
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The scalable growth of high-performance nanostructured heterojunction photoanodes for applications in tandem photoelectrochemical-photovoltaic solar water splitting devices†
Brian
Tam
 *ab,
Sebastian D.
Pike
c,
Jenny
Nelson
b and
Andreas
Kafizas
ad
*ab,
Sebastian D.
Pike
c,
Jenny
Nelson
b and
Andreas
Kafizas
ad
aDepartment of Chemistry, Molecular Science Research Hub, Imperial College London, White City, London, W12 0BZ, UK. E-mail: b.tam18@imperial.ac.uk
bDepartment of Physics, Imperial College London, South Kensington, London, SW7 2AZ, UK
cDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
dLondon Centre for Nanotechnology, Imperial College London, South Kensington, London, SW7 2AZ, UK
First published on 1st April 2025
Abstract
Due to their complementary absorption characteristics and band energy structure, the BiVO4-coated WO3 heterojunction architecture is commonly employed as a metal oxide photoanode for the water oxidation half-reaction. The energy level ordering results in a staggered heterojunction that can effectively separate photoexcited electrons into the WO3 layer towards the current collector and photoexcited holes into the BiVO4 layer towards the interface with the electrolyte. Chemical vapour deposition (CVD) is an upscalable technique for fabricating large-area thin films of a wide range of semiconductors with nanoscale control. The fluorine-doped tin oxide (FTO)-coated transparent conductive glass substrates used herein are mass-produced by the glass industry with atmospheric pressure CVD and so the entire photoelectrode could be produced in one production process on float glass panels. This work is a detailed study of the use of atmospheric pressure CVD to fully-fabricate high-performance BiVO4-coated WO3 nanostructures (500–2000 nm in length with 25–100 nm thick BiVO4 coatings) for photoelectrochemical (PEC) water splitting. Incident photon-to-current efficiency measurements were used to calculate optimal solar predicted photocurrents of 1.92 and 2.61 mA cm−2 (2.3% and 3.2% solar-to-hydrogen efficiency if coupled to a hypothetical photovoltaic providing 1.23 V) for WO3/BiVO4 heterojunction samples under front and back-illumination, respectively. The heterojunction showed more than additive improvements over the parent materials, with bare WO3 and BiVO4 samples showing 0.68 and 0.27 mA cm−2 and 0.50 and 0.87 mA cm−2 under front and back-illumination, respectively. Simulations of the current–voltage characteristics of tandem crystalline silicon photovoltaic modules coupled to the PEC devices were consistent with the solar predicted photocurrents. These promising results for BiVO4-coated WO3 nanoneedles fully-deposited by atmospheric pressure CVD enables future research into photoanodes amenable to large-area scale-up.
1. Introduction
Technologies that store renewable but intermittent solar energy as a chemical fuel will help society reduce its dependence on fossil fuel extraction. One such technology, photoelectrochemical (PEC) water splitting, uses sunlight to drive the reduction and oxidation of water into hydrogen fuel and oxygen gas at the surfaces of a (photo)cathode and a (photo)anode respectively.1 PEC water splitting, however, is still an immature technology, with both laboratory and prototype-scale research underway to improve efficiency, durability, and scalability.2,3 In contrast to how crystalline Si has become the archetypal low-cost photovoltaic solar cell,4 no single device design, light absorber nor catalytic material system has emerged as a defining example for efficient, low-cost and scalable PEC water splitting.5 The Solar Fuels Database6 collates data on reported PEC water splitting devices. The solar-to-hydrogen (STH) conversion efficiencies for PEC devices made using Si and III–V semiconductors have reached 19%,7 and the STH conversion efficiencies for devices made using lower-cost, earth-abundant metal oxides have reached ∼10%,6 although only for small-scale devices. Notably, a minimum of 10% is reported as a requirement for the overall performance of PEC water splitting to be comparable to PV-coupled electrolysis,8 with values as high as 20–26% targeted by the U.S. Department of Energy.9 Reaching these efficiencies with non-precious materials, whilst also demonstrating acceptable stability, scalability and cost targets, remains a significant challenge for the PEC water splitting research community.Si and III–V semiconductor-based devices typically use light absorbers known to be effective in photovoltaic (PV) devices and then add protective passivation layers to prevent their corrosion and promote catalysis in an electrolyte.10 This method of adding complexity to a traditional PV device is, however, unlikely to be cost-effective compared to PV-coupled electrolysis,8 which does not have to mitigate the effects of having the electrolyte in close proximity to the light absorbers. Transition metal oxides, in contrast, have higher durability, lower cost, and can be more readily produced using scalable synthesis, and thus are the most commonly studied class of material for PEC water splitting.2 One limitation to the water splitting performance of metal oxides is their often wide band gaps that can restrict their ability to harvest solar energy.11 Nevertheless, using multiple distinct light absorbing layers in tandem in a type II heterojunction with staggered conduction and valence bands can enable wider light absorption and maintain efficient transfer of electrons and holes through the layered architecture.12 The bismuth vanadate-coated tungsten trioxide (WO3/BiVO4) photoanode is one such example of a tandem type II heterojunction.13 WO3 has been shown to have a long hole diffusion length (∼150 nm) and good electron mobility (∼12 cm2 V−1 s−1), but suffers from slow charge transfer at the semiconductor/electrolyte interface14 and can only absorb blue and ultraviolet light with its 2.7–2.8 eV bandgap energy.15,16 BiVO4, with its indirect bandgap of 2.4 eV,17 absorbs up to green light, but is hindered by electron–hole recombination and poor charge transport and water oxidation kinetics.18 Together, however, photocatalysts with a WO3/BiVO4 sputter-fabricated core–shell architecture, when coupled with a double-junction GaAs/InGaAsP photovoltaic cell, have demonstrated a record 8.1% STH conversion efficiency for BiVO4-based photoanode water splitting devices.19 This heterojunction configuration of light absorbers benefits from improved charge separation across the interfacial region,20 while the nanostructuring allows for a simultaneously thick BiVO4 layer along the length of the WO3 and a radially thin BiVO4 layer that mitigates for the poor charge mobility through BiVO4.21,22 Metal oxide photocathodes, such as those based on Cu2O, have been demonstrated with up to approximately 7.6 mA cm−2 or 9.3% STH efficiency when measured at 0 V vs. RHE in a half-cell configuration.23 The challenge to using photocathodes is that they are typically sensitive to photocorrosion and so require complex capping layers to function. The remainder of this work is focused on photoanodes.
Future commercial PEC water splitting facilities may require photoabsorbers with large areas on the order of centimetres to meters in dimensions.24 The WO3/BiVO4 heterojunction photoabsorber literature to date have, however, primarily employed fabrication techniques such as sputtering under vacuum,25 or electrochemical26 and spin-coating methods27,28 that may be difficult to apply to large-area coating. Along with spray pyrolysis,29,30 chemical vapour deposition (CVD) is a route to making metal oxide thin films that can be scaled to large areas and has been demonstrated to be capable of producing a wide range of inorganic photoabsorbers.2 Seminal works on water splitting photocatalysts fabricated by CVD include the vapour transport deposition of BiVO4,31 aerosol-assisted chemical vapour deposition (AACVD) of BiVO4,32 and demonstration and optimization of WO3 nanoneedles by AACVD.33,34
This work demonstrates, to our knowledge, the first nanostructured WO3/BiVO4 heterojunction photoanode where both components are deposited sequentially by AACVD. BiVO4 is conformally coated onto WO3 nanoneedles on FTO-coated glass to form dense coatings 25–100 nm thick surrounding micron-length, 50–100 nm diameter nanoneedles, as seen by scanning electron microscopy. The characteristic X-ray crystal diffraction, X-ray photoemission spectroscopy, and Raman spectroscopy features for WO3 and BiVO4 are unchanged when the heterojunction is formed, indicating that each component retains its physical integrity, and that the heterojunction has a distinct interface. Additionally, we also investigate the importance of preventing the aging of the vanadium precursor solution (vanadium(III) acetylacetonate in acetone and methanol), where tests were conducted to help elucidate the mechanism for CVD deposition of BiVO4, adding to the emerging body of knowledge in this field.35 The WO3/BiVO4 heterojunction photoanode showed significantly enhanced activity for water oxidation and a superior calculated solar predicted photocurrent (SPP) compared to single component WO3 and BiVO4 photoanodes, validating the enhanced absorptance properties of the heterojunctions as measured by UV-visible spectroscopy. Modelling of a PV-PEC hybrid device, carried out herein, where a back-illuminated WO3/BiVO4 heterojunction is coupled to dual crystalline Si PVs placed electrically in series and optically in tandem, estimates that our photoanodes can achieve an unassisted STH of up to 3.4%. Overall, we demonstrate that AACVD can fabricate WO3/BiVO4 heterojunction photoanodes with promising performance for large-scale device applications.
2. Results & discussion
2.1 Synthesis of the WO3 and BiVO4 photoanodes
A wide range of WO3 nanoneedles (NN), BiVO4, and BiVO4-coated WO3 samples were fabricated in triplicate and are listed in Table 1 according to their deposition volumes for aerosol-assisted chemical vapour deposition and approximate layer thicknesses determined by side-on scanning electron microscopy (SEM). Fig. S1† shows representative photographs of the post-annealed FTO-glass substrates coated with WO3 NN, BiVO4, and BiVO4-coated WO3 along with a photograph of the CVD reactor that can deposit samples up to 16 cm × 5 cm in surface area. The FTO-coated substrates used herein are mass-produced by the glass industry with atmospheric pressure CVD36 and so there is the possibility for the entire photoelectrode to be produced in one production process on float glass panels. WO3 films change from a deep blue (sub-stoichiometric WO3−x) to white colour (near stoichiometric WO3) when annealed in air. Annealing removes trace contaminants, oxidizes the film, and a two-hour annealing time retains optimal ∼2% levels of oxygen vacancies37 enhancing the photoactivity of the WO3 photoanodes.38,39 Annealing BiVO4 films ensures complete conversion of the films to the more photoactive monoclinic crystal phase40 and oxidation to yellow BiVO4 with V in the 5+ oxidation state. The rough surface of the WO3 NN scatter light and cause the films to appear opaque. The BiVO4 on FTO is flat in comparison and appears transparent, while on WO3, the BiVO4 coats the NN conformally, so the final heterojunction sample also appears opaque.

|
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone and methanol) and 40 mL of BiVO4 precursor (5 mM triphenylbismuth and vanadium(III) acetylacetonate dissolved in 3:1 acetone and methanol) were fabricated and measured to more easily image the distinct layers on top of the FTO-coated glass substrate, Fig. 1a, in contrast to the thinner layers described in Table 1. Fig. 1b shows the top-down view of WO3 NN, which forms as sharp needles (50–100 nm in diameter) growing predominantly upwards in a range of angles. Fig. 1c shows the top-down view of WO3 NN that have become more globular upon coating with BiVO4. The final diameter of the BiVO4-coated WO3 needles are between 250 and 300 nm, corresponding to a radial coating of about 100 nm, herein referred to as the BiVO4 thickness. BiVO4 on bare FTO-coated glass, shown from side-on and top-down views in Fig. 1d, coats as a thicker film than on WO3 NN; 10 mL precursor solution is used to make the coating in Fig. 1d compared to 40 mL for the coatings in Fig. 1c and f. Fig. 1e and f, which are the side-on images of WO3 NN and NN coated with BiVO4, on FTO. The majority of NN are pointing upwards from the FTO. Notably, the AACVD coating process for the BiVO4 layer fully penetrates to the bottom of the host nanostructure, while preserving the needle-structure of the WO3.
:1 acetone and methanol) and 40 mL of BiVO4 precursor (5 mM triphenylbismuth and vanadium(III) acetylacetonate dissolved in 3:1 acetone and methanol) were fabricated and measured to more easily image the distinct layers on top of the FTO-coated glass substrate, Fig. 1a, in contrast to the thinner layers described in Table 1. Fig. 1b shows the top-down view of WO3 NN, which forms as sharp needles (50–100 nm in diameter) growing predominantly upwards in a range of angles. Fig. 1c shows the top-down view of WO3 NN that have become more globular upon coating with BiVO4. The final diameter of the BiVO4-coated WO3 needles are between 250 and 300 nm, corresponding to a radial coating of about 100 nm, herein referred to as the BiVO4 thickness. BiVO4 on bare FTO-coated glass, shown from side-on and top-down views in Fig. 1d, coats as a thicker film than on WO3 NN; 10 mL precursor solution is used to make the coating in Fig. 1d compared to 40 mL for the coatings in Fig. 1c and f. Fig. 1e and f, which are the side-on images of WO3 NN and NN coated with BiVO4, on FTO. The majority of NN are pointing upwards from the FTO. Notably, the AACVD coating process for the BiVO4 layer fully penetrates to the bottom of the host nanostructure, while preserving the needle-structure of the WO3.
 | ||
| Fig. 1 Exemplar SEM images of (a) FTO substrate, (b) top view of WO3 NN, (c) top view of WO3 NN coated with 100 nm thick BiVO4, (d) (top): side-on view of 100 nm thick BiVO4 on FTO, (bottom): top-down view of BiVO4, (e) side-on view of 4000 nm long WO3 NN on FTO and (f) side-on view of 100 nm thick BiVO4 coated on 4000 nm long WO3 NN on FTO. | ||
2.2 Hypothesis for the mechanism of BiVO4 deposition
Understanding the mechanism for CVD deposition of the ternary BiVO4 is important for ensuring high quality depositions. The deposition is more complex than for binary oxides like WO3,41 and has not previously been discussed in the literature.31,32,42,43 In this work, we use thermogravimetric analysis (TGA) and UV-vis of the precursor solutions along with a temperature-dependent X-ray diffraction study to help determine the decomposition dynamics of the BiVO4 precursors. The findings enable us to formulate the hypothesis that differences in activation energy between bismuth and vanadium metal–organic precursors leads to controlled product stoichiometry with changing deposition temperature.TGA in air coupled with mass spectrometry was conducted on the neat precursor powders, triphenylbismuth, Bi(Ph)3, and vanadium(III) acetylacetonate, V(acac)3, to investigate their decomposition at the temperatures used in our CVD reaction. Bi(Ph)3 is expected to decompose to Bi2O3, V(acac)3 to V2O5 and the 1:1 mixture to BiVO4 as these are the common fully oxidated forms. In Fig. 2a the TGA profiles show that Bi(Ph)3 appears to lose mass primarily between 200–300 °C and then 400–500 °C. The V(acac)3 loses mass during three temperature ranges between 150–200 °C, 200–250 °C, and 300–400 °C. A 1:1 mixture of Bi(Ph)3 and V(acac)3 also appears to lose mass in two major, albeit prolonged temperature ranges, between 150–250 °C and then 250–400 °C. The final residual mass of 22% for V(acac)3 decomposition to V2O5 was close to the expected value of 26%, while for Bi(Ph)3 decomposition to Bi2O3, the 30% final residual mass was less than the expected value of 53%, which may be caused by partial sublimation of Bi(Ph)3,44 although any Bi-containing species were too heavy to be measured in the TGA-MS. Importantly, the final residual mass for the decomposition of the 1:1 mixture to BiVO4 of 40% matches the expected value of 41%.
 | ||
| Fig. 2 TGA-mass spectrometry (MS) of the precursors used in the AACVD synthesis of BiVO4 (a) weight loss % as a function of temperature, and mass spectrometry of volatile materials for (b) Bi(Ph)3, (c) V(acac)3, and (d) 1:1 mixture of Bi(Ph)3 and V(acac)3. | ||
The MS of Bi(Ph)3 as a function of temperature, shown in Fig. 2b, shows a distinct release of benzene attributed to the release of the phenyl ligand between 200 and 300 °C, which corresponds to the first mass loss seen in the TGA. There is a brief pause in mass change before a second mass loss at 400 °C, corresponding to the release of CO2 until 500 °C, which may be attributed to the combustion of stray phenyl remaining in the material. In Fig. 2c, the MS of V(acac)3 shows an initial loss of H2O up to 100 °C which is likely surface adsorbed water from storage of the material in air. There are then three distinct peaks at 150 °C, 250 °C and 400 °C where characteristic fragments of the acetylacetone (m/z 43), water and CO2 are released, matching the temperatures for mass losses in the TGA. The area of the MS peaks also corresponds with the relative mass losses. In Fig. 2d, MS of the 1:1 mixture of Bi(Ph)3 and V(acac)3 shows all expected peaks from the individual materials with lower resolution, suggesting that decay of the solid-state mixture is generally a combination of the constituent individual compounds, and not from other (mixed-metal) intermediates. The V(acac)3 precursor loses mass, and the corresponding products are observed by mass spectroscopy at approximately 50 °C lower compared to the Bi(Ph)3 precursor, indicating that V(acac)3 has a lower activation energy for decomposition than Bi(Ph)3.45
The choice of precursors should have an influence on the reaction mechanism for the deposition of the ternary metal oxide. An alternative commonly studied vanadium precursor for CVD is vanadyl(IV) acetylacetonate, VO(acac)2.46 VO(acac)2 has been reported with TGA to fully decompose by about 300 °C,47 approximately 100 °C lower than for the complete decomposition of V(acac)3. If VO(acac)2 is used in AACVD with Bi(Ph)3, there may then be a wider range of temperatures in which the two precursors have differing levels of decomposition. Use of VO(acac)2 with Bi(Ph)3 should then be avoided when stoichiometric BiVO4 is desired. According to UV-vis studies shown in Fig. S2,† V(acac)3 decomposes and oxidises into VO(acac)2 under atmospheric conditions over time when dissolved in 3:1 acetone:methanol. The oxidation process occurs rapidly over the first few hours and is followed by further oxidation to generate primarily V(V) species over the course of two days. Therefore, we postulate that conducting AACVD using solutions containing V(acac)3 aged for even only a few hours, may also have the same effect of using the VO(acac)2 precursor directly, and, therefore, recommend the immediate use of V(acac)3 stored under inert gases upon dissolution in the 3:1 acetone:methanol solution for CVD synthesis (where herein, depositions were conducted within ∼15 min of dissolution).
Flat BiVO4 films prepared on FTO-coated glass at 400 °C show the XRD patterns for stoichiometric BiVO4 (Fig. 3). The as-deposited sample shows the (002) (101) (011) (112) (004) (200) (020) (211) (015) (204) (024) (220) (116) (017) & (132) peaks corresponding to polycrystalline BiVO4 (monoclinic I112/b15a = 5.1907 Å, b = 5.0912 Å, c = 11.6941 Å; α = β = 90°, γ = 90.360°; PDF no. 01-090-8670) along with the (110) (101) (200) (211) (220) (310) & (301) diffraction peaks for the FTO substrate (tetragonal P42/mnm (136); a = b = 4.7380 Å, c = 3.1865 Å; α = β = γ = 90°; PDF no. 01-071-0652). The effect of annealing the BiVO4 serves to eliminate the peak corresponding to the (101) crystalline plane, leaving only the (011) peak in the region near 19°. Peaks corresponding to Bi2VO5 (PDF no. 01-086-1181), Bi2O3 (PDF no. 01-080-9185) and V2O5 (PDF no. 01-090-7484) are not evident before or after annealing, indicating that the materials are deposited in a stoichiometric ratio and do not segregate into phases of the binary metal oxides.
 | ||
| Fig. 3 XRD patterns for BiVO4 films grown on FTO-coated glass by AACVD before annealing (as deposited) and after annealing at 500 °C for 2 hours in air. Data is plot alongside PDF card references patterns for SnO2 (i.e. the FTO substrate), BiVO4, Bi2VO5, Bi2O3 and V2O5. | ||
Next, flat BiVO4 films were grown by CVD on FTO-coated glass for reactor temperatures between 250 and 450 °C. In these experiments the vanadium precursor was aged for one month in the solid state and therefore a minute amount of Bi2VO5 was expected. Fig. S3† shows the XRD patterns for these films before and after annealing at 500 °C in air for two hours. For relatively low temperatures, at 250 °C neither precursor has begun to significantly decompose, so both species deposit on the substrate as amorphous solid solutions and the final film after annealing reflects the starting precursor stoichiometry. At relatively high deposition temperatures, 400 °C and 450 °C, both precursors rapidly decompose and crystallise together as-deposited, resulting in annealed films that also reflect the starting precursor stoichiometry. For moderate deposition temperatures at 300 °C and 350 °C, the V(acac)3 decomposes to a greater extent compared to the Bi(Ph)3. Under these conditions, the lighter V(acac)3 derivatives may be re-evaporated into the passing aerosol stream, increasing the relative amount of Bi in the film, with an additional Bi2VO5 phase identified, evidenced by the relatively large and broad peak corresponding to the Bi2VO5 (002) plane present after annealing.
These results suggest that differences in activation energy between the bismuth and vanadium metal–organic precursors allows for the control of BiVO4 stoichiometry with changing deposition temperature, with an optimal deposition temperature of 400 °C for fabricating BiVO4 for no observable sub-stoichiometric phases.
2.3 Chemical analysis of the WO3 and BiVO4 photoanodes
 | ||
| Fig. 4 (a) XRD patterns and (b) Raman spectra for 1000 nm long WO3 nanoneedles, BiVO4 and 50 nm thick BiVO4 coated on WO3 nanoneedles corresponding to monoclinic WO3 (ref. 34, 48 and 49) and monoclinic BiVO4.50,51 | ||
The Raman spectra shown in Fig. 4b show characteristic peaks matching literature spectra of WO3 (ref. 52 and 53) and BiVO4, with the peak at 828 cm−1 corresponding to the Ag symmetric vibrations of V–O bonds, the peaks at 366 and 327 cm−1 corresponding to the Bg asymmetric and Ag symmetric modes of VO43− respectively54 and the peak at 211 cm−1 being an external mode.55,56 For the heterojunction spectrum, the widened peak seen at 827 cm−1 is likely due to the combination of the Ag peak at 807 cm−1 for WO3 and the Ag peak at 828 cm−1 for BiVO4.57 The superimposed peaks of the BiVO4-coated WO3, therefore, suggest that distinct phases of BiVO4 and WO3 form in the heterostructure, and that a distinct interface is formed.
Peak fitting of high-resolution XPS spectra confirm near stoichiometric values for WO3 and BiVO4. Atomic% values of 25% for W6+, 2% for W5+, and 73% for O2− were observed and is commensurate with the expected stoichiometry of WO3 with approximately 2% oxygen vacancies. 14% Bi3+, 12% V5+, 6% V4+, 1% V3+, and 67% O2− were the at% values observed for BiVO4. The proportion of oxygen matches the expected stoichiometry, while the total amount of vanadium is similar to the bismuth. The mixture of vanadium reduced states may be induced by X-ray reduction during the XPS measurement. Survey scans and high-resolution peak fitting with elemental quantification are shown in Fig. S4 for WO3 and S5 for BiVO4.† The peak fit parameters are also detailed in Tables S2 and S3† for WO3 and BiVO4 respectively.
2.4 Optical characterisation of the WO3 and BiVO4 photoanodes
Fig. 5a compares the absorptance spectra calculated from measured transmittance and total reflectance spectra (Fig. S6a and S6b†) of representative photoanode films of 2000 nm thick WO3 nanoneedles (NN) and 100 nm of BiVO4 conformally coated on 2000 nm thick WO3 NN on FTO-coated glass. As each layer is added, the absorption onset of the samples shifts from 350 nm for FTO-coated glass to 450 nm for WO3-coated FTO, and finally to 510 nm for BiVO4-coated WO3 on FTO. These values are consistent with the expected bandgaps for SnO2, WO3 and BiVO4 of 3.6,58 2.8 and 2.4 eV,11 respectively. The overall optical properties of the layered materials are dominated by the absorptance of the constituent material with the smallest bandgap energy. For a more precise determination of the bandgap, a less scattering exemplar film was used, which consisted of a 400 nm thick BiVO4 layer coated onto a 100 nm thick flat (not nanostructured) WO3 underlayer. Flat WO3 is fabricated with a similar CVD procedure as for nanostructured WO3 using a lower substrate temperature.34 The measured transmittance and total reflectance spectra of this film was used to produce a Tauc plot, shown in Fig. 5b, which revealed a 2.5 eV indirect transition and 2.6 eV direct transition for the BiVO4 layer. The transmittance and reflectance spectra of the flat films are compared to the nanostructured films in Fig. S6a and S6b† showing the WO3 NN are less transmitting and more reflecting than flat WO3, but samples of BiVO4 coated on flat and NN WO3 have similar properties. Fig. S7a and S7b† show Tauc plots for WO3 and BiVO4 grown directly on FTO, indicating indirect bandgaps of approximately 2.8 eV and 2.5 eV, respectively. | ||
| Fig. 5 (a) UV-vis absorptance for 2000 nm thick WO3 NN and 100 nm BiVO4-coated NN WO3 compared to FTO-coated glass and (b) Tauc plots derived from measurements of transmittance and total reflectance of 400 nm planar BiVO4 on 100 nm planar WO3 on FTO-coated glass. | ||
 | ||
| Fig. 6 Calculated optical propagation of light through example heterojunction structures. Planar FTO/WO3/BiVO4 heterojunctions, with each respective layer being 320, 100 and 400 nm thick, for the case of (a) front illumination and (b) back illumination, and nanostructured FTO/WO3/BiVO4 heterojunctions with the FTO layer being 320 nm thick, coated with WO3 NN that are 100 nm wide and 2000 nm long, that are conformally coated with a 100 nm layer of BiVO4, for the case of (c) front illumination and (d) back illumination. Pastelle shading represents the layers of the heterojunction light is passing through; colourless = FTO, blue = WO3, green = BiVO4. Calculations were carried out for 250, 325, 400 and 475 nm light. | ||
The optical behaviour of a planar WO3/BiVO4 heterojunction system are shown in Fig. 6a and b, for back and front illumination, respectively. For such a system to be functional for water splitting, the planar layers cannot be too thick, or else both hole diffusion to the BiVO4 surface to drive water oxidation reactions and electron diffusion out of BiVO4 into WO3 would be inhibited. Thus, herein, we modelled the case for a 100 nm thick WO3 layer and a 400 nm thick BiVO4 layer. Under front illumination (Fig. 6a), UV light is effectively harvested by the BiVO4 layer, with ∼80% of 250 nm and ∼70% of 325 nm absorbed. Blue light is also strongly absorbed, with only ∼20% of 400 nm and ∼40% of 475 nm light remaining. Much of this light is then absorbed by the WO3 layer. Under back illumination (Fig. 6b), UV light of 250 nm is almost all absorbed by the FTO layer, and therefore cannot be harvested by the heterojunction. Similarly, ∼60% of UV light of 325 nm is absorbed by the FTO layer. This is then strongly absorbed by the proceeding WO3 layer. For the cases of 400 and 475 nm light, these are not significantly attenuated by the FTO layer. About 40% of 400 nm light and 20% of 475 nm light is absorbed by the WO3 layer, with only ∼20% of the remaining 400 and 475 nm light absorbed upon reaching the BiVO4 layer. Overall, back illumination of the planar heterojunction shows that UV light (i.e. 250 and 325 nm) cannot be harvested effectively due to parasitic absorption by the FTO layer, and that light near the band edge of BiVO4 (i.e. 475 nm) is not effectively harvested either.
For the case of a nanostructured heterojunction WO3/BiVO4 system, the nanostructured WO3 layer can decouple the limited electron and hole diffusion of the conformally coated BiVO4. Moreover, given the excellent electron transport found in WO3, one can use a microns-thick nanostructured layer. Thus, herein, we modelled the case for a nanostructured WO3 layer with rods 100 nm wide and 2000 nm long that are coated with a 100 nm conformal BiVO4 layer, like what we have produced experimentally herein (Fig. 1). A cross-section is modelled that transects the middle of the nanorod, where light encounters the WO3 layer one-third of the time and the BiVO4 layer two-thirds of the time. For the case of front illumination (Fig. 6c), all light from 250 to 475 nm is harvested within the WO3/BiVO4 nanostructure, which results in optimal charge carrier production. Under back illumination (Fig. 6d), significant absorption within the FTO layer is seen for 250 nm and 325 nm light, just as for the planar heterojunction (Fig. 6c). Different behaviour is, however, seen for longer wavelengths, with all 400 and 475 nm light absorbed by the nanostructured architecture because of the increased length of the absorber. The BiVO4 layer in the nanostructured architecture is more capable of harvesting light, particularly wavelengths at the band edge of the material where a more significant portion of the solar spectrum resides. The micron-scale distance that light must travel through the BiVO4 in the nanostructured heterojunction results in all the light being absorbed, whereas residual light is transmitted through the flat heterojunctions. Under back illumination, exciton electron–hole pairs are formed much closer to the FTO electron collector than for photoanodes under front illumination.
2.5 Photoelectrochemical (PEC) experiments
 | ||
| Fig. 7 J–V curves for 1000 nm thick WO3 NN (pH 1.1, 0.1 M H2SO4 (aq.)), 200 nm thick BiVO4 and 50 nm thick BiVO4-coated 1000 nm thick WO3 NN (pH 7, 0.1 M K2HPO4/KH2PO4 (aq.)) on TEC15 FTO-coated glass. Scan rate: 10 mV s−1, illuminated with a 75 W Xe lamp attenuated with a neutral density filter at approximately 1 sun intensity. Solid lines represent front illumination, and dashed lines represent back illumination. | ||
Current density losses along the length of the nanostructures and mass transport losses arising from the increased distance that reactants and products travel between the electrode surface and electrolyte bulk may limit the ultimate photocurrents. It was previously shown by Kafizas et al., however, that nanostructured WO3 had significantly increased photoanode performance compared to flat WO3.34 This result was attributed to the full utilisation of photon penetration depth that increases the light absorption through the length of the needles.34 The increase in surface area likely also contributes to mitigating current density and mass transport losses in highly crystalline, annealed WO3 nanostructures where oxygen vacancy defects are minimized to improve electron transport.60
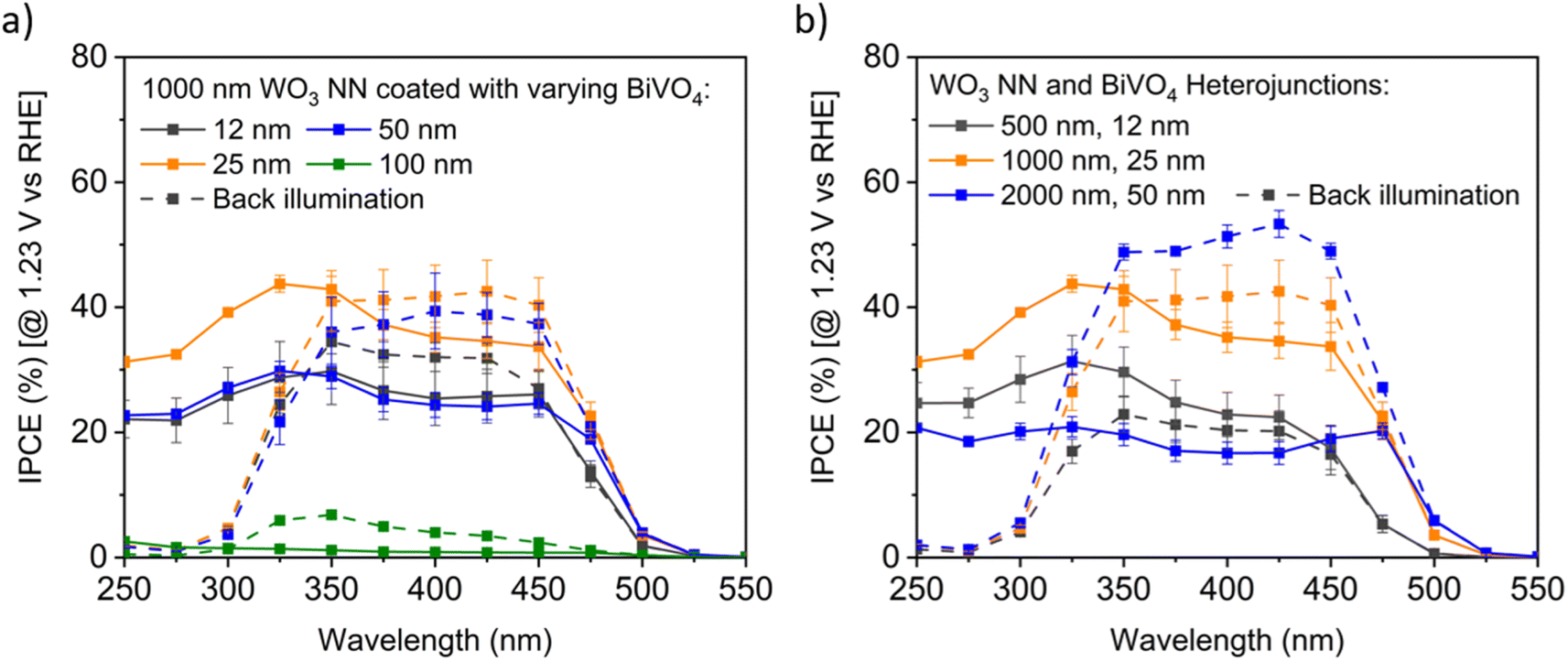 | ||
| Fig. 8 Exemplar IPCE profiles for BiVO4-coated WO3 on FTO, measured at pH 7 in 0.1 M KH2PO4/K2HPO4 (aq.) buffer electrolyte under front and back illumination (dashed lines), with 95% confidence intervals for triplicate measurements (a) varying BiVO4 on 1000 nm long WO3 nanoneedles, and (b) constant ratio of varying BiVO4 and WO3 nanoneedle thicknesses. | ||
Fig. 8b compares the IPCE profiles for samples with an aspect ratio of NN to BiVO4 thickness of 40:1. The thinnest samples with 500 nm long WO3 NN show the least IPCE under back illumination, the samples with 1000 nm long WO3 NN showed moderate efficiency for both front and back illumination, and the samples with 2000 nm long WO3 NN demonstrated the highest efficiency under back illumination. The difference in the photocurrents of the front and back-illuminated photoanodes for the thickest photoanode is likely due to more facile extraction of excited majority carrier electrons away into the circuit when the excitons are formed close to the substrate compared to when they are formed near the photoanode surface under front illumination. The optimal BiVO4-coated WO3 nanoneedle photoanodes show excellent visible light photoactivity with up to approximately 50% IPCE for visible light with wavelengths up to 450 nm.
 | ||
| Fig. 9 Chronoamperometry stability measurements of a representative 500 nm long WO3/50 nm thick BiVO4 heterojunction photoanode illuminated with a 75 W Xe lamp attenuated with a neutral density filter at approximately 1 sun intensity under (a) front and (b) back illumination. Electrolyte: 0.1 M, pH 7 KH2PO4/K2HPO4 (aq.), 1.23 V vs. RHE. X-axis scale varies as indicated. | ||
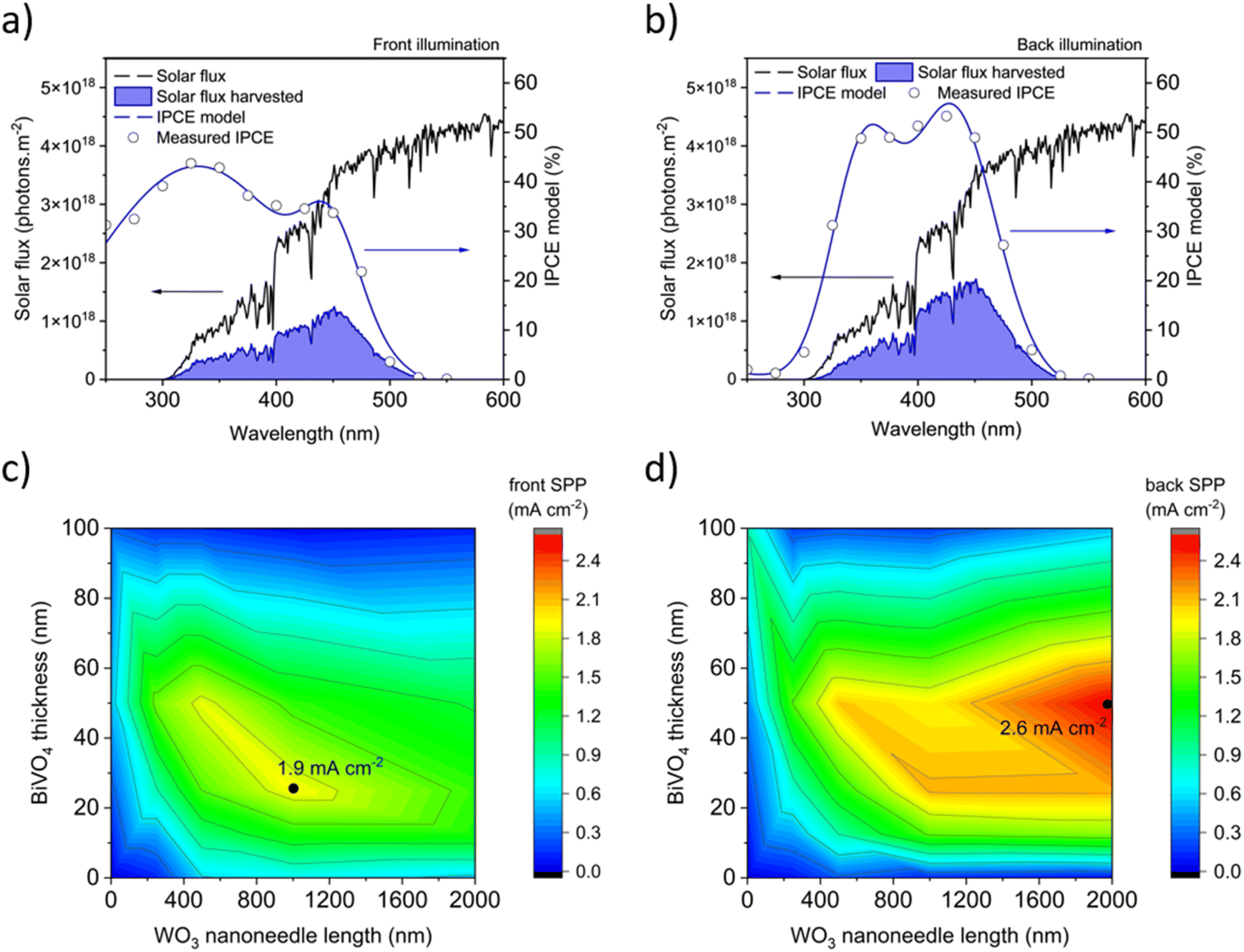 | ||
| Fig. 10 Calculations of solar predicted photocurrent (SPP) from the IPCE measured at 1.23 V vs. RHE for the nanostructured WO3/BiVO4 heterojunction samples showing the highest average performance under (a) front illumination (1000 nm thick WO3 NN coated with 25 nm of BiVO4) and (b) back illumination (2000 nm thick WO3 NN coated with 50 nm of BiVO4). Heat maps of the SPP for varying thicknesses of WO3 nanoneedles and BiVO4 in the heterojunction for the 22 unique samples examined for (c) front illumination and (d) back illumination. | ||
2.6 Model of a hypothetical PV-coupled PEC water splitting device
A hypothetical PV-coupled PEC water splitting (PV-PEC) device is arranged with a photoanode, metal cathode mesh, and silicon PV system optically in tandem as shown in Fig. 11a. The photoanode is illuminated from the backside to avoid being shaded by other device components such as the Pt cathode or electrolyte. The orientation of the cathode should be placed parallel to the photoanode for optimal mass transport of ions in solution and to reduce pH gradients in electrochemical set-ups.30 In our hypothetical device, the dual c-Si PV collects the transmitted light after passing through the whole PEC module. | ||
| Fig. 11 (a) Schematic of the hypothetical PV-PEC device with a dual c-Si PV electrically in series and optically in tandem with a back-illuminated PEC photoanode, (b) J–V curves for samples as indicated under back illumination as solid lines, and simulated J–V curves of corresponding dual c-Si PV modules behind the PEC module as dashed lines. Simulations and measured curves are calibrated to AM1.5G 1 sun illumination. | ||
In Fig. 11b, PV-PEC system J–V curves are shown for three cases of BiVO4-coated WO3 placed in front of dual c-Si PV modules as in Fig. 11a. The J–V curves for the PV module are simulated in MATLAB using eqn (1) considering the light transmitted though the PEC photoanode and other optical losses. Transmittance spectra for all the PEC photoanodes were measured and plotted in Fig. S10.†Jsc is the calculated short-circuit current based on the solar spectrum and a conservative external quantum efficiency of 90%. J0 is the calculated exchange current, Rs is the series resistance, 0.001 kΩ cm2,63 and Rsh is the shunt resistance, 10 kΩ cm2,64 taken for a typical c-Si PV cell. e is the elemental charge, n is the ideality factor, k is Boltzmann's constant, and T is the temperature, taken as 297 K.
 | (1) |
The operating points of the three tested PV-PEC systems in Fig. 11b are highlighted at the intersections between the three sets of PV and PEC J–V curves. The PV-PEC systems utilising a 2000 nm long WO3 NN photoanode coated with 100 nm thick BiVO4 and a 1000 nm long WO3 NN photoanode coated with 50 nm thick BiVO4 are simulated to have PV-PEC system operating currents of 0.5 mA cm−2 and 0.8 mA cm−2 respectively. The operating current for the simulated PV-PEC system with the 2000 nm long WO3 NN photoanode coated with 50 nm thick BiVO4 was an optimal value of 2.8 mA cm−2, which equates to an STH efficiency of 3.4%. This simulation shows the importance of increasing photocurrent from the PEC component and reflects the trends in system performance shown in Fig. 10 because the PV module in each case is providing an excess of photocurrent.
2.7 Comparison of similar unassisted PV-PEC systems in the literature
A wide range of unassisted PEC-based systems have been explored in the literature, including fully PEC65,66 and PV-PEC tandem devices. Herein, we focus exclusively on literature examples where WO3 and/or BiVO4 were applied as a photoanode in conjunction with a PV to achieve unassisted water splitting. Their performances are summarised in Table 2.| PEC – PV | PEC synthesis | Biased Ja (mA cm−2) | SPP (mA cm−2) | Unassisted PV-PEC STH (%) | Ref. |
|---|---|---|---|---|---|
| a PEC only at 1.23 V vs. RHE and 1 sun illumination using solar simulated light; n.a. = not applicable. | |||||
| WO3 – MAPI | AACVD | n.a. | 1.24 | 0.75 | 34 |
| WO3 – DSC | Sol–gel | 2.4 | 2.23 | 3.1 | 67 |
| BiVO4/CoPi – MAPI | MOD + PAE | 2.2 | n.a. | 2.0 | 68 |
| BiVO4/Ni:FeOOH – OPV | ED + PAE | 1.9 | n.a. | 2.2 | 69 |
| SiOx/BiVO4/Ni:FeOOH – MAPI | RIE + MOD + ED | 6.0 | n.a. | 6.2 | 70 |
| WO3/BiVO4/CoPi – GaAs/InGaAsP | GLAD + ED + ED | 6.7 | n.a. | 8.1 | 19 and 68 |
| TiO2/WO3/BiVO4/Fe(Ni)OOH – n-Si/SiO2 | AACVD + spin coating | 2.6 | n.a. | 0.31 | 27 |
| W:BiVO4/Co-Pi – thin-film amorphous Si | Spray pyrolysis | 4.0 | n.a. | 5.2 | 71 |
| H, 1% Mo:BiVO4/NiFeOx – crystalline silicon PV | Drop-casting & calcination | 4.5 | n.a. | 3.0 | 72 |
| WO3/BiVO4 – c-Si | AACVD + AACVD | n.a. | 2.61 | 3.2 | This work |
Kafizas et al. produced nanoneedle-structured WO3 photoanodes using an analogous AACVD method to that applied in this work.34 Their highest performing material was composed of a 300 nm flat WO3 seed layer with a 5 μm top layer of WO3 nanoneedles. Under back irradiation, this material showed an SPP of 1.24 mA cm−2 at 1.23 V vs. RHE, and when coupled in tandem with a photovoltaic device containing a methylammonium lead iodide perovskite (MAPI), an STH of 0.75% for unassisted water splitting was predicted. Sivula et al. produced 2.5 μm thick mesoporous WO3 photoanodes through a sol–gel process that showed 2.4 mA cm−2 at 1.23 V vs. RHE and 1 sun irradiance.67 When coupled to a high open-circuit voltage cobalt-based dye-sensitized solar cell (DSC), it showed an unassisted STH of 3.1%.67
Kamat et al. demonstrated a tandem BiVO4 photoanode coupled with a MAPI PV that achieved STH efficiencies of 2.0%.68 Their BiVO4 was produced using metal organic deposition (MOD) and was coated with a cobalt phosphate (CoPi) surface co-catalyst using photo-assisted electrochemical (PAE) deposition.68 It resulted in a photocurrent density of 2.2 mA cm−2 at 1.23 V vs. RHE and 1 sun irradiance.68 Andrew et al. produced 700 nm thick mesoporous BiVO4 photoanodes using electrochemical deposition (ED) that were coated with a nickel:iron oxyhydroxide (Ni:FeOOH) co-catalyst using PAE deposition.69 Their photoanodes showed a photocurrent density of 1.9 mA cm−2 at 1.23 V vs. RHE and 1 sun irradiance, and when coupled with a non-fullerene acceptor organic photovoltaic (OPV) showed an unassisted STH of 2.2%.69 Using SiOx nanocones, produced using a reactive ion etching process (RIE), Qiu et al. grew a conformal coating of Mo-doped BiVO4 on top using a MOD followed by a Ni:FeOOH surface co-catalyst using PAE deposition.70 Their photoanodes showed photocurrent density of 6.0 mA cm−2 at 1.23 V vs. RHE and 1 sun irradiance, and when coupled with a MAPI PV showed an unassisted STH of 6.2%.70 Lastly, the current state-of-the-art example, produced by Pihosh et al., is a nanostructured WO3/BiVO4/CoPi system.19 3 μm long WO3 nanorods were made using glancing angle deposition (GLAD) and were coated with BiVO4 and CoPi using ED methods.19 Their photoanodes showed a photocurrent density of 6.72 mA cm−2 at 1.23 V vs. RHE and 1 sun irradiance, and when coupled to a double-junction GaAs/InGaAsP PV showed an unassisted STH efficiency of 8.1%.19
Herein, we synthesised nanoneedle-structured WO3 with a conformal coating of BiVO4 using an AACVD method at ambient pressures (Fig. S1†), which simplifies the operation of and reduces the equipment and operating costs compared to many of the above-mentioned deposition techniques that require vacuum conditions. No surface co-catalyst as yet was applied in this work, which is often a pre-requisite for higher performance in this heterojunction system. Our best performing heterojunction was composed of 2000 nm long WO3 nanoneedles coated with a 50 nm thick conformal BiVO4 layer showing an SPP of 2.61 mA cm−2 under back irradiation, and when coupled to a dual c-Si PV in tandem, our modelling predicted an STH of 3.2%. Overall, the performance we observe is comparable to other examples from the literature. Notably, the higher performing examples use surface co-catalysts to achieve more favourable onset potentials and photocurrent densities in their photoanode, which was not explored herein but will be the subject of future work. Also, these examples that achieved higher performance used synthetic techniques that are not readily scalable (e.g. RIE, GLAD, etc.), and are therefore unlikely to be used in the commercial mass-production of this technology. Broadly, many of the PVs applied in these studies are either not commercially available (e.g. DSC, MAPI and OPV) or not cost effective (GaAs/InGaAsP). As such, the focus of this work was to use a scalable synthetic technique such as CVD, which is currently used to produce FTO-coated glass and can therefore be applied in line to produce photoanodes, alongside a PV material that is commercially available and low cost (i.e. c-Si). Fig. S1† shows a photograph of our CVD reactor which can deposit samples up to 16 cm × 5 cm surface area. A further publication currently in preparation will demonstrate the ability for our CVD method to produce arrays of photoanodes on a larger scale – increasing the size from the ∼1 cm2 to >50 cm2 scale – and couple these with c-Si PV in tandem to demonstrate unassisted water splitting at a scale more commensurate to its future commercial application.
3. Conclusions
BiVO4-coated WO3 nanostructured photoanodes for the water oxidation reaction were fabricated by aerosol-assisted chemical vapour deposition (AACVD) at atmospheric pressure, which is a considerable simplification and reduction in equipment costs compared to deposition under vacuum conditions. This work demonstrates the potential for AACVD to fabricate effective photoanodes for water splitting devices, despite not yet applying co-catalysts to our heterojunction system, which often results in higher performance. The mechanism of ternary BiVO4 deposition by CVD was investigated by thermogravimetric analysis of the metal–organic precursors used. TGA provided evidence of a difference in activation energy for decomposition between the precursors. XRD evidence of sub-stoichiometric phases of BiVO4 deposited at intermediate 300–350 °C temperatures allowed us to conclude that the stoichiometry of BiVO4 depends on the deposition temperature, rather than solely on the ratio of precursors in the starting deposition solution. CVD deposition of WO3/BiVO4 heterojunction photoanodes that showed photocurrents more than double compared to bare BiVO4 and several times that of bare WO3 photoanodes. The optimal solar predicted photocurrents of 1.9 mA cm−2 under front illumination and 2.6 mA cm−2 under back illumination for the samples tested represent promising STH conversion efficiencies of 2.3% and 3.2% respectively.PV modules that can provide the 1.23 V vs. RHE to drive a PEC device include dual crystalline silicon cells connected electrically in series because they can absorb the red and near-infrared light, not utilised by the photoelectrode and are commercially available for a relatively low cost. We thus developed a model that simulates the performance of tandem PV-PEC devices, where our WO3/BiVO4 heterojunction photoanodes were coupled to a dual c-Si PV in tandem, and a maximum STH performance of 3.2% was predicted. Further increases in the PEC photocurrent and in the photovoltage provided by the PV module would shift the operating current to higher values. A further publication currently in preparation will demonstrate the ability for our CVD method to produce arrays of photoanodes on a larger scale (>50 cm2) more commensurate to commercial application. There exists a need to examine larger scale, unassisted water splitting devices, so that technoeconomic and life-cycle analyses can be more accurately conducted. Furthermore, additional steps may be implemented using AACVD, such as the incorporation of surface co-catalysts, to beneficially cathodically shift the onset potential and increase the plateau photocurrents to achieve higher overall STH efficiency.
4. Experimental section
4.1 Metal oxide fabrication by chemical vapour deposition
The metal–organic precursors tungsten hexacarbonyl (97%, Aldrich), bismuth triphenyl (98+%, Alfa Aesar), and vanadium(III) acetylacetonate (97%, Aldrich) were used as received. FTO-coated glass substrates (1.33 × 2.5 cm2 × 0.22 cm thick) (TEC 15, Pilkington) were cleaned by successive sonication (VWR ultrasonic cleaner, 30 W, 45 kHz) in detergent with deionised water, deionised water, acetone, and isopropanol for 10 minutes each. The substrates were placed on a carbon block in a custom-built quartz tube reaction chamber and heated to the target deposition temperature. For WO3 deposition, varying volumes of 11.4 mM concentration precursor solution in 3:1 acetone and methanol was aerosolised by a humidifier (Liquifog®, Johnson Matthey, ∼1.6 MHz operating frequency) and carried into the reaction chamber by a 2 L min−1 flow of inert N2 gas. The carbon block was heated with an internal cartridge heater to 375 °C to grow nanoneedle films and to 325 °C to grow flat WO3 films as shown previously by Kafizas et al.34 Following complete transfer of precursor, the films were annealed in a 500 °C furnace (Nabertherm Electric Muffle Furnace, L 9/11/B410) in air for 2 hours (temperature ramp rate of 200 °C per hour), which leaves an optimal quantity of oxygen vacancies in the films.37 Deposition of BiVO4 occurred similarly with varying volumes of 5 mM concentration precursor solution aerosolised and carried into the reaction chamber by a 1 L min−1 flow of compressed air. The carbon block was heated to 400 °C during the deposition. Annealing of the BiVO4 was also done at 500 °C for 2 hours in air, which crystallises the BiVO4 with an optimal grain size while limiting vanadium vacancies that form trap states for recombination73 and reduces propensity to dissolve upon illumination in phosphate electrolyte solutions.74 Heterojunction samples were fabricated by sequentially depositing WO3, annealing at 500 °C for 2 hours, and depositing BiVO4 before a second annealing stage while adjusting the volumes of precursor solution transferred.
4.2 Physical characterisation
Scanning electron microscopy (SEM) images were taken with a Zeiss Auriga microscope with a 5 kV acceleration voltage, Inlens detector, and 5 mm working distance. Side-on SEM images of sample cross sections were used to determine that the WO3 nanoneedles deposited at a rate of 500 nm per 10 mL of precursor, BiVO4 deposited on nanoneedles at a rate of 25 nm per 10 mL of precursor, and 100 nm per 10 mL of BiVO4 precursor on flat surfaces. Thermo-gravimetry analysis with mass spectrometry (TGA-MS) was tested with a Mettler Toledo TGA/DSC 1LF/UMX, HIDEN ANALYTICAL HPR-20 QIC Evol. Precursor powders stored in air with initial masses of approximately 5 mg were tested under air gas environment at constant heating rate of 10 °C per minute. X-ray diffraction (XRD) patterns were measured with a Bruker D2 Phaser diffractometer with parallel beam optics equipped with a Lynx-Eye detector. X-rays were generated using a Cu source (V = 30 kV, I = 10 mA) through a 0.6 mm slit; with Cu Kα1 (λ = 1.54056 Å) and Cu Kα2 radiation (λ = 1.54439 Å) emitted with an intensity ratio of 2:1. Patterns were collected between 10° ≤ 2θ ≤ 70° with a step size of 0.0527° and a speed of 1 second per step. Patterns were compared to references from the Physical Sciences Data-Science (PSDS) database. Raman spectra were obtained using a Horiba LabRAM Infinity spectrometer equipped with a helium–neon laser (633 nm, 8 mW) from 100 to 1100 cm−1. X-ray photoelectron spectroscopy (XPS) between 0 and 1400 eV binding energy was measured with a Thermo Fisher K-Alpha+ automated system with Al Kα X-ray source and ion beam etch sputterer. Transmittance and reflectance of solid films were measured by a Shimadzu UV-2600 UV-vis spectrometer with integrating sphere in the range of 190 to 1200 nm with 1 nm resolution. Solution phase UV-vis spectroscopy was carried out on a Shimadzu 2600i spectrometer.
4.3 Photoelectrochemical experiments
A three-electrode set-up in a cappuccino-type cell75,76 was used in all experiments with a pH 7, 0.1 M aqueous potassium phosphate buffer electrolyte (potassium phosphate dibasic, ACS reagent ≥ 98%, Sigma-Aldrich & potassium phosphate monobasic, ACS reagent ≥ 99%, Sigma-Aldrich) made with 18.2 MΩ cm ultrapure water. Pt mesh and an Ag/AgCl (saturated KCl) electrode served as the counter and reference electrode respectively. Potentials versus the Ag/AgCl reference electrode were applied using a Metrohm Autolab PGSTAT 101 and converted to the reversible hydrogen electrode scale using the Nernst equation: VRHE = VAg/AgCl + 0.0592 × pH + V0Ag/AgCl, for V0Ag/AgCl = 0.197 V vs. RHE. Illumination was provided by a 75 W Xe lamp and monochromator (OBB-2001, Photon Technology International) and calibrated with a Thorlabs optical power meter with photodiode sensor (PM100D Power Energy Meter equipped with a S120VC sensor). The PEC performances are quantified by measuring the current–voltage (J–V) characteristic curves and the incident photon to current efficiency (IPCE) profiles. J–V Curves are obtained by linear sweep voltammetry at 10 mV s−1 under white light illumination by approximately 1 sun intensity as determined by measuring the photocurrent with a Si photodiode. IPCE values are obtained by changing the wavelength of calibrated monochromatic light while measuring the photocurrent by chronoamperometry under applied potential of 1.23 V vs. RHE. The IPCE was calculated using the following equation:| IPCE(%) = (Iph × 1239.8)/(Pmono × λ) × 100 | (2) |
 | (3) |
| STH(%) = SPP/81.3 × 100 | (4) |
Calculating AM1.5G photocurrents based on IPCE integration gives the same result as a photocurrent measured under solar simulated light when the measurements are conducted within a light intensity regime such that photocurrents change linearly with light intensity. For materials in which intensity-dependent recombination dominate, the photocurrent would increase non-linearly with light intensity. There is evidence in the literature that for moderate illumination intensities less than AM1.5G intensity, photocurrent densities do vary linearly with light intensity for BiVO4 and other metal oxides.77,78 This method additionally prevents adverse effects such as heating of the device and electrolyte. The measurement of IPCE profiles additionally gives the user insights into properties such as photoabsorber bandgap and losses for photons with short wavelengths due to illumination through the glass backing of photoelectrodes. IPCE integration is also important to highlight as an acceptable route to enabling the use of more inexpensive lighting (that otherwise would have a spectrum incongruent with the solar spectrum), to give results that then may be rigorously compared to AM1.5 photocurrents reported in the literature.
Data availability
The data that support the findings of this study are available in the tables presented in the study and in the ESI.†Author contributions
Conceptualization, B. T., J. N., and A. K.; investigation, B. T. and S. P.; project administration, B. T.; supervision, J. N. and A. K.; visualization, B. T.; writing – original draft, B. T.; writing – review & editing, B. T., S. P., J. N., A. K.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
AK thanks the EPSRC for a Programme Grant (EP/W017075/1). BT thanks Imperial College for support via the President's PhD Scholarship. SDP thanks the Royal Society for a University Research Fellowship (URF\R1\191458) and Research Grant (RG\R2\232264). JN thanks the Royal Society for award of a Research Professorship and the European Research Council for funding via an Advanced Grant (CAPaCITy, grant no: 742708). We wish to acknowledge the use of the EPSRC funded Physical Sciences Data-science Service hosted by the University of Southampton and STFC under grant number EP/S020357/1. Lee Tooley, Steve Atkins and Stefanos Karapanagiotidis are thanked for constructing and maintaining our chemical vapour deposition apparatus.References
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 CAS.
- B. Moss, O. Babacan, A. Kafizas and A. Hankin, Adv. Energy Mater., 2021, 11, 13 Search PubMed.
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W. H. Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K. S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper and F. Houle, J. Phys. D Appl. Phys., 2022, 55, 323003 CrossRef CAS.
- C. Battaglia, A. Cuevas and S. De Wolf, Energy Environ. Sci., 2016, 9, 1552–1576 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- SolarFuelsDB, EPFL, 2022available from, https://solarfuelsdb.epfl.ch.
- W.-H. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater and H.-J. Lewerenz, ACS Energy Lett., 2018, 3, 1795–1800 CrossRef CAS.
- K. Sivula, J. Phys. Chem. Lett., 2015, 6, 975–976 CrossRef CAS PubMed.
- Hydrogen and Fuel Cell Technologies Office, DOE Technical Targets for Hydrogen Production from Photoelectrochemical Water Splitting, U.S. Department of Energy, 2022, available from, https://www.energy.gov/eere/fuelcells/doe-technical-targets-hydrogen-production-photoelectrochemical-water-splitting.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 RSC.
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 15010–15026 CrossRef CAS.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z. X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- J. Choi, P. Sudhagar, J. H. Kim, J. Kwon, J. Kim, C. Terashima, A. Fujishima, T. Song and U. Paik, Phys. Chem. Chem. Phys., 2017, 19, 4648–4655 RSC.
- G. Zheng, J. Wang, H. Liu, V. Murugadoss, G. Zu, H. Che, C. Lai, H. Li, T. Ding, Q. Gao and Z. Guo, Nanoscale, 2019, 11, 18968–189694 CAS.
- B. Song, T. Wang, H. Sun, Q. Shao, J. Zhao, K. Song, L. Hao, L. Wang and Z. Guo, Dalton Trans., 2017, 46, 15769–15777 CAS.
- P. P. González-Borrero, F. Sato, A. N. Medina, M. L. Baesso, A. C. Bento, G. Baldissera, C. Persson, G. A. Niklasson, C. G. Granqvist and A. Ferreira da Silva, Appl. Phys. Lett., 2010, 96, 061909 CrossRef.
- J. K. Cooper, S. Gul, F. M. Toma, L. Chen, Y. S. Liu, J. Guo, J. W. Ager, J. Yano and I. D. Sharp, J. Phys. Chem. C, 2015, 119, 2969–2974 CAS.
- Y. Park, K. J. McDonald and K. S. Choi, Chem. Soc. Rev., 2013, 42, 2321–2337 RSC.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141–11150 CrossRef PubMed.
- C. Ràfols i Bellés, S. Selim, N. M. Harrison, E. A. Ahmad and A. Kafizas, Sustainable Energy Fuels, 2019, 3, 264–271 RSC.
- F. F. Abdi, T. J. Savenije, M. M. May, B. Dam and R. Van De Krol, J. Phys. Chem. Lett., 2013, 4, 2752–2757 CrossRef CAS.
- M. Lamers, S. Fiechter, D. Friedrich, F. F. Abdi and R. Van De Krol, J. Mater. Chem. A, 2018, 6, 18694–18700 RSC.
- L. Pan, J. H. Kim, M. T. Mayer, M. K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- R. Sathre, C. D. Scown, W. R. Morrow, J. C. Stevens, I. D. Sharp, J. W. Ager, K. Walczak, F. A. Houle and J. B. Greenblatt, Energy Environ. Sci., 2014, 7, 3264–3278 RSC.
- X. Yao, D. Wang, X. Zhao, S. Ma, P. S. Bassi, G. Yang, W. Chen, Z. Chen and T. Sritharan, Energy Technol., 2018, 6, 100–109 CrossRef CAS.
- K. Song, H. Hou, C. Gong, F. Gao, D. Zhang, Z. Fang, W. Yang and F. He, J. Mater. Chem. A, 2022, 10, 22561 RSC.
- I. Y. Ahmet, S. Berglund, A. Chemseddine, P. Bogdanoff, R. F. Präg, F. F. Abdi and R. van de Krol, Adv. Energy Sustainability Res., 2020, 1, 2000037 CrossRef.
- I. Grigioni, K. G. Stamplecoskie, E. Selli and P. V. Kamat, J. Phys. Chem. C, 2015, 119, 20792–20800 CrossRef CAS.
- I. Y. Ahmet, Y. Ma, J. W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. F. Abdi and R. van de Krol, Sustainable Energy Fuels, 2019, 3, 2366–2379 RSC.
- A. Hankin, F. E. Bedoya-Lora, C. K. Ong, J. C. Alexander, F. Petter and G. H. Kelsall, Energy Environ. Sci., 2017, 10, 346–360 RSC.
- E. Alarcón-Lladó, L. Chen, M. Hettick, N. Mashouf, Y. Lin, A. Javey and J. W. Ager, Phys. Chem. Chem. Phys., 2014, 16, 1651–1657 RSC.
- P. Brack, J. S. Sagu, T. A. Nirmal Peiris, A. McInnes, M. Senili, K. G. Upul Wijayantha, F. Marken and E. Selli, Chem. Vap. Deposition, 2015, 21, 41–45 CrossRef CAS.
- S. Vallejos, P. Umek and C. Blackman, J. Nanosci. Nanotechnol., 2011, 11, 8214–8220 CrossRef CAS PubMed.
- A. Kafizas, L. Francàs, C. Sotelo-Vazquez, M. Ling, Y. Li, E. Glover, L. McCafferty, C. Blackman, J. Darr and I. Parkin, J. Phys. Chem. C, 2017, 121, 5983–5993 CrossRef CAS.
- S. Byun, G. Jung, Y. Shi, M. Lanza and B. Shin, Adv. Funct. Mater., 2020, 30, 1806662 CrossRef CAS.
- Pilkington NSG, NSG TEC™: The original ITO replacement, 2025, available from, https://www.pilkington.com/en/powered-by-nsg-tec/overview#.
- S. Corby, L. Francàs, A. Kafizas and J. R. Durrant, Chem. Sci., 2020, 11, 2907–2914 RSC.
- A. Braun, S. Erat, X. Zhang, Q. Chen, T. W. Huang, F. Aksoy, R. Löhnert, Z. Liu, S. S. Mao and T. Graule, J. Phys. Chem. C, 2011, 115, 16411–16417 CrossRef CAS.
- M. Sachs, J. S. Park, E. Pastor, A. Kafizas, A. A. Wilson, L. Francàs, S. Gul, M. Ling, C. Blackman, J. Yano, A. Walsh and J. R. Durrant, Chem. Sci., 2019, 10, 5667–5677 RSC.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- M. Ling and C. Blackman, Phys. Status Solidi C, 2015, 12, 869–877 CrossRef CAS.
- S. N. F. Mohd-Nasir, M. A. Mat-Teridi, M. Ebadi, J. S. Sagu, M. Y. Sulaiman, N. A. Ludin and M. A. Ibrahim, Phys. Status Solidi A, 2015, 212, 2910–2914 CrossRef CAS.
- P. S. Archana, Z. Shan, S. Pan and A. Gupta, Int. J. Hydrogen Energy, 2017, 42, 8475–8485 CrossRef CAS.
- S. Saha, J. M. Amador, S. Goberna-Ferron, S. R. Decker, D. H. Park and D. A. Keszler, Solid State Sci., 2021, 113, 106451 CrossRef CAS.
- A. Galembeck and O. L. Alves, J. Mater. Sci., 2002, 37, 1923–1927 CrossRef CAS.
- K. R. Tolod, S. Hernández, M. Castellino, F. A. Deorsola, E. Davarpanah and N. Russo, Int. J. Hydrogen Energy, 2020, 45, 605–618 CrossRef CAS.
- R. N. Nenashev, N. E. Mordvinova, V. P. Zlomanov and V. L. Kuznetsov, Inorg. Mater., 2015, 51, 967–973 CrossRef.
- T. Thilagavathi, D. Venugopal, R. Marnadu, J. Chandrasekaran, R. Alshahrani and M. Shkir, J. Inorg. Organomet. Polym. Mater., 2021, 31, 1217–1230 CrossRef CAS.
- A. Ghatak, S. Roy Moulik and B. Ghosh, RSC Adv., 2016, 6, 31705–31716 RSC.
- R. Liu, J. Ren, D. Zhao, J. Ning, Z. Zhang and Y. Wang, Inorg. Chem. Front., 2017, 4, 2045–2054 RSC.
- S. Wang, P. Chen, J. H. Yun, Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2017, 56, 8500–8504 CrossRef CAS PubMed.
- M. Arai, S. Hayashi and K. Yamamoto, Solid State Commun., 1990, 75, 613–616 CrossRef CAS.
- S. Prabhu, L. Cindrella, O. J. Kwon and K. Mohanraju, Int. J. Hydrogen Energy, 2017, 42, 29791–29796 CrossRef CAS.
- V. I. Merupo, S. Velumani, K. Ordon, N. Errien, J. Szade and A. H. Kassiba, CrystEngComm, 2015, 17, 3366–3375 RSC.
- Y. Gao, Z. Tian, H. Zhu, H. Xue, L. Ma, Y. Dai, W. Zhao, X. Li, N. Li and L. Ge, ACS Appl. Energy Mater., 2021, 4, 14649–14661 CrossRef CAS.
- S. Saxena, A. Verma, K. Asha, N. K. Biswas, A. Banerjee, V. R. Satsangi, R. Shrivastav and S. Dass, Int. J. Hydrogen Energy, 2020, 45, 26746–26757 CrossRef CAS.
- H. Hassani, B. Partoens, E. Bousquet and P. Ghosez, Phys. Rev. B, 2022, 105, 014107 CrossRef CAS.
- J. A. Spencer, A. L. Mock, A. G. Jacobs, M. Schubert, Y. Zhang and M. J. Tadjer, Appl. Phys. Rev., 2022, 9, 011315 CAS.
- Photonic Solutions, 2025, Fully Reflective Solar Simulator, available from, https://photonicsolutions.co.uk/products/fully-reflective-solar-simulator/.
- D. K. Lee and K. S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- The global standard spectrum (AM1.5g), 2018, available from, https://www2.pvlighthouse.com.au/resources/courses/altermatt/The-Solar-Spectrum/The-global-standard-spectrum-(AM1-5g).aspx.
- D. S. Kim, K. W. Lee, J. H. Choi, H. H. Lee, H. W. Suh, H. S. Lee and H. K. Cho, J. Mater. Chem. A, 2022, 10, 21300 RSC.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat. Energy, 2017, 2, 17032 CrossRef CAS.
- O. Sow, M. L. Ba, H. Y. Ba, M. A. O. El Moujtaba, Y. Traore, M. S. Diop, H. Lemrabott, M. Wade and G. Sissoko, J. Electromagn. Anal. Appl., 2019, 11, 203–216 CAS.
- P. Bornoz, F. F. Abdi, S. D. Tilley, B. Dam, R. Van De Krol, M. Graetzel and K. Sivula, J. Phys. Chem. C, 2014, 118, 16959–16966 CrossRef CAS.
- X. Yin, Q. Liu, Y. Yang, Y. Liu, K. Wang, Y. Li, D. Li, X. Qiu, W. Li and J. Li, Int. J. Hydrogen Energy, 2019, 44, 594–604 CrossRef CAS.
- J. Brillet, J. H. Yum, M. Cornuz, T. Hisatomi, R. Solarska, J. Augustynski, M. Graetzel and K. Sivula, Nat. Photonics, 2012, 6, 824–828 CrossRef CAS.
- Y. S. Chen, J. S. Manser and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 974–981 CrossRef CAS PubMed.
- Y. Peng, G. V. Govindaraju, D. K. Lee, K. S. Choi and T. Andrew, ACS Appl. Mater. Interfaces, 2017, 9, 22449–22455 CrossRef CAS PubMed.
- Y. Qiu, W. Liu, W. Chen, W. Chen, G. Zhou, P. C. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, Sci. Adv., 2016, 2, 1501764 CrossRef PubMed.
- L. Han, F. F. Abdi, R. Van De Krol, R. Liu, Z. Huang, H. J. Lewerenz, B. Dam, M. Zeman and A. H. M. Smets, ChemSusChem, 2014, 7, 2832–2838 CrossRef CAS PubMed.
- J. H. Kim, S. M. Hwang, I. Hwang, J. Han, J. H. Kim, Y. H. Jo, K. Seo, Y. Kim and J. S. Lee, iScience, 2019, 19, 232–243 CrossRef CAS PubMed.
- M. Lamers, S. Fiechter, D. Friedrich, F. F. Abdi and R. Van De Krol, J. Mater. Chem. A, 2018, 6, 18694–18700 RSC.
- M. Favaro, I. Y. Ahmet, P. C. J. Clark, F. F. Abdi, M. J. Sear, R. Van De Krol and D. E. Starr, J. Phys. D: Appl. Phys., 2021, 54, 164001 CrossRef CAS.
- T. Lopes, L. Andrade, H. Aguilar Ribeiro and A. Mendes, Int. J. Hydrogen Energy, 2010, 35, 11601–11608 CrossRef CAS.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- Y. Ma, C. A. Mesa, E. Pastor, A. Kafizas, L. Francàs, F. Le Formal, S. R. Pendlebury and J. R. Durrant, ACS Energy Lett., 2016, 1, 618–623 CrossRef CAS.
- F. Le Formal, E. Pastor, S. D. Tilley, C. A. Mesa, S. R. Pendlebury, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2015, 137, 6629–6637 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08595g |
| This journal is © The Royal Society of Chemistry 2025 |