
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-poisoning of CO and carbonyl species over Pd catalysts during the electrooxidation of ethylene glycol to glycolic acid at elevated current density†
Jia
Cheng
a,
Yunchuan
Tu
 a,
Yang
Xiang
a,
Jingtian
Ni
a,
Tao
Guo
a,
Xun
Huang
*a,
Bin
Liu
*b and
Zidong
Wei
*a
a,
Yang
Xiang
a,
Jingtian
Ni
a,
Tao
Guo
a,
Xun
Huang
*a,
Bin
Liu
*b and
Zidong
Wei
*a
aCenter of Advanced Electrochemical Energy, State Key Laboratory of Advanced Chemical Power Sources, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 40004, P. R. China. E-mail: huangxun@cqu.edu.cn; zdwei@cqu.edu.cn
bDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong 999077, P. R. China. E-mail: bliu48@cityu.edu.hk
First published on 1st February 2025
Abstract
The electrocatalytic oxidation of ethylene glycol (EG) to produce valuable glycolic acid (GLYA) is a promising strategy to tackle EG overcapacity. Despite the good selectivity of Pd for EG oxidation, its performance is constrained by limited mass activity and toxicity of intermediates like CO or CO-analogues. This study reports the alloying of Pd with Ni and Mo metals to enhance the activity and durability of EG oxidation in alkaline media. Notably, the peak current density reached up to 2423 mA mg−1, double that of pristine Pd/C, accompanied by a GLYA Faraday efficiency up to 87.7%. Moreover, PdNiMo/C exhibited a 5-fold slower activity decline compared to Pd/C. In situ experiments and theoretical analysis reveal that Ni and Mo synergistically strengthen the oxygen affinity of the catalyst, facilitating the generation of *OH radicals at lower potentials, thereby accelerating EG oxidation kinetics. Additionally, Ni incorporation prevents C–C bond cleavage and weakens CO adsorption, effectively mitigating catalyst poisoning.
Introduction
Ethylene glycol (EG) is a large-scale chemical compound derived from oil through ethylene and from coal through syngas. It is predominantly used in the manufacture of polyethylene glycol terephthalate (PET). However, the proliferation of production facilities in recent years has resulted in a surplus of capacity and a notable decline in market prices.1 Additionally, the extensive use of PET also raises concerns about the sustainable reutilization of massive recycled EG.2,3 Consequently, the transformation of EG into high-value products has emerged as an urgent issue in the present day.4EG contains two hydroxyl groups and can be oxidized into various chemicals.3,5–7 There are two main pathways in the EG oxidation reaction (EGOR): the C1 pathway involving the breaking of the C–C bond, and the C2 pathway with the C–C bond remaining unbroken.8 As shown in Scheme 1, among C2 products in the EGOR, the final product oxalic acid is less valued while the products containing aldehyde like glycol aldehyde, glyoxal and glyoxylic acid are prone to overoxidation. Thus, glycolic acid (GLYA), a stable and valuable product of unilateral hydroxyl oxidation of EG, gains significant attention for upgrading EG. As it finds applications in various areas such as metal cleaning, adhesives, and as complexing agents, GLYA has seen increasing market demand in recent years.2,9
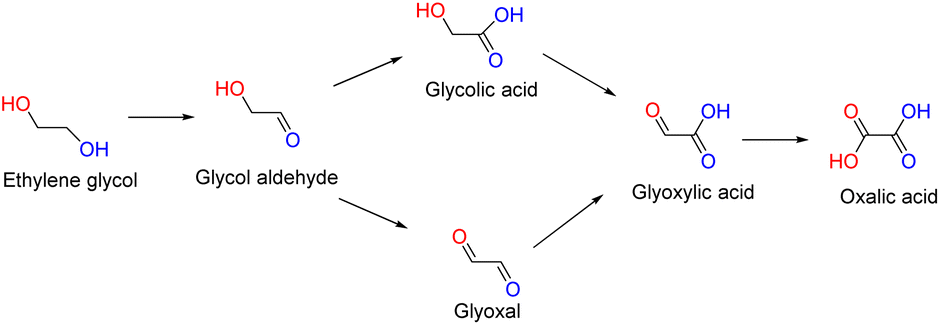 | ||
| Scheme 1 Reaction routes of ethylene glycol oxidation. | ||
Compared with conventional thermochemical methods, the electrocatalytic oxidation of EG for GLYA preparation shows greater promise due to its ability to utilize green electrical energy and avoid severe reaction conditions such as high temperature, high pressure, or strong acid. Previous studies have demonstrated that noble metals such as Pt,10 Pd,3,6,11,12 and Au13,14 display superior activity for the EGOR compared to the non-noble metal catalysts.15,16 In comparison with Pt, Pd and Au exhibit a higher d-band center and lower deprotonation ability under alkaline conditions. Consequently, the C–C bond is less readily broken, resulting in the predominant formation of GLYA.8,17–19 For example, Liu et al.3 and Yan et al.2 recently reported the use of Pd/Ni(OH)2 and Au/Ni(OH)2 as EGOR electrocatalysts, respectively, and demonstrated their ability to achieve industrial-level current densities.
In general, the electrochemical oxidation of EG begins with the stepwise dehydrogenation to form the 2-hydroxyacetyl intermediate (CH2OH-CO*), followed by its interaction with reactive OH to produce GLYA.20,21 Without sufficient OH, the intermediate could strongly occupy the active sites and temporarily deactivate the catalyst.9 This impeding effect on kinetic responses becomes more prominent under elevated-current conditions, resulting in a rapid decline in catalyst activity. Therefore, frequent electrolyte replacements are usually necessary to refresh the composition at the electrode interface. Furthermore, the ongoing oxidation process of the 2-hydroxyacetyl intermediate can also lead to the cleavage of the C–C bond and the formation of CO,22 which strongly poisons the active sites and causes current decay over time.8,17,23
Apparently, to improve the activity and prolong the lifetime of catalysts, it is vital to facilitate the generation of active OH on the anode and avoid the cleavage of the C–C bond in EG.24 It has been widely recognized that Pd–M alloys have higher catalytic activity and stability than Pd in alkaline medium. For example, in direct alcohol fuel cells (DAFCs), the alloying of Ni can introduce more OH on the active sites, which facilities the oxidation of CO to prevent poisoning.25,26 Mo can also act as a stable electron donor for Pd and provide extra active oxygenates for the catalytic process,27–29 which in turn enhances the catalytic activity and the tolerance to CO.30 Though the expected reaction pathway in the preparation of GLYA is different from the alcohol oxidation in DAFCs as the breaking of the C–C bond is not favored, tuning the electron structure and oxygen affinity of Pd is still a prospective strategy to intensify activity and stability.
In this work, oxyphilic Ni and Mo elements are incorporated into the Pd/C catalyst to improve its performance in EG oxidation. The synthesized PdNiMo/C catalyst attains a peak mass current density of 2423 mA mg−1 and a GLYA Faraday efficiency (FE) up to 87.7%, with a lifetime prolonged 5 fold compared to Pd/C. In situ infrared (IR) and in situ mass spectrometry, along with theoretical calculations, demonstrate that the presence of Ni and Mo enables the generation of *OH active species at relatively low potentials, which facilitates the oxidation of EG while simultaneously inhibiting the cleavage of C–C bonds, thereby favoring the generation of GLYA at elevated current density.
Results and discussion
Physical characterization of Pd-based catalysts
The as-prepared PdNiMo/C, PdMo/C, PdNi/C, and Pd/C catalysts were first characterized using X-ray diffraction (XRD) (Fig. 1a). The broad peaks observed before 30° are the characteristic diffraction peaks of carbon, while the peaks at 39.4°, 45.5°, 66.4°, and 79.8° can be attributed to the (111), (200), (220), and (311) crystal planes of Pd, respectively.31 The diffraction peaks of metals typically exhibit low intensity, possibly due to the low concentration of the metal present. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) measurements revealed that the Pd metal loading is less than 2%, with the loading of other metals being even lower (Fig. S1†). The emergence of an extra set of diffraction peaks at slightly higher angles for PdM/C (M = Ni or Mo) in comparison to Pd/C, especially at the (111) plane, can be attributed to the partial incorporation of the transition metal into the Pd lattice, leading to a reduction in the lattice parameter. This observation confirms the successful partial alloying of Pd. The X-ray photoelectron spectroscopy (XPS) analysis also indicates the presence of Pd, Ni, and Mo elements in the PdNiMo/C material (Fig. 1b–d). Pd is identified in the forms of Pd0 and Pd2+, with the corresponding peaks located at 335.41/340.67 eV and 336.7/342.4 eV, respectively.11,32 These peaks are shifted toward lower binding energies compared to the Pd/C peaks (335.73/341.03 eV and 336.9/342.5 eV), because the transition metals act as electron donors to increase the electron density of Pd.28,33 The dominance of Pd in PdNiMo/C can be ascribed to metallic Pd, whereas the presence of a minor quantity of Pd2+ can be attributed to the inevitable partial oxidation of the surface Pd when exposed to air. High-resolution Ni 2p and Mo 3d spectra revealed that both Ni and Mo are predominantly present as Ni0 and Mo0, respectively, in the PdNiMo/C samples, while the smaller part in higher valence states is due to the surface oxidation. Compared with the standard peaks, the XPS peaks of both Ni and Mo are shifted to higher binding energies. This is because the electrons of Ni and Mo are partially transferred to Pd, in accordance with the peak shift of Pd towards lower binding energies. | ||
| Fig. 1 (a) XRD spectra of different Pd-based catalysts; XPS spectra of (b) Pd 3d, (c) Ni 2p and (d) Mo 3d in PdNiMo/C; (e and f) TEM image and the corresponding elemental mapping images of PdNiMo/C catalysts. | ||
The morphology of PdNiMo/C was examined via transmission electron microscopy (TEM), revealing a spherical nanoparticle architecture on the carbon support, with an average diameter of about 5.8 nm (Fig. 1e). Elemental mapping confirms a uniform dispersion of Pd, Ni, and Mo elements, comprising 75%, 17%, and 8%, respectively (Fig. S4†), corroborating the findings from ICP-OES measurements (Fig. S1†). High-resolution TEM images (Fig. 1f) show a reduced lattice spacing compared to the (111) plane of pure Pd (0.226 nm), further substantiating alloy formation. The combination of XRD, TEM, and XPS results definitively validates the successful preparation of PdNiMo/C alloys.
Electrochemical evaluation of Pd-based catalysts
To assess the electrocatalytic performance of PdNiMo/C, a sequence of electrochemical tests was carried out utilizing a three-electrode configuration in an H-type electrolyzer. Fig. 2a illustrates the cyclic voltammetry (CV) curves of various catalysts in an alkaline solution under a N2-saturated atmosphere. The peak within 0.2 V to 0.6 V vs. RHE signifies the adsorption of *OH on the catalyst surface,3,34 while a negative peak around 0.5–0.7 V vs. RHE in the reverse scan reflects the reduction of PdO.35,36 Notably, PdNiMo/C outperforms other Pd-based catalysts, exhibiting the highest *OH adsorption current and PdO reduction current. Assuming a PdO monolayer reduction charge of 0.405 mC cm−2,5,37 the calculated electrochemical surface area (ECSA) for PdNiMo/C, PdMo/C, PdNi/C, and Pd/C is 941.0, 296.5, 452.6, and 39.0 m2 g−1, respectively. Fig. 2b depicts the linear sweep voltammetry (LSV) curves of PdNiMo/C, PdMo/C, PdNi/C, and Pd/C in a 1.0 M KOH solution containing 1.0 M EG, where PdNiMo/C stands out with a maximum mass current density of up to 2423 mA mgPd−1, 1.86 times higher than that of Pd/C. Besides, PdNiMo/C requires only 0.81 V vs. RHE to achieve a current density of 1000 mA mg−1, which is 90 mV lower than that of PdNi/C (0.90 V vs. RHE) and 100 mV lower than that of PdMo/C (0.91 V vs. RHE). This underscores the synergistic effect of Ni and Mo elements, enhancing EG oxidation catalysis beyond individual additives. As the potential increases, in addition to the occupation of active sites by CO-like intermediates, there is a competing reaction for Pd oxidation, leading to a decrease in Pd0 active sites,38 hindering further current increase. Nevertheless, as shown in Fig. 2c, PdNiMo/C attains its peak current density for EG oxidation at a higher potential, indicating a stronger resistance to deactivation. | ||
| Fig. 2 (a) CV curves of PdNiMo/C, PdMo/C, PdNi/C and Pd/C electrocatalysts in 1.0 M KOH solution with a scan rate of 20 mV s−1. (b) LSV curves in 1.0 M KOH + 1.0 M EG with a scan rate of 5 mV s−1. (c) Peak current densities obtained from the forward scans in (b). (d and e) LSV curves of PdNiMo/C catalysts with different alloy ratios with a scan rate of 5 mV s−1. (f) EGOR current variation over time in 1.0 M EG + 1.0 M NaOH solution at 0.83 V vs. RHE. (g) FE of GLYA produced by PdNiMo/C electrocatalytic oxidation of EG at different number of cycles. FE of GLYA (h), OA and FA (i) on different catalysts. | ||
The impact of Ni and Mo on enhancing the electrocatalytic activity of PdNiMo/C towards EG oxidation was further studied. Experimental findings demonstrate that the incorporation of both Ni and Mo could improve the catalytic activity of Pd-catalyzed EG oxidation, albeit with distinct contributions. As evident from Fig. 2d and e, an adequate augmentation in Mo loading can markedly elevate the catalytic activity, but it results in a slight decrease in the peak potential. Conversely, progressive augmentation of Ni doping shifts the peak potential to higher values while sustaining the current activity at low potentials. In short, Ni is more capable of oxidizing EG at elevated potentials, while Mo attained superior catalytic activity at comparable potentials.
The electrochemical stability of PdNiMo/C, PdNi/C, PdMo/C, and Pd/C in EG oxidation was assessed through chronoamperometry measurement (Fig. 2f). It is recognized that anodic deactivation is more pronounced in batch reactors than in continuous flow reactors, therefore the current density decreases significantly within several hours. One possible explanation is the existence of a desorption–re-adsorption-reaction mechanism for the reaction intermediates between the electrode surface and the local electrolyte.39 In an H-type electrolyzer, the diffusion is slow and the poisoning intermediates are accumulated on the surface, leading to the deactivation of active sites; in contrast, in a continuous flow reactor, the electrolyte adjacent to the electrode surface is rapidly refreshed and the key toxic intermediates can easily diffuse away from the active sites, leading to a lowered deactivation rate. After electrolysis at 0.83 V vs. RHE for 6 h, PdNiMo/C retains a mass current density of 259.8 mA mg−1, surpassing PdNi/C (120.2 mA mg−1), PdMo/C (32.1 mA mg−1), and Pd/C (71.9 mA mg−1). Notably, PdNiMo/C operated for over 6 h before its current density dropped to 24% of its initial level, five-fold longer than the 1.2 h performance of Pd/C. Comparing the results between Pd/C and PdNi/C or PdMo/C, it can be found that Mo addition decreases catalytic stability, whereas Ni incorporation extends the electrocatalytic lifetime of the EGOR process (Fig. S6†). It is worth mentioning that the deactivation is reversible and the catalytic activity can be recovered to the initial level by refreshing the electrolyte (Fig. 2g). HPLC analysis of the post-electrolysis electrolyte, following treatment with an anion-exchange resin to convert carboxylate into carboxylic acid, identifies GLYA as the primary product with oxalic acid (OA) and formic acid (FA) as byproducts. Based on the standard curve (Fig. S7 and 8†), the calculated Faraday efficiency (FE) of GLYA on PdNiMo/C is 87.7%, higher than that of the other catalysts. The experiments demonstrate that the alloying of Ni and Mo has a positive impact on enhancing not only the catalyst activity and stability, but also the product selectivity as well as the yield of GLYA.40 Among the evaluated catalysts, PdNiMo/C predominantly produces OA as the byproduct, while other catalysts favor C–C bond cleavage to generate FA (Fig. 2i).
Revealing the activity enhancement mechanism
DFT calculations were employed to investigate the mechanism of EG oxidation on different Pd-based catalysts. Pd (111) was selected to stand for Pd/C, whereas PdNi (111), PdMo (111), and PdNiMo (111) were constructed by replacing partial surface Pd atoms with Ni and/or Mo (Fig. S9†). As depicted in Fig. 3a and S10,† the partial density of states (PDOS) reveals a consistent negative shift in the d-band centers of Pd after alloying. For example, the d-band center of Pd in PdNiMo is decreased to −1.87 eV compared with pristine Pd (−1.70 eV), which can be attributed to the electron transfer from transition metal dopants to Pd atoms. | ||
| Fig. 3 (a) PDOS (d-band) of PdNiMo, PdMo, PdNi, and Pd. (b) Calculated adsorption energies of EG and *OH. (c) Charge density difference during EG adsorption onto PdNiMo and Pd (111); negative and positive charges are presented in green and yellow, respectively. (d) Gibbs free energy diagram and the optimized configurations of EG oxidation to GLYA on Pd and PdNiMo. The colored values are the Gibbs free energy changes in eV. | ||
Moreover, an analysis of EG and *OH adsorption energies (Eads) on electrode surfaces demonstrates the following trend: PdNiMo > PdMo > PdNi > Pd (Fig. 3b and S11†), suggesting that Mo and Ni doping enhanced the adsorption of *OH, a probable contributor to activity improvement.41,42 Specifically, *OH can be adsorbed on different metal sites on the PdNiMo surface, with adsorption energy being −3.92 eV on Mo, which is higher than those on the Ni site (−3.06 eV) and Pd site (−2.32 eV). On the other hand, EG is believed to interact with the Pd surface through the oxygen in the OH group (Fig. 3c and d).13,36 After alloying, the preferential adsorption site of EG shifts from Pd to Mo because the more vacant d orbitals of Mo promote oxygen affinity, favoring oxygen atom binding. Differential charge density analysis revealed a substantial intensification of charge transfer between the oxygen in EG and active sites upon the introduction of Mo, as is evident in Fig. 3c.
Gibbs free energy diagrams for EG oxidation to GLYA are compared on the surface of Pd and PdNiMo catalysts (Fig. 3d and S12†). With Pd, the rate-limiting step is the dehydrogenation of hydroxyl, with an energy barrier of 0.82 eV. In contrast, PdNiMo lowers this hydroxyl dehydrogenation barrier to 0.04 eV, shifting the rate-limiting step to the dehydrogenation of methylene, which exhibits a lower barrier of 0.72 eV. Fig. S13† visualizes the adsorption energies and M–O binding strength during the first dehydrogenation step on hydrogen of EG oxidation. Notably, the adsorption energies of both EG (b1) and CH2OH-CO* (b2) on the PdNiMo surface exceed those on the Pd surface. In particular, crystal orbital Hamilton population (COHP) analysis reveals that the binding of the metal site to the oxygen atom in CH2OH-CO* is significantly enhanced, which in turn stabilizes the CH2OH-CO* intermediate and reduces the energy barrier of the first dehydrogenation step.
Revealing the anti-toxicity mechanism of PdNiMo
CO and CH2OH-CO* produced during the EGOR are the prevalent toxic species, which hinder catalytic activity by occupying the active site.3 Accelerating CH2OH-CO* oxidation and mitigating CO generation were pivotal for enhancing the toxicity resistance of catalysts. Electrochemical in situ Fourier-transform infrared spectroscopy (FTIR) and in situ mass spectrometry were conducted on various Pd-based catalysts to understand the evolution of intermediates during EG oxidation. The IR vibrational bands of different bonds or groups are summarized in Table S1.† As depicted in Fig. 4a and b, the peaks at 1069 cm−1, 1322 cm−1, 1409 cm−1, and 1581 cm−1 are associated with GLYA,6,8,43 whereas the peak at 1631 cm−1 is attributed to CH2OH-CO* and interfacial water.43 Apparently, their signals are amplified on PdNiMo/C in comparison to Pd/C, indicating that the adsorption of oxygenates and carbonyls is enhanced, in accordance with the DFT results. Notably, the peak at 1889 cm−1 corresponding to multiple-bonded COM is clearly observed on Pd/C,5 whereas no characteristic peaks corresponding to CO and CO2 are detected on PdNiMo/C throughout the entire spectral range of 1800–2500 cm−1 (Fig. 4b).44,45 This finding suggests that PdNiMo is less prone to inducing C–C bond cleavage and the subsequent formation of C1 products. Moreover, the peaks of GLYA exhibit an increase with the potential on PdNiMo/C, but those on Pd/C diminish and even disappear at higher potentials, implying that CO intermediates have a tendency to persist on the Pd surface, thereby potentially deactivating the active sites.34 Likewise, the peaks of GLYA increase with the applied potential on PdNi/C, but they decrease with the applied potential on PdMo/C and eventually vanish at elevated potentials (Fig. S14†), highlighting the role of Ni in inhibiting Pd oxidation. Overall, no distinct peaks characteristic of CO or CO2 are observed for PdNiMo/C, PdNi/C, and PdMo/C. In situ mass spectrometry results also reveal the absence of C1 products, such as FA and CO2, on the PdNiMo/C anode (Fig. 4c, d and S15†). In contrast, Pd/C results in evident formation of FA and CO2. These results verify that the alloying of Pd with Ni and Mo can prevent C–C bond cleavage in EG oxidation. | ||
| Fig. 4 Electrochemical in situ FTIR spectra of (a) PdNiMo/C and (b) Pd/C in 1.0 M KOH + 1.0 M EG. The in situ mass spectra of (c) PdNiMo/C and (d) Pd/C. CO stripping of (e) PdNiMo/C and (f) Pd/C with a scan rate of 5 mV s−1. | ||
Additional CO stripping experiments were conducted to assess the CO poisoning resistance of the PdNiMo/C catalysts (Fig. 4e, f and S16†).46 The peak potential for CO dissolution shifts negatively by 70 mV on PdNiMo/C versus Pd/C, implying a better capacity to eliminate CO even when a trace amount is generated. This improved capacity originates from the stronger affinity to OH that oxidizes CO. In a word, PdNiMo/C not only reduces the formation of CO, but also accelerates the conversion of CO.
The excellent anti-poisoning ability of PdNiMo/C in the EGOR process was further investigated using DFT. As shown in Fig. S17,† C–C bond breaking encompasses multiple routes, including the direct breaking of EG, CH2OH-CO* and OA.20,22 The weaker deprotonation ability of the Pd-based catalysts and the unilateral adsorption of EG suggest that the immediate C–C bond breaking of EG upon adsorption is not the main pathway for the generation of C1 products. Meanwhile, as the major by-product is FA rather than OA (Fig. 2h and i), it can be assumed that the generation of FA and CO is not from OA via the over-oxidation of GLYA. Therefore, C–C breaking most probably occurs in CH2OH-CO*, as shown in Fig. 5a. The subsequent oxidation of the CH2OH-CO* intermediate can generate either GLYA via the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or FA via the cleavage of C–C, depending on the competitive activation of CO and C–C, which can be identified by their bond length variation. As shown in Fig. 5b and c, the M–O bond length between CH2OH-CO* and the active metal site decreases from 2.36 Å on Pd (111) to 2.15 Å on PdNiMo (111), as the binding energy increases from 2.69 eV to 3.05 eV, implying a stronger interaction between the metal site and oxygen atom. As a result, the bond length of the CO bond in CH2OH-CO* increases by 0.079 Å upon the introduction of Ni and Mo metals, whereas the bond length of the C–C bond experiences a slight reduction, which activates the CO bond while inhibiting the cleavage of the C–C bond. This observation is consistent with the experimental finding that the generation of FA and CO is significantly diminished on PdNiMo. Interestingly, the introduction of Mo alone primarily stretches the CO bond, while the introduction of Ni alone predominantly compresses the C–C bond. This outcome also agrees with the experimental findings that Mo improves the activity of EG oxidation while Ni enhances the resistance to deactivation.
O or FA via the cleavage of C–C, depending on the competitive activation of CO and C–C, which can be identified by their bond length variation. As shown in Fig. 5b and c, the M–O bond length between CH2OH-CO* and the active metal site decreases from 2.36 Å on Pd (111) to 2.15 Å on PdNiMo (111), as the binding energy increases from 2.69 eV to 3.05 eV, implying a stronger interaction between the metal site and oxygen atom. As a result, the bond length of the CO bond in CH2OH-CO* increases by 0.079 Å upon the introduction of Ni and Mo metals, whereas the bond length of the C–C bond experiences a slight reduction, which activates the CO bond while inhibiting the cleavage of the C–C bond. This observation is consistent with the experimental finding that the generation of FA and CO is significantly diminished on PdNiMo. Interestingly, the introduction of Mo alone primarily stretches the CO bond, while the introduction of Ni alone predominantly compresses the C–C bond. This outcome also agrees with the experimental findings that Mo improves the activity of EG oxidation while Ni enhances the resistance to deactivation.
 | ||
| Fig. 5 (a) Reaction pathways for C–C bond breaking in the EGOR process. (b) Calculated configuration of CH2OH-CO* and its C–C bond breaking products. Calculated adsorption energies of (c) CH2OH-CO* and (d) CO on Pd-based electrocatalyst surfaces. (e) Calculated Gibbs free energy change during C–C bond cleavage in CH2OH-CO* on Pd and PdNiMo surfaces. | ||
Subsequently, the adsorption of CO, a toxic intermediate from C–C bond cleavage, on different catalytic surfaces is calculated (Fig. 5d). It is found that PdNiMo exhibits the weakest CO interaction with an adsorption energy of −1.946 eV, whereas Pd shows the strongest CO binding with an adsorption energy of −2.246 eV, which implies that Ni and Mo can significantly inhibit the continuous adsorption of CO on the active sites. Furthermore, as shown in Fig. 5e, the Gibbs free energy of CH2OH-CO* C–C bond breaking on PdNiMo (−0.18 eV) was much higher than that on Pd (−0.86 eV). These calculations clearly illustrate that CH2OH-CO* was less susceptible to C–C bond breaking under PdNiMo catalysis and that PdNiMo was more resistant to toxicity.
Conclusions
We successfully developed high-performance anodic catalysts for the electrocatalytic oxidation of EG to produce GLYA by partially alloying Pd with two oxygen-affinity elements, Ni and Mo. The resulting PdNiMo/C catalyst exhibits remarkable enhancements in both activity and stability compared to pristine Pd/C. Specifically, the peak current density of PdNiMo/C reaches an impressive 2423 mA mg−1, double that of Pd/C, while maintaining a high GLYA FE of up to 87.7%. Furthermore, the PdNiMo/C catalyst demonstrates a five-fold slower activity decline compared to Pd/C, highlighting its improved durability.The in situ characterization and DFT calculation reveal that the synergistic effects of Ni and Mo play a crucial role in enhancing the catalytic performance. Ni incorporation suppresses C–C bond cleavage, thus preventing catalyst deactivation, while Mo contributes to the generation of *OH radicals at lower potentials, accelerating the EG oxidation kinetics. The combination of these two effects not only improves the catalytic activity but also promotes the selectivity towards GLYA production.
In summary, this study provides a promising strategy for the development of highly active and stable anodic catalysts for EG oxidation to GLYA. The findings of this work have significant implications for the efficient utilization of EG and the production of valuable chemicals, contributing to the advancement of sustainable energy conversion and storage technologies.
Data availability
The data that support the findings of this study are available in the paper and ESI.†Author contributions
Z. D. W., B. L., and X. H. conceived the project and supervised the research. J. C., Y. C. T., Y. X., J. T. N., and T. G. performed the experiments and drafted the manuscript. J. C. provided the EFT calculation support. All authors analysed the data and contributed to discussions.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22178033 and 22090030) and the Fundamental Research Funds for the Central Universities (No. 2024CDJXY010).References
- H. Yan, S. Yao, J. Wang, S. Zhao, Y. Sun, M. Liu, X. Zhou, G. Zhang, X. Jin, X. Feng, Y. Liu, X. Chen, D. Chen and C. Yang, Appl. Catal., B, 2021, 284, 119803 CrossRef CAS.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202301639 CrossRef.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D.-H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C.-T. Dinh, J. Li, T.-T. Zhuang, F. Li, T.-K. Sham, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 14–22 CrossRef CAS.
- W. Huang, X. Y. Ma, H. Wang, R. Feng, J. Zhou, P. N. Duchesne, P. Zhang, F. Chen, N. Han, F. Zhao, J. Zhou, W. B. Cai and Y. Li, Adv. Mater., 2017, 29, 1703057 CrossRef PubMed.
- R. G. Da Silva, A. Rodrigues de Andrade, K. Servat, C. Morais, T. W. Napporn and K. B. Kokoh, ChemElectroChem, 2020, 7, 4326–4335 CrossRef CAS.
- N. L. Chauhan, V. Dameera, A. Chowdhury, V. A. Juvekar and A. Sarkar, Catal. Today, 2018, 309, 126–132 CrossRef CAS.
- Y. Qin, W. Zhang, F. Wang, J. Li, J. Ye, X. Sheng, C. Li, X. Liang, P. Liu, X. Wang, X. Zheng, Y. Ren, C. Xu and Z. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200899 CrossRef CAS PubMed.
- D. Si, B. Xiong, L. Chen and J. Shi, Chem Catal., 2021, 1, 941–955 CrossRef CAS.
- K. Yin, Y. Chao, F. Lv, L. Tao, W. Zhang, S. Lu, M. Li, Q. Zhang, L. Gu, H. Li and S. Guo, J. Am. Chem. Soc., 2021, 143, 10822–10827 CrossRef CAS PubMed.
- Z. Wu, X. Liu, Y. Zhong and L. Li, ACS Appl. Nano Mater., 2022, 5, 15162–15171 CrossRef CAS.
- Y. Chen, A. Lavacchi, H. Miller, M. Bevilacqua, J. Filippi, M. Innocenti, A. Marchionni, W. Oberhauser, L. Wang and F. Vizza, Nat. Commun., 2014, 5, 4036 CrossRef CAS PubMed.
- Y. Kwon, S. C. S. Lai, P. Rodriguez and M. T. M. Koper, J. Am. Chem. Soc., 2011, 133, 6914–6917 CrossRef CAS PubMed.
- Z. Li, Y. Yan, S. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- T. Matsumoto, M. Sadakiyo, M. L. Ooi, T. Yamamoto, S. Matsumura, K. Kato, T. Takeguchi, N. Ozawa, M. Kubo and M. Yamauchi, Phys. Chem. Chem. Phys., 2015, 17, 11359–11366 RSC.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- R. M. Arán-Ais, E. Herrero and J. M. Feliu, Electrochem. Commun., 2014, 45, 40–43 CrossRef.
- W. Zhang, X. Ma, S. Zou and W. Cai, J. Electrochem., 2021, 27, 233–256 CAS.
- W. Chen, J. Shi, C. Xie, W. Zhou, L. Xu, Y. Li, Y. Wu, B. Wu, Y. Huang, B. Zhou, M. Yang, J. Liu, C. Dong, T. Wang, Y. Zou and S. Wang, Natl. Sci. Rev., 2023, 10(5), nwad099 CrossRef CAS PubMed.
- W. Chen, J. Shi, Y. Wu, Y. Jiang, Y. C. Huang, W. Zhou, J. Liu, C. L. Dong, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2023, 63(4), e202316449 CrossRef PubMed.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed.
- H. Xu, B. Huang, Y. Zhao, G. He and H. Chen, Inorg. Chem., 2022, 61, 4533–4540 CrossRef CAS PubMed.
- H. Lv, L. Z. Sun, D. Xu, S. L. Suib and B. Liu, Green Chem., 2019, 21, 2367 RSC.
- R. Kottayintavida and N. K. Gopalan, Int. J. Hydrogen Energy, 2020, 45, 8396–8404 CrossRef CAS.
- S. Y. Shen, T. S. Zhao, J. B. Xu and Y. S. Li, J. Power Sources, 2010, 195, 1001–1006 CrossRef CAS.
- Y. Pan, H. Li, J. Min, J. Xiong, Y. Qin, Z. Wang, Z. Wu, S. Feng, J. Lai and L. Wang, Chem. Eng. J., 2022, 446, 137178 CrossRef CAS.
- Y. Qin, H. Huang, W. Yu, H. Zhang, Z. Li, Z. Wang, J. Lai, L. Wang and S. Feng, Adv. Sci., 2021, 9, 2103722 CrossRef PubMed.
- H. Luo, K. Wang, F. Lin, F. Lv, J. Zhou, W. Zhang, D. Wang, W. Zhang, Q. Zhang, L. Gu, M. Luo and S. Guo, Adv. Mater., 2023, 35(29), 2211854 CrossRef CAS PubMed.
- W. J. Pech-Rodríguez, D. González-Quijano, G. Vargas-Gutiérrez, C. Morais, T. W. Napporn and F. J. Rodríguez-Varela, Appl. Catal., B, 2017, 203, 654–662 CrossRef.
- A. Dutta, R. Adhikary, P. Broekmann and J. Datta, Appl. Catal., B, 2019, 257, 117847 CrossRef CAS.
- J.-X. Tang, Q.-S. Chen, L.-X. You, H.-G. Liao, S.-G. Sun, S.-G. Zhou, Z.-N. Xu, Y.-M. Chen and G.-C. Guo, J. Mater. Chem. A, 2018, 6, 2327–2336 RSC.
- J. Li, Y. Chen, R. Bai, C. Chen, W. Wang, Y. Pan and Y. Liu, Chem. Eng. J., 2022, 435(3), 134932 CrossRef CAS.
- J. Zhu, L. Xia, R. Yu, R. Lu, J. Li, R. He, Y. Wu, W. Zhang, X. Hong, W. Chen, Y. Zhao, L. Zhou, L. Mai and Z. Wang, J. Am. Chem. Soc., 2022, 144, 15529–15538 CrossRef CAS PubMed.
- Y. Zhong, Z. Wu, X. Liu and L. Li, Colloids Surf., A, 2022, 652, 129672 CrossRef CAS.
- D. Fan, K. Guo, Y. Zhang, Q. Hao, M. Han and D. Xu, J. Colloid Interface Sci., 2022, 625, 1012–1021 CrossRef CAS PubMed.
- K. Guo, Y. Teng, R. Guo, Y. Meng, D. Fan, Q. Hao, Y. Zhang, Y. Li and D. Xu, J. Colloid Interface Sci., 2022, 628, 53–63 CrossRef CAS PubMed.
- E. Berretti, L. Osmieri, V. Baglio, H. A. Miller, J. Filippi, F. Vizza, M. Santamaria, S. Specchia, C. Santoro and A. Lavacchi, Electrochem. Energy Rev., 2023, 6, 30 CrossRef CAS.
- H. H. Heenen, H. S. Pillai, K. Reuter and V. J. Bukas, Nat. Catal., 2024, 7, 847–854 CrossRef CAS.
- P. A. Christensen and D. Linares-Moya, J. Phys. Chem. C, 2010, 114, 1094–1101 CrossRef CAS.
- X. Z. Lao, X. Lao, L. Yang, B. Zhang, W. Ye, A. Fu and P. Guo, J. Energy Chem., 2023, 79, 279–290 CrossRef CAS.
- Z. Zeng, S. Wu, X. Huang and Z. Wei, Small, 2023, 20, 2305462 CrossRef PubMed.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed.
- J.-L. Lin, J. Ren, N. Tian, Z.-Y. Zhou and S.-G. Sun, J. Electroanal. Chem., 2013, 688, 165–171 CrossRef CAS.
- G. L. Liangping Xiao, Z. Yang, K. Chen, R. Zhou, H. Liao, Q. Xu and J. Xu, Adv. Funct. Mater., 2021, 31, 2100982 CrossRef.
- B. Qiao, T. Yang, S. Shi, N. Jia, Y. Chen, X. Chen, Z. An and P. Chen, Small, 2021, 17, 2006534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08579e |
| This journal is © The Royal Society of Chemistry 2025 |