
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic investigation on cellular internalization triggering structure-induced conformational modulation of boron–nitrogen luminogens†
Retwik
Parui
 a,
Hirakjyoti
Roy
b,
Niranjan
Meher
a,
Siddhartha Sankar
Ghosh
bc and
Parameswar Krishnan
Iyer
*ab
a,
Hirakjyoti
Roy
b,
Niranjan
Meher
a,
Siddhartha Sankar
Ghosh
bc and
Parameswar Krishnan
Iyer
*ab
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India. E-mail: pki@iitg.ac.in
bCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India
cDepartment of Bioscience and Bioengineering, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India
First published on 28th February 2025
Abstract
Exploring the effects of electron donor (D) and acceptor (A) functional groups in tuning the condensed state properties has been a challenging yet efficient approach to reveal promising materials for cutting-edge applications. Herein, a series of boron–nitrogen (BN) incorporated organic congeners (NBNMe2, NBOMe, NBF, NBCl, NBBr, NBCN, NBPy) appended with functional groups having various degrees of D/A characteristics were developed and their potential in controlling supramolecular assembly and condensed state luminescence features (>90 nm redshift in λem,max) was explored. Despite the minor structural engineering in BN-based small molecules, they effectively modulated conformational orientation and molecular packing, leading to the directed growth of distinct and highly ordered self-assembly patterns, i.e., nanosheets, nanospheres, nanowires, and nanorods. The structure–property correlation investigation also highlighted the time-dependent fluorescence enhancement for NBPy owing to morphological growth via the fusion of nanospheres into nanowire conformation. Further, these nano-architectures with distinct conformations were employed to examine the mechanistic aspects as well as the influence of morphologies in cellular uptake and imaging, where all the nano-aggregates exhibited lysosomal localization following multiple endocytosis pathways and the nanorods possessed the highest uptakes (CTCF4h/0.5h = 3.11) with respect to other conformations. The in-depth inspection of the structural impact in single crystal X-ray diffraction (SCXRD) analysis disclosed the decisive role of boron atoms and functional group tuning that built a conceptual correlation between the molecular architecture and their photophysical characteristics, supramolecular assembly, and cellular internalization process, offering key insights on the development of rapid and effective drug delivery techniques.
Introduction
Functional group engineering in exploring novel luminogens always provides new vistas to fluorescence material-related research and accelerates scientific development in diverse research fields.1–5 The bottom-up strategy is one of the most natural and competent techniques employed for the supramolecular assembly of organic molecules utilizing weak hydrophobic interactions in an aqueous medium.6–8 However, the distinct noncovalent interactions produced by the organic building block and the anchored functional groups govern the assembly construction pathway and trigger various shape-defined assembly growth from nano to micro range.9–11 Apart from that, other external stimuli like light, temperature, solvent polarity, etc. are also realized to be effective parameters that can control the molecular motion in the aggregate phase and act as conclusive factors behind their assembly formation.12–15 The modulation in the self-assembly architecture of organic fluorophores to achieve distinct alteration in their optical and electronic features is an emerging strategy for utilizing them in multiple applications like biomedicine, optoelectronic devices, biosensors, and many other applications.16,17 In the past few years, numerous planar aromatic cores like perylenediimide (PDI) and naphthalimide (NI) have been established where the tailoring of different functional groups in their bay, imide, or peri position demonstrated several luminogens possessing nanoarchitectures like nanorods, nanoribbons, nanotubes, etc.6,18,19 Noteworthily, very few of these planar rigid cores exhibited aggregation-induced-emission (AIE)20,21 behavior as these planar π-cores encounter a high propensity of π–π stacking and poor solubility, leading to the unfavorable aggregation-caused-quenching (ACQ) nature in the condensed state and limiting their real-world applications.22,23 Hence, an efficient formula to trigger the self-assembly directed conformational control and develop new planar AIEgens with distinct time-dependent cellular internalization features remains very challenging and an untapped area.The inclusion of main block elements, like N, Si, O, B, etc., into an organic framework has made a promising impact on the development of versatile materials having broad applications.24–28 The formation of heteroatom-involved noncovalent interactions could assist in restricting the bond rotation via a ‘conformation lock’ mechanism and could produce a bright glow in the condensed phase of planar luminogens.29,30 Also, the proposed inclusion of BN group furnished a few planar luminogens, which were successful in overcoming the shortcomings of stacking-induced ACQ phenomena.31,32 Importantly, the B–N containing planar BODIPY core (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) has achieved huge success in the chemical and biological fields owing to its superior luminescence and outstanding biocompatibility.33–36 After its coining by Treibs and Kreuzer in 1968,37 numerous BODIPY derivatives have been employed as eminent fluorescent probes for bioimaging and theranostic purposes. The success of BODIPY dyes has inspired researchers to explore more BN-fused parent cores to expand the library of fluorescent materials.
Recently, the isosteric and isoelectronic B–N modification substituting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C has become an appealing approach to achieve unique optical and electronic properties.38–40 The bond polarizability of the B–N bond perturbs different electronic distributions or dipole moments, which plays a pivotal role in self-assembly in the condensed state. Alongside, the nucleophilic and electrophilic behavior of the heteroatoms also introduces various weak intermolecular interactions, which not only produce different molecular packing but also provide distinct emissive properties. However, the development of boron-containing organic molecules remains a matter of concern because of their complex synthetic route and ambient stability. Due to a lack of understanding of the luminescence mechanism, the development of organic luminogens through B–N functional group incorporation often gives unsuccessful outcomes. A facile strategy to tune ACQ to AIE phenomena via functional group engineering in a BN fused π conjugated backbone was attempted recently, which encouraged us to explore more BN embedded materials and obtain an in-depth understanding of the anatomy of those materials.41 We also established the role of aliphatic/aromatic functional groups on a BN-embedded polycyclic core that explains the importance of B–π interaction triggering the luminogenic behavior.42 However, the intriguing phenomena with the introduction of the donor/acceptor functionality are yet to be revealed. Herein, a simple, planar BN fused polycyclic core has been subjected to functional group engineering to tailor the aggregation, luminescence, and conformational control in their solution, aggregate, and solid state.
C has become an appealing approach to achieve unique optical and electronic properties.38–40 The bond polarizability of the B–N bond perturbs different electronic distributions or dipole moments, which plays a pivotal role in self-assembly in the condensed state. Alongside, the nucleophilic and electrophilic behavior of the heteroatoms also introduces various weak intermolecular interactions, which not only produce different molecular packing but also provide distinct emissive properties. However, the development of boron-containing organic molecules remains a matter of concern because of their complex synthetic route and ambient stability. Due to a lack of understanding of the luminescence mechanism, the development of organic luminogens through B–N functional group incorporation often gives unsuccessful outcomes. A facile strategy to tune ACQ to AIE phenomena via functional group engineering in a BN fused π conjugated backbone was attempted recently, which encouraged us to explore more BN embedded materials and obtain an in-depth understanding of the anatomy of those materials.41 We also established the role of aliphatic/aromatic functional groups on a BN-embedded polycyclic core that explains the importance of B–π interaction triggering the luminogenic behavior.42 However, the intriguing phenomena with the introduction of the donor/acceptor functionality are yet to be revealed. Herein, a simple, planar BN fused polycyclic core has been subjected to functional group engineering to tailor the aggregation, luminescence, and conformational control in their solution, aggregate, and solid state.
Further, a unified concept of regulating the luminogenic behavior along with their modulation in morphological conformation is proposed by developing a library of BN-based congeners via minor D/A functional group engineering. Despite performing only minor structural engineering, the transformation in the condensed state emission was highly pronounced. Hence, this aspect was carefully explored through elaborate computational and experimental studies, which suggested the dominant role that molecular packing orientations can provide over their electronic contribution. The SCXRD explained the crucial role of boron atoms and the functional group in breaking the detrimental π–π stacking and ensured variation in intermolecular interactions. The regulation in the molecular packing also resulted in a well-organized self-assembly pattern including nanosheets, nanospheres, nanowires, and nanorods, along with special highlights on the time-dependent morphological growth via fusion of nanospheres into nanowire conformation. Further, the highly emissive luminogens with distinct variations in self-assembly conformation show potential for bioimaging and mechanistic investigation on cell permeability, where different conformations influence the cellular uptake rate. Organic luminogens are recognized as potential candidates for biological applications bestowing an excellent signal-to-noise ratio in biological environments.43–45 Previous studies have demonstrated that cellular internalization of nanospheres depends on the surface charge and size of the nanospheres,46,47 but limits in-depth studies due to the nonfluorescent nature of the applied dye. Thus, most of the previously reported probes failed to provide mechanistic information behind the complex biological processes owing to the poor fluorescence signal inside the cell.48 Therefore, along with carrying out the investigation of the structure–property relationship, four different low-cytotoxic self-assemblies were employed for time-dependent cellular internalization to unveil the mechanistic aspect of the rate of internalization regarding their assembly conformation. The overall findings reveal significant insights on understanding the precise regulation of the condensed state emissions and supramolecular self-assembly induced conformational transformations through a simple D/A functional group engineering strategy in a BN-incorporated polycyclic core with a mechanistic investigation on cancer cell internalization and imaging applications (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of the donor/acceptor mediated regulation in luminogenic properties, fine-tuning in self-assembly structure, and morphology-dependent cellular uptake process. | ||
Results and discussion
In order to study the structure–property relationship in the condensed state, a library of seven BN doped derivatives was synthesized with different D/A functional groups through a facile catalyst-free one-step condensation reaction (Fig. 1a). A strong donor (NMe2, OMe), strong acceptor (CN, pyridine ring), and moderate acceptor (F, Cl, Br) were introduced strategically to explore their role in regulating electronic distribution and intermolecular interaction, which could potentially influence their condensed state properties. All seven congeners were thoroughly characterized by NMR (1H, 13C) and HRMS, presented in the ESI file.† | ||
| Fig. 1 (a) Chemical structures of the BN-incorporated organic luminogens varying the donor/acceptor functional groups; (b) calculated ground state HOMO–LUMO energy profile and distribution of electronic density in the gaseous phase for all the congeners using DFT with the B3LYP/6-31G set in the Gaussian 09 package. | ||
To validate the impact of the D/A functional group, the structures and electronic distribution were evaluated theoretically using density functional theory (DFT). A reduction in the HOMO–LUMO energy bandgap was observed from 4.2 eV to 3.19 eV in accordance with the increment in the donor-to-acceptor characteristics of the incorporated functional group where the HOMO electron cloud dispersed on the naphthalene core, and the LUMO electronic cloud lies on the introduced functional group differently depending on the donating/accepting aptitude of the functional groups (Fig. 1b). Interestingly, the introduction of different functional groups also varied the magnitude and direction of the resultant dipole moments (Fig. S1†). Additionally, the increment in accepting proficiency resulted in a conformational twist in the DFT optimized structures where the dihedral angle between the functional group anchored phenyl ring and the boron–nitrogen embedded naphthalene core was widened from 27.66° to 34.94° suggesting the profound influence of the functional group on the rigidity of the molecules (Fig. S2†).
To investigate the emissive nature in their aggregate state, the fluorescence emission of all the congeners was recorded by varying the water fraction (fw) in a methanol–water dual solvent system where methanol was selected as a good solvent, and water was considered as a poor solvent. NBNMe2 perceived a very weak emission at a 470 nm wavelength maximum in methanol [fluorescence quantum yield (QY) = 0.160], but a gradual rise in the fw displayed a blue-shifted emission enhancement owing to a dual hump emission spectrum at 50% fw reflecting the occurrence of vibrational relaxation during the phase transformation from the monomer to aggregate (Fig. 2a). A gradual rise in the emission intensity was observed on increasing the fw up to 99.9% (36-fold emission enhancement, QY = 3.344), indicating a higher extent of aggregate formation in the hydrophobic medium as a result of AIE phenomena. However, NBOMe showed dual-state emissive (DSE) characteristics where the moderate emission (QY = 0.770) in its solution phase (λmax = 433 nm) was slightly blue-shifted (λmax = 425 nm) with an enhancement in emission intensity (QY = 0.768) (Fig. 2b). NBF and NBCl exhibited excellent AIE behavior where maximum fluorescence intensity was obtained at 99.9% fw, but a sudden rise in the emission spectra was experienced after 70–80% fw signifying the initiation of nanoaggregate formation (Fig. 2c and d). Despite a similar electronic configuration, NBBr displayed a DSE nature owing to similar QY while the red-shifted maxima appeared with lower intensity in the assembly state (QY = 0.379) compared to their monomeric emission (QY = 0.337), suggesting a heavy atom effect in the presence of bulky bromine atoms (Fig. 2e).49,50 The presence of a strong accepting unit also induced strong intramolecular charge transfer (ICT) phenomena for NBCN and NBPy resulting in reduced fluorescence intensity in both solution and aggregate states. For NBCN, a large stokes shift was observed where the solution emission (λmax = 384 nm) was vastly redshifted (λmax = 515 nm) after aggregation, indicating the impact of intermolecular interaction on its emissive nature (Fig. 2f). Nevertheless, NBPy revealed a bathochromic shift in the emission spectra with a negligible change in the emission intensity (Fig. 2g).
 | ||
| Fig. 2 Photoluminescence (PL) spectra of (a) NBNMe2, (b) NBOMe, (c) NBF, (d) NBCl, (e) NBBr, (f) NBCN, and (g) NBPy at various fw in MeOH (50 μM) and the corresponding inset represents fluorescence intensity vs. fw in MeOH. (h) Fluorescence intensity of NBPy (50 μM) in 99.9% fw at different times. Inset in (h) shows the respective fluorescence kinetic data at λem = 433 nm up to 1 h. (i) Normalized PL spectra of all the congeners (50 μM) in 99.9% fw in MeOH at room temperature. All the parameters are mentioned in Table S1.† | ||
Nonetheless, for NBPy an abrupt change in the emission phenomena was observed where the emission intensity and corresponding fluorescence quantum yield increased gradually with time (QY = 0.986 to 6.321) (Fig. 2h and Table S1†). The enhancement in the fluorescence intensity was further confirmed with the fluorescence kinetic data recorded for up to one hour (Fig. 2h, inset). Additionally, to confirm the emission stability of the other congeners, the aggregate state emission intensity was checked for up to 1 hour. Notably, none of them showed any fluorescence enhancement with time, whereas all the congeners possessed significant stability within a few minutes of aggregate formation (Fig. S3†). The robust nature of these aggregates further offered additional potential during applications in cell imaging. Later the prominent role of assembly conformation was scrutinized during morphology analysis.
However, despite the reduction in the energy gap between HOMO and LUMO, the UV-Vis spectra revealed a minor shift in the absorption maxima in the solution phase, whereas the aggregate phase attained broad-range spectra owing to assembly formation (Fig. S4†). Particularly, the broadening of the UV-Vis spectrum for NBPY after 1 hour indicated a greater degree of aggregation with respect to the instantly formed aggregates (Fig. S5†). On the other hand, an insignificant bathochromic shift was observed with an increase in electron deficiency property; rather, it showed a contrary result where fluorescence emission of NBNMe2, NBOMe, and NBF was more redshifted in the solution state (Fig. S6†). It could be assumed that the planar conformation of the probes connected to the donor functionality facilitated excimer formation in the solution phase.51,52 Interestingly, a large shift in the emission maxima was noticed in the aggregate state. Besides, a reverse trend was noticed for NBF, NBCl, and NBBr, with respect to the electronegativity of the halogen atom, suggesting the insignificant contribution of electronic distribution in condensed phase emission (Fig. 2i and Table S1†). On the other hand, in the solid state, these luminogens covered a wide range of emission wavelength nearly identical to their aggregated phase (Fig. 3a, b and Table S2†). Comparably, the molecules with the donor functionality anchored to the phenyl ring displayed more intense emissions than those with the strong acceptor unit, which could be attributed to the extensive ICT in the presence of electron deficient functional groups and resulted in a quenched emission signal (Fig. 3c).53–55 Notably, the unusual shift in emission wavelength in the condensed phase compared to the solution phase indicated the significant role of molecular conformation and intermolecular interactions. The close proximity of the molecule in the hydrophobic medium also showed a marked change in the lifetime of the excitons, which was confirmed with time-resolved photoluminescence (TRPL) spectroscopy.56,57 In accordance with aggregate formation, all the congeners (NBNMe2, NBF, NBCl, NBCN, NBPy) achieved a stable excited state endowing long-lived exciton generation, while NBBr and NBOMe perceived short-lived exciton formation depending on their intramolecular interaction (Fig. S7 and Table S3†). For NBF and NBCl, the decay rate slowed down significantly, and a remarkable increment in lifetime was noticed, reflecting the role of the halogen atom in intermolecular interaction, whereas the heavy atom effect from the bromine atom presumably lowered the fluorescence intensity as well as fluorescence lifetime in their aggregate state.58 Distinct variation in the fluorescence decay rate in the assembly phase confirmed that the molecular packing interactions are much more dominating over their electronic configuration due to the presence of different functional groups in their closer proximity, which was further analyzed from the single crystal X-ray experiment.
 | ||
| Fig. 3 (a) PL spectra of all the congeners in the solid state. (b) Luminescence color of all the BN-incorporated congeners in the solid state plotted in a CIE chromatogram, with the coordinates listed in Table S2.† (c) Digital photographs of the solid powder captured in daylight and 365 nm UV light. | ||
To divulge the distinct luminescent behavior in the condensed state, single crystals of the congeners were grown by slow evaporation of volatile solvents. Well-diffracting crystals were collected from methanol and chloroform fractions and analyzed further to interpret the molecular conformations, packing orientation, and interactions (Fig. 4, S8, Tables S4 and S5†). NBNMe2 adopted a planar-like conformation constructing a uniform herringbone growth where a dimeric and monomeric unit was assembled alternatively along the c-axis (Fig. 4a). The dimeric unit was aligned in an antiparallel, head-to-tail orientation containing two B–π (3.448 Å, 3.609 Å) and one CH–π (2.803 Å) interaction and was connected to the monomeric component through CH–π (2.893 Å) and NH–π (2.743 Å) interactions (Fig. S9†). It could be assumed that the electrostatic potential difference between the phenyl ring and boron atom governed the antiparallel rigid dimeric arrangement in two planar motifs holding two B–π interactions. The donor dimethyl amine unit enhanced the electron density in the connected phenyl ring and developed negative electrostatic potential that facilitated a strong B–π interaction with the acceptor boron atom, regulating the intermolecular motion via rigid dimer formation. Thus, the employment of slip-stack packing through multiple interactions restricted the molecular motion and resulted in fluorescence emission enhancement in the aggregate state (QY = 3.344). Apart from that, the dimeric component revealed two different structural conformations where one unit attained rigid planar conformation with an angular deformation of 6.02° between two planes containing functionalized phenyl ring and naphthalene core. However, the other one formed a slightly bent structure with a 16.85° angular distortion for the corresponding angle, reflecting a bent to linear vibrational twist for the NBNMe2 congener as depicted in Fig. 4a. This vibrational relaxation also elucidated the dual hump emission peak during the monomer-to-aggregation phase transition at higher fw (≥60%) (Fig. 2a). Moreover, this conformational difference also implied that the occurrence of restriction in intramolecular vibration in their condensed state could be an additional reason for achieving the extensive emission intensity of the NBNMe2 molecule.
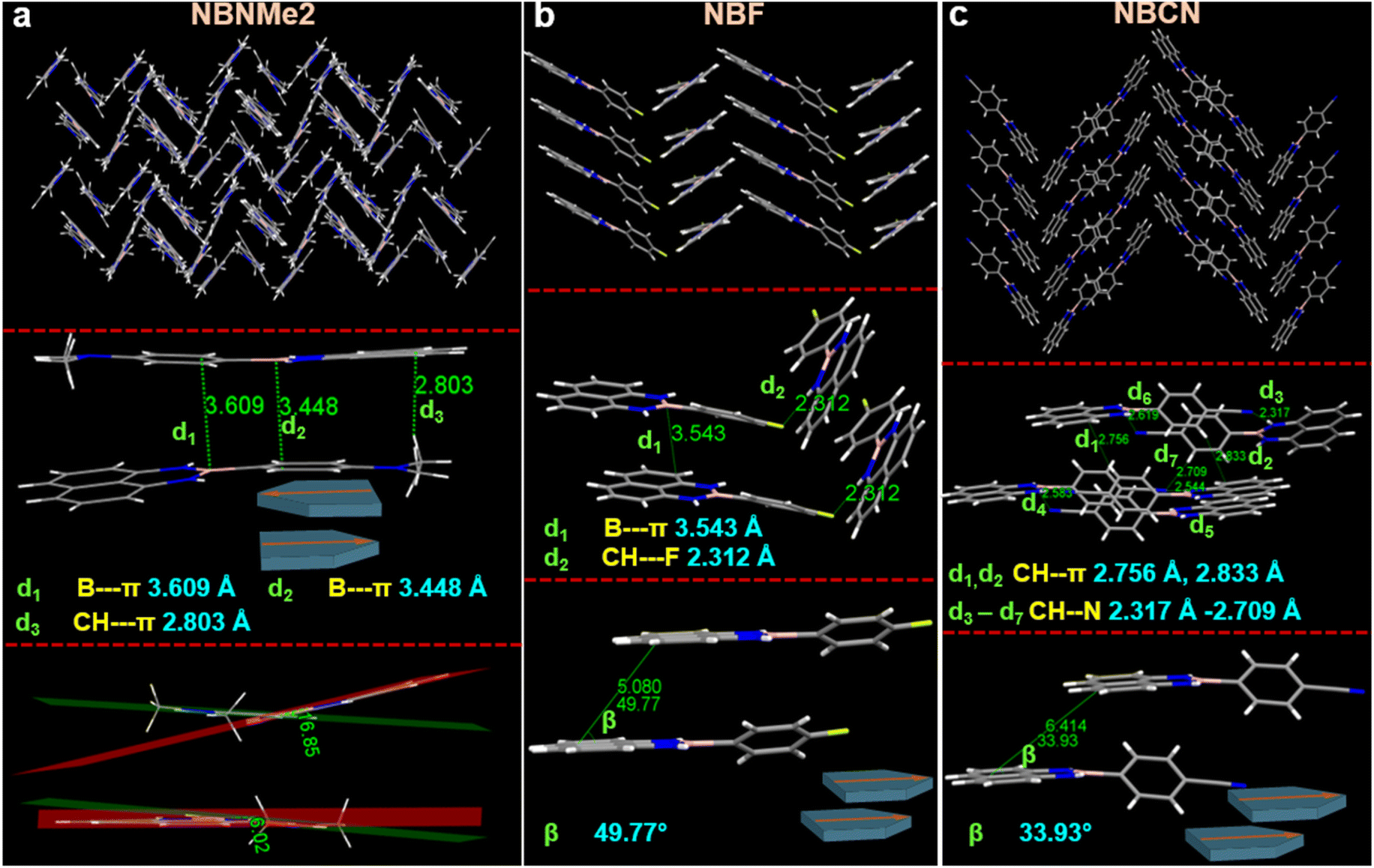 | ||
| Fig. 4 Molecular packing orientation, intermolecular interaction, molecular structural conformation for (a) NBNMe2, (b) NBF, and (c) NBCN obtained from the SCXRD experiment. ‘d’ and ‘β’signified the distance of a noncovalent interaction present between two moieties and the slippage angle present between two neighboring molecules respectively. The schematic illustration demonstrated the alignment of two congeners present in closer proximity. | ||
However, the incorporation of halogen atoms weakened the intramolecular NH–π interaction and caused a larger dihedral angle between the phenyl ring and BN-embedded naphthalene core. Importantly, angular deformation in the planar core also resulted in a higher extent of slip stack packing, which hindered face-to-face strong π–π overlap reasonable for quenched luminescence behavior. Compared to NBNMe2, NBF revealed a twisted structure with a dihedral angle of (22.71°) and assembled in a zig-zag pattern in bulk growth (Fig. 4b and S10b†). It was witnessed that two adjacent molecules attained a head-to-head packing comprising B–π (3.543 Å) interaction owing to a slippage distance and angle of 5.080 Å and 49.77° between two naphthalene cores, respectively. Similarly, for NBCl, comparably less electronegativity of the chlorine atom produced a smaller dihedral angle of (3.75°), endowing two comparatively stronger intramolecular NH–π interactions (Fig. S8a and S10c†). This rigidification helped to form two analogous B–π (3.427 Å) B–π (3.670 Å) interactions between the parallel motifs. Although NBCl followed a similar trend in terms of slippage distance (4.730 Å), it displayed a different staircase-like packing arrangement. In contrast, despite low polarizability of the bromine atom, NBBr exhibited a more twisted conformation with a dihedral angle of (19.41°). Along with that, it also adopted a herringbone pattern with more slip stack packing between two adjacent NBBr molecules comprising one B–π (3.440 Å), π–π (3.240 Å), and CH–π (2.793 Å) interaction (Fig. S8b and S10d†). Thus, it could be stated that steric constraints trigger the molecular conformation and packing arrangement regardless of the electronic effect of the anchoring functional group.
Furthermore, NBCN acquired a maximum twist of (36.37°) and configured a distinct herringbone orientation where two NBCN components are packed in a head-to-head manner with little overlap between the phenyl rings. The packing pattern was regulated by multiple weak interactions (d1–d7) (Fig. 4c), especially involving the cyano group. Interestingly, in addition to the twist, the slippage within the analogous parallel unit was too high to form the B–π interaction, which loosened the molecular packing and enhanced the nonradiative decay rate that could be attributed to its low fluorescence emission in the condensed state. On closer inspection, it was noticed that two kinds of conformations are present within the molecular packing where one holds a twist angle of 36.37° and another one is at 68.73° (Fig. S10e†). Notably, the flexible conformation having wide twist angle enhanced the plausibility of twisted intramolecular charge transfer (TICT) phenomena,59 resulting in more redshifted emission with low fluorescence intensity in the aggregate state (QY = 0.254). Overall, it was evidenced that the introduction of distinct D/A functional groups produced a significant change in the molecular arrangements, dipole moment, and steric constraint in the congeners, which effectively tuned their photophysical behavior in the condensed state. Moreover, to reveal the molecular arrangements in the bulk phase, powder X-ray diffraction (PXRD) of the solid powder and aggregates (generated in water) was performed for all the congeners between 2θ values of 5–50°. The multiple sharp peaks in the PXRD spectrum could be attributed to the excellent crystallinity of all the congeners in their powder form (Fig. S11†). The comparison of the PXRD patterns obtained for the powder, aggregates, and crystal simulated spectrum revealed excellent alignment across all phases, which suggested that all the molecules followed uniform crystalline phase orientation in their condensed phases. Besides, the complete disparity in the PXRD patterns also supported the significant variation in their packing orientation. Upon realizing the distinct contribution of the functional groups on molecular packing, we explored the influence of functional groups on self-assembly. Usually, aqueous media provide a hydrophobic repulsive force on organic molecules which drives them to aggregate at higher water percentages. At the same time, the functional group could regulate the assembly pattern into diverse structures, which might have a direct impact on their emissive behavior.60–62 To affirm the assembly pattern, the morphology of all the congeners was visualized using electron microscopy techniques (FESEM & TEM), where the aggregate solution bestowing the highest emission intensity was considered for further evaluation. The uniformly dispersed aggregate solution was drop-cast on the respective solid surface (silicon wafer for FESEM & copper grid for TEM) and dried under vacuum instantly at room temperature for microscopic visualization.
NBNMe2 with a donor functionality obtained a ‘nano-sheet’ structure, while the relatively less donating methoxy functionality (NBOMe) showed spherical conformation in FESEM, and both revealed similar sets of morphology in TEM analysis on a copper grid (Fig. 5a and b (a1 and b1)). Notably, a distinctive difference in the diameter was also noticed where NBNME2 and NBOMe exhibited an average size of 362 nm and 164 nm, respectively, validating the effect of dimerization occurring in case of NBNMe2. Surprisingly, with the introduction of the acceptor group, NBF perceived a flexible nanowire structure where a long-range growth was noticed containing a ∼200 nm hydrodynamic diameter (Fig. 5c and c1). With increasing acceptance properties, for NBCN, the growth range was reduced, and a rigid nanorod structure was spotted owing to an average size of ∼800 nm which aligns with its DLS spectra (Fig. 5d and d1). Compared to NBCN, despite the large growth of nanowires, NBF possessed less size distribution validating the flexibility of these wires in the dispersed phase that allowed shrinkage in the conformation whereas the rigidity of the rod structures impeded the shrinkage and displayed large hydrodynamic diameter in the dispersed phase. Meanwhile, the less electronegative chloro (NBCl) and bromo (NBBr) substituted motifs attained a spherical morphology confirmed by the same microscopic technique, and both the molecules displayed good agreement with the DLS data endowing an average diameter of 178 nm and 215 nm respectively (Fig. S12†).
 | ||
| Fig. 5 FESEM and FETEM images of the self-assemblies generated spontaneously in 99.9% water (50 μM) following a drop-casting technique on a silicon wafer and copper grid, respectively. The morphology of (a) NBNMe2, (b) NBOMe, (c) NBF and (d) NBCN possessed a distinct variation from nanosheet to nanorod conformation. Insets (i–iv) represent the corresponding DLS spectra with the average hydrodynamic diameter. TEM images of (a1) NBNMe2, (b1) NBOMe, (c1) NBF, and (d1) NBCN showed good agreement with the morphology obtained from the FESEM experiment. | ||
Interestingly, the NBPy molecule having a pyridine acceptor unit revealed a spherical shape with a diameter of approximately 100 nm initially, and a remarkable increment in the hydrodynamic size was observed from 129 nm to 317 nm within 1 hour, which possibly could be associated with its unusual luminescence behavior in the aggregate phase. To investigate the distinct nature and spontaneous growth of the self-assemblies, the dispersed solution was considered for microscopic analysis at time intervals of 15 minutes and 1 hour and compared with the instant growth. It was evidenced that initially formed similar spherical assemblies had closer proximity after 15 minutes and were connected to each other, generating a pearl necklace-like conformation that was confirmed from both SEM and TEM analysis (Fig. 6a, a1, b and b1). The elongation process continued further and a wire-like conformation was obtained after 1 hour, where the spherical bulbs (indicated with yellow arrows) in the inset FESEM image strongly authenticated the growth kinetics of the self-assemblies (Fig. 6c and c1). Therefore, it could be inferred that the surface of the spheres has the unique tenacity to join with each other via a weak attractive force leading to a linear bidirectional ‘supramolecular-linear growth’ type assembly process.9,63–65 Meanwhile, photoluminescence kinetics also reflected a linear increment in the fluorescence intensity up to 1 hour at 433 nm wavelength, which plausibly describes the orderliness in the molecular packing defining different morphology.66
 | ||
| Fig. 6 (A) Schematic illustration of the sample preparation technique for analyzing morphology at particular time intervals. (B) (a–c) FESEM and (a1–c1) FETEM images of the self-assembly growth for NBPy at different time intervals of 0 (instant), 15 and 60 minutes. (c) Inset yellow dotted box illustrates similar growth of a smaller portion at the same time period. Yellow arrows in (c) and (c1) indicate the assembly of spheres towards linear growth. Insets (i–iv) represent the corresponding DLS spectra with their average hydrodynamic diameter. | ||
Further, the plausible mechanism might be attributed to the intermolecular weak van der Waals interaction produced by the acceptor functionality present at the spherical surface and facilitated the release of the curvature strain via the fusion of these spheres into a rod- or wire-like structure, which also had uniformity with the single crystal analysis.
A significant change in the morphology was speculated starting from nanosheets to nanospheres, nanowires, and nanorod morphology by varying the D/A functionality to a simple BN-embedded naphthalene core which encouraged us to visualize their cellular internalization potential into cancerous cells. In this regard, four congeners, NBNMe2, NBOMe, NBF, and NBCN, endowing different morphologies, were selected for biological experiments. Initially, to know the cytotoxic behavior of the probes inside the cells, a concentration-dependent colorimetric MTT assay was conducted in two cancerous cell lines, MCF7 and HeLa (Fig. S13†). A substantial number of cells were viable up to 100 μM doses of each component, which indicated the less toxic nature of the probes inside the corresponding cell lines. Since 50 μM concentration was used for the aggregate phase experiment and insignificant cell death was observed at the same concentration, the same dose was finalized to conduct the cellular internalization process. To explore the morphology-dependent uptake efficiency, HeLa cells were treated with the nano-aggregates and a time-dependent incubation process was tracked up to four hours (Fig. 7A, B and S14†). From the CLSM images, a periodic enhancement in the fluorescence intensity was monitored for all probes and the similar emissive nature of the probes inside the cells displayed good agreement with the emission of nanoaggregates formed in 99.9% fw, signifying intactness of nanoaggregates inside the cell lines. Moreover, the spontaneous increment in the fluorescence intensity and homogeneous dispersion inside the cells suggested that the aggregates are efficient at crossing the cell membrane. Although a regular increment in the fluorescence intensity was witnessed for all four, the quantified corrected total cell fluorescence (CTCF) value inside the cells indicated deviation in uptake rates with the variation in the shapes of the aggregates [Fig. 7C (a–e)]. After 4 hours, NBOMe (nanospheres) displayed a CTCF increment of 55%, while NBCN (nanorods) acquired the highest increment of 68% with respect to the uptake after 30 min, which was ascribed to the significant efficacy of the rod-shape structure on membrane penetration owing to the large surface area for multiple interactions with the cell membrane.67,68 Moreover, their distinct geometry plausibly lowered the membrane bending energy barrier and might facilitate superior uptake.69 Interestingly, the longer length of the nanowires might reduce the uptake potential compared to the nanorods.70
 | ||
| Fig. 7 Time-dependent confocal laser scanning microscopy (CLSM) images to evaluate the cellular uptake process of the (A) spherical nano-assemblies of NBOMe and (B) nanorod assemblies of NBCN incubated in a HeLa cell up to 4 hours. (C) Bar diagram demonstrates CTCF values for (a) NBNMe2, (b) NBOMe, (c) NBF, and (d) NBCN after incubation for a specific time interval. (e) The increment in the CTCF values after 4 hours with respect to the preliminary value indicates the uptake efficiency of the assemblies inside the HeLa cell. (f) Percentage uptake in the presence of sodium azide inhibitor, MβCD, and dynasore. (n = 3, mean ± SD). | ||
To investigate the intracellular localization of these nano-aggregates, a cellular colocalization study was performed using LysoTracker Deep Red. HeLa cells were treated with these congeners for 3 h and then stained with LysoTracker for another 1 h. Following incubation, cells were fixed with 4% formaldehyde and visualized under confocal microscopy. The results showed a clear overlap between the LysoTracker signal and the compounds, suggesting lysosomal localization (Fig. S15†). Additionally, the Pearson correlation coefficient (PCC) values [NBNMe2 (0.851), NBOMe (0.856), NBF (0.745), and NBCN (0.763)] confirmed significant overlap, due to predominant localization within the lysosome.
The study on cellular internalization in the presence of multiple endocytosis inhibitors revealed distinct uptake behaviors of the tested compounds [Fig. 7C (f) and S16†]. To explore the energy-dependent endocytosis mechanism, the cellular internalization process was assessed after treating the cells with sodium azide, an ATPase inhibitor.71 The compounds NBOMe, NBF, and NBNMe2 showed significantly reduced uptake in the presence of sodium azide, indicating internalization is predominantly energy-dependent endocytic pathways. In contrast, the uptake of NBCN remained unaffected, suggesting that its internalization might occur through an energy-independent mechanism. However, at 4 °C, a temperature that usually suppresses active endocytosis, no inhibition was observed. This could suggest that ATP production was not entirely halted at this temperature, allowing the cells to utilize residual ATP for active transport.72,73 Later, to evaluate the involvement of clathrin-mediated endocytosis, cells were treated with dynasore, a dynamin inhibitor74–76 where no change in the uptake was observed, indicating that the dynamin-dependent pathway was not involved in the internalization of these compounds. Furthermore, the uptake of NBNMe2 and NBCN was inhibited by methyl-β-cyclodextrin, a caveolae-mediated endocytosis inhibitor, indicating possible involvement of lipid raft-mediated pathways. These findings provide valuable insights into the mechanisms underlying the cellular uptake of these compounds, highlighting the complexity and diversity of the endocytosis pathways. Thus, the systematic evaluation highlighted the intriguing role of the D/A functional group in molecular assemblies that are capable of modulating their biological activity, presenting an encouraging strategy to develop highly efficient nanodrugs for biomedical purposes.
Conclusion
Rationally designed BN-embedded organic luminogens have been developed through donor–acceptor functional group engineering, offering excellent tunability in the condensed state emissive and self-assembly properties. This simple strategy endows unique luminescence characteristics in solution and aggregate states, where excellent redshift (>90 nm) in the condensed state emission was witnessed by incorporating functional groups with electron deficient properties. Along with the redshift, a significant change in the fluorescence quantum yield [3.344% in NBNMe2 to 0.254% in NBCN] in the condensed state was also observed with functional group modifications. The SCXRD analysis confirmed that the functional group can induce conformational twist [DA 3.32° (NBNMe2) to 68.73° (NBCN)] to attain distinct molecular packing. Nevertheless, variations in the intermolecular interactions proficiently tailored the growth kinetics of the molecular assemblies, leading to an alteration in morphological shapes like nanosheets, nanospheres, nanowires, and nanorods. Importantly, a thorough investigation of the time-dependent enhancement (QY = 0.986 to 6.321) in the fluorescence intensity of NBPy revealed the morphological change via linear fusion of the instantly formed nanospheres (Zave = 129 nm) into a spontaneously elongated nanowire-like (Zave = 317 nm) conformation, plausibly enhancing their aggregate state emission. Moreover, four entirely different conformations exhibited distinct magnitudes of internalization in the cancerous cell line, where the superior uptake of nanorods (CTCF4h/0.5h = 3.11) indicated the influence of morphology in the cellular internalization process. Overall, a simple strategy to decorate a BN-incorporated organic core with a D/A functional group has demonstrated an effective way to regulate condensed state emission with systematic transformation in a self-assembly pattern, which could potentially benefit the development of novel luminogens for biomedical applications.Data availability
All the data relevant to this work have been uploaded as the ESI file.†Author contributions
RP designed the project, synthesized all the probes, carried out the experiments to find the structure–property relationship, performed computational calculations, data analysis, and prepared the manuscript. HR performed biological experiments and assisted with manuscript writing and revisions. NM contributed to data analysis and revisions to the manuscript. PKI and SSG supervised the project, obtained funding and were involved in planning, analysis, manuscript preparation and revisions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank DST-India (No. DST/SERB/EMR/2014/000034), DST/CRG/2019/002164, DeitY-India Project No. 5(1)/2022-NANO, ICMR, Grant No. 5/3/8/20/2019-ITR and DST-Max Planck Society, Germany (No. IGSTC/MPG/PG(PKI)/2011A/48) for financial support. RP thanks DST for the INSPIRE fellowship (DST/INSPIRE Fellowship/2017/IF170271). The Department of Chemistry (FIST: SR/FST/CS-II/2017/23C), Centre for Nanotechnology and Central Instruments Facility, IIT Guwahati, are acknowledged for providing instrument facilities.References
- A. von Hippel, Science, 1956, 123, 315–317 CrossRef CAS PubMed.
- N. Meher and P. K. Iyer, Nanoscale, 2017, 9, 7674–7685 RSC.
- B. Shao, H. Qian, Q. Li and I. Aprahamian, J. Am. Chem. Soc., 2019, 141, 8364–8371 CrossRef CAS PubMed.
- D. Barman, K. Narang, R. Parui, N. Zehra, M. N. Khatun, L. R. Adil and P. K. Iyer, Aggregate, 2022, 3, e172 CrossRef CAS.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- S. Chen, P. Slattum, C. Wang and L. Zang, Chem. Rev., 2015, 115, 11967–11998 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill, M. V. Lee, A. Vinu, R. Charvet and S. Acharya, Sci. Technol. Adv. Mater., 2008, 9, 014109 CrossRef PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- Q. Liu, Q. Xia, S. Wang, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 4807–4816 RSC.
- C.-A. Shen, D. Bialas, M. Hecht, V. Stepanenko, K. Sugiyasu and F. Würthner, Angew. Chem., Int. Ed., 2021, 60, 11949–11958 CrossRef CAS.
- H.-Q. Peng, X. Zheng, T. Han, R. T. K. Kwok, J. W. Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 10150–10156 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- H. Liu, Y. Shen, Y. Yan, C. Zhou, S. Zhang, B. Li, L. Ye and B. Yang, Adv. Funct. Mater., 2019, 29, 1901895 CrossRef.
- Y. Shen, Z. An, H. Liu, B. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202214483 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- D. Barman, M. Annadhasan, A. P. Bidkar, P. Rajamalli, D. Barman, S. S. Ghosh, R. Chandrasekar and P. K. Iyer, Nat. Commun., 2023, 14, 6648 CrossRef CAS PubMed.
- P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281–13291 CrossRef.
- Y. Tani and T. Ogawa, J. Mater. Chem. C, 2021, 9, 4281–4288 RSC.
- D. Srideep, K. Sriram, S. Kotha, D. J. Babu, S. K. Singh and K. V. Rao, Chem.–Asian J., 2022, 17, e202200099 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- N. Meher and P. K. Iyer, Angew. Chem., Int. Ed., 2018, 57, 8488–8492 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- M.-C. Giel and Y. Hong, Aggregate, 2023, 4, e336 CrossRef CAS.
- H. Narita, H. Choi, M. Ito, N. Ando, S. Ogi and S. Yamaguchi, Chem. Sci., 2022, 13, 1484–1491 RSC.
- Y.-g. Shi, J.-w. Wang, H. Li, G.-f. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC.
- L. Yang, P. Ye, W. Li, W. Zhang, Q. Guan, C. Ye, T. Dong, X. Wu, W. Zhao, X. Gu, Q. Peng, B. Tang and H. Huang, Adv. Opt. Mater., 2018, 6, 1701394 CrossRef.
- H. Li, H. Shu, Y. Liu, X. Wu, H. Tian, H. Tong and L. Wang, Adv. Opt. Mater., 2019, 7, 1801719 CrossRef.
- J.-S. Ni, T. Min, Y. Li, M. Zha, P. Zhang, C. L. Ho and K. Li, Angew. Chem., Int. Ed., 2020, 59, 10179–10185 CrossRef CAS.
- H. Ji, W. Liu, Y. Huo, M. Han, Q. Yao, S. Gong and W. Duan, Dyes Pigm., 2023, 209, 110915 CrossRef CAS.
- N. Dorh, S. Zhu, K. B. Dhungana, R. Pati, F.-T. Luo, H. Liu and A. Tiwari, Sci. Rep., 2015, 5, 18337 CrossRef CAS PubMed.
- S. Qi, N. Kwon, Y. Yim, V.-N. Nguyen and J. Yoon, Chem. Sci., 2020, 11, 6479–6484 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- A. Treibs and F.-H. Kreuzer, Adv. Cycloaddit., 1968, 718, 208–223 CAS.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS.
- R. Parui, N. Meher and P. K. Iyer, Mater. Adv., 2022, 3, 5980–5986 RSC.
- H. Huang, L. Liu, J. Wang, Y. Zhou, H. Hu, X. Ye, G. Liu, Z. Xu, H. Xu, W. Yang, Y. Wang, Y. Peng, P. Yang, J. Sun, P. Yan, X. Cao and B. Z. Tang, Chem. Sci., 2022, 13, 3129–3139 RSC.
- R. Parui, N. Zehra and P. K. Iyer, J. Mater. Chem. C, 2023, 11, 11243–11251 RSC.
- N. Meher, A. P. Bidkar, D. Barman, S. S. Ghosh and P. K. Iyer, Chem. Commun., 2020, 56, 14861–14864 RSC.
- S. Liu, G. Feng, B. Z. Tang and B. Liu, Chem. Sci., 2021, 12, 6488–6506 RSC.
- D. Barman, P. Rajamalli, A. P. Bidkar, T. Sarmah, S. S. Ghosh, E. Zysman-Colman and P. K. Iyer, Small, 2025, 21, 2409533 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- E. C. Cho, L. Au, Q. Zhang and Y. Xia, Small, 2010, 6, 517–522 CrossRef CAS.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- J. C. Koziar and D. O. Cowan, Acc. Chem. Res., 1978, 11, 334–341 CrossRef CAS.
- M. Rae, F. Perez-Balderas, C. Baleizão, A. Fedorov, J. A. S. Cavaleiro, A. C. Tomé and M. N. Berberan-Santos, J. Phys. Chem. B, 2006, 110, 12809–12814 CrossRef CAS PubMed.
- V. C. Wakchaure, Goudappagouda, T. Das, S. Ravindranathan and S. Santhosh Babu, Nanoscale, 2021, 13, 10780–10784 RSC.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS PubMed.
- Z. Yang, W. Qin, J. W. Y. Lam, S. Chen, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725–3730 RSC.
- D. C. Santra, M. K. Bera, P. K. Sukul and S. Malik, Chem.–Eur. J., 2016, 22, 2012–2019 CrossRef CAS PubMed.
- L. He, X. Yang, K. Xu, X. Kong and W. Lin, Chem. Sci., 2017, 8, 6257–6265 RSC.
- J. Heo, D. P. Murale, H. Y. Yoon, V. Arun, S. Choi, E. Kim, J.-S. Lee and S. Kim, Aggregate, 2022, 3, e159 CrossRef CAS.
- D. Chaudhuri, D. Li, Y. Che, E. Shafran, J. M. Gerton, L. Zang and J. M. Lupton, Nano Lett., 2011, 11, 488–492 CrossRef CAS PubMed.
- Y. Xiang, Y. Zhao, N. Xu, S. Gong, F. Ni, K. Wu, J. Luo, G. Xie, Z.-H. Lu and C. Yang, J. Mater. Chem. C, 2017, 5, 12204–12210 RSC.
- S. Sasaki, G. P. C. Drummen and G.-I. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- P. Singh, L. S. Mittal, V. Vanita, K. Kumar, A. Walia, G. Bhargava and S. Kumar, J. Mater. Chem. A, 2016, 4, 3750–3759 CAS.
- W. Su, Y. Zhang, C. Zhao, X. Li and J. Jiang, ChemPhysChem, 2007, 8, 1857–1862 CrossRef CAS PubMed.
- Y. Huang, L. Fu, W. Zou, F. Zhang and Z. Wei, J. Phys. Chem. C, 2011, 115, 10399–10404 CrossRef CAS.
- J. Zhang, Q. Liu, W. Wu, J. Peng, H. Zhang, F. Song, B. He, X. Wang, H. H. Y. Sung, M. Chen, B. S. Li, S. H. Liu, J. W. Y. Lam and B. Z. Tang, ACS Nano, 2019, 13, 3618–3628 CrossRef CAS PubMed.
- S. Sarkar, A. Sarkar and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 19841–19845 CrossRef CAS PubMed.
- B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681–16689 CrossRef CAS PubMed.
- I. Javed, T. Zhou, F. Muhammad, J. Guo, H. Zhang and Y. Wang, Langmuir, 2012, 28, 1439–1446 CrossRef CAS PubMed.
- J. Zhang, B. Xu, W. Tian and Z. Xie, Chem. Sci., 2018, 9, 2620–2627 RSC.
- D. Li, Z. Tang, Y. Gao, H. Sun and S. Zhou, Adv. Funct. Mater., 2016, 26, 66–79 CrossRef CAS.
- A. M. Thamizhchelvan, H. Ma, T. Wu, D. Nguyen, J. Padelford, T. J. Whitworth, Y. Li, L. Yang and H. Mao, Nanoscale, 2024, 16, 21398–21415 RSC.
- J. R. Finnegan, L. I. FitzGerald, M. Z. Chen, N. M. Warne, D. Yuen, T. P. Davis, A. P. R. Johnston and K. Kempe, Nano Lett., 2024, 24, 89–96 CrossRef CAS PubMed.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
- N. L. Chanaday and E. T. Kavalali, FEBS Lett., 2018, 592, 3606–3614 CrossRef CAS PubMed.
- H. Tomoda, Y. Kishimoto and Y. C. Lee, J. Biol. Chem., 1989, 264, 15445–15450 CrossRef CAS PubMed.
- G. Preta, J. G. Cronin and I. M. Sheldon, Cell Commun. Signaling, 2015, 13, 24 CrossRef PubMed.
- E. Macia, M. Ehrlich, R. Massol, E. Boucrot, C. Brunner and T. Kirchhausen, Dev. Cell, 2006, 10, 839–850 CrossRef CAS PubMed.
- T. Kirchhausen, E. Macia and H. E. Pelish, in Methods in Enzymology, Academic Press, 2008, vol. 438, pp. 77–93 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization spectra (1H & 13C NMR, HRMS), theoretical data, SCXRD, PXRD, TRPL data, NMR, and additional supplementary figures. CCDC 2336090, 2336092, 2336105, 2336104 and 2336103. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08296f |
| This journal is © The Royal Society of Chemistry 2025 |