
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning-driven global optimization reveals nanometre-scale mixed phases of borophene on Ag(100)†
Yunlei
Wang
,
Haifeng
Lv
 * and
Xiaojun
Wu
*
* and
Xiaojun
Wu
*
State Key Laboratory of Precision and Intelligent Chemistry, School of Chemistry and Materials Sciences, Key Laboratory of Materials Sciences for Energy Conversion, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), CAS Center for Excellence in Nanoscience, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: hflv@ustc.edu.cn; xjwu@ustc.edu.cn
First published on 14th March 2025
Abstract
Metal-supported borophene exhibits significant polymorphism and an inherently complex potential energy landscape, posing challenges to exploring its structural diversity. In this study, we integrate a neural network-driven machine learning potential with stochastic surface walking global optimization and an active learning framework to comprehensively map the potential energy surface (PES) of large-size borophene on an Ag(100) substrate. Our exhaustive search identifies 59![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 857 local minima across 556 distinct supercells, revealing a PES segmented into multiple energy basins and three major funnels. Among the low-energy configurations, 1391 low-energy structures extend to the nanometre scale, showcasing a diverse array of mixed-phase borophene architectures, including monolayer ribbons (β12 and χ3) and bilayer fragments (BL-α5, BL-α7, BL-α1, BL-α6, and BL-α1α6). Notably, the global minimum structures feature monolayers composed of alternating χ3 and β12 ribbons and bilayers formed from BL-α5, BL-α1α6, and BL-α1 fragments. All mixed-phase borophenes exhibit metallic properties, and their simulated scanning tunneling microscopy (STM) images are provided to facilitate future experimental validation. These findings highlight the extraordinary structural complexity and rich polymorphism of borophene on extended metal surfaces, offering valuable insight into their formation, stability, and potential for tunable electronic properties.
857 local minima across 556 distinct supercells, revealing a PES segmented into multiple energy basins and three major funnels. Among the low-energy configurations, 1391 low-energy structures extend to the nanometre scale, showcasing a diverse array of mixed-phase borophene architectures, including monolayer ribbons (β12 and χ3) and bilayer fragments (BL-α5, BL-α7, BL-α1, BL-α6, and BL-α1α6). Notably, the global minimum structures feature monolayers composed of alternating χ3 and β12 ribbons and bilayers formed from BL-α5, BL-α1α6, and BL-α1 fragments. All mixed-phase borophenes exhibit metallic properties, and their simulated scanning tunneling microscopy (STM) images are provided to facilitate future experimental validation. These findings highlight the extraordinary structural complexity and rich polymorphism of borophene on extended metal surfaces, offering valuable insight into their formation, stability, and potential for tunable electronic properties.
Introduction
Borophene has attracted extensive attention due to its structural diversity1,2 and intriguing physical properties,3–5 suggesting potential applications in nanoelectronics. Owing to its electron-deficiency, borophene often adopts frameworks composed of mixed triangles and hexagons6,7 resulting in a high degree of polymorphism. Unlike many two-dimensional (2D) materials, borophene does not have a bulk counterpart and generally requires a metallic substrate for stabilization.8,9 In experiment, various polymorphs of borophene have been synthesized on different substrates, with the atomic arrangements highly influenced by experimental conditions, highlighting the potential to achieve specific polymorphs with tailored functionalities.8,10 However, the identification of synthesized borophene often relies on comparing experimental scanning tunneling microscopy (STM) images with simulated ones. This method is limited in capturing the detailed structural features of large superlattices, such as mixed phases,11,12 ribbon dislocation,13 and twin boundaries.14Accurately predicting the structures of these large superlattices is essential to capture the complex atomic patterns in borophene.15–17 So far, theoretically reported phases are limited to be single-phase at the Angstrom scale. This is because of the exponential increase of local minima with the number of atoms and the rapid growth of computational cost with the number of atoms.18,19 Machine learning methods are powerful tools to overcome these limitations, enabling data-driven exploration of broader configurational spaces at a fraction of the computational cost.18,20 Machine learning-driven simulations have offered valuable insights into realistic structural motifs, properties,21–24 and complex structure–property relationships in a variety of systems.25–27 Specifically, the realization of mixed-ribbon monolayer phases on Ag(100) motivates us to fully explore the configurational space. While machine learning-based global structure prediction has successfully discovered new phases of bulk boron28–30 and boron clusters,31 applying these strategies to borophene on metal substrates remains challenging due to the intricate interplay between boron–boron and boron–metal bonding.
In this work, we develop an on-the-fly neural network (NN) potential combined with stochastic surface walking (SSW) global optimization and an active learning framework to comprehensively explore the PES of borophene on an Ag(100) substrate. Our global dataset comprises 177023 data points, revealing a complex energy landscape with multiple basins and three distinct energy funnels. By examining 59857 local minima across 556 diverse supercells, we identify 1391 low-energy configurations extending to nanometre-scale lattice constants. Detailed structural analysis shows that the first two energy funnels predominantly host monolayer phases, while the third funnel is characterized by bilayers. Notably, both the first and third funnels display a range of mixed phases, whereas the second funnel remains predominantly single-phase borophene. The first funnel reproduces experimentally observed monolayer structures, validating our computational approach. In particular, the global minimum mixed phases feature monolayers composed of alternating χ3 and β12 ribbons and bilayers formed from BL-α5, BL-α1α6, and BL-α1 fragments. All these mixed-phase borophenes exhibit metallic properties, and their simulated STM images are provided to support future experimental investigations. This study establishes a general framework for leveraging ML-driven global optimization to uncover the structural characteristics of experimental mixed phases, aiming to achieve the targeted polymorph of borophene.
Methods and models
First-principles calculations
All first-principles calculations were performed using the Vienna Ab initio Simulation Package (VASP)32,33 with the projector augmented wave (PAW)34,35 method to describe ionic cores and valence electrons. The exchange-correlation functional is treated within the generalized gradient approximation (GGA), in the Perdew–Burke–Ernzerhof (PBE) functional.36,37 The D3 correction38 was employed to describe the van der Waals interaction between substrates and borophenes. The kinetic energy cutoff for the plane wave basis is 400 eV and the first Brillouin zone is sampled using the Γ-centered Monkhorst–Pack scheme with a k-space density of 2π × 0.04 Å−1.Structure search driven by the neutral network potential
The stochastic surface walking (SSW) global optimization39,40 implemented in the Large-scale Atomistic Simulation with neural network Potential (LASP) software41 is used to sample and search the global potential energy surface (PES). The high dimensional neural network (HDNN) scheme in LASP software is employed to train the global dataset. For more detailed information about SSW methods and HDNN architecture, refer to the ESI.†Structural models
The Ag(100) substrate model contains two layers with the bottom layer fixed during the SSW optimization. To evaluate the stability of borophene on Ag(100), formation energy (Eform) is used, which is defined as Eform = (Etot − Esub)/NB. Etot and Esub denote the energy of the total system and Ag(100) substrate, respectively, and NB is the number of boron atoms. For additional details, refer to the ESI.†Results and discussion
Construction of the neutral network potential and global dataset
Fig. 1 illustrates the workflow for constructing the NN potential used to identify global minima of borophene on an Ag(100) surface. This procedure involves three main steps. First, we generate metal substrate supercells of varying sizes and shapes by redefining a matrix based on the primitive cell, with the supercell size denoted by M. To ensure balanced cell dimensions and avoid extremely small angles, only those supercells meeting the criteria |a − b| ≤ M and 40° ≤ γ ≤ 140° are selected. In total, 556 supercells with M values ranging from 2 to 30 are chosen (Table S1†). Then, boron atoms are deposited onto these supercells at coverage (BC) ranging from 2.3 to 7.3, as detailed in Fig. S1.† | ||
| Fig. 1 Workflow for building the on-the-fly neural network (NN) potential of the borophene/Ag(100) system. M denotes the determinant of the redefined matrix and represents the multiple of the supercell relative to the primitive cell. a, b and γ are lattice parameters. The boron coverage on the Ag(100) surface is defined as the number of boron atoms/M. During iterative refinement of the global dataset, the energy convergence criterion is ΔE ≤ 5 meV per atom, in which ΔE = |ENN − EDFT|/N. ENN and EDFT denote the energies calculated by the NN potential and DFT method, respectively, N is the number of total atoms. The screening of minima is conducted based on the formation energy (Eform). | ||
In the second step, we iteratively build a global dataset that captures the diverse bonding environments encountered in borophene on Ag(100). We initiate this by performing stochastic surface walking (SSW) pathway sampling on 174 smaller systems (2 ≤ M ≤ 5) containing up to 47 atoms, each starting from random configurations. This comprehensive sampling generates approximately 7 million raw structure data points, from which around 120000 are selected to form the initial global dataset (Fig. S2†).
The initial global dataset is used to train the NN potential. Once trained, the NN potential is applied to predict 59857 larger systems (6 ≤ M ≤ 30, BC = 2.3 to 7.3). If inaccuracies are detected in predicting minima, additional structures are extracted from SSW trajectories and subjected to ab initio calculations. These newly calculated structures are then added to the global dataset, and the NN potential is retrained. This iterative, active learning-like approach27,42 ensures the robustness and reliability of the NN potential for exploring the global PES. Performance metrics of the resulting NN potential are provided in Fig. S3, S4 and ESI.†
Overview of the global dataset
The final global dataset includes 177023 structures, with the trained NN potential achieving root-mean-square errors (RMSE) of 11.281 meV per atom for energies and 0.456 eV Å−1 for forces, demonstrating high accuracy (Fig. S3†). Fig. 2a presents the formation energy (Eform) versus Steinhardt-type order parameter (OP2), spanning from −6.5 to −1 eV per atom and 0 to 6, respectively. The broad distribution of Eform and OP2 values (Fig. S5†) highlights the dataset's extensive diversity and the NN potential's flexibility. Additional details regarding M, BC, and OP2 are provided in Fig. S6.†
 | ||
| Fig. 2 (a) Illustration of the global dataset from first principles for borophene on Ag(100) systems shown as a contour map of formation energy (Eform) vs. OP2. The density is indicated by color. The OP2 parameter reflects the averaged geometrical environment of bonded boron atoms. (b) Schematic diagram of the potential energy surface (PES) for borophene on Ag(100) systems scaled by BC and OP2 dimensionality. | ||
Focusing on the low-energy region (Eform < −6.0 eV per atom), which comprises approximately 130000 structures, we utilize BC and OP2 as descriptors and apply dimensionality reduction techniques43,44 to visualize the multidimensional PES (Fig. 2b). This analysis uncovers three distinct energy funnels at BC ranges of 2.7 to 2.9, 3.4 to 3.5, and 6.3 to 6.8, alongside numerous energy basins across different BC values, indicating the presence of multiple stable structural motifs in borophene on Ag(100). An energy funnel represents regions where the lowest-energy local minimum, including the global minimum, is most likely to occur, facilitating the steepest descent pathways on the PES. In contrast, the energy basin indicates clusters of local minima.43,45 The first two funnels exhibit similar energy levels, while the third energy funnel is slightly deeper, suggesting that borophene configurations with varying boron coverage in the first two funnels can coexist with comparable stability and the configurations in the third funnel are inherently more stable. OP2 values for funnels and basins primarily lie between 1 and 2 (the most stable and ordered structures), with OP2 > 4 corresponding to high-energy ridges (less stable and more disordered configurations). These findings provide a conceptual map of the global PES, aiding in the identification of stable minima from highly disordered starting configurations and enhancing the predictive power for discovering new and experimentally relevant phases.
Identified local minima and low-energy configurations
Using the established PES, we identified 59857 local minima from random initial structures, as shown in Fig. 3a. The distribution of these minima in terms of OP2 and BC is color-coded by Eform. The corresponding minima exhibit mean absolute errors of 3.278 meV per atom for energies and 0.299 eV Å−1 for forces, indicating the reliability of our NN potential.
 | ||
| Fig. 3 The OP2–BC distribution of (a) minima and (b) lowest-energy minima borophenes on Ag(100) colored by the formation energy (Eform). (c) The proportion of different coordination numbers (CN) ranging from 3 to 8 and (d) Eform of low-energy minima structures as a function of boron coverage (BC). In (c), the transparent colored lines represent raw data, and the solid lines are the result of applying an adjacent-averaging filter. The ML, MixL, and BL denote the monolayer, mixed-layer, and bilayer, respectively. The black dashed line located at BC of 3.6 separates the ML from the MixL, and that at BC of 6 separates the MixL from the BL. Each data point under specific BC value represents the most stable configuration. The luminance of light blue spheres denotes the magnitude of the M value, and the higher luminance corresponds to the smaller M value. The (e) statistical count and (f) the maximal lattice constant (L) of the low-energy minima under different M values ranging from 6 to 30. | ||
To isolate the most stable low-energy configurations, we screened these minima by selecting, at each BC value, the structure with the lowest Eform across all supercell shapes and sizes. This screening yielded 1391 low-energy minima, whose OP2–BC distribution and Eform data are shown in Fig. 3b. Three prominent low-energy regions emerge at OP2 values between 1.6 and 2.0, consistent with the three energy funnels identified in the global PES. Additionally, the overall OP2–BC distribution of these minima also aligns with the previously mapped low-energy landscape, confirming that our global dataset effectively captures the key atomic environments of borophene on Ag(100).
To elucidate the bonding environments within these low-energy minima, we calculated the boron coordination number (CN) using a 2.3 Å f boron–boron distance cutoff. Fig. 3c shows the relative percentages of each CN value as a function of BC. The presence of CN values ranging from 2 to 8 underscores the complex multi-center bonding, akin to bulk boron.29 While CN = 2 and CN = 8 are relatively rare, CN = 3 briefly appears at low BC values (2.33 to 2.5) before disappearing. In contrast, CN values of 4, 5, 6, and 7 become increasingly prominent across the entire BC range, indicating evolving coordination patterns in these low-energy minima.
As BC increases from 3.6 to 6, the CN of 7 gradually rises, indicating the formation of borophene structures beyond a monolayer. Distinct shifts in CN distribution are observed around BC = 3.6 and BC = 6, as evident from both the average CN (ave_CN) and average bond length (ave_bond) (Fig. S7a and b†). Consequently, we define BC = 3.6 as the boundary between monolayer (ML) and mixed-layer (MixL) phases, and BC = 6 as the boundary between MixL and bilayer (BL) phases. In this classification, the ML refers to a single-atomic-layer sheet, MixL denotes an ML with attached boron nanostructures, and the BL signifies two covalently bonded MLs, following our previous work.46 Although some structures may not conform strictly to these categories, this classification provides a useful framework for understanding the structural evolution with increasing BC.
Fig. 3d correlates these structural transitions with stability by plotting Eformversus BC. Three distinct energy valleys emerge, corresponding to the three PES funnels. Energy valley 1 (BC ≈ 2.7 to 2.85) and energy valley 2 (BC ≈ 3.45 to 3.6) correspond to ML configurations, while energy valley 3 (BC ≈ 6.3 to 6.45) pertains to BL configurations. The first two valleys exhibit comparable stability, whereas the third valley is approximately 57 meV per atom deeper, reflecting the enhanced structural complexity and stronger bonding networks in bilayers. Within energy valley 1, CN values of 4, 5, and 6 appear in a 25:50:25 ratio, while energy valley 2's minima exclusively feature CN = 6. Energy valley 3 displays a mix of CN = 5, 6, and 7 (40%, 52%, and 8%, respectively), resulting in a higher average CN (5.5 to 5.75) and increased stability.
The influence of supercell size M is also evident. As shown in Fig. 3e, the vast majority (88.7%) of low-energy minima are found in supercells with M values between 15 and 30. Indeed, most structures with Eform < −6.50 eV per atom have M > 15 (Fig. S7c†). Fig. 3f further demonstrates that the maximal lattice constants (L) of these low-energy minima range from 8.667 Å up to 38.75 Å, underscoring the critical importance of large supercells in accurately capturing the global minima of borophene on Ag(100). These results highlight the necessity of the extended-scale supercells to reflect the inherent structural complexity of borophene phases.
Structural analysis of energy valleys
To gain deeper insight into the structural motifs within each of three energy valleys, we selected ten lowest-energy configurations from each valley. The corresponding Eform–BC plots are shown in Fig. 4a to c, and detailed information about these structures is listed in Tables S3 to S5.† For convenience, we label the structures in energy valleys 1, 2, and 3 as ML1 to ML10, ML11 to ML20, and BL1 to BL10, respectively, based on their Eform values. Within these sets, the top ten structures in valleys 1, 2, and 3 lie within energy windows of 19.4, 11.3, and 19.7 meV per atom, respectively, and their M value ranges from 19 to 30, 16 to 29, and 16 to 27.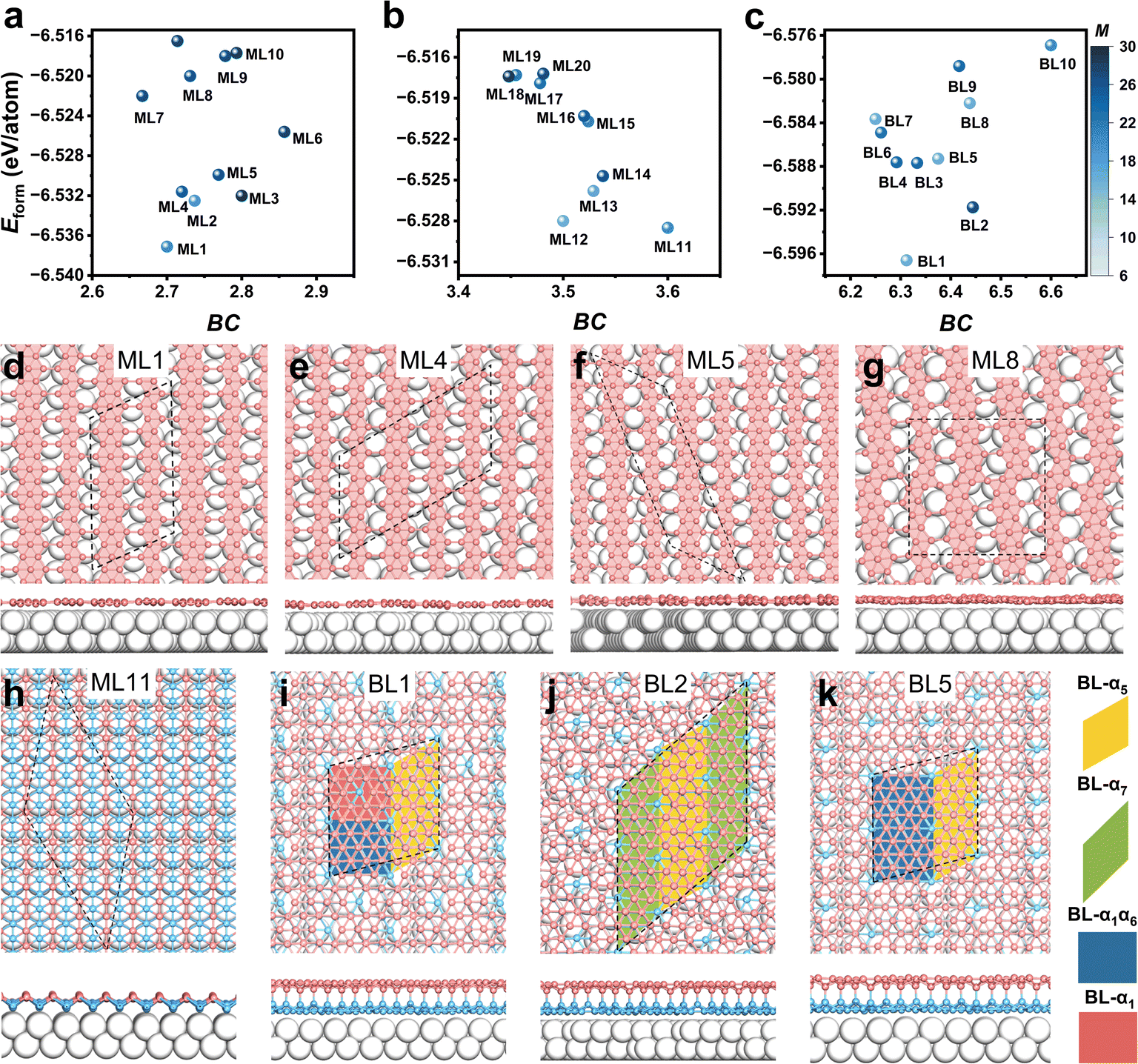 | ||
| Fig. 4 The formation energy (Eform) varying with boron coverage (BC) for the top ten most stable configurations from energy valleys (a) 1, (b) 2, and (c) 3. The structures from energy valleys 1, 2, and 3 are numbered by ML1–ML10, ML11–ML20, and BL1–BL10, respectively. The luminance of light blue spheres denotes the magnitude of the M value, and the higher luminance corresponds to the smaller M value. (d)–(k) The atomic patterns of several representative structures (ML1, ML4, ML5, ML8, ML11, BL1, BL2, and BL5) of (a)–(c), in which the black dashed lines denote the unit cell. For ML1, ML4, ML5, and ML8, only top view is provided. For ML11, BL1, BL2, and BL5, both top and side views are provided. The light gray spheres denote Ag atoms, and the pink colors denote boron atoms in which the lower boron atoms of ML11 and the bottom sheet of BL structures are colored by light cyan. Different bilayer borophene fragments are depicted by different shapes with different colors. | ||
To unveil the atomic patterns in different energy valleys, several representative structures are displayed in Fig. 4d to k, with additional structures shown in Fig. S8 to S10.† The names, BC, Eform, interfacial charge transfer (C), M values, and structural descriptions of these selected structures are summarized in Table 1 (see more details in Tables S3 to S5†). Structures in the energy valleys 1 and 3 exhibit mixed phases. In energy valley 1, monolayers (MLs) feature combinations of different borophene ribbons, such as χ3, β12, and so on (Fig. 4d–g and S8†). In energy valley 3, bilayers (BLs) are characterized by mixtures of various borophene phases, including BL-α5, BL-α7, BL-α1, BL-α6, and BL-α1α6 (Fig. 4i–k and S10†). Note that BL-α5 denotes the bilayer covalently stacked by two α5 monolayers. Interestingly, energy valley 2 shows a high degree of structural degeneracy, with ML10 to ML20 all being buckled triangular lattices (δ6) (Fig. 4h and S9†). The simulated STM images of all these structures are provided in Fig. S11 and S12,†Fig. 5a–h. The nomenclature of all borophene structures follows the rules proposed in previous work.1 The structures of MLs in this work, including new structures (α6 and α7) and borophene ribbons (χ3, β12, and R1), are shown in Fig. S13.† Fig. S14† provides detailed atomic patterns of BL-α5, BL-α7, BL-α1, BL-α6 and BL-α1α6. Here, BL-αi or BL-αiαj denotes the bilayer covalently stacked by two αi monolayers or αi and αj monolayers, respectively.
| Name | BC | E form (eV per atom) | Structures | C (e per atom) |
|---|---|---|---|---|
| ML1 | 2.700 | −6.5371 | 1χ31β12 | 0.0265 |
| ML4 | 2.720 | −6.5316 | 3χ31β12 | 0.0274 |
| ML5 | 2.769 | −6.5299 | 1χ31β12 | 0.0290 |
| ML8 | 2.731 | −6.5200 | 1χ32β12-dislocation | 0.0268 |
| ML11 | 3.600 | −6.5285 | δ6 | 0.0108 |
| BL1 | 6.312 | −6.5966 | 2BL-α51BL-α11BL-α1α6 | 0.0103 |
| BL2 | 6.444 | −6.5917 | 3BL-α53BL-α7 | 0.0095 |
| BL5 | 6.375 | −6.5870 | 2BL-α52BL-α1α6 | 0.0110 |
 | ||
| Fig. 5 Simulated images of (a) ML1, (b) ML4, (c) ML5, (d) ML8, (e) ML11, (f) BL1, (g) BL2, and (h) BL5 on Ag(100). The red dashed lines denote the primitive cell. The (i)–(l) corresponding projected density of states, in which the gray and cyan lines denote Ag and B atoms, respectively. The fermi energy level is set as 0 eV. | ||
In energy valley 1, MLs feature combinations of different borophene ribbons, such as χ3 and β12 with varying ratios (Fig. 4d–g, S8 and Table S3†). ML1 comprises one χ3 and one β12 ribbon in the unit cell (Fig. 4d), and its simulated STM image is shown in Fig. 5a. It is more stable than ML2 (Fig. S8a†) and ML3 (Fig. S8b†) by 4.6 and 5.1 meV per atom, respectively (Table 1). ML2 and ML3 comprise two χ3 and one β12 ribbon and one χ3 and two β12 ribbons, respectively, which closely match experimental phases C and A based on simulated STM images (Fig. S11a and b†).12 Additionally, the simulated STM images of the pure β12 sheet (ML6, Fig. S8c†) are consistent with phase B (Fig. S11c†). Minor discrepancies, such as the position of a less bright stripe in the simulated STM images, may arise from undulation fluctuations due to substrate interactions. ML4 (Fig. 4e), composed of three χ3 and one β12 ribbon, and ML5 (Fig. 4f), composed of one χ3 and one β12, possess comparable stability to ML2 and ML3. Note that ML5 shares the same atomic pattern as ML1, but ML1 has a lower lattice mismatch, enhancing its stability (Table S3†). The pure β12 (ML6, Fig. S8c†) and χ3 (ML7, Fig. S8d†) phases, which have been experimentally synthesized, are less stable than the mixed ribbons (ML1 to ML5) according to their Eform values. Therefore, the newly identified ML1 and ML4 are thermodynamically feasible under experimental conditions. Additionally, structures featuring ribbon dislocation (ML8, Fig. 4g) and hybridized χ3-β12 ribbons (ML10, Fig. S8f†) have been predicted. These structural defects are inevitable in as-grown borophene,11 and can only be captured in large supercells.
In energy valley 2, all structures from ML11 to ML20 adopted a buckled δ6 lattice (Fig. 4h and S9†), despite being observed in different supercells (Table S4†). The simulated STM image of ML11 is provided in Fig. 5e. In energy valley 3, BLs are characterized by mixtures of various borophene phases, including BL-α5, BL-α7, BL-α1, BL-α6, and BL-α1α6 (Fig. 4i–k and S10†). BL1 (Fig. 4i), BL3, and BL4 are composed of varying proportions of BL-α5, BL-α1α6, and BL-α1. BL2 (Fig. 4j), composed of BL-α5 and BL-α7, is more stable than the single-phase BL-α7 (BL6) and BL-α5 (BL10) by 3.8 and 19.7 meV per atom, respectively. This indicates a preference for mixed-phase bilayer borophenes on Ag(100). Additionally, BL5 (Fig. 4k) consists of BL-α5 and BL-α1α6, while BL7 comprises BL-α5 and BL-α1. Similarly, BL8 and BL9 are mixtures of BL-α5, BL-α1α6, and BL-α6 in different proportions. The simulated STM images of BL1, BL2, and BL5 are provided in Fig. 5f–h, and those of the other BLs are shown in Fig. S12.† Notably, the pattern of BL10 matches well with the experimental CO-AFM images of bilayer borophene on Ag(111) in the literature.47 All aforementioned BLs are stacked in an AB pattern, while AA-stacking variants exhibit lower stability (Fig. S15†) and their corresponding atomic arrangements are displayed in Fig. S16.†
Electronic properties of representative structures
We conducted projected density of states (PDOS) calculations for representative structures (ML1, ML4, ML5, ML8, ML11, BL1, BL2, and BL10) on the Ag(100) substrates, as shown in Fig. 5f–i. Detailed PDOS information, including those for free-standing borophene, is presented in Fig. S17 and S18.† These calculations reveal that all analyzed structures exhibit metallicity when supported on Ag(100). Notably, there is a significant increase in the density of states (DOS) near the Fermi level for ML1, ML4, ML5, and ML8, while ML11, BL1, BL2, and BL10 show a decrease in DOS in this region. These trends can be directly compared with the future experimental dI/dV spectra to validate our computational predictions.Furthermore, all examined structures, except for BL-α5, retain their metallicity in free-standing forms and demonstrate good conductivity. This suggests that the mixed-phase borophenes not only maintain their electronic properties when supported on metal substrates but also exhibit promising conductivity characteristics in isolation, enhancing their potential applications in nanoelectronics.
Conclusions
In summary, we established a machine learning-driven framework to resolve the complex potential energy landscape of borophene on Ag(100), revealing unprecedented structural diversity at the nanometer scale. By integrating active learning with SSW global optimization, we systematically identified 1391 low-energy configurations across three distinct energy funnels, including mixed-phase monolayers (χ3/β12 ribbons) and bilayers (BL-α5/BL-α1α6/BL-α1 assemblies). These findings not only reconcile experimental observations of borophene polymorphism but also unveil previously unexplored metastable phases stabilized by substrate interactions. The methodology developed here, combining scalable neural network potentials with adaptive sampling, transcends borophene systems, offering a generalized strategy for exploring polymorphism in substrate-supported 2D materials (e.g., silicene, phosphorene). Furthermore, the metallic nature of these mixed phases and their tunable electronic properties suggest promising avenues for nanoelectronic applications.Data availability
Data for this article, including the NN potential file and the top ten most stable structures in the three energy valleys are available at GitHub at https://github.com/yunleiw/Mixed-Phases-of-Borophene-on-Ag-100-.git.Author contributions
Y. W., H. L., X. W. designed the study. Y. W. performed the theoretical simulations. Y. W., H. L., X. W. analyzed the data and wrote the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work is supported by the National Natural Science Foundation for Distinguished Young Scholars (grant no. 22225301), the National Natural Science Foundation of China (grant no. 22303092 and 22321001), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB0450101), the Fundamental Research Funds for the Central Universities (20720220009 and WK2490000001), and Super Computer Center of USTCSCC and SCCAS.References
- X. Wu, J. Dai, Y. Zhao, Z. Zhuo, J. Yang and X. C. Zeng, Two-Dimensional Boron Monolayer Sheets, ACS Nano, 2012, 6, 7443–7453 CrossRef CAS PubMed.
- E. S. Penev, S. Bhowmick, A. Sadrzadeh and B. I. Yakobson, Polymorphism of two-dimensional boron, Nano Lett., 2012, 12, 2441–2445 CrossRef CAS PubMed.
- B. Mortazavi, O. Rahaman, A. Dianat and T. Rabczuk, Mechanical responses of borophene sheets: a first-principles study, Phys. Chem. Chem. Phys., 2016, 18, 27405–27413 RSC.
- E. S. Penev, A. Kutana and B. I. Yakobson, Can Two-Dimensional Boron Superconduct?, Nano Lett., 2016, 16, 2522–2526 CAS.
- X.-F. Zhou, A. R. Oganov, Z. Wang, I. A. Popov, A. I. Boldyrev and H.-T. Wang, Two-dimensional magnetic boron, Phys. Rev. B, 2016, 93, 085406 Search PubMed.
- H. Tang and S. Ismail-Beigi, Novel Precursors for Boron Nanotubes: The Competition of Two-Center and Three-Center Bonding in Boron Sheets, Phys. Rev. Lett., 2007, 99, 115501 Search PubMed.
- T. R. Galeev, Q. Chen, J.-C. Guo, H. Bai, C.-Q. Miao, H.-G. Lu, A. P. Sergeeva, S.-D. Li and A. I. Boldyrev, Deciphering the mystery of hexagon holes in an all-boron graphene α-sheet, Phys. Chem. Chem. Phys., 2011, 13, 11575–11578 RSC.
- Z. Zhang, Y. Yang, G. Gao and B. I. Yakobson, Two-Dimensional Boron Monolayers Mediated by Metal Substrates, Angew. Chem., Int. Ed., 2015, 54, 13022–13026 CrossRef CAS PubMed.
- N. Karmodak and E. D. Jemmis, The Role of Holes in Borophenes: An Ab Initio Study of Their Structure and Stability with and without Metal Templates, Angew. Chem., Int. Ed., 2017, 56, 10093–10097 CAS.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Experimental realization of two-dimensional boron sheets, Nat. Chem., 2016, 8, 563–568 CAS.
- X. Liu, Z. Zhang, L. Wang, B. I. Yakobson and M. C. Hersam, Intermixing and periodic self-assembly of borophene line defects, Nat. Mater., 2018, 17, 783–788 CAS.
- Y. Wang, L. J. Kong, C. Y. Chen, P. Cheng, B. J. Feng, K. H. Wu and L. Chen, Realization of Regular-Mixed Quasi-1D Borophene Chains with Long-Range Order, Adv. Mater., 2020, 32, 2005128 CAS.
- R. T. Wu, S. Eltinge, I. K. Drozdov, A. Gozar, P. Zahl, J. T. Sadowski, S. Ismail-Beigi and I. Bozovic, Micrometre-scale single-crystalline borophene on a square-lattice Cu(100) surface, Nat. Chem., 2022, 14, 377–383 CAS.
- L. R. Liu, Z. H. Zhang, X. L. Liu, X. Y. Xuan, B. I. Yakobson, M. C. Hersam and W. L. Guo, Borophene Concentric Superlattices via Self-Assembly of Twin Boundaries, Nano Lett., 2020, 20, 1315–1321 CAS.
- R. Wu, I. K. Drozdov, S. Eltinge, P. Zahl, S. Ismail-Beigi, I. Bozovic and A. Gozar, Large-area single-crystal sheets of borophene on Cu(111) surfaces, Nat. Nanotechnol., 2019, 14, 44–49 CAS.
- P. Sutter and E. Sutter, Large-Scale Layer-by-Layer Synthesis of Borophene on Ru(0001), Chem. Mater., 2021, 33, 8838–8843 CAS.
- C. Y. Chen, H. F. Lv, P. Zhang, Z. W. Zhuo, Y. Wang, C. Ma, W. B. Li, X. G. Wang, B. J. Feng, P. Cheng, X. J. Wu, K. H. Wu and L. Chen, Synthesis of bilayer borophene, Nat. Chem., 2022, 14, 25–31 CAS.
- J. Wang, H. Gao, Y. Han, C. Ding, S. Pan, Y. Wang, Q. Jia, H.-T. Wang, D. Xing and J. Sun, MAGUS: machine learning and graph theory assisted universal structure searcher, Natl. Sci. Rev., 2023, 10, nwad128 CAS.
- A. R. Oganov and C. W. Glass, Crystal structure prediction using ab initio evolutionary techniques: principles and applications, J. Chem. Phys., 2006, 124, 244704 Search PubMed.
- S. Kang, W. Jeong, C. Hong, S. Hwang, Y. Yoon and S. Han, Accelerated identification of equilibrium structures of multicomponent inorganic crystals using machine learning potentials, npj Comput. Mater., 2022, 8, 108 Search PubMed.
- X. Shi, D. Cheng, R. Zhao, G. Zhang, S. Wu, S. Zhen, Z.-J. Zhao and J. Gong, Accessing complex reconstructed material structures with hybrid global optimization accelerated via on-the-fly machine learning, Chem. Sci., 2023, 14, 8777–8784 CAS.
- Y. Shen, S. I. Morozov, K. Luo, Q. An and W. A. Goddard Iii, Deciphering the Atomistic Mechanism of Si(111)-7 × 7 Surface Reconstruction Using a Machine-Learning Force Field, J. Am. Chem. Soc., 2023, 145, 20511–20520 CAS.
- C. Schran, F. L. Thiemann, P. Rowe, E. A. Müller, O. Marsalek and A. Michaelides, Machine learning potentials for complex aqueous systems made simple, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110077118 CAS.
- P. Mignon, A. R. Allouche, N. R. Innis and C. Bousige, Neural Network Approach for a Rapid Prediction of Metal-Supported Borophene Properties, J. Am. Chem. Soc., 2023, 145, 27857–27866 CrossRef CAS PubMed.
- L. H. Luo, S. D. Huang, C. Shang and Z. P. Liu, Resolving Activation Entropy of CO Oxidation under the Solid-Gas and Solid-Liquid Conditions from Machine Learning Simulation, ACS Catal., 2022, 12, 6265–6275 CrossRef CAS.
- D. Chen, L. Chen, Q.-C. Zhao, Z.-X. Yang, C. Shang and Z.-P. Liu, Square-pyramidal subsurface oxygen [Ag4OAg] drives selective ethene epoxidation on silver, Nat. Catal., 2024, 7, 536–545 CAS.
- Y. Han, J. Xu, W. Xie, Z. Wang and P. Hu, Comprehensive Study of Oxygen Vacancies on the Catalytic Performance of ZnO for CO/H2 Activation Using Machine Learning-Accelerated First-Principles Simulations, ACS Catal., 2023, 13, 5104–5113 CAS.
- V. L. Deringer, C. J. Pickard and G. Csányi, Data-Driven Learning of Total and Local Energies in Elemental Boron, Phys. Rev. Lett., 2018, 120, 156001 CrossRef CAS PubMed.
- S. D. Huang, C. Shang, P. L. Kang and Z. P. Liu, Atomic structure of boron resolved using machine learning and global sampling, Chem. Sci., 2018, 9, 8644–8655 CAS.
- E. V. Podryabinkin, E. V. Tikhonov, A. V. Shapeev and A. R. Oganov, Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning, Phys. Rev. B, 2019, 99, 064114 CrossRef CAS.
- Q. C. Tong, L. T. Xue, J. Lv, Y. C. Wang and Y. M. Ma, Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface, Faraday Discuss., 2018, 211, 31–43 RSC.
- G. Kresse and J. Hafner, Ab-Initio Molecular-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- P. E. Blochl, P. Margl and K. Schwarz, Ab initio molecular dynamics with the projector augmented wave method, ACS Symp. Ser., 1996, 629, 54–69 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Li, P. Jin, D.-e. Jiang, L. Wang, S. B. Zhang, J. Zhao and Z. Chen, B80 and B101-103 clusters: remarkable stability of the core-shell structures established by validated density functionals, J. Chem. Phys., 2012, 136, 074302 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Shang and Z. P. Liu, Stochastic Surface Walking Method for Structure Prediction and Pathway Searching, J. Chem. Theory Comput., 2013, 9, 1838–1845 CAS.
- C. Shang, X. J. Zhang and Z. P. Liu, Stochastic surface walking method for crystal structure and phase transition pathway prediction, Phys. Chem. Chem. Phys., 2014, 16, 17845–17856 CAS.
- S.-D. Huang, C. Shang, P.-L. Kang, X.-J. Zhang and Z.-P. Liu, LASP: fast global potential energy surface exploration, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1415 CAS.
- H. W. Jung, L. Sauerland, S. Stocker, K. Reuter and J. T. Margraf, Machine-learning driven global optimization of surface adsorbate geometries, npj Comput. Mater., 2023, 9, 114 Search PubMed.
- A. R. Oganov and M. Valle, How to quantify energy landscapes of solids, J. Chem. Phys., 2009, 130, 104504 Search PubMed.
- A. R. Oganov, A. O. Lyakhov and M. Valle, How Evolutionary Crystal Structure Prediction Works-and Why, Acc. Chem. Res., 2011, 44, 227–237 CAS.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Structure prediction drives materials discovery, Nat. Rev. Mater., 2019, 4, 331–348 Search PubMed.
- Y. Wang, H. Lv, X. Wu and J. Yang, First-Principles Particle-Swarm Structure Calculations on Metal Substrate Modulated Monolayer to Bilayer Borophene Crystalline Structures toward Nanoelectronics, ACS Appl. Nano Mater., 2023, 6, 15871–15878 CAS.
- X. L. Liu, Q. C. Li, Q. Y. Ruan, M. S. Rahn, B. I. Yakobson and M. C. Hersam, Borophene synthesis beyond the single-atomic-layer limit, Nat. Mater., 2022, 21, 35–40 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08616c |
| This journal is © The Royal Society of Chemistry 2025 |