
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective electrocatalytic reduction of nitrate to ammonia over a copper–cobalt bimetallic catalyst†
Daopeng Liab,
Shengbo Zhang *ab,
Zhixian Maoab,
Min Liua,
Kui Hua,
Dongnan Zhaoa,
Zhengguo Qva,
Li Zhoua and
Tongfei Shi*ab
*ab,
Zhixian Maoab,
Min Liua,
Kui Hua,
Dongnan Zhaoa,
Zhengguo Qva,
Li Zhoua and
Tongfei Shi*ab
aKey Laboratory of Materials Physics, Centre for Environmental and Energy Nanomaterials, Anhui Key Laboratory of Nanomaterials and Nanotechnology, CAS Center for Excellence in Nanoscience Institute of Solid State Physics, HFIPS, Chinese Academy of Sciences, Hefei 230031, China. E-mail: tfshi@issp.ac.cn; shbzhang@issp.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, China
First published on 28th March 2025
Abstract
The electrocatalytic nitrate reduction reaction (NitRR) is a promising alternative to the traditional Haber–Bosch process. However, the competitive hydrogen evolution reaction results in poor NH3 selectivity (SNH3). Here, a Cu–Co bimetallic catalyst supported on biomass-derived porous carbon (Cu–Co/BPC) is designed and synthesized. Interestingly, the catalyst presents a high NH3 yield rate of 9114.1 ± 244.8 μg h−1 cm−2 at −1.4 V (vs. RHE) and a high faradaic efficiency (FE) of 84.5 ± 1.6% at −1.0 V (vs. RHE). Notably, the SNH3 of Cu–Co/BPC catalyst is kept above 94.2% under a broad range from −1.0 to −1.4 V (vs. RHE), indicating the high NitRR-to-NH3 selectivity of Cu–Co/BPC. The combination of in situ characterization and experimental results indicates that the electron transfer occurs between Cu and Co, and many active sites are generated for adsorption and activation of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds, and hydrogenation reactions occur with adjacent H protons to improve the selectivity of NH3.
O double bonds, and hydrogenation reactions occur with adjacent H protons to improve the selectivity of NH3.
Ammonia (NH3) is a chemical product widely used in industries such as fuel, pharmaceuticals, and fertilizer manufacturing.1–3 Currently, the Haber–Bosch method is the main method used in industry to produce NH3.4,5 Different from the Haber–Bosch technology which requires high temperatures (400–500 °C) and pressures (200–300 atm),6–8 the mild approaches for nitrogen (N2) fixation such as homogeneous catalysis with organic or reductive agents,9 electrocatalysis,10 nitrogenase catalysis,11 and photocatalysis12 are promising alternatives. Amongst these, the electrochemical nitrogen reduction reaction (NRR) has gained much attention, since it takes water and air as the synthetic source and can be powered by renewable electricity.13 However, NRR is a representative gas–liquid–solid reaction system in aqueous electrolyte systems, in which the system exhibits poor N2 solubility, high N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonding energy, and sluggish kinetic process resulting in low NH3 yields and Faraday efficiencies (FE), leading to the limited activity and selectivity in NRR.13–15 To overcome the bottleneck, the electrochemical nitrate reduction reaction (NitRR) is widely recognized for its advantages of mild and environmentally friendly reaction conditions.16 The dissociation energy of the NO bond (204 kJ mol−1) is lower than that of the NN bond (941 kJ mol−1).17 The solubility of nitrate is also much greater than that of N2.18,19 These two advantages emphasize the research value of NitRR. In addition, electrochemical NitRR from various industrial wastewater and municipal sewage discharges can efficiently remove polluted nitrate and obtain high-value-added chemical products, affording a popular approach for nitrate-involving wastewater treatment and clean energy regeneration.20–22
N bonding energy, and sluggish kinetic process resulting in low NH3 yields and Faraday efficiencies (FE), leading to the limited activity and selectivity in NRR.13–15 To overcome the bottleneck, the electrochemical nitrate reduction reaction (NitRR) is widely recognized for its advantages of mild and environmentally friendly reaction conditions.16 The dissociation energy of the NO bond (204 kJ mol−1) is lower than that of the NN bond (941 kJ mol−1).17 The solubility of nitrate is also much greater than that of N2.18,19 These two advantages emphasize the research value of NitRR. In addition, electrochemical NitRR from various industrial wastewater and municipal sewage discharges can efficiently remove polluted nitrate and obtain high-value-added chemical products, affording a popular approach for nitrate-involving wastewater treatment and clean energy regeneration.20–22
Transition metals (TM) have been widely considered in the design of various catalysts owing to their wide distribution, lower cost, and unsaturated d-orbitals, which are capable of inducing a rearrangement between electrons occurring between the adsorbed molecules and the catalysts, leading to orbitals hybridization with the reactants and following activation.23–25 Until now, most of the catalysts that were reported for NitRR are popular by using transition metal as the dominant catalytic center.26,27 In particular, Cu or Co based catalysts are promising and widely implemented in NitRR.26 For example, Zhu et al. reported a nanoporous Cu/CoOOH catalyst that exhibited enhanced *NO2 affinity and reduces the energy barrier in the rate-determining *NO2H formation step, effectively facilitating NO3− reduction to NH3.26 Song and co-workers demonstrated that the bimetallic cMOF (CuxCoyHHTP) is proposed for efficient NitRR and the synergy effect between different types of single-metal sites is also revealed. Both theoretical and experimental results reveal that the Co sites can affect the electron structure of Cu sites in Cu1Co1HHTP slab and decrease the ΔG of potential determining step in NitRR process. Moreover, the Co sites bring a higher selectivity to Cu active sites for reducing *NO2 to *NO, rather than the desorption of NO2−.28 Additionally, theoretical calculation reveals that the surface oxygen on cobalt sites can stabilize the adsorbed hydrogen on cobalt oxide, which hampers the evolution of hydrogen and leads to an enhanced NitRR activity.27 These findings hold significant implications for electrocatalysis research focused on studying TMs.
Herein, regarding the advantage of Co species in the adsorption of NO3− and the formation of active H intermediate, we construct the Cu–Co bimetallic nanoparticles supported on biomass-derived porous carbon (Cu–Co/BPC) as an effective electrocatalyst for a high-efficient NH3 production via NO3−electro-reduction at ambient conditions. As a result, the as-synthesized Cu–Co/BPC affords an excellent RNH3 of 9114.1 ± 244.8 μg h−1 cm−2 at −1.4 V (vs. RHE) and a maximum FE of 84.5 ± 1.6% at −1.0 V (vs. RHE), with persistent cycle stability of 16 consecutive recycling tests in 0.1 M K2SO4 + 0.1 M KNO3 electrolyte at −1.25 V (vs. RHE). Furthermore, the differential electrochemical mass spectrometry (DEMS) was adopted to monitor the electrochemical separation of the NitRR products on the surface of the Cu–Co/BPC. Accordingly, the possible reaction sites and electrocatalytic NitRR mechanism of Cu–Co/BPC have been proposed.
Fig. 1a shows the synthetic illustration of the Cu–Co/BPC catalyst supported on coconut powder-derived biomass carbon. X-ray diffraction (XRD) was carried out to confirm the crystal structure of as-synthesized Cu–Co/BPC. As evidenced in Fig. 1b, the broad diffraction peak observed at 26.4° correspond to the (002) plane of graphitic carbon (PDF no. 41-1487), indicating the presence of graphitic carbon substrate in the catalyst. Additionally, the main diffraction peaks at 32.5°, 35.5°, 38.9° and 48.7° correlate well with the (110), (11−1), (200) and (20−2), planes of copper oxide (PDF no. 48-1548). Furthermore, peaks at 36.5°, 42.4° and 61.5°, correspond to the (111), (200) and (220) planes of CoO (PDF no. 48-1719). The above results indicate that Cu and Co elements in the Cu–Co/BPC catalyst mainly exist in the form of oxidation state. The microstructure and elemental distribution of the as-prepared Cu–Co/BPC catalyst were also investigated. The transmission electron microscopy (TEM) image suggests that the Cu–Co/BPC is composed of nanoparticles with a size of ∼20 nm (Fig. 1c). From the high-resolution transmission electron microscopy (HR-TEM) image of Cu–Co/BPC in Fig. 1c, the crystal lattice distance of d = 0.21 nm corresponds to the (111) crystal plane of CuO as recorded. Also, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) along with elemental mapping images confirmed the uniform distribution of Cu, Co, O and C elements in the Cu–Co/BPC catalyst, as illustrated in Fig. 1d, e.
 | ||
| Fig. 1 (a) Schematic for diagram illustrating synthetic procedure of Cu–Co/BPC. (b) XRD pattern and (c) TEM image (inset is the HR-TEM image) of Cu–Co/BPC. (d) HAADF-STEM and (e) corresponding elemental mapping images of Cu–Co/BPC. | ||
The N2 physisorption isotherm and pore size distribution in Fig. S1 (ESI†) demonstrate its mesopore structure, which are beneficial to exposure of the active sites and the mass transport of electrolytes during electrolysis process.29 The survey XPS spectrum of the Cu–Co/BPC catalyst is illustrated in Fig. S2 (ESI†). From the spectrum, it is evident that four elements-C, O, Cu and Co-are distributed on the surface of the Cu–Co/BPC catalyst (Fig. S2, ESI†). Fig. 2a presents the high-resolution XPS spectrum of C 1s, which displays a peak at 284.8 eV, indicating the presence of C–C/CO chemical bonds in the catalyst. Additionally, a peak at 286.6 eV correlates to the existence of C–O bonds.30–32 In addition, the high-resolution XPS spectrum of O 1s exhibits two characteristic peaks located at 532.9 eV and 538.2 eV (Fig. 2b). These two peaks are respectively representative of lattice oxygen and oxygen vacancies.33,34 The high-resolution XPS spectrum of Cu 2p (Fig. 2c) shows that the peaks for Cu 2p3/2 appear at 934.1 eV and 932.3 eV. These two characteristic peaks are respectively caused by Cu2+ and Cu0/Cu1+. The peaks for Cu 2p1/2 appear at 954.2 eV and 952.1 eV. The Cu satellite peaks are located at 941.9 eV, 944.3 eV, and 962.8 eV. These two characteristic peaks are respectively caused by Cu2+ and Cu0/Cu1+ (Fig. 2c). The high-resolution XPS spectrum of Cu 2p cannot distinguish between Cu0 and Cu1+, so the Auger Cu LMM spectrum is used to determine whether both Cu0 and Cu1+ are present in the sample. As shown in Fig. S3 (ESI†), the Auger kinetic energy peaks in the range of 906–924 eV are broad and asymmetric. Two asymmetric peaks exist at 912.8 eV and 917.3 eV, where 912.8 eV is attributed to Cu1+ and the peak at 917.3 eV is attributed to Cu0. Moreover, the Co element in the Cu–Co/BPC catalyst exhibits two main peaks in the high-resolution XPS spectrum: Co 2p3/2 and Co 2p1/2. The Co 2p3/2 peak is observed at binding energies of 780.6 eV (Co3+) and 783 eV (Co2+). Similarly, the Co 2p1/2 peak is found at 795.2 eV (Co3+) and 797 eV (Co2+) (Fig. 2d). In Cu–Co/BPC, the Cu LMM (Fig. S3, ESI†) indicates that copper primarily exists as Cu0 within the catalyst at Fig. S6.† Additionally, small amounts of CuO and Cu2O are also present in Cu–Co/BPC. From the Co 2p XPS spectrum (Fig. 2d), it can be observed that cobalt mainly exists in the Co3+ oxidation state. We analyzed the electronic properties of Cu/BPC, Co/BPC, and Cu–Co/BPC using high-resolution Cu 2p and Co 2p XPS spectra (Fig. 2c, d and S4 ESI†). We were surprised to find that after the electrocatalytic reaction, the binding energy of Cu 2p significantly shifted toward higher energy, while the characteristic peaks of Co 2p shifted toward lower energy. This phenomenon indicates electron transfer between copper and cobalt, leading to the generation of active sites.
 | ||
| Fig. 2 High-resolution (a) C 1s, (b) O 1s, (c) Cu 2p and (d) Co 2p spectra of Cu–Co/BPC. | ||
The electronic and coordinative structure of Cu–Co/BPC is further investigated by the X-ray absorption spectroscopy (XAS). The Cu K edge XANES spectra reveal that the near-edge absorption of Cu–Co/BPC is close to that of CuO, suggesting that the valence state of Cu in Cu–Co/BPC is close to +2 (Fig. S5a, ESI†). Notably, the characteristic peak at ∼1.50 Å is shown in the Fourier-transformation k3-weighted Cu K edge EXAFS spectrum of Cu–Co/BPC (Fig. S5b, ESI†), confirming the absence of Cu–O bonds, while the presence of the peak at ∼2.65 Å suggests the existence of the Cu–Co/Cu–Cu bonds. According to the Co K edge XANES spectra (Fig. S5c, ESI†), the valence states of Co species in Cu–Co/BPC is close to the valence states of Co species in CoO. As unveiled by Fig. S5d (ESI†), the presented peak at ∼1.41 Å in the EXAFS spectrum of Cu–Co/BPC confirm the existence of Co–O bonds. Additionally, the peaks at ∼2.54 Å obtained from the spectra of Cu–Co/BPC are assignable to the Co–Cu/Co–Co bonds, which is significantly inconsistent with that of Co foil, indicating the increased Co valence state of Cu–Co/BPC, ascribed to the electron transfer from Co to Cu due to lower electronegativity of Co (1.88) than Cu (1.90). This observation underscores the strong Cu–Co electronic interaction in Cu–Co/BPC.
To investigate the electrochemical activity of Cu–Co/BPC catalyst for NO3− electrochemical reduction, NitRR measurements were performed using a two-chamber cell (h cell) separated by a proton exchange membrane (Nafion 117). The cell consists of a Cu–Co/BPC catalyst coated on a carbon paper as a working electrode, Ag/AgCl (saturated KCl solution) as a reference electrode, and a Pt mesh as a counter electrode.35 Fig. 3a shows linear sweep voltammetry (LSV) curves of a mixed solution containing 0.1 M K2SO4 + 0.1 M KNO3 and 0.1 M K2SO4 solution. Without KNO3, the onset potential of the hydrogen evolution reaction (HER) was approximately −0.5 V (vs. RHE). With the addition of KNO3, the potential in the mixed solution shifted to −357 mV (vs. RHE), compared to −721 mV (vs. RHE) in the solution without KNO3, at a current density of 10 mA cm−2. This indicates an increase in potential of 360 mV (vs. RHE) with the addition of KNO3 at a current density of 10 mA cm−2. Furthermore,the solution with nitrate added always had a higher current density when the same potential was applied, which diaplays that the catalyst has a high NitRR activity. The LSV results showed two peaks of reduction. The reduction peak at S1 near 0.08 V is attributed to the reduction of NO3− to NO2−, while the main reaction at the peak S2 involves the reduction of NO2− to NH3. Since the chemical reaction at peak S2 is predominant, the onset potential is determined near the vicinity of the S2 peak. The as-synthesized Cu–Co/BPC electrocatalyst was operated at six different potentials, which were selected in the range of −1 V to −1.4 V (vs. RHE). Each potential was tested for two hours of it in a mixture of 0.1 M KNO3 and 0.1 M K2SO4 (Fig. S6a, ESI†). The ultraviolet–visible (UV–Vis) absorption spectra of electrolytes obtained for each potential colored with indophenol blue reagent were measured (Fig. S6b, ESI†). The electrolysis product of NH3 at a given potential was quantitatively determined by the indophenol blue method (Fig. S7, ESI†). Fig. 3b show the calculated NH3 yield rate and corresponding FE of Cu–Co/BPC at diverse potentials. It shows that the maximum NH3 yield rate of 9114.1 ± 244.8 μg h−1 cm−2 is achieved at −1.4 V (vs. RHE) while the highest FE of 84.5 ± 1.6% is obtained at −1.0 V (vs. RHE). Obviously, the FE decreases with the applied potentials shifts to more negative values, mainly due to the competition of the HER on catalyst surface.36 After the TEM characterization (Fig. S8, ESI†), we found that the structure of the catalyst remained largely unchanged during the reaction. That indicate the morphology and structural stability of the catalyst post-reaction are relatively well. We also tested the performance of Cu–Co/BPC, Cu/BPC and Co/BPC at −1.25 V (vs. RHE), which demonstrated the superiority of Cu–Co/BPC (Fig. S9, ESI†). In addition, the few by-product NO2− is detected in the electrolytes (Fig. S10, ESI†), and the selectivity of NH3 (SNH3) of Cu–Co/BPC at different applied potentials were calculated as displayed in Fig. 3c. The SNH3 of Cu–Co/BPC can reach the maximum FE of 97.8 ± 0.65% at −1.0 V (vs. RHE) and excellent FE of NH3 over 94.0% under a broad range from −1.0 to −1.4 V (vs. RHE) (Fig. 3c), demonstrating the high selective reduction of nitrate to NH3.
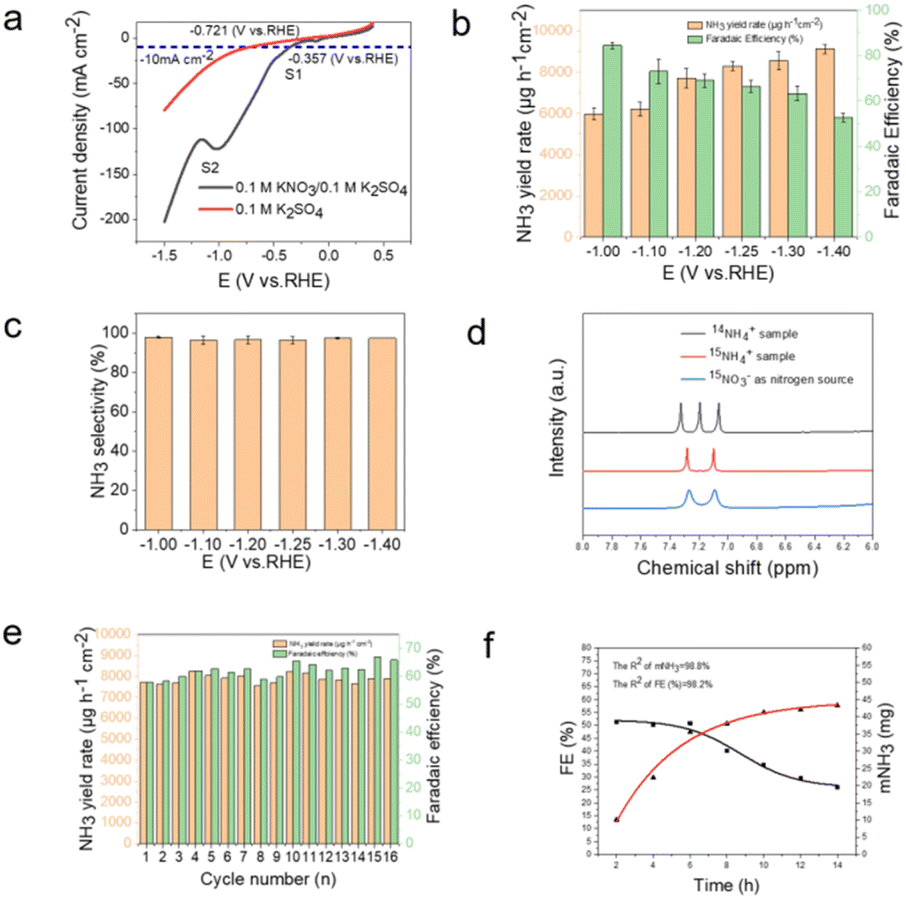 | ||
| Fig. 3 (a) LSV curves of Cu–Co/BPC catalyst in 0.1 M K2SO4 and 0.1 M K2SO4 + 0.1 M KNO3 solution. (b) RNH3 and FE of Cu–Co/BPC catalyst obtained at different potentials for 2 h NitRR measurement. (c) SNH3 of Cu–Co/BPC catalyst obtained at different potentials. (d) 1H NMR spectra of Cu–Co/BPC catalyst using 15NO3− and 14NO3− as nitrogen source for NitRR and standards (15NH4)2SO4. (e) Recycling tests for Cu–Co/BPC catalyst during NitRR at −1.25 V (vs. RHE). (f) The FE and NH3 yield were monitored over a 14 h NitRR conducted at −1.25 V (vs. RHE). | ||
To eliminate potential experimental errors caused by the self-disintegration of catalyst and environmental interference, series control experiments were carried out. As shown in Fig. S11 (ESI†), the negligible enhancing of signals associated with ammonia is observed in electrolysis at −1.25 V (vs. RHE) for 2 h in 0.1 M K2SO4 electrolyte without NO3− or carbon paper (CP) without Cu–Co/BPC catalyst, and Cu–Co/BPC electrode under open circuit potential (OCP) for 2 h in 0.1 M K2SO4 + 0.1 M KNO3 electrolyte. Furthermore, 15N isotope labelling experiments are also conducted. The 1H nuclear magnetic resonance (1H NMR) spectra were measured to verify the origin of produced NH3. As exhibited in Fig. 3d, doublet coupled peaks of 15NH4+ is observed in the 1H NMR spectrum when using 15NO3− as the nitrogen source for NitRR, confirming the generated NH3 is produced from NitRR over Cu–Co/BPC catalyst.
The stability of electrochemical NH3 production is an important indicator of the electrochemical performance of electrocatalysts. Therefore, the as-synthesized Cu–Co/BPC electrocatalyst was conducted out for 16 consecutive recycling tests in 0.1 M K2SO4 + 0.1 M KNO3 solution at −1.25 V (vs..RHE). After recycling tests, it can be clearly seen in Fig. 3e that no remarkable decay in NH3 yield rate and FE during the NitRR process. During the two-hour electrochemical reaction, the current gradually increased to 180 mA cm−2 (Fig. S12, ESI†). Besides superior stability, the durability of the catalyst is another crucial parameter for practical applications. Then a 50 ml electrolysis cell was used for the 10 h NitRR experiments in our study. The Cu–Co/BPC catalyst was electrolyzed in a mixed solution of 0.1 M K2SO4 + 0.1 M KNO3 at −1.25 V (vs..RHE) for 10 h with samples of the solution taken every 2 h. As shown in Fig. 3f, NH3 production can be steadily increased with a slight decrease in FE, further demonstrating the superior stability of the Cu–Co/BPC catalyst. Notably, After electrolysis, the high-resolution Cu 2p and Co 2p XPS spectra were carried out to analyze the electronic properties of Cu–Co/BPC before and after NitRR process (Fig. S13, ESI†). Interestingly, after electrocatalytic reaction, the Cu 2p significantly shifted towards higher binding energies (Fig. S13a, ESI†), while the characteristic peaks of Co2p shifted to the lower binding energy (Fig. S13b, ESI†), indicating the existence of charge transfer from Cu atoms to Co atoms during electrocatalytic NitRR process, which results in high electrocatalytic selectivity and FE of Cu–Co/BPC for NH3 synthesis. The Nyquist plot (Fig. S14, ESI†) shows that the catalyst exhibits the strongest corrosion resistance at this potential of −1.25 V vs. RHE. The improvement in corrosion resistance promotes the stability of the conversion of nitrate to ammonia. However, when the potential is −1.25 V vs. RHE, the reaction activity is relatively low. The reaction activity initially decreases with the rise in potential, reaches a maximum, and then increases.
Moreover, to further investigate the process of the NitRR reaction of the Cu–Co/BPC catalyst, the in situ Differential Electrochemical Mass Spectrometer (DEMS) was performed. The in situ DEMS is an instrument capable of analyzing the intermediates of electrochemical reactions as well as the final products in milliseconds. With the applied voltage is −1.25 V (vs..RHE), the m/z signals of 17, 30, 31, 32, 33 and 46, corresponding to NH3, NO, HNO, NH2O, NH2OH and NO2, respectively, appeared during ten continuous cycles. As shown in the Fig. 4, the amount of NH3 gradually increased with the increase of reaction time, and the data were finally stable, indicating the catalytic reaction catalyzed by the catalyst has a relatively good stability. The other intermediate products show a decreasing trend with increasing reaction time, a phenomenon that supports the high FE of the reaction. The other intermediate products were significantly less than NH3, indicating that NH3 was the main product (Fig. 4). This above result suggests that the main reaction pathway of NitRR on Cu–Co/BPC catalyst may be NO3− → *NO3 → *NO2 → *NO → *NHO → *NH2O → *NH2OH → *NH3 → NH3(g). Furthermore, we also utilized in situ attenuated total reflection surface-enhanced infrared adsorption spectroscopy (ATR-SEIRAS) to monitor the evolution of NitRR intermediates. Fig. S15 (ESI†) shows the infrared signals when the in situ electrocatalytic NitRR on Cu–Co/BPC during the negative scan from OCP V to −1.4 V (vs. RHE). At the applied potential, the significantly enhanced infrared peaks at 1390 cm−1 were assigned to N–O symmetric stretching vibration of NO3−, indicates the activation and consumption of NO3− catalyzed by Cu–Co/BPC. Meanwhile, the vibration bands of adsorbed NO in bridge adsorption mode were detected at 1525 cm−1. The infrared bands, centred at 1647 cm−1, are assigned to the bending vibrations H–O–H loosely bound water molecules. In addition, the gradually enhanced infrared peaks at 1458 cm−1, which can be ascribed to N–H bending vibration of NH4+. Based on the in situ ATR-SEIRAS analysis, we proposed the following pathway for the NitRR on Cu–Co/BPC surface: NO3− → *NO3 → *NO → *NH3. This observation agrees with the in situ DEMS results.
 | ||
| Fig. 4 In situ DEMS measurements of NitRR over Cu–Co/BPC. | ||
In summary, the Cu–Co bimetallic nanoparticles supported on biomass-derived porous carbon (Cu–Co/BPC) have been successfully synthesized as an effective electrocatalyst for electrocatalytic NitRR in 0.1 M K2SO4 + 0.1 M KNO3 electrolyte. As a result, the as-synthesized Cu–Co/BPC affords an excellent RNH3 of 9114.1 ± 244.8 μg h−1 cm−2 at −1.4 V (vs. RHE) and a maximum FE of 84.5 ± 1.6% at −1.0 V (vs. RHE), with outstanding stability during 10 h NitRR. We compared the performance of Cu–Co/BPC with other catalysts, demonstrating the superior performance of Cu–Co/BPC (Table S1, ESI†). The 15N isotope labelling experiments confirmed that the produced NH3 indeed originated from the nitrate reduction reaction catalyzed by Cu–Co/BPC. Combining electrochemical experiments with in situ characterization, the possible reaction sites and electrocatalytic NitRR mechanism of Cu–Co/BPC have been proposed.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Daopeng Li: conceptualization, investigation, visualization, writing – original draft. Shengbo Zhang: data curation, resources, supervision, writing – review & editing. Zhixian Mao: investigation, resources, formal analysis. KuiHu: investigation, resources, formal analysis. Dongnan Zhao: investigation, resources, formal analysis. Min Liu: software. Zhengguo Qu: software. Li Zhou: software. Tongfei Shi: funding acquisition, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Anhui Provincial Natural Science Foundation (Grant No. 2408085MB021).Notes and references
- H. Zhang, H. Wang, X. Cao, M. Chen, Y. Liu, Y. Zhou, M. Huang, L. Xia, Y. Wang, T. Li, D. Zheng, Y. Luo, S. Sun, X. Zhao and X. Sun, Adv. Mater., 2024, 36, 2312746 CrossRef CAS PubMed.
- C. Yu, X. Huang, H. Chen, H. C. J. Godfray, J. S. Wright, J. W. Hall, P. Gong, S. Ni, S. Qiao, G. Huang, Y. Xiao, J. Zhang, Z. Feng, X. Ju, P. Ciais, N. C. Stenseth, D. O. Hessen, Z. Sun, L. Yu, W. Cai, H. Fu, X. Huang, C. Zhang, H. Liu and J. Taylor, Nature, 2019, 567, 516–520 CrossRef CAS PubMed.
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrochim. Acta, 2017, 227, 77–84 CrossRef.
- Y. Wang, S. Wang, Y. Fu, J. Sang, P. Wei, R. Li, D. Gao, G. Wang and X. Bao, Nat. Commun., 2025, 16, 897 CrossRef CAS PubMed.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 14131–14137 CrossRef CAS PubMed.
- J.-Y. Fang, Q.-Z. Zheng, Y.-Y. Lou, K.-M. Zhao, S.-N. Hu, G. Li, O. Akdim, X.-Y. Huang and S.-G. Sun, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, ACS Catal., 2021, 11, 15135–15140 CAS.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Rüscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Angew. Chem., Int. Ed., 2022, 61, e202202556 CrossRef CAS PubMed.
- G. Zhang, Q. Li, X. Wang, L. Jin and Q. Liao, J. Am. Chem. Soc., 2025, 147(4), 3747–3757 CrossRef CAS PubMed.
- Y. Sun, S. Ding, B. Xia, J. Duan, M. Antonietti and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202115198 CrossRef CAS PubMed.
- C. C. Lee, W. Kang, A. J. Jasniewski, M. T. Stiebritz, K. Tanifuji, M. W. Ribbe and Y. Hu, Nat. Catal., 2022, 5, 443–454 CrossRef CAS PubMed.
- P. Xia, X. Pan, S. Jiang, J. Yu, B. He, P. M. Ismail, W. Bai, J. Yang, L. Yang and H. Zhang, Adv. Mater., 2022, 34, 2200563 Search PubMed.
- X. Mao, X. Bai, G. Wu, Q. Qin, A. P. O'Mullane, Y. Jiao and A. Du, J. Am. Chem. Soc., 2024, 146, 18743–18752 Search PubMed.
- G.-F. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L.-X. Ding and H. Wang, Small Methods, 2019, 3, 1800337 Search PubMed.
- Q. Huang, B. Xia, M. Li, H. Guan, M. Antonietti and S. Chen, Nat. Commun., 2024, 15, 4157 Search PubMed.
- N. C. Kani, J. A. Gauthier, A. Prajapati, J. Edgington, I. Bordawekar, W. Shields, M. Shields, L. C. Seitz, A. R. Singh and M. R. Singh, Energy Environ. Sci., 2021, 14, 6349–6359 Search PubMed.
- C. Liu, G. Zhang, W. Zhang, Z. Gu and G. Zhu, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2209979120 CAS.
- Y. Huang, C. He, C. Cheng, S. Han, M. He, Y. Wang, N. Meng and B. Zhang, Nat. Commun., 2023, 14, 7368 CAS.
- J. Zhou, M. Wen, R. Huang, Q. Wu, Y. Luo, Y. Tian, G. Wei and Y. Fu, Energy Environ. Sci., 2023, 16, 2611–2620 CAS.
- Y. Xi, Y. Xiang, T. Bao, Z. Li, C. Zhang, L. Yuan, J. Li, Y. Bi, C. Yu and C. Liu, Angew. Chem., Int. Ed., 2024, 63, e202409163 CAS.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Elucidating the activity, Energy Environ. Sci., 2022, 15, 760–770 CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CAS.
- J. Wang, S. Zhang, C. Wang, K. Li, Y. Zha, M. Liu, H. Zhang and T. Shi, Inorg. Chem. Front., 2022, 9, 2374–2378 CAS.
- S. Wang, Y. Pi, Y. Song, Y. Jiang and L. Zhou, Water Res., 2020, 173, 115539 CAS.
- K. Yang, M. Li, T. Gao, G. Xu, D. Li, Y. Zheng, Q. Li and J. Duan, Nat. Commun., 2024, 15, 7060 CAS.
- X. Zhou, W. Xu, Y. Liang, H. Jiang, Z. Li, S. Wu, Z. Gao, Z. Cui and S. Zhu, ACS Catal., 2024, 14, 12251–12259 CAS.
- Y. Liu, W. Qiu, P. Wang, R. Li, K. Liu and K. M. Omer, Appl. Catal. B: Environ., 2024, 340, 123228 CrossRef CAS.
- P. Liu, J. Yan, H. Huang and W. Song, Chem. Eng. J., 2023, 466, 143134 CrossRef CAS.
- O. Šolcová, L. Matějová and P. Schneider, Appl. Catal. A: Gen., 2006, 313, 167–176 CrossRef.
- S. Zhang, J. Wu, M. Zheng, X. Jin, Z. Shen, Z. Li, Y. Wang, Q. Wang, X. Wang, H. Wei, J. Zhang, P. Wang, S. Zhang, L. Yu, L. Dong, Q. Zhu, H. Zhang and J. Lu, Nat. Commun., 2023, 14, 3634 CrossRef CAS PubMed.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- Y. Zha, M. Liu, J. Wang, J. Feng, D. Li, D. Zhao, S. Zhang and T. Shi, RSC Adv., 2023, 13, 9839–9844 RSC.
- K. Liu, H. Li, M. Xie, P. Wang, Z. Jin, Y. Liu, M. Zhou, P. Li and G. Yu, J. Am. Chem. Soc., 2024, 146, 7779–7790 CrossRef CAS PubMed.
- D. Liu, X. Li, S. Chen, H. Yan, C. Wang, C. Wu, Y. A. Haleem, S. Duan, J. Lu, B. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Nat. Energy, 2019, 4, 512–518 CrossRef CAS.
- F. Zhu, J. Ge, Y. Gao, S. Li, Y. Chen, J. Tu, M. Wang and S. Jiao, Exploration, 2023, 3, 20210186 CrossRef PubMed.
- D. Liu, L. Qiao, Y. Chen, P. Zhou, J. Feng, C. C. Leong, K. W. Ng, S. Peng, S. Wang, W. F. Ip and H. Pan, Appl. Catal. B: Environ., 2023, 324, 122293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00860c |
| This journal is © The Royal Society of Chemistry 2025 |