
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing transient CAAC-stabilized mesitylborylenes for chalcogen activation†
Maximilian
Michel
ab,
Lukas
Endres
ab,
Felipe
Fantuzzi
 c,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
c,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cSchool of Chemistry and Forensic Science, University of Kent, Park Wood Rd, Canterbury CT2 7NH, UK
First published on 27th February 2025
Abstract
Newly synthesized adducts of CAAC-bound mesitylborylene with carbon monoxide (CO) and trimethylphosphine (PMe3) are established as efficient precursors for the in situ generation of the dicoordinate borylene [(CAAC)BMes] (CAAC = cyclic(alkyl)(amino)carbene), as demonstrated by their ability to activate elemental chalcogens. Upon thermal or photolytic activation, these precursors readily react with sulfur and selenium, yielding boron chalcogenides characterized by terminal boron–chalcogen double bonds. In contrast, the reaction with tellurium leads to the formation of an unusual diradical ditelluride species with a Te–Te bond. Quantum chemical calculations of its electronic structure indicate an open-shell singlet ground state characterized by significant diradical character. Further investigations into the redox behavior of these boron chalcogenides reveal intriguing transformations, including the redox-induced formation and cleavage of E–E bonds.
Introduction
In contrast to carbenes (CR2),1 their group 13 analogues, borylenes (BR),2 which possess only one substituent (R), two vacant orbitals, and a lone pair of electrons, have yet to be isolated in their free form. While established as reactive intermediates with carbene-like reactivity patterns,3,4 borylenes have found only limited applications in synthetic chemistry due to their inherent high reactivity and transient nature. Strategies using transition metals and,5 more recently, Lewis bases have enabled greater control over their reactivity,6 facilitating more selective transformations with substrates. The transfer of borylenes from transition metal complexes to alkynes, forming borirenes,5a,7 and the activation of dinitrogen by Lewis-base-stabilized borylenes exemplify the synthetic potential of these reactive species.8Despite Lewis-base coordination, borylene adducts of the type LBR (L = Lewis base) retain the ambiphilic behavior characteristic of free borylenes, showing reactivity with reagents via both their occupied and vacant orbitals.6,9 A widely adopted strategy for the synthesis of such borylenes involves the chemical reduction of dihaloboranes or haloboryl radicals stabilized by cyclic (alkyl)(amino)carbenes (CAACs).8,10 This approach has already demonstrated its effectiveness in enabling dicoordinate borylenes to activate dinitrogen, as referenced earlier.8 Alternatively, a milder approach to these traditional chemical reduction methods is the in situ generation of CAAC-stabilized borylenes from both CO- and phosphine-bound borylenes under mild conditions.11–13 As illustrated in Scheme 1, the CO ligand in [(CAAC)B(CO)Ar] (A), where Ar is an aryl group, dissociates upon photolysis,11,12 whereas in [(CAAC)B(PMe3)B(NMe2)2] (B), the PMe3 ligand dissociates thermally to release the corresponding borylene.13 Although our group has demonstrated the generation of CAAC-stabilized borylenes using these approaches, their synthetic potential remains largely unexplored.12,13 Notably, the generation of dicoordinate borylenes (LBR) under non-reducing conditions may offer advantages for selective intermolecular transformations. A related approach recently reported by the Cummins and Gilliard groups mirrors the well-established diazo decomposition pathway for generating carbenes.14 The thermal loss of dinitrogen from a diazoborane (C) hereby enables the transfer of a reactive N-heterocyclic carbene (NHC)-stabilized mesitylborylene fragment to suitable substrates. However, in addition to serving as a borylene precursor, the 1,3-dipole character of diazoborane C allows for further reactivity pathways, including cycloaddition reactions with unsaturated substrates such as phenylacetylene.14 The same groups published another thermal approach, involving the release of transient CAAC-stabilized haloborylenes from boranorbornadienes (D, Scheme 1).15 While there is evidence for their in situ generation, their reactivity with substrates has yet to be explored.
 | ||
| Scheme 1 Thermal and photolytic generation of base-stabilized borylenes (L = Lewis base; Ar = Tip (2,4,6-triisopropylphenyl) or Dur (2,3,5,6-tetramethylphenyl); Dipp = 2,6-diisopropylphenyl; X = Br or Cl). | ||
To further illustrate the potential of our approach in transferring the transient dicoordinate borylenes from doubly base-stabilized precursors, we set out to synthesize new phosphine and CO adducts featuring mesityl substituents on the boron center. In this study, we detail new procedures for their preparation and explore their use as borylene precursors for chalcogen activation. Our borylene precursors were shown to readily react with elemental sulfur and selenium, forming stable boron chalcogenides with a boron–chalcogen double bond, proving their ability to act as a source for the elusive dicoordinate species. The unsaturated products formed in these reactions were previously postulated as intermediates in the formation of heterocyclic ring systems in related borylene classes, although isolation and high-yield synthesis have proven challenging.16–19 The reaction with elemental tellurium produced a diradical ditelluride species with a Te–Te single bond, representing the first activation of elemental tellurium by a metal-free borylene. Its unusual electronic structure was thoroughly investigated using multiconfigurational quantum chemical methods. Additionally, analysis of these diverse boron chalcogenides reveals their remarkable redox versatility, characterized by both structural integrity across multiple redox states and participation in redox-induced bond rearrangements at chalcogen centers.
Results and discussion
Synthesis of borylene precursors
The synthesis of the carbon monoxide (CO) and phosphine (PMe3) adducts of the borylene [(CAAC)BMes] (1) follows established methods, beginning with the reduction of CAAC-coordinated dibromo(mesityl)borane (1-Br2, CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene).8e Treating 1-Br2 with 5 equivalents of lithium under CO atmosphere yielded the CO-bound CAAC-mesitylborylene (1-CO) in an excellent yield of 88% (Fig. 1). Its spectroscopic data (δ(11B) = −14.3 ppm; CO stretching frequency = 1947 cm−1) are comparable to that of the duryl derivative (δ(11B) = −13.4 ppm; ν(CO) = 1942 cm−1), indicating a similar bonding situation with significant π-backbonding between the borylene and CO moieties.11 Crystallographic data of 1-CO support this, revealing a short B–CO (1.466(2) Å) and a significantly elongated C–O bond (1.163(2) Å). In contrast to the generation of the closely related duryl derivative using a borylene transfer from an iron complex, the synthesis of transient dicoordinate borylenes through reduction of carbene-stabilized haloboranes is now a well-established method.8,10,17,19 The use of CAAC as a ligand can lead to the formation of varying amounts of the corresponding boryl radical,20 necessitating purification of the crude product, as observed in our case with [(CAAC)B(Mes)Br] (1-Br). | ||
| Fig. 1 Synthesis of the CO (1-CO) and PMe3 (1-PMe3) adducts of CAAC-bound mesitylborylene. Molecular structures are shown with thermal ellipsoids at 50% probability. The thermal ellipsoids for the aryl and alkyl substituents, along with hydrogen atoms, are omitted. Selected bond lengths (Å) and angles (°): 1-CO: C2–O1 1.163(2), B1–C1 1.500(2), B1–C2 1.466(2), N1–C1 1.358(2); O1–C2–B1 171.3(1); 1-PMe3: B1–P1 1.934(1), B1–C1 1.462(2), N1–C1 1.433(1); C1–B1–P1 124.14(9). | ||
Using a similar protocol with potassium graphite (KC8) as the reducing agent, the trimethylphosphine-bound CAAC-mesitylborylene (1-PMe3) can be synthesized in an isolated yield of 44% (Fig. 1). It exhibits two broad 11B (δ = 5.0 and 2.8 ppm) and 31P NMR signals (δ = −27.3, −29.6 ppm), suggesting the formation of two conformers in solution in a ratio of ca. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.13 Attempts to separate the two species were unsuccessful. In our efforts to shift the equilibrium to the thermodynamically favored isomer, we found that 1-PMe3 is thermally labile, noticeably decomposing at temperatures above 40 °C. The thermodynamic product was identified as the PMe3 adduct of the expected intramolecular C–H activation product (δ(11B) = −28.3 ppm, see ESI† for NMR details), likely formed via PMe3 dissociation and the intermediacy of the highly reactive borylene.11 Due to hindered rotation around the boron–carbene bond, the phosphine ligand in solution likely adopts both syn and anti conformations relative to the C–N bond. In the solid state, the structure reveals an anti configuration for the phosphine group. The observed B–P bond length of 1.934(1) Å and the B–CCAAC bond length of 1.462(2) Å, indicative of partial multiple bond character, reflect an electronic structure similar to that of other phosphine-stabilized borylenes.13,21–23 Interestingly, while phosphine-supported borylenes with (pseudo)halide21,22 and boryl ligands13 are known, arylborylene analogues of the form [L2BR] have not been previously reported.
:1.13 Attempts to separate the two species were unsuccessful. In our efforts to shift the equilibrium to the thermodynamically favored isomer, we found that 1-PMe3 is thermally labile, noticeably decomposing at temperatures above 40 °C. The thermodynamic product was identified as the PMe3 adduct of the expected intramolecular C–H activation product (δ(11B) = −28.3 ppm, see ESI† for NMR details), likely formed via PMe3 dissociation and the intermediacy of the highly reactive borylene.11 Due to hindered rotation around the boron–carbene bond, the phosphine ligand in solution likely adopts both syn and anti conformations relative to the C–N bond. In the solid state, the structure reveals an anti configuration for the phosphine group. The observed B–P bond length of 1.934(1) Å and the B–CCAAC bond length of 1.462(2) Å, indicative of partial multiple bond character, reflect an electronic structure similar to that of other phosphine-stabilized borylenes.13,21–23 Interestingly, while phosphine-supported borylenes with (pseudo)halide21,22 and boryl ligands13 are known, arylborylene analogues of the form [L2BR] have not been previously reported.
Chalcogen activation
The growing availability of low-valent boron species in recent years has revealed their rich and diverse reactivity towards chalcogens, leading to the isolation of numerous ring systems, as well as multiply bonded species with boron–chalcogen double bonds.16–18,24 Consequently, we probed the reactivity of 1-CO and 1-PMe3 with chalcogens, extending through to tellurium. Both compounds were anticipated to display reactivity characteristic of the CAAC-stabilized borylene [(CAAC)BMes] (1).The phosphine adduct (1-PMe3) undergoes facile reaction in benzene at room temperature with excess elemental sulfur, yielding thioborane 1-S featuring a boron–sulfur double bond in 44% isolated yield (Scheme 2). Given the mild reaction conditions, an insertion-type mechanism can be envisioned alongside a free borylene intermediate pathway. The CO adduct (1-CO) exhibits similar reactivity under photolysis, also affording the thioborane 1-S in 64% isolated yield. The 11B NMR spectrum of 1-S shows a peak at 63.2 ppm, consistent with chemical shifts of closely related NHC-stabilized derivatives (e.g. [(IMe)(tBu)B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S] δ = 66.4 ppm).16 Single-crystal X-ray diffraction reveals a molecular geometry that is consistent with its thioborane formulation, as evidenced by the short boron–sulfur bond length of 1.735(1) Å, which is comparable to that in analogous compounds (e.g., 1.739(2) Å in [(IMe)(tBu)BS]; Fig. 2).16,25–29 Compound 1-S is remarkably stable in solution, likely due to steric protection and electronic stabilization provided by the CAAC ligand. We pursued alternative pathways for its formation using other sulfur reagents and found that 1-S can be synthesized in two additional ways: from the CO-adduct 1-CO using trimethylphosphine sulfide (Me3PS) and from the dibromo(mesityl)borane precursor 1-Br2 using sodium disulfide (Na2S2, Scheme 2). Both protocols produce compound 1-S in approximately 60% yield.
S] δ = 66.4 ppm).16 Single-crystal X-ray diffraction reveals a molecular geometry that is consistent with its thioborane formulation, as evidenced by the short boron–sulfur bond length of 1.735(1) Å, which is comparable to that in analogous compounds (e.g., 1.739(2) Å in [(IMe)(tBu)BS]; Fig. 2).16,25–29 Compound 1-S is remarkably stable in solution, likely due to steric protection and electronic stabilization provided by the CAAC ligand. We pursued alternative pathways for its formation using other sulfur reagents and found that 1-S can be synthesized in two additional ways: from the CO-adduct 1-CO using trimethylphosphine sulfide (Me3PS) and from the dibromo(mesityl)borane precursor 1-Br2 using sodium disulfide (Na2S2, Scheme 2). Both protocols produce compound 1-S in approximately 60% yield.
 | ||
| Scheme 2 Synthetic routes to the thio- and selenoboranes 1-S and 1-Se. | ||
 | ||
| Fig. 2 Molecular structures of 1-S and 1-Se. Thermal ellipsoids set at 50% probability. For clarity, thermal ellipsoids of the aryl and alkyl substituents, as well as hydrogen atoms, are omitted. Selected bond lengths (Å) and angles (°): 1-S: B1–S1 1.735(1), B1–C1 1.613(2), N1–C1 1.314(1); C1–B1–S1 104.50(9); 1-Se: B1–Se1 1.882(4), B1–C1 1.600(6), N1–C1 1.322(5); C1–B1–Se1 106.8(3). | ||
Encouraged by literature reports of heavier selenium analogs of thioboranes,16,25–27 we adapted the synthetic strategies used for the sulfur derivatives to their synthesis. Gratifyingly, all three synthetic methods extend to the selenium analog (1-Se), with photolysis of the CO adduct (1-CO) in the presence of trimethylsilyl selenide (Me3PSe) affording the highest isolated yield (72%, Scheme 2). Interestingly, the phosphine adduct 1-PMe3 activates elemental selenium in benzene solution at room temperature, affording 1-Se in a moderate yield of 44% after 2 d. The activation process is likely facilitated by the formation of Me3PSe, which is produced as a byproduct of the reaction. Compound 1-Se, a red solid, shows a 11B NMR chemical shift of 70.7 ppm, consistent with a closely related NHC derivative, [(IMe)(tBu)BSe] (δ = 73.5 ppm).16 The 77Se{1H,11B} NMR spectrum displays a singlet at δ = 716.1 ppm. The B–Se bond length (1.882(4) Å) in the solid state aligns well with values typically observed for compounds containing terminal BSe double bonds, which range from 1.871(5) to 1.909(2) Å.16,19,25–27
In previous studies, the activation of chalcogen bonds with borylenes has yielded various outcomes, including BE2 ring structures (E = S and Se) with N-heterocyclic carbene-supported borylenes16 and B2E2 cyclic dimers with CAAC ligands, likely formed via a boron–chalcogen double-bond intermediate.17 While a boron–selenium double bond was successfully formed with a CAAC-stabilized aminoborylene, the analogous sulfur-containing product proved unstable.19
Starting from the compounds 1-CO and 1-PMe3, we also attempted the synthesis of a telluro derivative.26 Stirring a suspension of 1-PMe3 and elemental tellurium in benzene at room temperature for 7 d yielded a blue-green solid in 57% yield. The same product also formed from the UV irradiation (390 nm) of a mixture of 1-CO and tri-n-butylphosphine telluride (nBu3PTe) in benzene for 16 h (Scheme 3). In contrast to our expectations, no 11B NMR signal was observed, and only broad resonances appeared in the 1H NMR spectrum, suggesting the presence of a paramagnetic species. High-resolution mass spectrometry (HRMS) uncovered a species formally derived from the dimerization of the expected terminal telluro derivative. X-ray diffraction analysis revealed that the product comprises two [(CAAC)BMes] units connected by a ditelluride bridge, with a Te–Te distance of 2.7251(3) Å, consistent with a single bond between the tellurium atoms (Fig. 3),30 as observed, for example, in the manganese-coordinated boron ditelluride by our group (2.7541(4) Å).16 The Te–Te bond length and the geometry around the tellurium atoms (Te–Te–B bond angle of 96.74(8)°) are comparable to those observed in diaryl ditellurides R–Te–Te–R.31 The B–Te–Te–B torsion angle of 180.0(1)° reveals a planar arrangement of all four atoms, with the boryl groups positioned trans to each other relative to the Te–Te bond. The B–Te bond length of 2.191(3) Å is comparable to that of boron-centered tellurium radicals, such as in (CAAC)BTePh(B(NMe2)2) (2.199(4)/2.196(4) Å),13 while being notably longer than typical BTe double bonds, as exemplified by [CpMn(CO)2BTe(tBu)(IMe)] (2.100(4) Å).18 The B–CCAAC bond length of 1.507(4) Å indicates significant double bond character and is comparable to that in the radical (CAAC)BTePh(B(NMe2)2), which features bond lengths of 1.504(5) and 1.514(5) Å.13 The structural data of (1-Te)2 thus aligns well with a diradical character of the molecule, where the two unpaired electrons are primarily delocalized across the BCN framework at both sites. Despite its expected radical character, the compound remains virtually EPR silent in both solution and solid state over the temperature range of 10 to 300 K. In the solution EPR spectra of (1-Te)2 in toluene, detectable but very weak resonances are present, suggesting they likely originate from degradation or impurities. In the solid state, only a weak resonance is observed, centered around a g-value of approximately 2.007, which may indicate a thermally populated triplet state (see ESI† for details). However, zero-field splittings or half-field signals, typically associated with such a state, were not observed. To gain deeper insights into the electronic properties of (1-Te)2, we conducted quantum-chemical calculations. Calculations at the DLPNO-NEVPT2/Def2-TZVP//ωB97X-D/Def2-SVP level of theory were performed on the full molecular model of the dimer (1-Te)2 (see ESI† for details). According to these calculations, compound (1-Te)2 has an open-shell singlet ground state, with an energy difference of only 0.34 kcal mol−1 from the corresponding triplet state. The weak antiferromagnetic coupling (2J = −119 cm−1) between the radical sites is associated with a pronounced diradical character, as evidenced by the y0 parameter of 0.88. The spin density is delocalized across the nitrogen (24.0%), carbon (42.7%), and boron atoms (18.9%) on both sides of the molecule, resembling the spin distribution seen in other CAAC-stabilized boryl radicals,20,32 and further extends to include the bridging tellurium atoms (9.7%; see Fig. 3 and SI for details). Although significant triplet population might be expected at room temperature, EPR spectroscopy of (1-Te)2 revealed no conclusive evidence for it. We further investigated the stability of the dimer, particularly in relation to the homolytic cleavage of the tellurium–tellurium bond. Our computations indicate that the rupture of the Te–Te bond and the formation of a terminal boron–tellurium double bond are highly unfavorable. They show that the monomeric form, 1-Te, is thermodynamically significantly higher in energy than the dimer, by approximately 26.3 kcal mol−1. In solution, dimer (1-Te)2 is therefore not expected to dissociate into its monomer 1-Te. For the lighter homologues, the energy relations differ, aligning with the expected group trend of higher π-bond energies. This is reflected in the increasing Mayer bond orders for the boron–chalcogen bond from heavier to lighter: 1.56 for 1-Te, 1.65 for 1-Se, and 1.74 for 1-S.
 | ||
| Scheme 3 Synthesis of (1-Te)2. | ||
 | ||
| Fig. 3 (a) Molecular structure of (1-Te)2. Thermal ellipsoids set at 50% probability. For clarity, thermal ellipsoids of the aryl and alkyl substituents, as well as hydrogen atoms, are omitted. Symmetry-related atoms are labeled with an apostrophe. Selected bond lengths (Å) and angles (°) of (1-Te)2: B–Te 2.191(3), Te–Te 2.7251(3), B–C 1.507(4), N–C 1.384(3); B–Te–Te’ 96.74(8). (b) Spin density plot of (1-Te)2, illustrating regions of positive (green) and negative (blue) spin density. | ||
We further aimed to expand this chemistry by attempting the synthesis of the corresponding oxoborane (i.e.1-O), the lightest congener in the series. To this end, we subjected 1-PMe3 to a series of common oxygenation reagents. Treatment of 1-PMe3 with oxygen (O2), pyridine-N-oxide, nitrous oxide (N2O), and iodosobenzene yielded a single product: trimesityl boroxine (δ(11B) = ca. 31 ppm).33 Neutral compounds featuring a terminal BO double bond, analogous to 1-S and 1-Se, are exceedingly rare in the literature, with only a handful of examples reported.26,29,34
Redox chemistry
Beyond neutral chalcogenoboranes featuring BE double bonds, both anionic and cationic derivatives have been identified.35–38 We therefore sought to determine if charged derivatives of 1-E (E = S, Se, and Te) could also be obtained.
We first investigated the redox chemistry of 1-S. Treatment of 1-S with an excess of lithium (11 equiv.) in THF resulted in a rapid color change from orange to yellow within minutes (Scheme 4). In the 11B NMR spectrum, the signal for the boron atom appears at δ = 28.0 ppm, which is significantly shifted to lower frequency compared to 1-S (δ = 63.2 ppm). Yellow single crystals were obtained in 52% yield from the reaction, enabling the elucidation of the molecular structure of Li2[1-S] (Fig. 4). The solid-state structure reveals a dimeric motif with a Li2S2 core. The two bridging lithium cations exhibit distinct coordination environments: one is solvated by a THF molecule, whilst the other is coordinated to two ipso carbons of the mesityl groups (Li⋯C 2.409(4) and 2.410(4) Å). The remaining two Li cations of the dimer bind to both sulfur and nitrogen atoms and are additionally solvated by a THF molecule. The boron–sulfur bonds are elongated, approaching typical values observed for single bonds (1.905(2) and 1.901(2) Å). In THF solution at −40 °C, two different environments are observed for the lithium cations, with peaks at δ = 1.01 and δ = 0.07. The data suggests a more symmetrical structure in solution, potentially indicative of a solvated monomer (cf.Li2[1-Se]).
 | ||
| Scheme 4 Synthesis of the dilithium salts Li2[1-E] (E = S, Se, and Te) by two-electron reduction of the neutral boron chalcogenides. The Lewis structures of the dianions illustrate their valence electrons. | ||
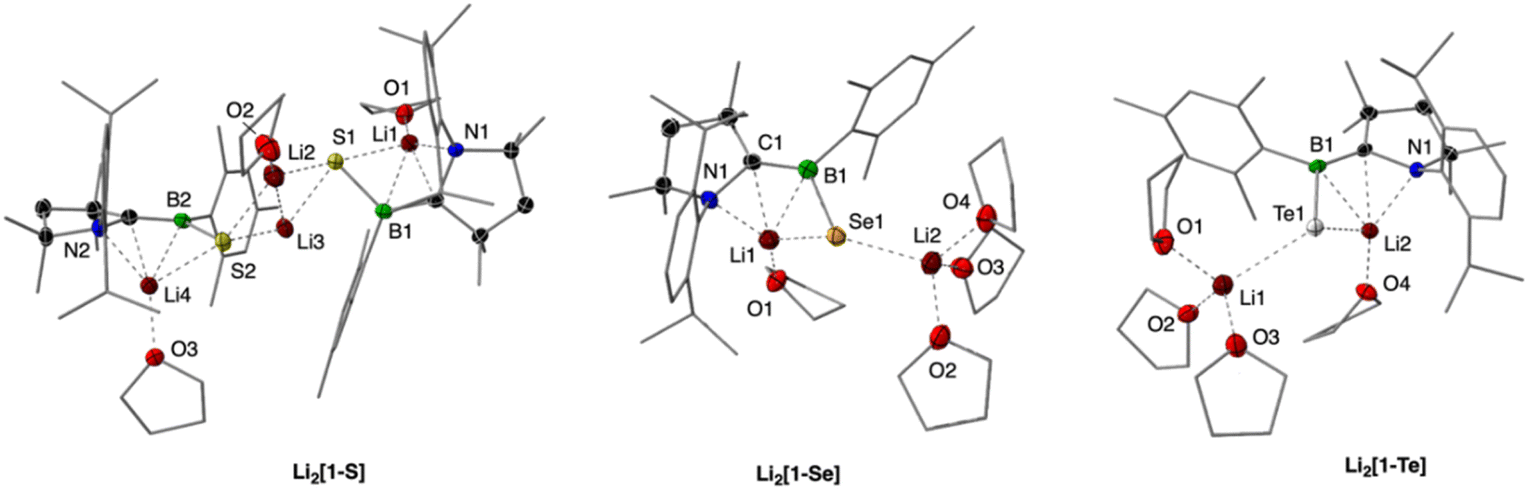 | ||
| Fig. 4 Molecular structures of the dianions Li2[1-S], Li2[1-Se], and Li2[1-Te]. Thermal ellipsoids set at 50% probability. For clarity, hydrogen atoms are omitted, along with the thermal ellipsoids for the substituents and thf molecules. For a comparison of key bond parameters, refer to Table S2 in the ESI.† | ||
By reducing the amount of lithium to five equivalents and shortening the reaction time, we were able to isolate and identify the corresponding radical anion, Li[1-S] (see ESI† for details). Stirring 1-S and lithium in THF for 10 min yielded a red solution, which upon workup afforded red crystals in 30% yield. The lack of NMR signals and the presence of an EPR signal at a g-value of 2.0039 confirmed its radical character. The EPR spectrum of Li[1-S] in toluene exhibits a 1:1:1 triplet, resulting from the interaction of the unpaired electron with the nitrogen atom of the CAAC ligand (a(14N) = 18.1 MHz). Based on the linewidth, the 11B coupling can be approximated to be rather small (a(11B) = 2 MHz), consistent with other CAAC-stabilized boryl radicals.20,32 The solid-state structure of the radical anion shares similar features to Li2[1-S]. Compound Li[1-S] adopts a dimeric structure featuring a central Li2S2 unit, with each lithium cation solvated by two THF molecules (see ESI† for details). At 1.804(2) Å, the B–S bond length is intermediate between the neutral (1.735(1) Å) and dianionic form (1.905(2) and 1.901(2) Å). Comparisons across the three oxidation states reveal a systematic progression of bond lengths within the CAAC unit: the N–C bond elongates from 1.314(1) to 1.394(3) to 1.503(5) Å upon reduction, while the C–B bond conversely contracts from 1.613(2) to 1.535(3) to 1.468(6) Å. For a comprehensive comparison, refer to Table S1 in the ESI.†
Extending the investigation to selenium, reduction of 1-Se with an excess of lithium (12 equiv.) resulted in the formation of the dianionic species Li2[1-Se] (Scheme 4). The reaction yielded yellow crystals of the product in 74% yield. 11B NMR spectroscopy in THF revealed a signal at 24.8 ppm and two distinct 7Li NMR resonances at 1.42 and 0.33 ppm, similar to those observed for the sulfur derivative. The 11B- and 1H-decoupled 77Se NMR spectrum of Li2[1-Se] exhibits a singlet at −428.8 ppm. X-ray diffraction analysis of Li2[1-Se] showed a solvated monomer, in contrast to the dimeric structure of the sulfur derivative (Fig. 4). Both lithium cations coordinate to the selenium atom, but their environments differ: one lithium ion further interacts with the CAAC nitrogen atom and a THF molecule, while the second ion coordinates solely to three THF molecules. The B–Se bond (2.028(3) and 2.033(3) Å for the two independent molecules in the asymmetric unit) is significantly longer than in the neutral species. The observed monomeric structure for Li2[1-Se] is likely adopted by the sulfur derivative in THF solution.
The neutral tellurium compound (1-Te)2, existing as a dimer with a Te–Te single bond, was also exposed to reducing conditions using lithium (Scheme 4). Treatment with an excess of lithium afforded a yellow solution, following an initial blue-green color. From the reaction mixture, yellow crystals were obtained in 58% yield. X-ray crystallography revealed a Te–Te bond cleavage, resulting in a monomeric structure isostructural to the sulfur and selenium derivative (Fig. 4). Compared to the neutral dimer, the B–Te bond in Li2[1-Te] (2.259(2) and 2.260(2) Å for the two independent molecules in the asymmetric unit) is elongated but remains characteristic of a single bond. In solution, the 11B NMR resonance at 17.1 ppm in conjunction with the 125Te NMR chemical shift at −1183.1 ppm support the monomeric structure with a terminal B–Te bond, analogous to the sulfur and the selenium derivatives (see Table S2 in the ESI† for more details).
To gain a deeper understanding of the charge distribution within the dianions Li2[1-E], we employed quantum chemical methods, specifically analyzing natural bond orbital (NBO) charges (see ESI† for details). The computed charge distribution is consistent across all derivatives, with the chalcogen atoms bearing the majority of negative charge (−0.82 to −0.86) and the remaining negative charge distributed evenly between the carbon and nitrogen atoms of the CAAC ligand. Meanwhile, the boron atoms maintain a positive charge, with values between +0.31 and +0.35. This charge distribution is corroborated by the observed lithium coordination environment, where the cations exhibit contacts not only with the chalcogen atoms but also with the carbon and nitrogen atoms of the CAAC ligand (Fig. 4).
Our investigations into the oxidation chemistry of the boron chalcogenides 1-S, 1-Se, and (1-Te)2 yielded intriguing transformations. Unexpectedly, the sulfur derivative (1-S) resisted clean oxidation with ferrocenium hexafluorophosphate, instead forming CAAC-coordinated difluoro(mesityl)borane (1-F2) as a side product. In contrast, the reactions with 1-Se and (1-Te)2 proceeded smoothly, affording the corresponding monocations in good yields (Scheme 5). Treatment of 1-Se with ferrocenium hexafluorophosphate in 1,2-difluorobenzene resulted in an immediate color change to deep purple. The selenium compound [(1-Se)2]PF6 was isolated as purple crystals (70% yield) following crystallization by diffusion of pentane into a 1,2-difluorobenzene solution at −30 °C. X-ray diffraction analysis revealed a dimeric structure with a Se–Se bond, reminiscent of the neutral tellurium compound (see ESI† for details). However, the poor quality of the X-ray diffraction data precludes a detailed structural discussion, restricting the conclusions to atomic connectivity. The dimeric structure of [(1-Se)2]PF6 is further supported by the observed molecular ion peak at m/z = 990.5167 in the high-resolution mass spectrum, exhibiting the characteristic selenium isotope pattern. The EPR spectrum exhibits a broad signal centered at g = 2.0053, devoid of resolved hyperfine couplings.
 | ||
| Scheme 5 Products resulting from one-electron oxidation of 1-Se and (1-Te)2. | ||
Oxidation of the tellurium derivative (1-Te)2 with one equivalent of ferrocenium hexafluorophosphate yielded the blue-green solid [(1-Te)2]PF6 in 62% yield (Scheme 5). The EPR spectrum, recorded in a mixture of toluene and 1,2-difluorobenzene, exhibits a weak, broad signal centered around a g-value of approximately 1.9997 (see ESI† for details). In the solid-state structure, the tellurium–tellurium bond distance of 2.7148(5) Å is only slightly affected by the positive charge, being slightly more contracted than in the neutral form (see ESI† for details). The bond distances around the two boron sites differ significantly, indicating an unequal charge distribution. One unit remains almost unchanged compared to the neutral species, while the boron–tellurium and boron–carbon bond distances in the other unit reflect the effect of one-electron oxidation. The B2–Te2 bond undergoes contraction to 2.152(3) Å, accompanied by a lengthening of the B2–C2 bond to 1.567(4) Å. Furthermore, the C2–N2 bond (1.322(3) Å) in this unit exhibits significant shortening upon oxidation. It can therefore be concluded that spin delocalization between the two radical units through the ditelluride bridge is minimal, despite the nearly planar arrangement of the atoms. This observation is consistent with the pronounced diradical character calculated for (1-Te)2.
Conclusions
This study demonstrates the effectiveness of both CO- and phosphine-bound mesitylborylene adducts as precursors for the generation of the reactive [(CAAC)BMes] borylene. Upon photolytic or thermal activation, these precursors readily react with elemental sulfur and selenium, yielding boron chalcogenides featuring a terminal boron–chalcogen double bond. The reaction with elemental tellurium produced an unusual diradical ditelluride species containing a Te–Te bond, representing a formal dimer of the products obtained with the lighter chalcogens. Its formation is likely due to the strong ability of CAAC to stabilize unpaired electrons and the relatively weaker boron–tellurium double bond strength compared to its lighter homologues. Quantum-chemical methods (i.e. DLPNO-NEVPT2) indicate an open-shell singlet ground state with a pronounced diradical character. Investigations into the redox behavior of these boron chalcogenides revealed intriguing transformations. Notably, while the Te–Te bond remains intact upon oxidation, reduction triggers its cleavage, leading to the formation of a dianionic species featuring a terminal B–Te bond. In contrast, oxidation of the boron selenide results in dimerization, forming a new Se–Se bond and adopting a structure analogous to the tellurium derivative. Overall, the findings highlight the significant potential of CAAC-stabilized borylenes, which are released under mild thermal or photolytic conditions, as versatile reagents for the synthesis of new boron-containing compounds, motivating further investigation.Data availability
The data supporting this article has been included as part of the ESI.† Crystallographic data has been deposited at the CCDC under 2392197–2392206.Author contributions
H. B. conceived and designed the research program. M. M. designed the studies and conducted the research, including crystallographic investigations. L. E. and F. F. performed the DFT calculations. I. K. performed the EPR studies and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Julius-Maximilians-Universität of Würzburg and the Deutsche Forschungsgemeinschaft (grant numbers BR1149/22-2 and 466754611) are acknowledged for financially supporting this work.Notes and references
- For general reviews, see for instance: (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef; (b) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- See, for instance: M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef PubMed.
- (a) P. L. Timms, J. Am. Chem. Soc., 1967, 89, 1629–1632 CrossRef; (b) P. L. Timms, J. Am. Chem. Soc., 1968, 90, 4585–4589 CrossRef CAS; (c) P. L. Timms, Acc. Chem. Res., 1973, 6, 118–123 CrossRef CAS; (d) B. Pachaly and R. West, Angew. Chem., Int. Ed. Engl., 1984, 23, 454–455 CrossRef; (e) W. J. Grigsby and P. P. Power, J. Am. Chem. Soc., 1996, 118, 7981–7988 CrossRef CAS; (f) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS; (g) P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed; (h) D. P. Curran, A. Boussonnière, S. J. Geib and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 1602–1605 CrossRef CAS PubMed; (i) M. Krasowska and H. F. Bettinger, Chem.–Eur. J., 2016, 22, 10661–10670 CrossRef CAS PubMed; (j) K. Edel, M. Krieg, D. Grote and H. F. Bettinger, J. Am. Chem. Soc., 2017, 139, 15151–15159 CrossRef CAS.
- M. Ito, N. Tokitoh, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1999, 40, 5557–5560 CrossRef CAS.
- (a) D. Vidovic, G. A. Pierce and S. Aldridge, Chem. Commun., 2009, 1157–1171 RSC; (b) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 Search PubMed.
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; M. He, C. Hu, R. Wei, X.-F. Wang and L. L. Liu, Chem. Soc. Rev., 2024, 53, 3896–3951 RSC.
- (a) C. E. Anderson, H. Braunschweig and R. D. Dewhurst, Organometallics, 2008, 27, 6381–6389 Search PubMed; (b) J. Wang and Q. Ye, Chem.–Eur. J., 2024, 30, e202303695 CrossRef CAS.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 Search PubMed; (b) M. A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef; (c) M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 1076–1080 CrossRef; (d) R. D. Dewhurst, M.-A. Légaré and H. Braunschweig, Commun. Chem., 2020, 3, 131 CrossRef CAS PubMed; (e) A. Gärtner, U. S. Karaca, M. Rang, M. Heinz, P. D. Engel, I. Krummenacher, M. Arrowsmith, A. Hermann, A. Matler, A. Rempel, R. Witte, H. Braunschweig, M. C. Holthausen and M.-A. Légaré, J. Am. Chem. Soc., 2023, 145, 8231–8241 CrossRef PubMed; (f) M. Rang, M. Heinz, A. Halkić, M. Weber, R. D. Dewhurst, A. Rempel, M. Härterich, M. C. Holthausen and H. Braunschweig, J. Am. Chem. Soc., 2024, 146, 11048–11053 CAS.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef.
- See, for instance: (a) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS; (b) S. Hagspiel, D. Elezi, M. Arrowsmith, F. Fantuzzi, A. Vargas, A. Rempel, M. Härterich, I. Krummenacher and H. Braunschweig, Chem. Sci., 2021, 12, 7937–7942 RSC.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS.
- R. Witte, S. Kar, K. Radacki, M. Härterich, M. Rang, M. Michel, C. Mihm, C. Czernetzki, T. Brückner, E. Beck, S. Lutz, R. D. Dewhurst and H. Braunschweig, Chem. Commun., 2024, 60, 8629–8632 RSC.
- C. Pranckevicius, M. Weber, I. Krummenacher, A. K. Phukan and H. Braunschweig, Chem. Sci., 2020, 11, 11055–11059 RSC.
- C. Zhang, C. C. Cummins and R. J. Gilliard Jr, Science, 2024, 385, 327–331 CrossRef CAS.
- C. Zhang, R. J. Gilliard Jr and C. C. Cummins, Chem. Sci., 2024, 15, 17873–17880 RSC.
- S. Liu, M.-A. Légaré, A. Hofmann and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 11223–11226 CrossRef CAS PubMed.
- D. Auerhammer, M. Arrowsmith, R. D. Dewhurst, T. Kupfer, J. Böhnke and H. Braunschweig, Chem. Sci., 2018, 9, 2252–2260 RSC.
- S. Liu, M.-A. Légaré, D. Auerhammer, A. Hofmann and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 15760–15763 CrossRef CAS PubMed.
- R. Witte, M. Arrowsmith, A. Lamprecht, F. Schorr, I. Krummenacher and H. Braunschweig, Chem.–Eur. J., 2023, 29, e202203663 CrossRef CAS PubMed.
- See, for instance: P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grüne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464–14468 CrossRef CAS PubMed.
- M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Härterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019, 10, 5095–5103 RSC.
- See, also: (a) T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098–4102 CrossRef CAS PubMed; (b) J. Fan, M.-C. Yang, M.-D. Su and C.-W. So, Inorg. Chem., 2023, 62, 863–870 CrossRef CAS.
- (a) H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, C. Schneider and S. Ullrich, Angew. Chem., Int. Ed., 2015, 54, 10271–10275 CrossRef CAS; (b) H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS; (c) S. R. Wang, M. Arrowsmith, J. Böhnke, H. Braunschweig, T. Dellermann, R. D. Dewhurst, H. Kelch, I. Krummenacher, J. D. Mattock, J. H. Müssig, T. Thiess, A. Vargas and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 8009–8013 CrossRef CAS; (d) J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. I. S. Röhr, R. Mitrič, B. Engels, F. Meyer and H. Braunschweig, Nat. Commun., 2018, 9, 1197 CrossRef; (e) A. Stoy, M. Arrowsmith, M. Eyßelein, T. Dellermann, J. Mies, K. Radacki, T. Kupfer and H. Braunschweig, Inorg. Chem., 2021, 60, 12625–12633 CrossRef CAS.
- H. Wang, J. Zhang, H. Hu and C. Cui, J. Am. Chem. Soc., 2010, 132, 10998–10999 CrossRef CAS PubMed.
- H. Dolati, L. Denker, B. Trzaskowski and R. Frank, Angew. Chem., Int. Ed., 2021, 60, 4633–4639 CrossRef CAS.
- K. Jaiswal, B. Prashanth, S. Ravi, K. R. Shamasundar and S. Singh, Dalton Trans., 2015, 44, 15779–15785 RSC.
- C. Chen, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, J. Am. Chem. Soc., 2020, 142, 19763–19771 CrossRef CAS.
- R. Nakano, R. Yamanashi and M. Yamashita, Chem.–Eur. J., 2023, 29, e202203280 CrossRef CAS PubMed.
- The Te single bond covalent radius is 1.36 Å, see: P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef.
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725–1739 RSC.
- See, also: (a) Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS; (b) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC; (c) T. Taniguchi, Chem. Soc. Rev., 2021, 50, 8995–9021 RSC.
- R. Anulewicz-Ostrowska, S. Luliński, J. Serwatowski and K. Suwiñska, Inorg. Chem., 2000, 39, 5763–5767 CrossRef CAS.
- H. Wang, J. Zhang, J. Yang and Z. Xie, Angew. Chem., Int. Ed., 2021, 60, 19008–19012 CrossRef CAS PubMed.
- Y. K. Loh, K. Porteous, M. A. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CrossRef CAS PubMed.
- D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS.
- R. Liu, F. Gao, J. Liu, J. Wei, L. Hou, G. Xie, S. Chen, F. Zeng, A. Li and W. Wang, Dalton Trans., 2021, 50, 6797–6801 RSC.
- T. Heitkemper, L. Naß and C. P. Sindlinger, Angew. Chem., Int. Ed., 2021, 60, 20055–20060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2392197–2392206. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00154d |
| This journal is © The Royal Society of Chemistry 2025 |