
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of ultrafast flavin photoreduction in the active site of flavoenzyme LSD1 histone demethylase†
Bo
Zhuang
 *a,
Rivo
Ramodiharilafy
b,
Alexey
Aleksandrov
*b,
Ursula
Liebl
b and
Marten H.
Vos
*b
*a,
Rivo
Ramodiharilafy
b,
Alexey
Aleksandrov
*b,
Ursula
Liebl
b and
Marten H.
Vos
*b
aBeijing National Laboratory for Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, 100871 Beijing, China. E-mail: zhuangbo@pku.edu.cn
bLOB, CNRS, INSERM, École Polytechnique, Institut Polytechnique de Paris, 91120 Palaiseau, France. E-mail: alexey.aleksandrov@polytechnique.edu; marten.vos@polytechnique.edu
First published on 25th November 2024
Abstract
Photoreduction of oxidized flavins has a functional role in photocatalytic and photoreceptor flavoproteins. In flavoproteins without light-dependent physiological functions, ultrafast, reversible flavin photoreduction is supposedly photoprotective by nature, and holds potential for nonnatural photocatalytic applications. In this work, we combine protein mutagenesis, ultrafast spectroscopy, molecular dynamics simulations and quantum mechanics calculations to investigate the nonfunctional flavin photoreduction in a flavoenzyme, lysine-specific demethylase 1 (LSD1) which is pivotal in DNA transcription. LSD1 harbors an oxidized flavin adenine dinucleotide (FAD) cofactor and multiple electron-donating residues in the active site. Upon photoexcitation, the FAD cofactor is photoreduced in <200 fs by electron transfer (ET) from nearby residue(s), and the charge pairs recombine in ca. 2 ps. Site-directed mutagenesis pinpoints a specific tryptophan residue, W751, as the primary electron donor, whereas a tyrosine residue, Y761, despite being located closer to the flavin ring, does not effectively contribute to the process. Based on a hybrid quantum-classical computational approach, we characterize the W751–FAD and Y761–FAD charge-transfer states (CTW751 and CTY761, respectively), as well as the FAD locally excited state (LEFAD), and demonstrate that the coupling between LEFAD and CTW751 is larger than those involving CTY761 by an order of magnitude, rationalizing the experimental observations. More generally, this work highlights the role of the intrinsic protein environment and details of donor–acceptor molecular configurations on the dynamics of short-range ET involving a flavin cofactor and amino acid residue(s).
Introduction
Flavins are arguably the most versatile cofactors in nature. Proteins containing flavin cofactors, named flavoproteins, are key components in a wide range of biological processes essential to life,1–5 They have also emerged as promising tools for biotechnological applications,3,6–9 including as photobiocatalytic tools.10–16 Flavins can adopt multiple redox and protonation states, with the oxidized form being the most common resting state in flavoproteins.17 Oxidized flavins absorb near-ultraviolet and blue light, and photoexcitation of flavoproteins usually leads to photoreduction of protein-bound flavins, a process that has a physiological function in some photocatalytic and photoreceptor flavoproteins. For instance, in fatty acid photodecarboxylase,18,19 cryptochrome20,21 and blue light using flavin (BLUF) domain,22,23 photoreduction of a flavin cofactor by electron transfer (ET) from substrates or protein residues constitutes the primary step in their photocycles.Photoreduction of oxidized flavins is also ubiquitous among flavoproteins that do not perform light-dependent physiological functions. In these systems, absorption of a photon induces ultrafast ET from nearby residues, typically tryptophan and tyrosine, to flavins, on a femtosecond-picosecond timescale, followed by efficient recombination of the separated charges on a picosecond timescale.24–40 Such an ultrafast, reversible process may serve as natural photoprotective self-quenching to dissipate the photon energy, preventing undesirable photochemical reactions that may result in harmful radical species.30,41 At the same time, this process will also impede any potential photocatalytic applications of these enzymes, as ultrafast flavin photoreduction by protein residues may outcompete productive photoreaction with external substrates. Identifying the key factors that regulate flavin photoreduction by ET from internal protein residues, as well as finding the balance between the photostability of the enzymes and their photocatalytic ability toward external substrates, is therefore crucial for the development of new oxidized-flavin-based photobiocatalysts.30
In this work, we focus on the non-functional photoinduced ET processes in a flavin-dependent amine oxidase, human lysine-specific demethylase 1 (LSD1, Fig. 1A), which has multiple essential roles in mammalian biology. It harbors an oxidized flavin adenine dinucleotide (FAD) cofactor, and specifically demethylates histone lysine, thus being pivotal in regulating DNA transcription.43–45 LSD1 is a promising molecular target for cancer therapy,43,45 and has been found to be relevant to neurodegenerative diseases.46,47
 | ||
| Fig. 1 (A) X-ray crystallographic structure of WT LSD1 (PDB entry: 2DW4).42 (B) Electron-donating residues in the active site of WT LSD1 in the crystal structure (distances given in Å). (C) Steady-state absorption spectra of WT, W751F and Y761F LSD1. (D) Distributions of the edge-to-edge distances between FAD and close-by tyrosine and tryptophan residues in 200 ns MD simulations. (E) Dynamics of the angles between the ring planes of the isoalloxazine moiety of FAD and the indole or phenol moiety of Y761 or W751, during the MD simulations of WT, W751F and Y761F LSD1, as indicated. | ||
The active site of LSD1 is characterized by remarkable conformational flexibility and versatile substrate- and inhibitor-binding properties, including for large compounds encompassing multiple aromatic groups.43,48–51 This versatility in the accommodation of peptides and other large molecules may make the enzyme a suitable template for engineering other biocatalysts, including photobiocatalysts given the presence of flavin in the active site. Yet, multiple tryptophan and tyrosine residues reside in the vicinity (<8 Å) of the FAD cofactor, among which W751 and Y761 are located within van der Waals contact of FAD (Fig. 1B). As mentioned above, photoinduced ET from these residues can potentially compete with photocatalytic reactions involving external molecules in the active site.
In this context, based on femtosecond time-resolved fluorescence and transient absorption measurements on wild-type and genetically modified proteins, the present work unravels the photoinduced ET dynamics and identifies the transiently formed radical intermediates upon photoexcitation of LSD1. This approach allows us to pinpoint W751 as the primary electron donor, despite a greater donor–acceptor separation distance compared to Y761. Employing molecular dynamics (MD) simulations and hybrid quantum mechanics/molecular mechanics (QM/MM) calculations, we explore the active-site conformational dynamics of LSD1 and characterize the flavin locally excited (LE) state and the charge-transfer (CT) states involving W751 and Y761, to obtain detailed insights into the mechanism of ultrafast photoreduction of the protein-bound flavin. The implications for possible photobiocatalytic applications will be discussed.
Results and discussion
We prepared wild-type (WT) LSD1, as well as its W751F and Y761F variants, where W751 or Y761 was genetically modified to redox-inactive phenylalanine (F). The mutations slightly perturb the absorption spectrum of FAD (Fig. 1C), reflecting the changes in the electrostatic environment around FAD.52 To investigate the configurations of FAD and nearby electron-donating residues in detail, we performed MD simulations for the LSD1 proteins. In the simulations, although the edge-to-edge distance (the shortest distance between any non-hydrogen atoms from two aromatic rings) from residues Y571 and W695 to FAD displays broader distributions in the mutated proteins, the distances from W751 and Y761 to FAD in W751F and Y761F LSD1 remain similar to those of WT LSD1, with average distances of 3.4 and 4.5 Å, respectively (Fig. 1D and S1†). The orientations of W751 or Y761 are largely unaffected by the mutations as well (Fig. 1E), although modest increases in the angles between the ring planes were observed (Fig. S2†). These results indicate that, although the mutations introduce changes in the protein active site, W751 and Y761 remain in close interaction with FAD in the corresponding variants. Notably, Y761 exhibits higher flexibility than W751, which can be associated with the functional role of Y761 in the accommodation of substrates.53To examine the excited-state dynamics, we performed time-resolved fluorescence measurements with femtosecond time resolution on the LSD1 proteins. As shown in Fig. 2, the characteristic emission spectra of FAD centered at ca. 525 nm were clearly resolved. In WT and Y761F LSD1, the fluorescence decays predominantly in ca. 180 and 270 fs, respectively (Fig. 2A, C, and Table S1†). Such short lifetimes of the singlet excited state of FAD (FAD*, i.e., the LE state of FAD) are indicative of quenching by ultrafast ET from nearby residue(s). On the other hand, an overall much slower, biexponential, fluorescence decay was detected in W751F LSD1, with time constants of 9 ps and 200 ps (Fig. 2B), an order of magnitude slower than WT and Y761F LSD1.
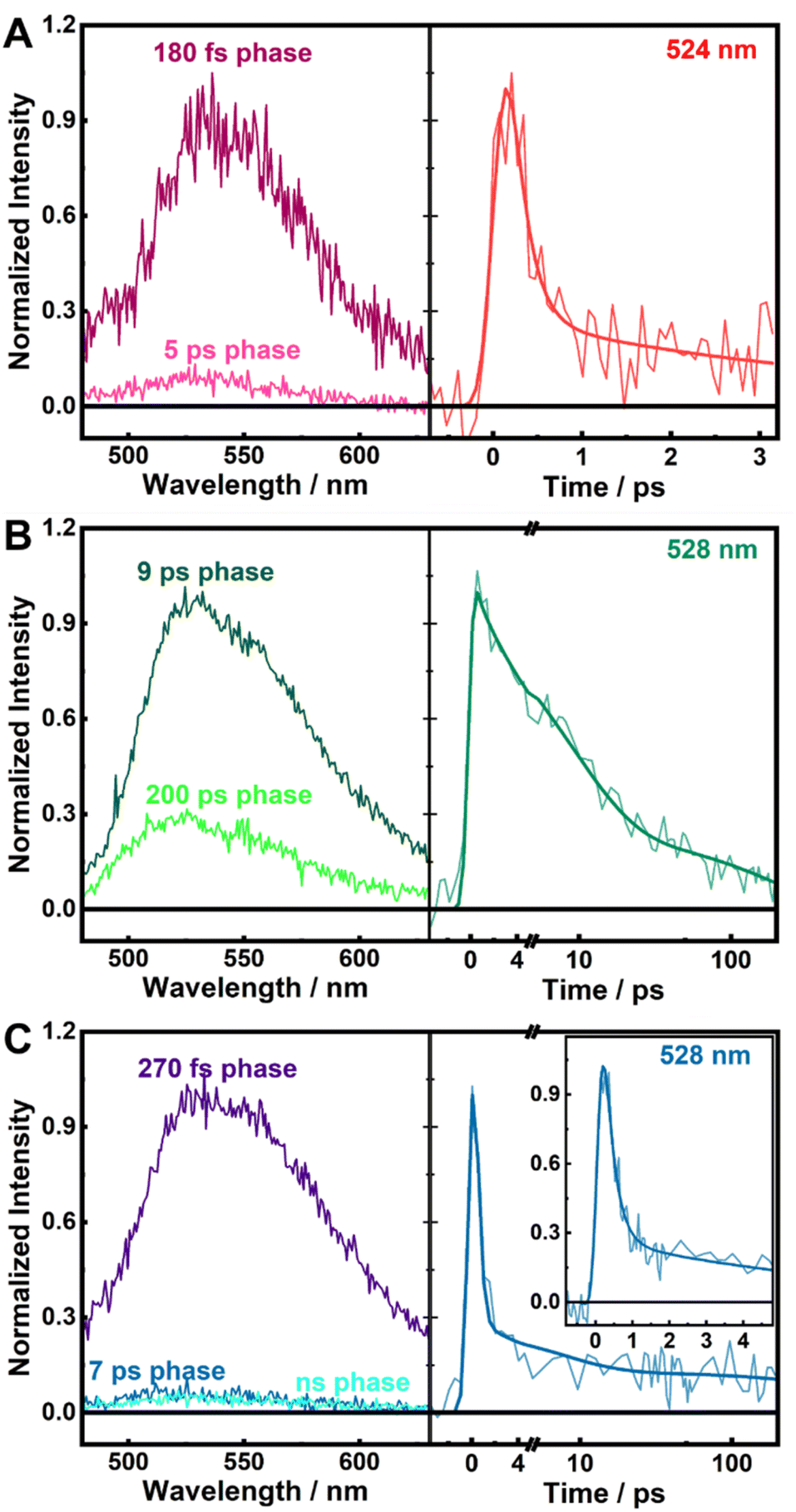 | ||
| Fig. 2 Decay associated spectra (DAS) from the global analysis of fluorescence decays (left) and kinetic traces at selected wavelengths (right) of WT (A), W751F (B, linear-logarithmic time scale), and Y761F (C, linear-logarithmic time scale; inset shows a zoom of the time range of a few picoseconds) LSD1, with 390 nm excitation. | ||
To characterize the formation of photoproducts and the subsequent charge recombination processes, we further performed transient absorption (TA) measurements on the LSD1 proteins. Fig. 3A shows the TA spectra of WT, W751F, and Y761F LSD1 recorded 3 ps after excitation. The evolution-associated spectra (EAS), obtained from global analysis of the TA data assuming linear reaction schemes, are given in Fig. S3.† These results can be compared with the reference TA spectra of the transient species involved in photochemical processes of FAD obtained in FNR proteins from Bacillus subtilis (BsFNR; Fig. S4†).27
 | ||
| Fig. 3 Transient absorption spectra of LSD1 variants measured at 3 ps after excitation (A), and normalized kinetic traces at 460 nm (B), with 390 nm excitation. | ||
In all three LSD1 proteins, the negative bands at ca. 450 nm are dominated by the ground-state bleaching of FAD (cf.Fig. 1B). The TA spectra of WT LSD1 and Y761F LSD1 are further characterized by induced absorption bands peaking at ca. 600 nm, which correspond well to the spectrum of transient W˙+ species in an unrelaxed protein environment, and the spectral features of FAD* are clearly absent (Fig. S4†).27 Additionally, the TA spectrum of WT LSD1 does not contain apparent contributions of Y˙+.27,38 Altogether, this demonstrates the ultrafast flavin photoreduction and the formation of a W˙+/FAD˙− radical pair as the photoproduct. The TA spectrum of W751F LSD1, by contrast, is characterized by a marked dip at ca. 570 nm, which resembles a stimulated emission (SE) feature, but is superimposed on a broad induced absorption band. Further spectral analysis suggests that a mixture of FAD* and a W˙+/FAD˙− radical pair can best reproduce the observed spectral features. (Fig. S5†). The persistence of FAD* for up to hundreds of picoseconds upon the excitation of W751F LSD1 is consistent with the time-resolved fluorescence measurement (Fig. 2B).
The TA kinetic traces probed at 460 nm (Fig. 3B) represent the recovery of ground-state FAD through charge recombination. In WT and Y761F LSD1, the separated charges mainly recombine in ca. 2 ps, with additional minor slower components (Fig. 3B and S3a and c†), which may be due to protein heterogeneity or the presence of a small fraction of unbound FAD, but were too small to be reliably analyzed (<1% of the initial bleaching signals). In W751F LSD1, the GSB signal displays a slow, multiphasic recovery, with fitted lifetimes of 6 and 250 ps (similar to those found in the fluorescence decay), as well as a long-lived component that was most likely due to the FAD triplet state (Fig. S3b†).
Taken together, our spectroscopic data point to W751 as a more effective electron donor than Y761, for the following reasons. First, in W751F LSD1, in the absence of W751, FAD* decays on a much longer, picosecond timescale compared to WT LSD1, and a more distant tryptophan, most likely W695 (Fig. 1B, D and S5†) is involved in the quenching process. Second, in Y761F LSD1, W751 quenches FAD* predominantly in 270 fs, close to the main fluorescence decay of WT LSD1 (Fig. 2A, C, and Table S1†), and in both systems, the W˙+/FAD˙− radical pair accumulates as the photoproduct (Fig. 3A). Additionally, In WT and Y761F, sequential ET beyond W751 is unlikely to occur, as other tryptophan and tyrosine residues (including Y761) are all located too far away (>8 Å) from W751 to allow further ET that can outcompete the picosecond charge recombination process (Fig. 3B).
Despite being a less effective quencher, Y761 is located closer to the flavin ring of FAD than W751 in LSD1 (ca. 3.5 Å versus 4.5 Å; Fig. 1B and D). Reportedly in other flavoprotein systems, such as BsFNR27 and flavodoxin from Helicobacter pylori (HpFd),37 a tyrosine residue located at a similar distance (ca. 3.5 Å) from the flavin cofactor quenches the flavin excited state in ca. 200 fs, comparable to those in WT and Y761F LSD1. It is intriguing to note that Y761 does not effectively participate in the quenching process in competition with W751. To obtain a rationale behind this observation, we adopted a QM/MM approach to investigate the photoinduced ET in LSD1, by estimating the energy levels of and the coupling between relevant electronic states, i.e., the FAD LE state and CT states involving W751 and Y761.
Specifically, the QM region, which was described at a density functional theory (DFT) level, included the flavin (isoalloxazine) ring of FAD, as well as the side chains of W751 and Y761, while the remaining part of the protein and water molecules were described by classical force fields. As the dynamics of photoinduced short-range ET in proteins are assumed to be faster than the local relaxation, the protein environment can be considered ‘frozen’ during the ET process, with a heterogeneous distribution of electrostatics that can reasonably be sampled from ground-state MD simulations.27,35,54 Therefore, we carried out time-dependent DFT (TDDFT) calculations along the ground-state MD trajectories after DFT geometry optimizations of the QM regions. Hole–electron analysis was further performed to reveal the underlying characteristics of the excited state, which described the changes in electron density upon the corresponding electronic excitation;55 in a typical LE state, the hole and electron are both localized on the flavin ring of FAD, and in a well-defined CT state the transferred electron is localized on the flavin ring, and the hole is localized on the donor (i.e., W751 or Y761 in LSD1).
As shown in Fig. 4A–C, two excited states with distinct CT characters and energy levels lower than that of the FAD LE state (corresponding to the S0 → S1 transition of FAD; hereafter referred to as LEFAD) were identified. CTY761 involves ET from Y761 to FAD and CTW751 involves ET from W751 to FAD, as evident from the corresponding hole–electron distributions. The oscillator strengths of LEFAD have values of ca. 0.19 (Table S2†), indicating that they can be populated directly by photoexcitation. By contrast, the CT states cannot be accessed directly, as the oscillator strengths of these transitions are virtually zero. These findings are consistent with the ultrafast experiments in which photoinduced charge separation in LSD1 is achieved following the decay of FAD* (Fig. 2 and 3). Notably, the energy levels of the CTY761 are slightly lower than CTW751 (Fig. 4D; with averaged values of 2.02 and 2.25 eV respectively), contradicting the general assumption that ET from tryptophan is energetically favorable compared to tyrosine in flavoproteins.56 As the CT states are close in energy, the ET dynamics should be predominantly determined by the electronic coupling matrix (Hab) between the LE and CT states, which depends on the overlap of the wave functions of relevant electronic states, and is sensitive to the relative molecular configuration and distance of the donor and acceptor molecules. Specifically, we estimated Hab with the two-state generalized Mulliken–Hush (GMH) approximation (eqn (S1†)).54,57–59 As shown in Fig. 4E and Table S2,† the Hab between LEFAD and CTW751 are an order of magnitude larger than those between LEFAD and CTY761. As larger coupling leads to a higher ET rate (which approximately scales with |Hab|2), this rationalizes our experimental finding that W751 is a more effective electron donor in ultrafast flavin photoreduction in WT LSD1 than Y761. Additionally, in the simulations of W751F and Y761F LSD1, LEFAD, CTW751 and CTY761 states with hole–electron distributions similar to those in WT LSD1 were identified (Fig. S6†). It is worth noting that in Y761F, on average a slightly smaller Hab between LEFAD and CTW751 was observed compared to that in WT LSD (5.92 and 6.42 meV, respectively). This finding is fully in line with the slightly faster quenching of FAD* observed in WT LSD1 compared to Y761F LSD1 (Fig. 2A, C, and Table S1†).
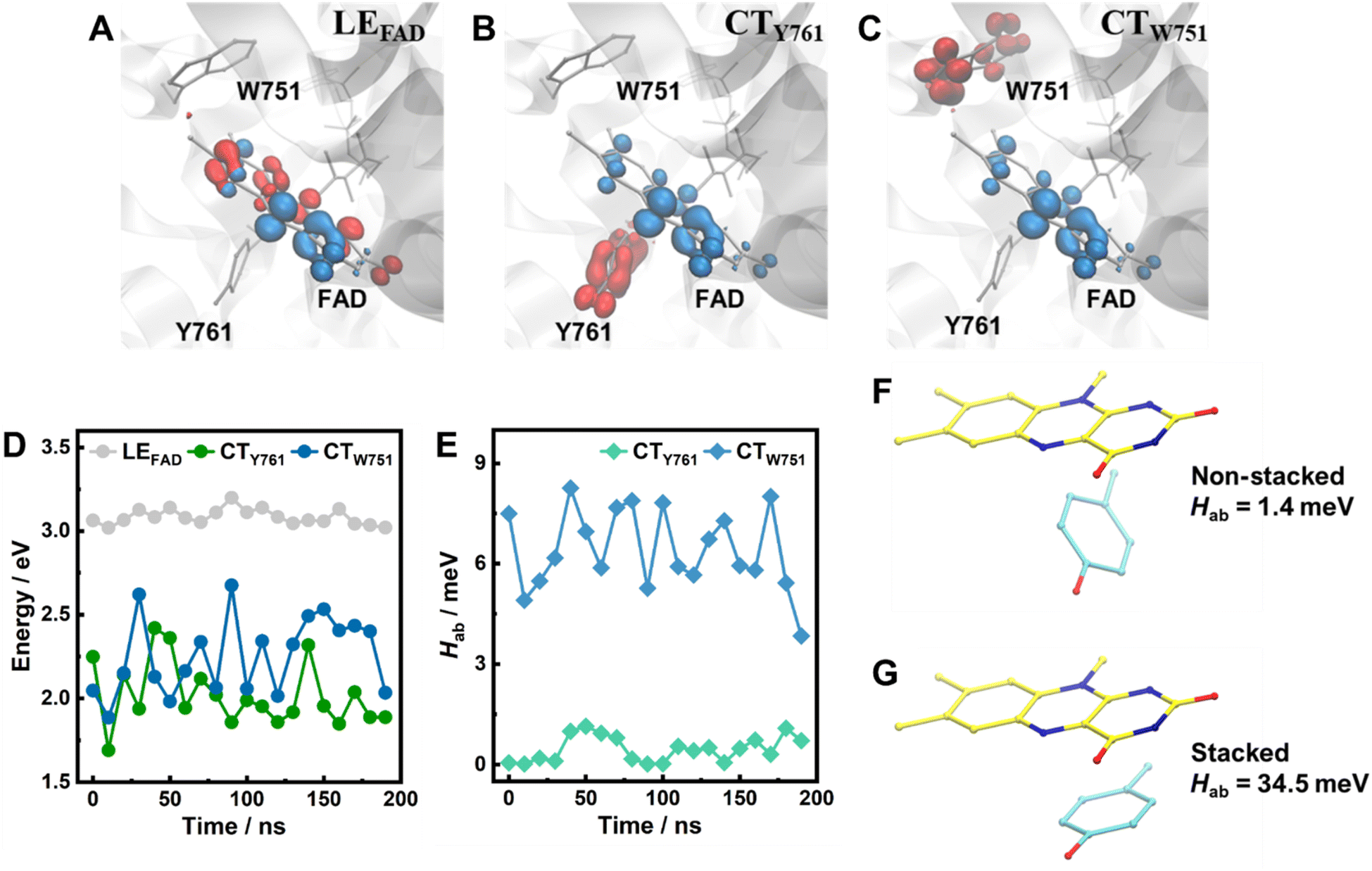 | ||
| Fig. 4 (A–C) Hole–electron distributions of the FAD LE state (A), the lowest-lying CT states involving Y761 (B) and W751 (C). Red and blue regions denote the hole and electron distributions, respectively (isovalue = 0.004). Hydrogen atoms are not shown for clarity. (D and E) The energy levels of these states (D), with respect to the electronic ground state, as well as the electronic couplings between the LE and CT states in WT LSD1 (E), along the MD trajectories. (F and G) Calculated electronic coupling between flavin LE state and tyrosine–flavin CT state in non-stacked (F) and stacked (G) donor–acceptor configurations but with similar separation distance (ca. 4 Å). Hydrogen atoms are not shown for clarity. | ||
We also found that subtle changes in the donor–acceptor configurations (Fig. 1D and E) can lead to considerable differences in Hab (Fig. 4E, and S8†). Specifically, we found that for Y761 in WT LSD1, shorter distances and smaller angles correlate well with larger Hab values (Fig. S9a and S10†). For W751, however, while there appears to be a similar trend, the correlation is less evident (Fig. S9b and S10†). This may be due to the fact that W751 is located farther away from the flavin ring (Fig. 1B), and considerably less flexible than Y761 (Fig. 1D, E, S1 and S2†), with the distances and angles fluctuating in relatively small ranges (Fig. S10†), possibly in a regime where the effects of distances and angles, as well as the contributions from surrounding protein environment, are coupled in a complex manner. The fluctuations in Hab explain the multiphasic fluorescence decay kinetics (Fig. 2). Moreover, the results for Tyr761 (Fig. S9a and S10†) suggest that a smaller angle between the ring planes corresponds to a larger Hab value, in line with the observation that, in BsFNR and HpFd, the sub-picosecond quenching events involve tyrosine residues that are stacked with the flavin rings.27,37 It has been demonstrated in various systems that a cofacial arrangement of an aromatic donor–acceptor pair can enhance the coupling and consequently expedite ET,60–62 Here, we applied TDDFT calculations on a simplified tyrosine–flavin model system (Fig. 4F and G) to further examine such an assessment. The result demonstrates that when a tyrosine residue is present in a stacking configuration, Hab is greater than that obtained for a non-stacking configuration by more than an order of magnitude (1.4 versus 34.5 meV), confirming that a stacking donor–acceptor configuration is more favorable for short-range ET between a flavin and a tyrosine residue.
Finally, our findings have implications for potential use of modified LSD1 as a photobiocatalyst. Flavoprotein-based photocatalysts often (but not always) are active in the fully reduced state of the protein. They can be brought into this quasi-stable state by flavin photoreduction in the presence of sacrificial electron donors.63 The very efficient (ca. 180 fs), reversible intra-protein ET between W751 and the flavin competes with such a reaction in WT LSD1. This competing flavin photoreduction reaction is slowed down by almost two orders of magnitude upon replacement of W751 with phenylalanine. At this point, quasi-stable photoreduction by external electron donors, which has been reported to be able to occur as fast as ca. 1 ps when accommodated in the active site of flavoproteins,64 is likely to become sizeable. Interestingly, and a priori unexpectedly, the Y761 residue, also a potential electron donor to excited flavin, does not significantly contribute to the intra-protein photoinduced ET despite its closeness. As this residue plays an important role in accommodation of the peptidic substrate,53 its photochemical inertness may be favorable for peptide photobiocatalysis. Future work will explore these issues experimentally.
Conclusions
Combining comprehensive experimental and computational approaches, we delved into the dynamics and mechanism of the ultrafast flavin photoreduction in LSD1. Based on ultrafast spectroscopic measurements on wild-type and genetically modified proteins, we demonstrated that FAD* in WT LSD1 was quenched in <200 fs by short-range ET from nearby residues, where W751 emerged as the dominant electron donor despite a greater donor–acceptor separation distance than that of a tyrosine residue, Y761, located closer to the flavin ring. Calculations based on a QM/MM approach allowed us to characterize the LE and CT states of the FAD/W/Y system in the protein active sites, where we found that the electronic coupling between the FAD LE state and the CT state involving W751 is greater than that involving Y761 by an order of magnitude, corroborating our experimental findings. By examining the effects of donor–acceptor configurations on the electronic coupling between the LE and CT states, we further showed that a stacking configuration of the tyrosine–flavin pair was required for short-range photoinduced ET to efficiently occur. Taken together, our results provide detailed insights into the ultrafast flavin photoreduction in an intricate protein environment, which helps understand similar features in many other flavoprotein systems and may guide potential photobiocatalytic applications.Data availability
The data supporting this study are available within the article and ESI.† The raw data are available from the corresponding authors upon reasonable request.Author contributions
B. Z.: methodology, investigation, data analysis, visualization, writing – original draft preparation, writing – reviewing and editing; R. R.: methodology; A. A.: methodology, investigation, writing – reviewing and editing; U. L.; conceptualization, methodology, writing – reviewing and editing; M. H. V.: conceptualization, methodology, investigation, data analysis, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partly supported by Beijing Natural Science Foundation (Grant Number 2244070) and China Postdoctoral Science Foundation (Grant Number 2024M750080).Notes and references
- S. Ghisla and V. Massey, Eur. J. Biochem., 1989, 181, 1–17 CrossRef CAS.
- C. T. Walsh and T. A. Wencewicz, Nat. Prod. Rep., 2012, 30, 175–200 RSC.
- W. P. Dijkman, G. De Gonzalo, A. Mattevi and M. W. Fraaije, Appl. Microbiol. Biotechnol., 2013, 97, 5177–5188 CrossRef CAS.
- W. Buckel and R. K. Thauer, Chem. Rev., 2018, 118, 3862–3886 CrossRef CAS PubMed.
- B. J. Henriques, R. Katrine Jentoft Olsen, C. M. Gomes and P. Bross, Gene, 2021, 776, 145407 CrossRef CAS PubMed.
- S. Chapman, C. Faulkner, E. Kaiserli, C. Garcia-Mata, E. I. Savenkov, A. G. Roberts, K. J. Oparka and J. M. Christie, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20038–20043 CrossRef CAS.
- J. M. Christie, J. Gawthorne, G. Young, N. J. Fraser and A. J. Roe, Mol. Plant, 2012, 5, 533–544 CrossRef PubMed.
- A. P. Ryumina, E. O. Serebrovskaya, M. V. Shirmanova, L. B. Snopova, M. M. Kuznetsova, I. V. Turchin, N. I. Ignatova, N. V. Klementieva, A. F. Fradkov, B. E. Shakhov, E. V. Zagaynova, K. A. Lukyanov and S. A. Lukyanov, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5059–5067 CrossRef CAS.
- C. E. Paul, D. Eggerichs, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Biotechnol. Adv., 2021, 51, 107712 CrossRef CAS PubMed.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 Search PubMed.
- L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 Search PubMed.
- L. E. Meyer, B. E. Eser and S. Kara, Curr. Opin. Green Sustain. Chem., 2021, 31, 100496 CrossRef CAS.
- A. Taylor, D. J. Heyes and N. S. Scrutton, Curr. Opin. Struct. Biol., 2022, 77, 102491 CrossRef CAS PubMed.
- W. Harrison, X. Huang and H. Zhao, Acc. Chem. Res., 2022, 55, 1087–1096 Search PubMed.
- M. A. Emmanuel, S. G. Bender, C. Bilodeau, J. M. Carceller, J. S. DeHovitz, H. Fu, Y. Liu, B. T. Nicholls, Y. Ouyang, C. G. Page, T. Qiao, F. C. Raps, D. R. Sorigué, S. Z. Sun, J. Turek-Herman, Y. Ye, A. Rivas-Souchet, J. Cao and T. K. Hyster, Chem. Rev., 2023, 123, 5459–5520 CrossRef CAS PubMed.
- W. Z. Ng, E. S. Chan, W. Gourich, C. W. Ooi, B. T. Tey and C. P. Song, Renew. Sustain. Energy Rev., 2023, 184, 113548 Search PubMed.
- R. Miura, Chem. Rec., 2001, 1, 183–194 CAS.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier and F. Beisson, Science, 2017, 357, 903–907 Search PubMed.
- D. Sorigué, K. Hadjidemetriou, S. Blangy, G. Gotthard, A. Bonvalet, N. Coquelle, P. Samire, A. Aleksandrov, L. Antonucci, A. Benachir, S. Boutet, M. Byrdin, M. Cammarata, S. Carbajo, S. Cuiné, R. B. Doak, L. Foucar, A. Gorel, M. Grünbein, E. Hartmann, R. Hienerwadel, M. Hilpert, M. Kloos, T. J. Lane, B. Légeret, P. Legrand, Y. Li-Beisson, S. L. Y. Moulin, D. Nurizzo, G. Peltier, G. Schirò, R. L. Shoeman, M. Sliwa, X. Solinas, B. Zhuang, T. R. M. Barends, J.-P. Colletier, M. Joffre, A. Royant, C. Berthomieu, M. Weik, T. Domratcheva, K. Brettel, M. H. Vos, I. Schlichting, P. Arnoux, P. Müller and F. Beisson, Science, 2021, 372, eabd5687 CrossRef.
- F. Lacombat, A. Espagne, N. Dozova, P. Plaza, P. Müller, K. Brettel, S. Franz-Badur and L. O. Essen, J. Am. Chem. Soc., 2019, 141, 13394–13409 CrossRef CAS PubMed.
- Q. Wang and C. Lin, Annu. Rev. Plant Biol., 2020, 71, 103–129 CrossRef CAS PubMed.
- A. Lukacs, R. Brust, A. Haigney, S. P. Laptenok, K. Addison, A. Gil, M. Towrie, G. M. Greetham, P. J. Tonge and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 4605–4615 CrossRef CAS PubMed.
- A. Lukacs, P. J. Tonge and S. R. Meech, Acc. Chem. Res., 2022, 55, 402–414 CrossRef CAS.
- N. Mataga, H. Chosrowjan, Y. Shibata, F. Tanaka, Y. Nishina and K. Shiga, J. Phys. Chem. B, 2000, 104, 10667–10677 CrossRef CAS.
- D. Zhong and A. H. Zewail, Proc. Natl. Acad. Sci. U.S.A., 2001, 98, 11867–11872 CrossRef CAS PubMed.
- B. Kudisch, D. G. Oblinsky, M. J. Black, A. Zieleniewska, M. A. Emmanuel, G. Rumbles, T. K. Hyster and G. D. Scholes, J. Phys. Chem. B, 2020, 124, 11236–11249 CrossRef CAS PubMed.
- B. Zhuang, D. Seo, A. Aleksandrov and M. H. Vos, J. Am. Chem. Soc., 2021, 143, 2757–2768 CrossRef CAS PubMed.
- J. Yang, Y. Zhang, T. F. He, Y. Lu, L. Wang, B. Ding and D. Zhong, Phys. Chem. Chem. Phys., 2021, 24, 382–391 RSC.
- B. Zhuang, L. Nag, P. Sournia, A. Croitoru, R. Ramodiharilafy, J.-C. Lambry, H. Myllykallio, A. Aleksandrov, U. Liebl and M. H. Vos, Photochem. Photobiol. Sci., 2021, 20, 663–670 CrossRef CAS PubMed.
- B. Zhuang, U. Liebl and M. H. Vos, J. Phys. Chem. B, 2022, 126, 3199–3207 CrossRef CAS.
- B. Zhuang, M. H. Vos and A. Aleksandrov, ChemBioChem, 2022, 23, e202200227 CrossRef CAS PubMed.
- Y. Zhang, J. Yang, N. Liu, L. Wang, F. Lu, J. Chen and D. Zhong, J. Phys. Chem. Lett., 2023, 14, 10657–10663 CrossRef CAS.
- N. Mataga, H. Chosrowjan, S. Taniguchi, F. Tanaka, N. Kido and M. Kitamura, J. Phys. Chem. B, 2002, 106, 8917–8920 CrossRef CAS.
- H. Chosrowjan, S. Taniguchi, N. Mataga, F. Tanaka, D. Todoroki and M. Kitamura, Chem. Phys. Lett., 2008, 462, 121–124 CrossRef CAS.
- T. F. He, L. Guo, X. Guo, C. W. Chang, L. Wang and D. Zhong, Biochemistry, 2013, 52, 9120–9128 CrossRef CAS PubMed.
- S. P. Laptenok, L. Bouzhir-Sima, J. C. Lambry, H. Myllykallio, U. Liebl and M. H. Vos, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8924–8929 CrossRef CAS.
- K. Lugsanangarm, S. Pianwanit, A. Nueangaudom, S. Kokpol, F. Tanakaa, N. Nunthaboot, K. Ogino, R. Takagi, T. Nakanishic, M. Kitamura, S. Taniguchi and H. Chosrowjan, J. Photochem. Photobiol. A Chem., 2013, 268, 58–66 CrossRef CAS.
- L. Nag, P. Sournia, H. Myllykallio, U. Liebl and M. H. Vos, J. Am. Chem. Soc., 2017, 139, 11500–11505 CrossRef CAS.
- L. Nag, A. Lukacs and M. H. Vos, ChemPhysChem, 2019, 20, 1793–1798 CrossRef CAS.
- N. Dozova, F. Lacombat, C. Bou-Nader, D. Hamdane and P. Plaza, Phys. Chem. Chem. Phys., 2019, 21, 8743–8756 RSC.
- S. Ernst, S. Rovida, A. Mattevi, S. Fetzner and S. L. Drees, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- S. Mimasu, T. Sengoku, S. Fukuzawa, T. Umehara and S. Yokoyama, Biochem. Biophys. Res. Commun., 2008, 366, 15–22 CrossRef CAS.
- Y. C. Zheng, J. Ma, Z. Wang, J. Li, B. Jiang, W. Zhou, X. Shi, X. Wang, W. Zhao and H. M. Liu, Med. Res. Rev., 2015, 35, 1032–1071 CrossRef CAS.
- A. Maiques-Diaz and T. C. P. Somervaille, Epigenomics, 2016, 8, 1103–1116 CrossRef CAS PubMed.
- X. Zhang, X. Wang, T. Wu, W. Yin, J. Yan, Y. Sun and D. Zhao, Pharmacol. Res., 2022, 175, 105958 CrossRef CAS.
- A. K. Engstrom, A. C. Walker, R. A. Moudgal, D. A. Myrick, S. M. Kyle, Y. Bai, M. Jordan Rowley and D. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29133–29143 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, X. Li, L. Zhai, H. Zheng, Y. Yan, Q. Fu, J. Ma, H. Fu, Z. Zhang and Z. Li, Front. Pharmacol, 2022, 13, 1020556 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, F. Wang, K. Wan, K. Yamane, Y. Zhang and M. Lei, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13956–13961 CrossRef CAS PubMed.
- F. Forneris, C. Binda, M. A. Vanoni, E. Battaglioli and A. Mattevi, J. Biol. Chem., 2005, 280, 41360–41365 CrossRef CAS.
- X. J. Dai, Y. Liu, L. P. Xue, X. P. Xiong, Y. Zhou, Y. C. Zheng and H. M. Liu, J. Med. Chem., 2021, 64, 2466–2488 CrossRef CAS PubMed.
- B. Noce, E. Di Bello, R. Fioravanti and A. Mai, Front. Pharmacol., 2023, 14, 1120911 CrossRef CAS PubMed.
- M. P. Kabir, D. Ouedraogo, Y. Orozco-Gonzalez, G. Gadda and S. Gozem, J. Phys. Chem. B, 2023, 127, 1301–1311 CrossRef CAS PubMed.
- M. Yang, J. C. Culhane, L. M. Szewczuk, C. B. Gocke, C. A. Brautigam, D. R. Tomchick, M. MacHius, P. A. Cole and H. Yu, Nat. Struct. Mol. Biol., 2007, 14, 535–539 CrossRef CAS PubMed.
- F. Cailliez, P. Müller, M. Gallois and A. De La Lande, J. Am. Chem. Soc., 2014, 136, 12974–12986 CrossRef CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon N. Y., 2020, 165, 461–467 CrossRef CAS.
- F. Tanaka, R. Rujkorakarn, H. Chosrowjan, S. Taniguchi and N. Mataga, Chem. Phys., 2008, 348, 237–241 CrossRef CAS.
- R. J. Cave and M. D. Newton, Chem. Phys. Lett., 1996, 249, 15–19 CrossRef CAS.
- R. J. Cave and M. D. Newton, J. Chem. Phys., 1997, 106, 9213–9226 CrossRef CAS.
- T. Kastinen, D. A. Da Silva Filho, L. Paunonen, M. Linares, L. A. Ribeiro Junior, O. Cramariuc and T. I. Hukka, Phys. Chem. Chem. Phys., 2019, 21, 25606–25625 RSC.
- D. M. Guldi, C. Luo, M. Prato, A. Troisi, F. Zerbetto, M. Scheloske, E. Dietel, W. Bauer and A. Hirsh, J. Am. Chem. Soc., 2001, 123, 9166–9167 CrossRef CAS.
- A. Gouloumis, D. González-Rodríguez, P. Vázquez, T. Torres, S. Liu, L. Echegoyen, J. Ramey, G. L. Hug and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 12674–12684 CrossRef CAS.
- C. Jiang, J. Miao, D. Zhang, Z. Wen, C. Yang and K. Li, Research, 2022, 2022, 9892802 CAS.
- F. C. Raps, A. Rivas-Souchet, C. M. Jones and T. K. Hyster, Nature, 2024 DOI:10.1038/s41586-024-08138-w.
- M. Speirs, S. J. O. Hardman, A. I. Iorgu, L. O. Johannissen, D. J. Heyes, N. S. Scrutton, I. V. Sazanovich and S. Hay, J. Phys. Chem. Lett., 2023, 14, 3236–3242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods; additional results of time-resolved spectroscopic measurements and theoretical calculations. See DOI: https://doi.org/10.1039/d4sc06857b |
| This journal is © The Royal Society of Chemistry 2025 |