
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailored design of an oxygen-rich stable Co-MOF integrated with MXene nanofibers as an advanced heterostructure for high-performance ammonium-ion supercapacitors†
Nissar
Hussain
a and
Shaikh M.
Mobin
 *ab
*ab
aDepartment of Chemistry, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, India. E-mail: xray@iiti.ac.in
bCentre for Advanced Electronics (CAE), Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, India
First published on 14th April 2025
Abstract
Ammonium ions (NH4+) are promising non-metallic charge carriers for sustainable and cost-effective advanced electrochemical energy storage. However, the development of electrode materials with well-defined structural features to facilitate rapid NH4+ diffusion kinetics remains a significant challenge. In this study, we demonstrate the design of a novel oxygen-rich cobalt-based metal–organic framework (Co-MOF) showcasing unique (O4–CoN2) coordination geometry. This distinctive structure of Co-MOF contributes to high stability, abundant active sites, and enhanced electrochemical performance. To further boost performance, Co-MOF nanoflowers were uniformly integrated with Ti3C2Tx MXene carbonized nanofibers (MXCNF), forming advanced Co-MOF@MXCNF heterostructures. These heterostructures exhibit a highly porous, nanofibrous morphology, delivering a notable specific capacitance of 980 F g−1 at a current density of 1 A g−1 and excellent cycling stability, retaining 91.1% capacitance after 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. When paired with a porous MXCNF anode, the ammonium-ion hybrid supercapacitors (AIHSCs) delivered an impressive energy density of 41.5 mW h kg−1 with the corresponding power density of 800 mW kg−1, retaining 87% of their capacitance after 16000 cycles. This study highlights the synergistic advantages of integrating stable MOFs with MXene nanofibers for remarkable ammonium-ion storage. It establishes a framework for designing high-performance energy storage materials, paving the way for next-generation sustainable energy storage devices.
000 cycles. When paired with a porous MXCNF anode, the ammonium-ion hybrid supercapacitors (AIHSCs) delivered an impressive energy density of 41.5 mW h kg−1 with the corresponding power density of 800 mW kg−1, retaining 87% of their capacitance after 16000 cycles. This study highlights the synergistic advantages of integrating stable MOFs with MXene nanofibers for remarkable ammonium-ion storage. It establishes a framework for designing high-performance energy storage materials, paving the way for next-generation sustainable energy storage devices.
1 Introduction
Advanced supercapacitors (SCs) demonstrate significant potential for energy storage applications, attributed to their remarkable power density, long cycling lifespan, and fast charge/discharge capabilities.1 The growing global demand for efficient and sustainable energy storage technologies has propelled research into hybrid supercapacitors, which combine an electrode of the battery type with an electrode of the capacitor type.2,3 Nevertheless, the limited energy density of supercapacitors (SCs) remains a prevailing challenge, often addressed by innovating hybrid supercapacitors (HSCs). Metallic cations (Li+, Na+, K+, Mg2+, and Zn2+) acting as charge carriers are extensively employed in hybrid supercapacitors (HSCs).4 On the other hand, non-metallic ions (e.g., H+, NH4+, F−, and Cl−) as charge carriers are less explored and present distinct advantages, including lower molar mass, smaller hydrated ionic radii, reduced corrosiveness, and improved sustainability.5,6 Among the various ionic species, ammonium ions (NH4+) have emerged as particularly attractive options due to their small hydration radius (3.31 Å), low molar mass (18 g mol−1), abundance, environmental compatibility, and favourable electrochemical properties.7 Utilizing ammonium ions as the charge carrier can enhance the performance of supercapacitors, as they offer rapid ion transport and reversible intercalation into host materials, leading to significant improvements in energy density. These distinctive properties position NH4+ as an exceptional candidate for next-generation high-performance HSCs.Substantial efforts have focused on the development of optimized electrode material-driven energy storage systems, with the primary objectives of achieving high capacity or capacitance and ensuring stable cycling performance.8 To date, vanadium-based oxides (V2O5, NH4V4O10, defective VO2, etc.),9,10 Prussian blue analogs (PBAs), including CuHCF, Na–FeHCF, FeFe(CN)6, etc.,11–13 sulfide-based composites (MoS2/PANI),14 MXene-based composites (NH4V4O10/Ti3C2Tx, PTCDA/Ti3C2Tx MXene, and MXene:CPE (CPE: conjugated oolyelectrolyte)),15–17 MOFs,18,19 and redox-active polymers/COFs, have been employed as electrode materials.20 Despite recent advancements, most reported electrode materials still exhibit limited capacity or capacitance and insufficient structural stability. Consequently, designing novel cathode materials capable of reversible NH4+ storage with enhanced electrochemical performance remains an urgent and critical challenge. Oxygen-rich materials, such as metal oxides (MnO2, MoO3, NH4V4O10, defective VO2, etc.)21–25 and related porous structures (PTCDA, COFs, redox-active polymers, etc.),26–28 have garnered significant attention as advanced electrode materials for ammonium-ion energy storage owing to their unique structural and electrochemical characteristics. These materials feature oxygen-functionalized surfaces that enhance ion–electrode interactions, leading to a high density for ammonium ion (NH4+) adsorption. The oxygen-rich environment facilitates surface redox processes, significantly boosting charge storage capacity and promoting strong pseudocapacitive behavior. The high porosity and large surface area inherent to oxygen-rich materials enable efficient ion diffusion and electrolyte penetration, ensuring fast charge–discharge kinetics while minimizing transport resistance. Additionally, their tunable pore architectures allow precise control over ion accessibility and storage dynamics, providing versatility to meet various energy storage demands. Notably, these materials demonstrate exceptional cycling stability, maintaining their structural integrity and electrochemical performance over extended use.
Herein, we report the design and synthesis of a new oxygen-rich Co-MOF with unique (O4–Co–N2) coordination geometry that enhances the stability of this MOF. Further benefiting from the structural features of the Co-MOF, versatile Co-MOF@MXCNF heterostructure electrodes comprising Co-MOF nanoflowers anchored on Ti3C2Tx MXene carbonized nanofibers (MXCNF) were systematically constructed through a combination of electrospinning, carbonization, and hydrothermal methods. Notably, the well-aligned MXene nanofibers served as a highly conductive core, while the uniformly distributed Co-MOF nanoflowers functioned as the shell layer, providing abundant electrochemically active sites, which encouraged us to develop ammonium-ion hybrid supercapacitors (AIHSCs) via assembly of Co-MOF@MXCNF as the cathode and MXCNF as the anode for excellent performance.
2 Results and discussion
In the present work, Co-MOF was synthesized through a hydrothermal technique at 90 °C using Co(NO3)2·6H2O and 2,2′-bipyridine-4,4′-dicarboxylic acid (DCBPy) as the ligand (Scheme 1). The resulting block-shaped crystal of Co-MOF was characterized by single-crystal X-ray analysis (SCXRD) to investigate the spatial structure and coordination sphere. | ||
| Scheme 1 Schematic representation for the synthesis of Co-MOF. | ||
The single-crystal X-ray diffraction (SC-XRD) studies reveal the formation of metal–organic frameworks, designated as Co-MOF, crystallizing in the trigonal crystal system with the P3121 space group (Table S1†). The asymmetric unit (Fig. S1a†) contains one half-occupied cobalt atom, one half of the DCBPy ligand, and one coordinated water molecule. The Co atom exhibits six-coordination in a N2O4 environment, involving two oxygen atoms from monodentate carboxylate groups of DCBPy, two oxygen atoms from two coordinated water molecules, and two nitrogen atoms from one unique chelating DCBPy ligand. The carboxylate groups are almost co-planar with the connected pyridyl ring and coordinate monodentate to two neighbouring cobalt ions. A higher-symmetry structure is formed due to this symmetrical coordination mode and the more balanced coordination environment surrounding the cobalt centre. This arrangement results in an octahedral coordination geometry, as illustrated in Fig. 1a.
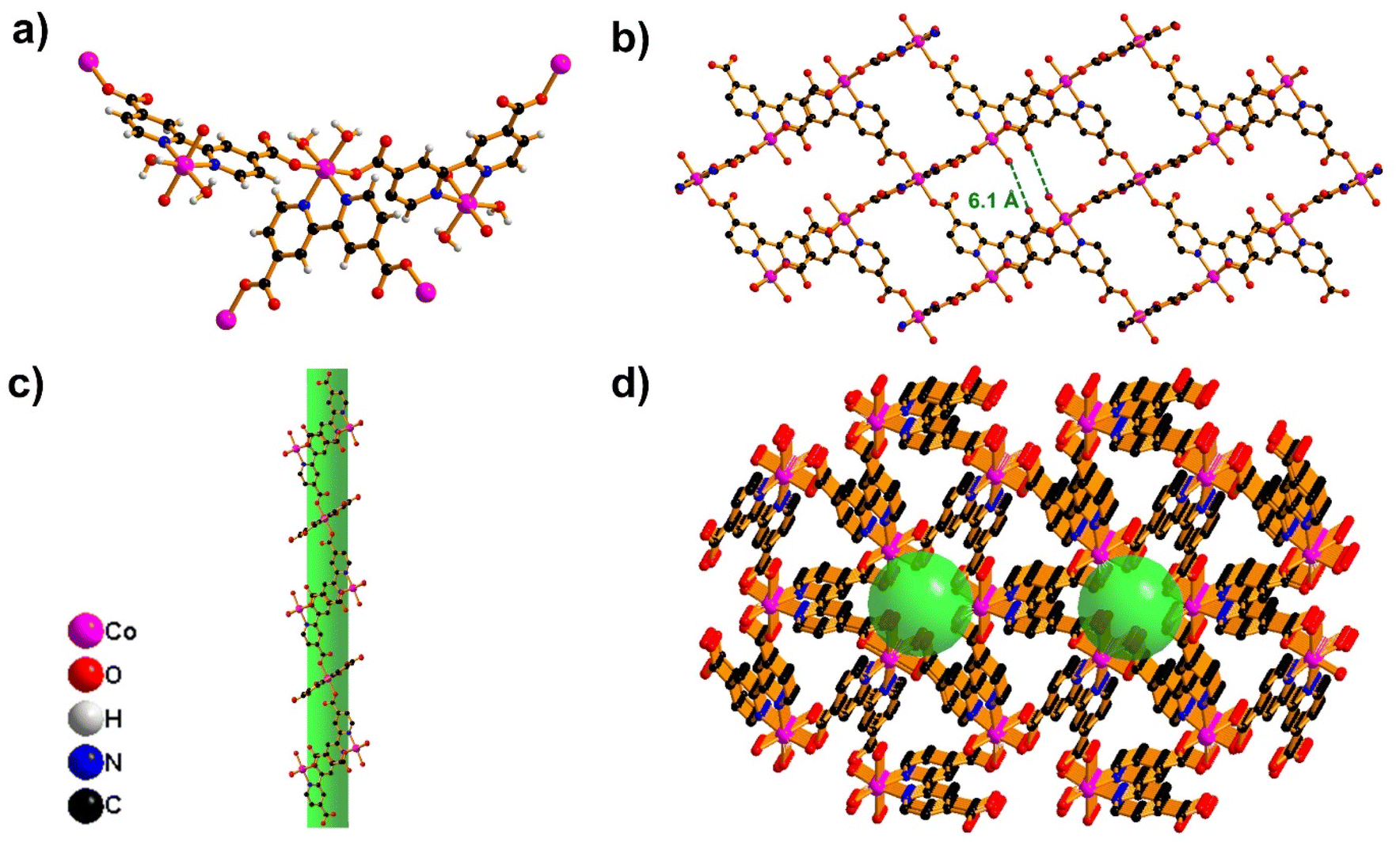 | ||
| Fig. 1 (a) Coordination environment. (b) View of interpenetrated 2D sheets along the b-axis. (c) 1D channels along the c-axis. (d) View showing the decorated cages in the 3D framework. Hydrogen atoms are omitted for clarification. | ||
The DCBPy ligand forms a triangular unit through tris-chelation with three Co atoms, which interacts with neighboring triangular units to create a unique building block characterized by equilateral triangle geometry (Fig. S1b†). These building blocks are organized in an inclined fashion, self-entangling to form an intriguing self-catenated network. As shown in Fig. 1b, four oxygen atoms are present in pores having a 6.1 Å distance that can be used to form hydrogen bonding with the NH4+ ions of electrolytes. The detailed structural analysis of 1-D reveals two distinct types of channels running along the c-axis, marking the first notable structural feature (Fig. 1c). The channel has a cross-section made up of three Co atoms and three 4-carboxy pyridine units from three ligands and is linked to adjacent channels through the sharing of bipyridine moieties. This interlinked channel-by-channel arrangement generates the overall 3D porous network. Fig. 1d, which shows a pore size of 4.1 Å, shows six oxygens from the H2O molecule coordinated to the cobalt ion, which is responsible for the insertion/extraction mechanism of NH4+ ion storage.
The well-arrayed Co-MOF@MXCNF heterostructure, illustrated in Scheme 2, was fabricated using a consecutive three-step procedure. Initially, Ti3C2Tx MXene sheets were prepared using the HF etching and delamination process.9 Further, optimized electrospinning solution was prepared by dissolving Ti3C2Tx MXene (20 wt%) and PAN polymer (10 wt%) in DMF. Ti3C2Tx MXene nanofibers (MXNF) were prepared using the electrospinning technique at a flow rate of 1 mL h−1 and 18 kV voltage. After that, the MXNF were stabilized in the air at 280 °C and carbonized at 800 °C to form stable Ti3C2Tx MXene carbonized nanofibers (MXCNF) leveraging their excellent electrical conductivity and mechanical flexibility.29 In the last step, Co-MOF nanoflowers were synthesized on the MXCNF surface via a hydrothermal method. Further, the synthesis of Co-MOF with MXCNF heterostructures is a promising strategy for enhancing the energy storage performance of ammonium-ion asymmetric supercapacitors. To assess the structural and morphological characteristics of the synthesized materials, a comprehensive suite of characterization techniques was employed.
 | ||
| Scheme 2 Schematic illustration for the synthesis procedures for Co-MOF@MXCNF. | ||
X-ray Diffraction (XRD) analysis was performed to assess the phase composition and crystallinity of the Co-MOF@MXCNF heterostructures. The XRD patterns (Fig. 2a) exhibited sharp peaks characteristic of both the MXene and the Co-MOF, indicating the successful formation of the heterostructures. The prominent peaks associated with MXene indicate that the structural integrity of the MXene framework was preserved throughout the synthesis process. No significant shift in the (00l) peaks was observed between the electrospun fiber mat and powder form of Ti3C2Tx MXene. The (002) peak is centered at 2θ of 6.31° and 6.21° for the fibers and powder, respectively. Notably, the appearance of new diffraction peaks corresponding to the Co-MOF signifies the successful integration of the Co-MOF onto the MXene nanofibers. These peaks were indexed to the specific crystal structure of Co-MOF, further validating the successful synthesis. The absence of significant peak shifts suggests that the MOF synthesis did not adversely affect the MXene structure, which is crucial for maintaining the conductive properties of the electrode material. In the ATR-FTIR (Fourier Transform Infrared-Attenuated Total Reflectance) spectrum, as shown in Fig. S2,† the coordination of the cobalt atom to the DCBPy linker in Co-MOF is evident from the emergence of two distinct bands corresponding to the asymmetric and symmetric stretching vibrations of the coordinated carboxylate at 1610 cm−1 and 1375 cm−1, respectively. Additionally, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of bipyridine at 1550 cm−1 confirms chelation through the nitrogen atom.30 The presence of metal–ligand interactions is further supported by the appearance of stretching vibrations at 400–600 cm−1, providing direct evidence of the coordination environment within the Co-MOF structure.31 In the spectrum of the Co-MOF@MXCNF heterostructure, all characteristic peaks of Co-MOF remain well-matched with those in the pristine Co-MOF, confirming its structural retention within the heterostructures. Importantly, no significant shifts or disappearance of functional group vibrations occur, indicating that the integration of Co-MOF with MXCNF does not alter their intrinsic chemical structures. Instead, a slight broadening and lower intensity of peaks suggest possible interfacial interactions between Co-MOF and MXCNF. This confirms the successful formation of the heterostructure while preserving the integrity of Co-MOF.
N stretching vibration of bipyridine at 1550 cm−1 confirms chelation through the nitrogen atom.30 The presence of metal–ligand interactions is further supported by the appearance of stretching vibrations at 400–600 cm−1, providing direct evidence of the coordination environment within the Co-MOF structure.31 In the spectrum of the Co-MOF@MXCNF heterostructure, all characteristic peaks of Co-MOF remain well-matched with those in the pristine Co-MOF, confirming its structural retention within the heterostructures. Importantly, no significant shifts or disappearance of functional group vibrations occur, indicating that the integration of Co-MOF with MXCNF does not alter their intrinsic chemical structures. Instead, a slight broadening and lower intensity of peaks suggest possible interfacial interactions between Co-MOF and MXCNF. This confirms the successful formation of the heterostructure while preserving the integrity of Co-MOF.
 | ||
| Fig. 2 Characterization of Co-MOF@MXCNF. (a) PXRD patterns. FESEM images of (b) MXCNF and (c–e) Co-MOF@MXCNF at high and low magnification. (f) TEM image of Co-MOF@MXCNF. (g) HRTEM images of Co-MOF@MXCNF (inset: IFFT image). XPS high-resolution spectra of (h) Co 2p and (i) Ti 2p. EDS mapping of Co-MOF@MXCNF: (j) cobalt (Co), (k) oxygen (O), (l) carbon (C) (red), (m) titanium (Ti), and (n) nitrogen (N) elements. | ||
The surface morphology was primarily examined by employing Field Emission Scanning Electron Microscopy (FE-SEM). The morphology of MXene is shown in Fig. S3a and b† exhibiting a sheet-like structure. Fig. 2b confirms the formation of MXCNF by demonstrating that MXene is fully immersed in the PAN nanofibers. Fig. S3c and d† depict the morphology of Co-MOF at different magnifications. Fig. 2c–e reveal a uniform distribution of Co-MOFs on the electrospun MXene nanofiber surface, confirming the successful in situ growth of Co-MOF during synthesis. The electrospun MXene fibers exhibited a distinct fibrous morphology with diameters ranging from 100 to 200 nm, characteristic of electrospinning processes. The Co-MOFs were observed as flower-like nanostructures, which effectively anchored onto the MXene fibers, thereby creating a highly interconnected network. This morphology is particularly advantageous for facilitating ion transport and increasing the performance of the heterostructures. The dense aggregation of MOFs on the fiber surface provides a larger active surface area, which is essential for improving the interaction with ammonium ions during charge–discharge processes. To gain insights into the nanoscale architecture of the synthesized heterostructures, Transmission Electron Microscopy (TEM) analysis was employed for further confirmation of the morphology observed in the SEM analysis, illustrating the intricate arrangement of the flower-like MOF structures on the MXCNF, as shown in Fig. 2f. High-resolution TEM images (Fig. 2g) confirmed the presence of Co-MOF on the MXCNF, illustrating distinct boundaries between the two materials. The lattice fringes observed in the TEM images corresponded to the (100) and (200) planes of the Co-MOF, demonstrating high crystallinity and confirming the successful synthesis of the targeted materials. This intimate contact between the Co-MOF and MXCNF supports efficient charge transfer and ion diffusion pathways, which are vital for improving the overall electrochemical performance of the heterostructure material.
X-ray photoelectron spectroscopy (XPS) was employed to examine the electronic states and elemental composition of the synthesized heterostructures. The XPS spectrum survey (Fig. S4†) of the Co-MOF@MXCNF heterostructure revealed six different peaks at binding energies of 284 eV, 400 eV, 458 eV, 530 eV, 687 eV, and 781 eV, attributed to the C 1s, N 1s, Ti 2p, O 1s, F 1s, and Co 2p elements, respectively. Particularly, the presence of Co 2p and N 1s peaks signifies the characteristic elements of Co-MOF, whereas the Ti 2p, F 1s and O 1s peaks are attributed to MXene, verifying the successful synthesis of the heterostructures. In Fig. 2h, the deconvolution of the Co 2p3/2 spectrum revealed a dominant peak at 781.1 eV, accompanied by a satellite at 785.6 eV. In the Co 2p1/2 region, a primary peak at 797.0 eV was identified, along with a corresponding satellite at 802.6 eV. The notable satellite peaks at 785.6 eV and 802.6 eV are characteristic of high-spin Co2+ ions, confirming the existence of Co2+ in the Co-MOF structure of the heterostructures.32 Deconvoluting the N 1s XPS spectra (Fig. S5a†) revealed peaks for graphitic-nitrogen and pyridinic-nitrogen, with binding energies of 400.2 eV and 399.5 eV, respectively. The deconvolution of four peaks was seen in the C 1s spectra; these peaks corresponded to the C–Ti, C–C, C–N, and CO functions, with their respective binding energies of 281.4 eV, 284.7 eV, 286.2 eV, and 288.1 eV, respectively (Fig. S5b†). Three deconvoluted peaks were visible in the O 1s spectrum (Fig. S5c†) for O–Ti, O2−, C–O, and H2O/OH−, with their respective binding energies of 529.6 eV, 531.3 eV, and 533.0 eV. Ti 2p spectra were deconvoluted into Ti–C, Ti2+, Ti3+, Ti–O, and satellite peaks, as seen in Fig. 2i. The Ti 2p1/2 satellite peaks were located at 454.8 eV, 455.9 eV, 459.2 eV, 461.8 eV, and 464.0 eV, indicative of effective interaction between the MXene nanofibers and MOFs.33 Collectively, the results confirm the growth of Co-MOF on the MXene nanofibers, forming Co-MOF@MXCNF heterostructures. Finally, the chemical composition of the synthesized materials was examined using energy-dispersive X-ray spectroscopy (EDX) for elemental mapping. Fig. 1j–n depict the uniform distribution of cobalt (Co), titanium (Ti), carbon (C), and nitrogen (N) elements in the Co-MOF@MXCNF heterostructures. To elucidate the specific surface area and porosity of the Co-MOF@MXCNF heterostructures, Brunauer–Emmett–Teller (BET) surface area and Barrett–Joyner–Halenda (BJH) pore size distribution analysis was performed (Fig. S6a and b†). The formation of MOF nanoflowers uniformly anchored onto the MXCNF framework resulted in a hierarchically porous three-dimensional (3D) architecture, as confirmed by nitrogen sorption measurements. The specific surface area of Co-MOF@MXCNF was determined to be 106.3 m2 g−1, with an average pore diameter of 3.8 nm, facilitating rapid ion transport and enhanced electrolyte accessibility. In contrast, pristine Co-MOF and MXCNF exhibited lower surface areas of 57.13 and 44.4 m2 g−1, with pore diameters of 3.4 nm and 17.6 nm, respectively. The notable enhancement in surface area arises from the effective dispersion of Co-MOF nanoflowers on MXCNF, which prevents MOF aggregation and introduces additional porosity. Moreover, the decrease in pore size can be attributed to the partial infiltration of MOF nanoparticles into the larger pores of MXCNF, leading to a more uniform and interconnected mesoporous network. Additionally, the synergistic effect of the intrinsic porosity MOF and the conductive MXCNF matrix ensures a high electroactive surface area, maximizing ammonium-ion storage sites while maintaining rapid ion diffusion kinetics. The well-integrated MOF@MXCNF heterostructure thus provides a structurally robust, high-surface-area platform conducive to efficient ion intercalation, electrochemical stability, and high-rate charge storage performance.
3 Electrochemical studies
The Co-MOF@MXCNF heterostructure, exhibiting abundant redox-active sites and a large specific surface area, is a favorable candidate for energy storage applications. The electrochemical characteristics were studied in 2 M (NH4)2SO4 electrolytes using a three-electrode system. Cyclic voltammetry (CV) tests were conducted to assess electrochemical kinetics as shown in Fig. S7a.† The CV curve of the Co-MOF electrode exhibited a distinct redox peak pair having a potential range of 0 to 0.8 V. In comparison, the Co-MOF@MXCNF electrode displayed redox peaks with greatly increased current responses, which are explained by the high utilization efficiency and better electronic conductivity. On increasing scan rates from 2 to 100 mV s−1, the distinctive CV curves remained consistent, indicating a quick electrochemical response and superior electrochemical redox performance of the Co-MOF@MXCNF heterostructure (Fig. 3a). The redox peaks observed in the CV curves of the MOF@MXCNF electrode arise due to the intercalation and deintercalation of ammonium ions (NH4+), leading to reversible redox transitions.34,35 The redox peaks seen in the CV curves closely correspond to the virtually symmetrical shapes with subtle plateaus seen in the galvanostatic charge–discharge (GCD) curves of the Co-MOF@MXCNF electrode. When compared to the individual Co-MOF electrode GCD curves (Fig. S7b†), the Co-MOF@MXCNF electrode exhibits a markedly longer discharge time, signifying improved capacitance that arises from the synergistic effects of its two constituent components. The energy storage capacity of the Co-MOF@MXCNF electrode was further confirmed by GCD experiments carried out at different current densities between 0 and 0.8 V (Fig. 3b). The observed low coulombic efficiency at a low current density of 1 A cm−2 and the large irreversible capacity loss in the Co-MOF@MXCNF heterostructures can be attributed to the deep intercalation and partial trapping of NH4+ ions within the MOF structure, which hinders complete deintercalation during discharge, leading to significantly lower coulombic efficiency. Furthermore, water molecules from the (NH4)2SO4 electrolyte can also intercalate into the MOF, contributing to structural stabilization but not directly participating in charge storage.36Fig. 3c presents the relationship between specific capacitance and current density for the synthesized electrodes. Notably, the specific capacitance of the Co-MOF@MXCNF electrode from the GCD curves reaches 980 F g−1 at a current density of 1 A g−1. Additionally, as the current density is increased to 30 A g−1, the Co-MOF@MXCNF electrode demonstrates remarkable rate capability, exhibiting a capacitance retention of 64.5%. The observed high capacitance in our material surpasses that of recently reported electrode materials as shown in Table S2.† This substantial enhancement in capacitance can largely be attributed to harnessing the synergetic effects in Co-MOF@MXCNF. The integrated structure facilitates efficient ion transport, allowing rapid ammonium ion intercalation and desorption during charge and discharge cycles. The Nyquist plots further reveal negligible resistance, with a low charge transfer resistance (Rct) of 12.7 Ω in Co-MOF@MXCNF and 22.3 Ω in the Co-MOF electrodes (Fig. 3d). | ||
| Fig. 3 Electrochemical performance of Co-MOF@MXCNF: (a) CV curves of the Co-MOF@MXCNF electrode were recorded at various scan rates. (b) GCD curves of the Co-MOF@MXCNF electrode at different current densities. (c) The specific capacitance of the Co-MOF@MXCNF electrode vs. current density. (d) EIS. (e) logi plot of current (i) versus scan rate (v) for the Co-MOF@MXCNF electrode within the 2 to 100 mV s−1 scan rate range. (f) Deconvolution of the CV curve of the Co-MOF electrode at a scan rate of 40 mV s−1 into capacitive (pink) and diffusion-controlled contributions. (g) The variation of normalized capacitive (pink) and diffusion-controlled (blue) contributions as a function of scan rate. (h) Long-term cycling performance measured at 10 A g−1 for 16000 cycles, with the inset presenting the GCD profiles for the initial and final ten cycles. | ||
To further interpret the storage mechanism of the Co-MOF@MXCNF heterostructures in the ammonium-comprising electrolyte, the b-values for the two redox peaks were determined using eqn (1) and (2).37,38
| i = avb | (1) |
| logi = logi + blogv | (2) |
i versus logv plot, and i and v stand for the peak current and scan rate, respectively, in this context. A diffusion-controlled process is indicated by a b-value of 0.5, whereas a redox reaction governed by surface interactions is suggested by a b-value of 1.
As illustrated in Fig. 3e, the obtained b-value is 0.71 for the Co-MOF@MXCNF electrode, suggesting a dominant capacitive behavior. In the ammonium ion-containing electrolyte, the Co-MOF@MXCNF electrode exhibits a hybrid mechanism that combines diffusion and capacitive effects. Using eqn (3), the contribution ratios of the diffusion-controlled and capacitive components are then calculated.
| i = k1v + k2v1/2 | (3) |
Furthermore, at a current density of 10 A g−1, the cycle stability of the Co-MOF@MXCNF electrode was evaluated (Fig. 3h). With minimally appreciable coulombic efficiency loss, the specific capacitance retained 91.2% of its initial value after 16000 charge/discharge cycles. This performance highlights the exceptional cycling stability of the Co-MOF@MXCNF electrode, which surpasses that of most cathode materials already reported. The synthesis of Co-MOF on electrospun MXene nanofibers presents a promising approach to enhancing the performance of ammonium ion hybrid supercapacitors. To understand the NH4+ ion storage mechanism, ex situ ATR-FTIR (Fourier Transform Infrared-Attenuated Total Reflectance) spectroscopy was recorded to analyze the interactions between NH4+ ions and the Co-MOF@MXCNF heterostructure. The spectrum shown in Fig. S8a† provides insights into the interaction of NH4+ with the MOF@MXCNF heterostructure, confirming the structural stability and charge storage behavior. NH4+ forms strong hydrogen bonds with the oxygen-containing functional groups of the Co-MOF@MXCNF heterostructure, stabilizing the intercalation process and ensuring high reversibility during charge–discharge cycles.39 The spectrum in the 2900–3500 cm−1 region clearly shows peaks corresponding to the N–H–O and N–H stretching vibrations. During charging, NH4+ ions intercalate into Co-MOF@MXCNF, forming hydrogen bonds with oxygen sites, as evidenced by the N–H stretching vibration at 3250 cm−1, indicative of hydrogen bonding interactions. A broad peak centered at 2930 cm−1 (Fig. S8b†), corresponding to H-bonded N–H, exhibits increased intensity, further confirming the intercalation process. Upon discharging, NH4+ ions partially deintercalate, while a fraction remains within the structure, as indicated by the persistence of N–H–O and NH-stretching vibrations in the range of 2900–3500 cm−1 with low intensity. The incomplete deintercalation suggests a stabilizing effect of residual NH4+ ions, similar to previous observations in multivalent ion storage systems.40,41 Further confirmation is provided by the bending vibrations of δ(OH) at 1420 cm−1 and δ(NH) at 1610 cm−1, which exhibit a peak shift and intensity variation, suggesting enhanced hydrogen bonding due to ammonium incorporation. Similarly, δ(NH) and δ(NH) (Fig. S8c and d†) show a distinct shift, indicating strong interactions between NH4+ and the host framework. After discharging, the peak remains almost the same as in the initial structure, demonstrating the structural stability of the host material even after ion intercalation and deintercalation.42,43 This behavior underscores the importance of understanding the electrochemical and structural dynamics of NH4+ storage in the Co-MOF@MXCNF electrode.
Finally, a hybrid supercapacitors (HSCs) utilizing ammonium ions was assembled with a Co-MOF@MXCNF cathode and MXCNF anode in a 1 M (NH4)2SO4/PVA gel electrolyte, as illustrated in Fig. 4a. Considering the potential windows of the Co-MOF@MXCNF electrode (0 to 0.8 V) and the MXCNF electrode (−0.8 to 0 V), the maximum voltage range achievable is 0 to 1.6 V for the Co-MOF@MXCNF//MXCNF AIHSCs (Fig. 4b). To ensure charge balance between the Co-MOF@MXCNF cathode and the MXCNF anode, the optimal mass ratio was determined to be 1:3. Fig. 4c exhibits the CV curves of the Co-MOF@MXCNF//MXCNF asymmetric ion hybrid supercapacitors (AIHSCs) at 5 to 100 mV s−1 scan rates. Remarkably, even at elevated scan rates, minimal polarization is observed up to 1.6 V, underscoring the superior electrochemical reversibility of the Co-MOF@MXCNF//MXCNF AIHSCs. Additionally, the GCD profiles exhibit distinct plateaus at current densities from 1 to 15 mA g−1, exhibiting closely matching features in the cyclic voltammetry curves (Fig. 4d). At 1 mA g−1 current density, the Co-MOF@MXCNF//MXCNF asymmetric ion hybrid supercapacitors (AIHSCs) achieve a high specific capacitance of 116.8 mF g−1. Fig. 3d illustrates the relationship between capacitance and current density for Co-MOF@MXCNF//MXCNF AIHSCs. Additionally, when the current density increases to 10 mA g−1, the fabricated AIHSCs exhibit commendable rate capability, retaining 62.2% of their capacitance (Fig. 4e) indicating rapid ion diffusion and charge transfer. Interestingly, fibrous and nanoflower morphologies of MXene and Co-MOF greatly improve ammonium ion diffusion, specifically within the interconnected pore channels, which act as the primary conduits for ammonium ion insertion and efficient diffusion across the layered networks. To investigate the effectiveness of the Co-MOF@MXCNF//MXCNF asymmetric ion hybrid supercapacitors (AIHSCs), Fig. 4f presents the Ragone plots in comparison to those of other reported AIHSCs. The device achieves a maximum energy density of 41.5 mW h kg−1 at a power density of 800 mW kg−1, outperforming many currently reported AHSCs. Remarkably, even at a power density of 12000 mW g−1, the full device retains an energy density of 20.6 mW h g−1. When compared to other previously reported supercapacitors (Table S3†), such as δ-MnO2//ACC (861.2 μW h cm−2),44 α-MnO2//(NH4)xWO3 (1010.1 μW h cm−2),45 MnO2/CNTF//MoS2@TiN/CNTF (195.1 μW h cm−2),46 ACC@VPP//PTCDI (320 μW h cm−2),47 Cu-HHB/I2//P-MXene (31.5 mW h cm−2),48 PMO//MnO2 (2.48 mW h cm−2),49 MnO2@ACC//WO3@ACC (508.1 μW h cm−2),50 and h-Ti3C2 MXene//MnO2 CNTs (394.59 μW h cm−2),51 the Co-MOF@MXCNF//MXCNF AIHSCs demonstrate superior energy and power characteristics. At 10 mA g−1 current density, the longstanding cycling stability of the Co-MOF@MXCNF//MXCNF AIHSCs was evaluated. Surprisingly, the devices maintained 87.0% of their original capacitance after 16000 cycles, and their coulombic efficiency remained close to 90% during the tests (Fig. 4g inset: first ten and last ten GCD cycles). Furthermore, to demonstrate practical significance, two asymmetric devices were connected in series and utilized to power a 2 V red light-emitting diode (LED) for five minutes (Fig. 4g, inset). This application highlights the practical viability of the assembled AIHSCs device, and the compelling electrochemical results indicate the potential use of Co-MOF@MXCNF//MXCNF for next-generation electronics.
 | ||
| Fig. 4 Electrochemical performance of Co-MOF@MXCNF//MXCNF AIHSCs. (a) Schematic illustration of the Co-MOF@MXCNF//MXCNF AHSCs. (b) CV curves of the Co-MOF@MXCNF cathode and MXCNF, at a scan rate of 100 mV s−1. (c) CV profiles at scan rates ranging from 5 to 100 mV s−1. (d) GCD profiles at various current densities between 2 and 15 mA g−1. (e) Specific capacitances as a function of current density. (f) Ragone plots comparing the performance of Co MOF@MXCNF//MXCNF AHSCs with that of other reported hybrid supercapacitors (HSCs). (g) Long-term cycling performance of Co-MOF@MXCNF//MXCNF AHSCs at a current density of 10 mA g−1 over 16000 cycles, with the inset showing GCD profiles for the initial and final ten cycles, along with a glowing LED demonstration. | ||
4 Conclusion
In summary, we designed and synthesized a novel oxygen-rich Co-MOF featuring a unique coordination geometry, which ensures high structural stability and abundant active sites for ammonium-ion storage. By integrating Co-MOF nanoflowers with MXCNF, we constructed advanced heterostructures that exhibit exceptional electrochemical properties, including a high specific capacitance of 980 F g−1 at 1 A g−1 and outstanding cycling stability, retaining 91.1% of the initial capacitance after 16000 cycles. When paired with a porous MXCNF anode, the Co-MOF@MXCNF heterostructures enabled the development of ammonium-ion hybrid supercapacitors (AIHSCs) with a high energy density of 41.5 mW h kg−1 and excellent long-standing performance, retaining 87% capacitance after extensive cycling. These results demonstrate the synergy of MOF structural stability and heterostructural integration with MXene for enhanced ammonium-ion storage. This work highlights the immense potential of MOF-based materials as cutting-edge candidates for advanced energy storage applications and provides a strategic approach for designing high-performance hybrid supercapacitors, paving the way for sustainable, next-generation energy storage technologies.
5 Experimental section
5.1 Synthesis of Co-MOF
24.4 mg (0.01 mmol) of 2,2′-bipyridine-4,4′-dicarboxylic acid (DCBPy) and 58.2 mg (0.02 mmol) of cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) were dissolved in 3 mL DMAc/H2O (2:1) in a 20 mL Teflon-lined autoclave, which was sealed and maintained at 100 °C for 48 h in an isothermal oven before cooling to ambient temperature. The resultant Co-MOF crystals were thoroughly washed with methanol and deionized water and dried at 80 °C for 12 h under vacuum.
5.2 Preparation of MXCNF mats
By dissolving 1 g of polyacrylonitrile (PAN, MW 150000) in dimethylformamide (DMF), the electrospinning solution was prepared. MXene (Ti3C2Tx) was incorporated at 20 wt% relative to the weight of polyacrylonitrile (PAN). This mixture underwent 10 hours of combined ultrasonic agitation and stirring to make uniform dispersion and complete dissolution of MXene within the PAN solution. Electrospinning was then performed under optimized conditions, applying an 18 kV voltage, a flow rate of 1 mL h−1, and a 15 cm distance between the spinneret and collector to produce a uniform Ti3C2Tx MXene/PAN fiber membrane. The as-spun MXene/PAN membrane was then subjected to a two-step thermal treatment: initial pre-oxidation in the air at 250 °C for 2 h to stabilize the fibers, followed by carbonization under an argon atmosphere at 800 °C for 2 h, yielding carbonized MXene nanofibers (MXCNF).
5.3 Synthesis of Co-MOF@MXCNF membranes
To prepare Co-MOF@MXCNF membranes, 58.2 mg (0.1 mmol) of cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) and 24.4 mg (0.01 mmol) of 2,2′-bipyridine-4,4′-dicarboxylic acid (DCBPy) were dissolved in 6 mL DMAc and 3 mL H2O in a 20 mL Teflon-lined autoclave. A 1 × 2 cm2 MXCNF membrane was then immersed in the above solution, which was sealed and maintained at 100 °C for 18 h in an isothermal oven before cooling to ambient temperature. To get rid of any unreacted precursors, the resulting Co-MOF@MXCF membranes were extensively cleaned using methanol and deionized water. The Co-MOF loading on the MXCNF membranes averaged about 2 mg cm−2, which suggests uniform coating and sufficient material coverage.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
N. H. conceptualized the work, performed all the experiments, and wrote the original draft of the manuscript. S. M. M. supervised the project and revised the manuscript. Both authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. M. acknowledges the SERB-DST Project (CRG/2020/001769), NTTM, Ministry of Textile, New Delhi, India, and IIT Indore for financial support. The authors gratefully acknowledge the Sophisticated Instrumentation Centre (SIC), IIT Indore. We sincerely acknowledge AMRC, IIT Mandi, for the XPS facility, Sophisticated Analytical Instrument Facility, and IIT Bombay for HR-TEM analysis. N. H. expresses gratitude to MOTA for providing a fellowship.References
- S. V. Sadavar, S.-Y. Lee and S.-J. Park, Advanced Science, 2024, 11, 2403172 CrossRef PubMed
.
- D. P. Chatterjee and A. K. Nandi, J. Mater. Chem. A, 2021, 9, 15880–15918 RSC
- A. Muzaffar, M. B. Ahamed, K. Deshmukh and J. Thirumalai, Renewable Sustainable Energy Rev., 2019, 101, 123–145 CrossRef CAS
- Y. Wang, R. Chen, T. Chen, H. Lv, G. Zhu, L. Ma, C. Wang, Z. Jin and J. Liu, Energy Storage Mater., 2016, 4, 103–129 CrossRef
- M. S. Javed, T. Najam, I. Hussain, M. Idrees, A. Ahmad, M. Imran, S. S. A. Shah, R. Luque and W. Han, Adv. Energy Mater., 2023, 13, 2202303 CrossRef CAS
- J. Xu, Y. Liu, C. Xu, J. Li, Z. Yang, H. Yan, H. Yu, L. Yan, L. Zhang and J. Shu, Coord. Chem. Rev., 2023, 474, 214867 CrossRef CAS
- J. Han, A. Varzi and S. Passerini, Angew. Chem., Int. Ed., 2022, 61, e202115046 CrossRef CAS PubMed
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Angew. Chem., 2022, 134, e202204351 CrossRef
- S. Dong, W. Shin, H. Jiang, X. Wu, Z. Li, J. Holoubek, W. F. Stickle, B. Key, C. Liu, J. Lu, P. A. Greaney, X. Zhang and X. Ji, Chem, 2019, 5, 1537–1551 CAS
- H. Li, J. Yang, J. Cheng, T. He and B. Wang, Nano Energy, 2020, 68, 104369 CrossRef CAS
- L. Chen, W. Sun, K. Xu, Q. Dong, L. Zheng, J. Wang, D. Lu, Y. Shen, J. Zhang, F. Fu, H. Kong, J. Qin and H. Chen, ACS Energy Lett., 2022, 7, 1672–1678 CrossRef CAS
- Z. Zhao, W. Zhang, M. Liu, S. J. Yoo, N. Yue, F. Liu, X. Zhou, K. Song, J.-G. Kim, Z. Chen, X.-Y. Lang, Q. Jiang, C. Zhi and W. Zheng, Nano Lett., 2023, 23, 5307–5316 CrossRef CAS PubMed
- X. Zhang, M. Xia, H. Yu, J. Zhang, Z. Yang, L. Zhang and J. Shu, Nano-Micro Lett., 2021, 13, 139 CrossRef CAS PubMed
- J. Dai, C. Yang, Y. Xu, X. Wang, S. Yang, D. Li, L. Luo, L. Xia, J. Li, X. Qi, A. Cabot and L. Dai, Adv. Mater., 2023, 35, 2303732 CrossRef CAS PubMed
- S. G. Krishnan, C. Padwal, X. Wang, H. D. Pham, S. Aberoumand, N. Motta, K. (Ken) Ostrikov and D. Dubal, J. Mater. Chem. A, 2024, 12, 8244–8253 RSC
- K. Niu, J. Shi, L. Zhang, Y. Yue, S. Mo, S. Li, W. Li, L. Wen, Y. Hou, L. Sun, S. Yan, F. Long and Y. Gao, Adv. Sci., 2024, 11, e2305524 CrossRef PubMed
- C. Chen, G. Quek, H. Liu, L. Bannenberg, R. Li, J. Choi, D. Ren, R. J. Vázquez, B. Boshuizen, B. Fimland, S. Fleischmann, M. Wagemaker, D. Jiang, G. C. Bazan and X. Wang, Adv. Energy Mater., 2024, 14, 2402715 CrossRef CAS
- M. Gao, Z. Wang, Z. Liu, Y. Huang, F. Wang, M. Wang, S. Yang, J. Li, J. Liu, H. Qi, P. Zhang, X. Lu and X. Feng, Adv. Mater., 2023, 35, 2305575 CrossRef CAS PubMed
- Y. Huang, M. Gao, Y. Fu, J. Li, F. Wang, S. Yang, M. Wang, Z. Qian, X. Lu, P. Zhang and R. Wang, Energy Storage Mater., 2024, 70, 103522 CrossRef
- Y. Zhang, Y. An, B. Yin, J. Jiang, S. Dong, H. Dou and X. Zhang, J. Mater. Chem. A, 2019, 7, 11314–11320 RSC
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, 2107992 CrossRef CAS PubMed
- J. Dai, X. Qi, L. Xia, Q. Xue, L. Luo, X. Wang, C. Yang, D. Li, H. Xie, A. Cabot, L. Dai and Y. Xu, Adv. Funct. Mater., 2023, 33, 2212440 CrossRef CAS
- Q. Chen, H. Li, X. Lou, J. Zhang, G. Hou, J. Lu and Y. Tang, Adv. Funct. Mater., 2023, 33, 2214920 CrossRef CAS
- Y. Wu, S. Dong, N. Lv, Z. Xu, R. Ren, G. Zhu, B. Huang, Y. Zhang and X. Dong, Small, 2022, 18, 2204888 CrossRef CAS PubMed
- M. Shi, Y. Zhang, X. Zhou, Y. Li, S. Xiao, S. Liu, R. Yao, H. Tan, Y. Wang and Y. Li, Chem. Eng. J., 2023, 464, 142600 CrossRef CAS
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204351 CrossRef CAS PubMed
- Q. Liu, D. Zhang, Y. Yang, Y. Gu, Z. Liang, W. Chen, Y. Wu and L. Hu, Adv. Energy Mater., 2024, 2402863 Search PubMed
- Z. Tian, V. S. Kale, Y. Wang, S. Kandambeth, J. Czaban-Jóźwiak, O. Shekhah, M. Eddaoudi and H. N. Alshareef, J. Am. Chem. Soc., 2021, 143, 19178–19186 CrossRef CAS PubMed
- H. Xu, B. Li, Z. Wang, Q. Liao, L. Zeng, H. Zhang, X. Liu, D.-G. Yu and W. Song, J. Mater. Chem. A, 2024, 12, 22346–22371 RSC
- M.-H. Zeng, Z. Yin, Y.-X. Tan, W.-X. Zhang, Y.-P. He and M. Kurmoo, J. Am. Chem. Soc., 2014, 136, 4680–4688 CrossRef CAS PubMed
- G. Kumar, R. Haldar, M. Shanmugam and R. S. Dey, J. Mater. Chem. A, 2023, 11, 26508–26518 RSC
- S. Gutiérrez-Tarriño, J. L. Olloqui-Sariego, J. J. Calvente, G. M. Espallargas, F. Rey, A. Corma and P. Oña-Burgos, J. Am. Chem. Soc., 2020, 142, 19198–19208 CrossRef PubMed
- T. Kshetri, D. D. Khumujam, T. I. Singh, Y. S. Lee, N. H. Kim and J. H. Lee, Chem. Eng. J., 2022, 437, 135338 CrossRef CAS
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204351 CrossRef CAS PubMed
- Y. Sun, B. Yin, J. Yang, Y. Ding, M. Li, H. Li, J. Li, B. Jia, S. Zhang and T. Ma, Energy Environ. Sci., 2023, 16, 5568–5604 RSC
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, 2107992 CrossRef CAS PubMed
- Q. Yang, Q. Wang, Y. Long, F. Wang, L. Wu, J. Pan, J. Han, Y. Lei, W. Shi and S. Song, Adv. Energy Mater., 2020, 10, 1903193 CrossRef CAS
- N. Hussain, Z. Abbas, K. Nabeela and S. M. Mobin, J. Mater. Chem. A, 2024, 12, 17642–17650 RSC
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, 2107992 CrossRef CAS PubMed
- G. Liang, Y. Wang, Z. Huang, F. Mo, X. Li, Q. Yang, D. Wang, H. Li, S. Chen and C. Zhi, Adv. Mater., 2020, 32, 1907802 CrossRef CAS PubMed
- Q. Chen, Z. Tang, H. Li, W. Liang, Y. Zeng, J. Zhang, G. Hou and Y. Tang, ACS Appl. Mater. Interfaces, 2024, 16, 18824–18832 CrossRef CAS PubMed
- Y.-Z. Zhang, J. Liang, Z. Huang, Q. Wang, G. Zhu, S. Dong, H. Liang and X. Dong, Advanced Science, 2022, 9, 2105158 CrossRef CAS PubMed
- T. Xiao, C. Tang, H. Lin, X. Li, Y. Mei, C. Xu, L. Gao, L. Jiang, P. Xiang, S. Ni, Y. Xiao and X. Tan, Inorg. Chem., 2024, 63, 17714–17726 CrossRef CAS PubMed
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, 2107992 CrossRef CAS PubMed
- Q. Chen, M. Song, X. Zhang, J. Zhang, G. Hou and Y. Tang, J. Mater. Chem. A, 2022, 10, 15614–15622 RSC
- L. Han, J. Luo, R. Zhang, W. Gong, L. Chen, F. Liu, Y. Ling, Y. Dong, Z. Yong, Y. Zhang, L. Wei, X. Zhang, Q. Zhang and Q. Li, ACS Nano, 2022, 16, 14951–14962 CrossRef CAS PubMed
- X. Chen, P. Wang, Z. Feng, C. Meng and Y. Zhang, Chem. Eng. J., 2022, 445, 136747 CrossRef CAS
- M. Gao, Z. Wang, Z. Liu, Y. Huang, F. Wang, M. Wang, S. Yang, J. Li, J. Liu, H. Qi, P. Zhang, X. Lu and X. Feng, Adv. Mater., 2023, 35, 2305575 CrossRef CAS PubMed
- Q. Chen, H. Li, X. Lou, J. Zhang, G. Hou, J. Lu and Y. Tang, Adv. Funct. Mater., 2023, 33, 2214920 CrossRef CAS
- Z. Han, X. Zhang, G. Tang, J. Liang and W. Wu, J. Energy Storage, 2024, 91, 112023 CrossRef
- J. Shi, K. Niu, L. Zhang, Q. Chen, M. Deng, L. Sun, S. Cheng and Y. Gao, ACS Energy Lett., 2024, 9, 2000–2006 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2402755. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00171d |
| This journal is © The Royal Society of Chemistry 2025 |