
172.5, C |
| 19 |
1.48 s |
27.6 CH3 |
0.95 d (6.6) |
20.3, CH3 |
| 20 |
|
170.5 C |
1.69 s |
16.6, CH3 |
 |
| | Fig. 5 Selected NOE correlations observed for 4. | |
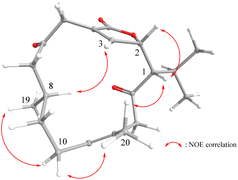 |
| | Fig. 6 Selected NOE correlations observed for 7. | |
The five obtained known cembrane diterpenoids were identified as sarsolilide A (2) and B (3),6,7 sarcophytolide (5),8 (4Z,8S,9R,12E,14E)-9-hydroxy-1-isopropyl-8,12-dimethyl oxabicyclo[9.3.2]-hexadeca-4,12,14-trien-18-one (6)8 and sarsolenone (8)7 by comparing the obtained spectroscopic data with published values. The absolute configurations of 2 and 6 were confirmed by single-crystal X-ray diffraction analysis with Cu Kα radiation [Flack parameters = 0.08(6) and 0.07(7)] for the first time (Fig. 3).
Previous studies indicated that cembranoids 1, 2, and 4–8 can be used to treat osteoclastogenic diseases. Using MG-63 human mesenchymal stem cells, we conducted an alkaline phosphatase (ALP) ELISA assay (Table 3). The results indicated that all tested compounds significantly increased ALP activity, with compound 8 showing the most significant effect, achieving an ALP activity of 139.41 ± 8.06% without cytotoxic effects. Moreover, these findings suggested that the tested compounds have potential biological activity in promoting ALP activity, and tested compounds did not affect cell the viability of MG63 cells.
Table 3 Evaluation of the ALP activity after the treatment of MG63 cells with 1, 2, and 4–8 at a concentration of 10 μM for 72 ha
| Compounds |
ALP activity (%) |
Cell viability (%) |
| The data are expressed as the standard error of the mean (n = 3). The significance was determined using Student's t-test (*p < 0.05, **p < 0.01, ***p < 0.001) and by comparison of the results with those of the untreated cells. 17β-Estradiol was utilized as a positive control at a concentration of 10 μM. |
| Control |
100.00 ± 0.65 |
|
100.00 ± 1.03 |
|
| 1 |
109.10 ± 3.38 |
*** |
105.34 ± 4.69 |
* |
| 2 |
113.44 ± 4.80 |
*** |
103.00 ± 2.27 |
* |
| 4 |
110.75 ± 3.90 |
*** |
106.18 ± 3.81 |
** |
| 5 |
110.98 ± 4.74 |
** |
96.72 ± 9.71 |
|
| 6 |
107.06 ± 3.48 |
** |
106.88 ± 6.10 |
* |
| 7 |
128.06 ± 3.04 |
*** |
93.58 ± 6.73 |
|
| 8 |
139.41 ± 8.06 |
*** |
107.29 ± 6.83 |
* |
| 17β-Estradiolb |
146.46 ± 2.34 |
*** |
68.23 ± 9.49 |
|
3 Conclusions
A diverse series of cembrane diterpenoids was isolated from the wild-type soft coral Sarcophyton glaucum. This study identified three new diterpenoids—glacunoids A (1), B (4), and C (7)—alongside five known compounds (2, 3, 5, 6, and 8). The absolute configurations of compounds 1, 2, and 6 were unambiguously confirmed by single-crystal X-ray diffraction analysis. Notably, metabolites 1–3 are rare examples of capnosane-based cembranoids featuring an α,β-unsaturated ε-lactone moiety. This work represents the first report of three distinct structural types of cembrane diterpenoids (normal-, capnosane-, and sarsolenane-based) isolated from a single soft coral species. All the isolated compounds enhanced alkaline phosphatase (ALP) activity, with sarsolenone (8) demonstrating the highest activity (139.41 ± 8.06%) and no cytotoxicity against MG-63 human mesenchymal stem cells.
4 Experimental
4.1 General experimental procedures
Infrared (IR) spectra were obtained on a Fourier-transform IR spectrophotometer (model: JASCO P-2000). 1H and 13C NMR spectra were recorded on a 600R NMR spectrometer (JEOL, Tokyo, Japan) with CDCl3 (Sigma-Aldrich, St. Louis, MO, USA) as the deuterated solvent. The detected signals in 1H and 13C NMR spectra were corrected at 7.26 ppm (singlet) and 77.0 ppm (triplet), respectively. The coupling constants (J) were converted to Hz. MS data, including ESIMS and HRESIMS, were obtained using a Bruker 7 Tesla Solera FTMS system (Bruker, Bremen, Germany). Optical rotations were determined by a digital polarimeter (Jasco P-1010). Single-crystal X-ray analyses were performed on a Bruker D8 Venture diffractometer. Thin-layer chromatography was performed on plates precoated with silica gel 60 F254 (0.25 mm-thick, MERCK); the plates were then sprayed with 10% (v/v) H2SO4 in methanol, followed by heating to visualize the spots. A normal-phase (NP) HPLC was performed using a system comprised of a HITACHI 5110 pump, a RHEODYNE 7725i injection port, and a NP column (YMC pack SIL, 5 μm, 12 nm, 250 × 20 mm, YMC group). Reverse-phase (RP) HPLC was performed using a system comprised of a HITACHI L-2130 pump, a HITACHI L-2455 photodiode array detector, a RHEODYNE 7725i injection port and an RP column (Luna 5 μm C18(2) 100 Å, 250 × 21.2 mm, Phenomenex).
4.2 Animal material
The soft coral S. glaucum (specimen no. NMMBA-SC-2024-1) samples were collected by scuba divers at a depth of around 10–15 m off the coast of Pingtung County, Taiwan, in February 2024. The samples were frozen immediately after collection. A voucher sample was deposited at the National Museum of Marine Biology and Aquarium.
4.3 Extraction and isolation
The frozen S. glaucum material (1.7 kg, wet wt) was minced and treated with an equal mixture of methanol (MeOH) and dichloromethane (CH2Cl2) at ambient temperature. This procedure resulted in a crude extract weighing 29.7 g. Liquid–liquid partition was performed to separate the mixture into ethyl acetate (EtOAc) and water layers. The ethyl acetate phase was evaporated under reduced pressure to afford a residue (15.9 g), and the residue was subjected to column chromatography on silica gel, using n-hexane, n-hexane and EtOAc mixture of increasing polarity, and finally pure acetone to yield 11 fractions: Fr-1 (eluted by n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc, 50:1), Fr-2 (eluted by n-hexane:EtOAc, 10:1), Fr-3 (eluted by n-hexane:EtOAc, 8:1), Fr-4 (eluted by n-hexane:EtOAc, 5:1), Fr-5 (eluted by n-hexane:EtOAc, 3:1), Fr-6 (eluted by n-hexane:EtOAc, 2:1), Fr-7 (eluted by n-hexane:EtOAc, 1:1), Fr-8 (eluted by n-hexane:EtOAc, 2:1), Fr-9 (eluted by n-hexane:EtOAc, 5:1), Fr-10 (eluted by EtOAc) and Fr-11 (eluted by acetone). Fraction 2 was further purified with normal-phase HPLC (n-hexane:EtOAc, 4:1) to afford seven subfractions (2A − 2G). Subfraction 2E was then separated by normal-phase HPLC (n-hexane:EtOAc, 4:1) to obtain 4 (30.5 mg) and 5 (680 mg). Fraction 3 was separated by normal-phase HPLC using n-hexane:EtOAc (5:1) to afford 1 (20.6 mg) and 2 (80.6 mg). Fraction 4 was further separated by silica gel column chromatography with gradient elution (n-hexane:EtOAc, 4:1 to 2:1) to afford ten subfractions (4 A-4J). Subfraction 4B was separated by normal-phase HPLC using n-hexane:EtOAc (7:2) to afford 7 (2.8 mg). Subfraction 4E (20.5 mg) was also separated by normal-phase HPLC using n-hexane:EtOAc (2:1) to afford 3 (1.5 mg), 6 (49.5 mg) and 8 (30.6 mg).
:EtOAc, 50:1), Fr-2 (eluted by n-hexane:EtOAc, 10:1), Fr-3 (eluted by n-hexane:EtOAc, 8:1), Fr-4 (eluted by n-hexane:EtOAc, 5:1), Fr-5 (eluted by n-hexane:EtOAc, 3:1), Fr-6 (eluted by n-hexane:EtOAc, 2:1), Fr-7 (eluted by n-hexane:EtOAc, 1:1), Fr-8 (eluted by n-hexane:EtOAc, 2:1), Fr-9 (eluted by n-hexane:EtOAc, 5:1), Fr-10 (eluted by EtOAc) and Fr-11 (eluted by acetone). Fraction 2 was further purified with normal-phase HPLC (n-hexane:EtOAc, 4:1) to afford seven subfractions (2A − 2G). Subfraction 2E was then separated by normal-phase HPLC (n-hexane:EtOAc, 4:1) to obtain 4 (30.5 mg) and 5 (680 mg). Fraction 3 was separated by normal-phase HPLC using n-hexane:EtOAc (5:1) to afford 1 (20.6 mg) and 2 (80.6 mg). Fraction 4 was further separated by silica gel column chromatography with gradient elution (n-hexane:EtOAc, 4:1 to 2:1) to afford ten subfractions (4 A-4J). Subfraction 4B was separated by normal-phase HPLC using n-hexane:EtOAc (7:2) to afford 7 (2.8 mg). Subfraction 4E (20.5 mg) was also separated by normal-phase HPLC using n-hexane:EtOAc (2:1) to afford 3 (1.5 mg), 6 (49.5 mg) and 8 (30.6 mg).
4.4 Structural characterization of compounds 1, 4, and 7
4.4.1 Glacunoid A (1). Colorless needless; mp 151–153 °C; [α]25D +133 (c 0.05, CHCl3); IR (KBr) νmax 3448, 2956, 2923, 1669, 1460 and 1378 cm−1; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data (Table 1); ESIMS: m/z 339 [M + Na]+; HRESIMS: m/z 339.1928 (calcd for C20H28O3 + Na, 339.1930).
4.4.2 Glacunoid B (4). Colorless oil; [α]25D +17 (c 0.19, CHCl3); IR (KBr) vmax 2963, 2932, 1712, 1450 and 1375 cm−1; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data (Table 2); ESIMS: m/z 339 [M + Na]+; HRESIMS: m/z 339.1928 (calcd for C20H28O3 + Na, 339.1930).
4.4.3 Glacunoid C (7). Colorless oil; [α]25D −157(c 0.04, CHCl3); IR (KBr) vmax 2958, 2924, 1763, 1703, 1458 and 1374 cm−1; 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data (Table 2); ESIMS: m/z 355 [M + Na]+; HRESIMS: m/z 355.1878 (calcd for C20H28O4 + Na, 355.1879).
4.5 Single-crystal X-ray analysis of 1, 2, and 6
4.5.1 Glacunoid A (1). The ethyl acetate solution was allowed to slowly evaporate to generate a suitable colorless crystal. Diffraction intensity data were obtained on a Bruker D8 Venture diffractometer using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Crystal data for this compound: C20H28O3 (formula weight 316.42), approximate crystal size, 0.330 × 0.289 × 0.110 mm3, monoclinic, space group, P21 (# 4), T = 200(2) K, a = 10.9787 (4) Å, b = 11.8275 (4) Å, c = 13.8005 (5) Å, α = β = γ = 90°, V = 1792.00 (11) Å3, Z = 4, Dcalcd = 1.173 Mg m−3, F(000) = 688, μ(MoKα) = 0.609 mm−1. A total of 18753 reflections were collected in the range 4.924 < θ < 74.417, with 3646 independent reflections [R(int) = 0.0333], completeness to theta was 99.8%; semi-empirical from equivalents absorption correction applied; refinement method: full-matrix least-squares on F2, the data/restraints/parameters were 3646/0/216; goodness-of-fit on F2 = 1.046; final R indices [I > 2 sigma (I)], R1 = 0.0317, wR2 = 0.0859; R indices (all data), R1 = 0.0326, wR2 = 0.0866, largest difference peak and hole, 0.207 and −0.145 e Å3; absolute structure parameter, 0.16(5).
4.5.2 Sarsolilide A (2). The ethyl acetate solution was allowed to slowly evaporate to generate a suitable colorless crystal. Diffraction intensity data were obtained on a Bruker D8 Venture diffractometer using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Crystal data for this compound: C20H28O3 (formula weight 316.42), approximate crystal size, 0.171 × 0.142 × 0.109 mm3, monoclinic, space group, P21 (# 4), T = 200(2) K, a = 10.9227 (3) Å, b = 11.4366 (3) Å, c = 14.0387 (4) Å, α = β = γ = 90°, V = 1573.69 (8) Å3, Z = 4, Dcalcd = 1.198 Mg m−3, F(000) = 688, μ(MoKα) = 0.622 mm−1. A total of 15840 reflections were collected in the range 4.988 < θ < 74.394, with 3550 independent reflections [R(int) = 0.0292], completeness to theta was 100%; semi-empirical from equivalents absorption correction applied; refinement method: full-matrix least-squares on F2, the data/restraints/parameters were 3550/6/212; goodness-of-fit on F2 = 1.031; final R indices [I > 2 sigma (I)], R1 = 0.0296, wR2 = 0.0799; R indices (all data), R1 = 0.0302, wR2 = 0.0804, largest difference peak and hole, 0.167 and −0.165 e Å3; absolute structure parameter, 0.08(6).
4.5.3 3β,11-dihydroxy-5α,6α-epoxy-24-methylene-9,11-secocholestan-9-one (6). The ethyl acetate solution was allowed to slowly evaporate to generate a suitable colorless crystal. Diffraction intensity data were obtained on a Bruker D8 Venture diffractometer using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Crystal data for this compound: C20H30O3 (formula weight 318.44), approximate crystal size, 0.332 × 0.118 × 0.100 mm3, monoclinic, space group, P21 (# 4), T = 200(2) K, a = 7.6524 (2) Å, b = 13.8446 (3) Å, c = 17.0676 (4) Å, α = β = γ = 90°, V = 1808.22 (7) Å3, Z = 4, Dcalcd = 1.170 Mg m−3, F(000) = 696, μ(MoKα) = 0.604 mm−1. A total of 14037 reflections were collected in the range 4.111 < θ < 74.422, with 3676 independent reflections [R(int) = 0.0327], completeness to theta was 99.9%; semi-empirical from equivalents absorption correction applied; refinement method: full-matrix least-squares on F2, the data/restraints/parameters were 3676/0/213; goodness-of-fit on F2 = 1.035; final R indices [I > 2 sigma (I)], R1 = 0.0296, wR2 = 0.0777; R indices (all data), R1 = 0.0315, wR2 = 0.0790, largest difference peak and hole, 0.149 and −0.141 e Å3; absolute structure parameter, −0.07(7).Crystallographic data for compounds 1, 2, and 6 were deposited at the Cambridge Crystallographic Data Centre (CCDC No. 2401636, 2401635, and 2401634 for compounds 1, 2, and 6, respectively).
4.6 ALP activity assay
In this study, we conducted assays to evaluate ALP activity. For each assay, MG63 cells were exposed to one of the 1, 2, and 4–8 compounds at a 10 μM concentration for three days. After treatment, the MG63 cells in each well were washed with phosphate-buffered saline (PBS) twice, and then lysis buffer with 0.1% Triton X-100 was added. After sonication, the cell lysate samples were subjected to ALP activity measurement with p-nitrophenyl phosphate in 0.2 M Tris hydrochloride magnesium chloride hexahydrate buffer (pH 9.5). The ALP activity of each sample was normalized by the protein levels determined with a protein assay kit (BCA kit; Thermo Fischer Scientific). The enzymatic reaction was halted by adding 0.1 N NaOH to the solution as soon as a noticeable color 8 change occurred, thereby ceasing the conversion of p-nitrophenyl phosphate to p-nitrophenol. The absorbance at 450 and 562 nm was measured via a spectrophotometer, and the ALP activity was subsequently quantified by relating these absorbance measurements to a standard curve derived from known concentrations of p-nitrophenol.
4.7 Cell viability assay
Cell viability assays were conducted by seeding 1 × 103 cells into each well of a 96-well plate, and after 24 hours, a mixture of culture medium supplemented with either 10.0 μM of a specific drug or 10.0 μM alendronate sodium hydrate was added to the wells. After incubation for 72 hours at 37 °C, the cells in each well were rinsed and treated with a solution composed of 10 μL of MTT solution (5 mg mL−1) and 90 μL of culture medium. After another incubation at 37 °C for 4 hours, this process led to the formation of formazan crystals, which were subsequently dissolved by adding 100 μL of DMSO to each well. Upon complete dissolution, the optical density of the solution was measured at a wavelength of 570 nm via an ELISA reader (Thermo Fischer Scientific).
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Mohamed El-Shazly and Jui-Hsin Su conceived and designed the experiments; Hsiao-Ling Chung, and Li-Guo Zheng performed the sample collections, extraction, isolation, structures determination, and qualitative HPLC analysis; the pharmacological experiments were carried out by You-Ying Chen and Jui-Hsin Su contributed reagents and analysis tools; Mohamed El-Shazly and Jui-Hsin Su participated in data interpretation, wrote the manuscript and revised the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Ms. Hsiao-Ching Yu and Chao-Lien Ho, of the High Valued Instrument Center, National Sun Yat-sen University, for the mass (MS 006500) and NMR (NMR 001100) spectra (NSTC 113-2740-M-110-002), and to the Instrumentation Center, National Taiwan University, for providing X-ray facilities (NSTC 113-2740-M-002-007, XRD 000200). This research has been principally supported by grants from the National Museum of Marine Biology & Aquarium, the National Science and Technology Council (NSTC 113-2320-B-291-002), in Taiwan, awarded to Jui-Hsin Su; and by National Science and Technology Council of Taiwan (MOST 111-2320-B-038-040-MY3, 113-2628-B-038-009 -MY3, and 113-2321-B-255-001), awarded to Kuei-Hung Lai.
Notes and references
- N. Zhang, W. Xu, Y. Yan, M. Chen, H. Li and L. Chen, Cembrane diterpenoids: Chemistry and pharmacological activities, Phytochemistry, 2023, 212, 113703 CrossRef CAS PubMed.
- Y. A. Elkhawas, M. E. Ahmed, S. E. Mohamed, M. M. Nada, Al-S. Eman, M. B. Mokhtar, N. B. S. Abdel and M. S. Osama, Chemical diversity in species belonging to soft coral genus Sarcophyton and its impact on biological activity: A review, Mar. Drugs, 2020, 18, 41 CrossRef CAS PubMed.
- C.-F. Dai and C.-H. Chin, Octocoral Fauna of Kenting National Park, Kenting National Park Headquarters, Kenting, Pingtung, 2019, pp. 270–271 Search PubMed.
- C.-F. Dai, Octocoral Fauna of Taiwan, Ocean Center, National Taiwan University, Taipei, Taiwan, 2019, pp. 346–347 Search PubMed.
- C.-F. Dai, Corals of Taiwan: Octocorallia, Owl Publishing House Co., LTD, Taipei, 2022, vol. 2, p. 220 Search PubMed.
- M. Zhang, K.-H. Long, S.-H. Huang, K.-L. Shi and T. C. W. Mak, A novel diterpenolide from the soft coral Sarcophyton solidun, J. Nat. Prod., 1992, 55, 1672–1675 CrossRef CAS.
- L.-F. Liang, T. Kurtán, A. Mándi, L.-X. Gao, J. Li, W. Zhang and Y.-W. Guo, Sarsolenane and capnosane diterpenes from the Hainan soft coral Sarcophyton trocheliophorum Marenzeller as PTP1B inhibitors, Eur. J. Org Chem., 2014, 1841–1847 CrossRef CAS.
- H. Gross, A. D. Wright, W. Beil and G. M. Konig, Two new bicyclic cembranolides from a new Sarcophyton species and determination of the absolute configuration of sarcoglaucol-16-one, Org. Biomol. Chem., 2004, 2, 1133e1138 RSC.
- H. Peng, Y. Zeng, H. Wang, W. Chang, H. Chen, F. Zhou, H. Dai and X. Wang, Six Undescribed Capnosane-Type Macrocyclic Diterpenoids from South China Sea Soft Coral Sarcophyton crassocaule: Structural Determination and Biological Evaluation, Mar. Drugs, 2023, 21, 645 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Hsiao-Ling Chungde,
You-Ying Chende,
Li-Guo Zheng
abc,
Hsiao-Ling Chungde,
You-Ying Chende,
Li-Guo Zheng




