
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation, total synthesis and structure determination of antifungal macrocyclic depsipeptide, tetraselide†
Hiroki
Nakahara
 ,
Goh
Sennari
,
Haruki
Azami
,
Hayama
Tsutsumi
,
Yoshihiro
Watanabe
,
Yoshihiko
Noguchi
,
Yuki
Inahashi
,
Masato
Iwatsuki
,
Tomoyasu
Hirose
* and
Toshiaki
Sunazuka
*
,
Goh
Sennari
,
Haruki
Azami
,
Hayama
Tsutsumi
,
Yoshihiro
Watanabe
,
Yoshihiko
Noguchi
,
Yuki
Inahashi
,
Masato
Iwatsuki
,
Tomoyasu
Hirose
* and
Toshiaki
Sunazuka
*
Ōmura Satoshi Memorial Institute, Graduate School of Infection Control Sciences, Kitasato University, 5-9-1 Shirokane, Minato-ku, Tokyo 108-8641, Japan. E-mail: thirose@lisci.kitasato-u.ac.jp; sunazuka@lisci.kitasato-u.ac.jp
First published on 4th March 2025
Abstract
Macrocyclic peptides, including depsipeptides, are an emerging new modality in drug discovery research. Tetraselide, an antifungal cyclic peptide isolated from a marine-derived filamentous fungus, possesses a unique amphiphilic structural feature consisting of five consecutive β-hydroxy-amino acid residues and fatty acid moieties. Because the structure elucidation of the naturally occurring product left six stereocenters ambiguous, we implemented bioinformatic analyses, chemical degradation studies and chiral pool fragment synthesis to identify two of the undetermined stereocenters. Convergent total synthesis of the four remaining plausible isomers of tetraselide was accomplished via liquid-phase peptide synthesis (LPPS) using soluble hydrophobic tag auxiliaries. The key advances involve fragment coupling by the serine/threonine ligation (STL) reaction and head-to-tail macrolactamization of the carrier-supported precursors that enabled systematic elaboration of the amphiphilic cyclic peptides. Ultimately, we determined the absolute structure of this natural product.
Introduction
Over the last decade, cyclic peptides have drawn much attention in drug discovery efforts as an attractive modality due to their unique properties such as high binding affinity and selectivity targeting protein–protein interactions (PPIs) with low toxicity.1 Thus, cyclic peptides can potentially serve as advantageous therapeutics complementary to antibodies and small drug molecules.2 Although cell permeability and oral bioavailability are generally considered to be challenging,3 there is emerging research that N-methyl amides4 and/or depsipeptides5 enhance membrane permeation.6 Despite chemical accessibility of this class of peptide molecules, the vast majority of therapeutic cyclic peptides have been developed based on the structures of natural products. Therefore, exploration of new cyclic peptides and depsipeptides from natural sources plays an important role as a foundation of drug leads in medicinal chemistry.Our research group has been on a quest for novel secondary metabolites of microorganisms from underexplored natural sources.7 During the course of our screening program to explore antifungal natural products,8 we isolated a macrocyclic depsipeptide, tetraselide (1, Fig. 1A), from a culture broth of Trichoderma sp. FKJ-0225, a marine-derived filamentous fungus. As a result of NMR spectroscopic and LC-MS/MS analyses (see the ESI†), we identified six polar amino acids, Orn, Thr, Ser, Ser, Ser, Ser, two hydrophobic Ala and Gly residues, and a β-hydroxy-γ-methyl fatty acid in the structure of 1.9 The five consecutive β-hydroxy-amino acid residues represent a unique structural feature that has rarely been seen in natural products.10 The advanced Marfey method11 to analyze the absolute configuration of amino acids revealed the presence of L-Ala, L-Orn, and D-allo-Thr. The four consecutive Ser moieties were found to be a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of L- and D-isomers, although the position of the D-Ser residue in the sequence remained ambiguous. Consequently, because six stereocenters of 1 including the C3 and C4 positions of the β-hydroxy-γ-methyl fatty acid moiety could not be determined, chemical synthesis was required to fully characterize the absolute structure of the natural product 1.
:1 mixture of L- and D-isomers, although the position of the D-Ser residue in the sequence remained ambiguous. Consequently, because six stereocenters of 1 including the C3 and C4 positions of the β-hydroxy-γ-methyl fatty acid moiety could not be determined, chemical synthesis was required to fully characterize the absolute structure of the natural product 1.
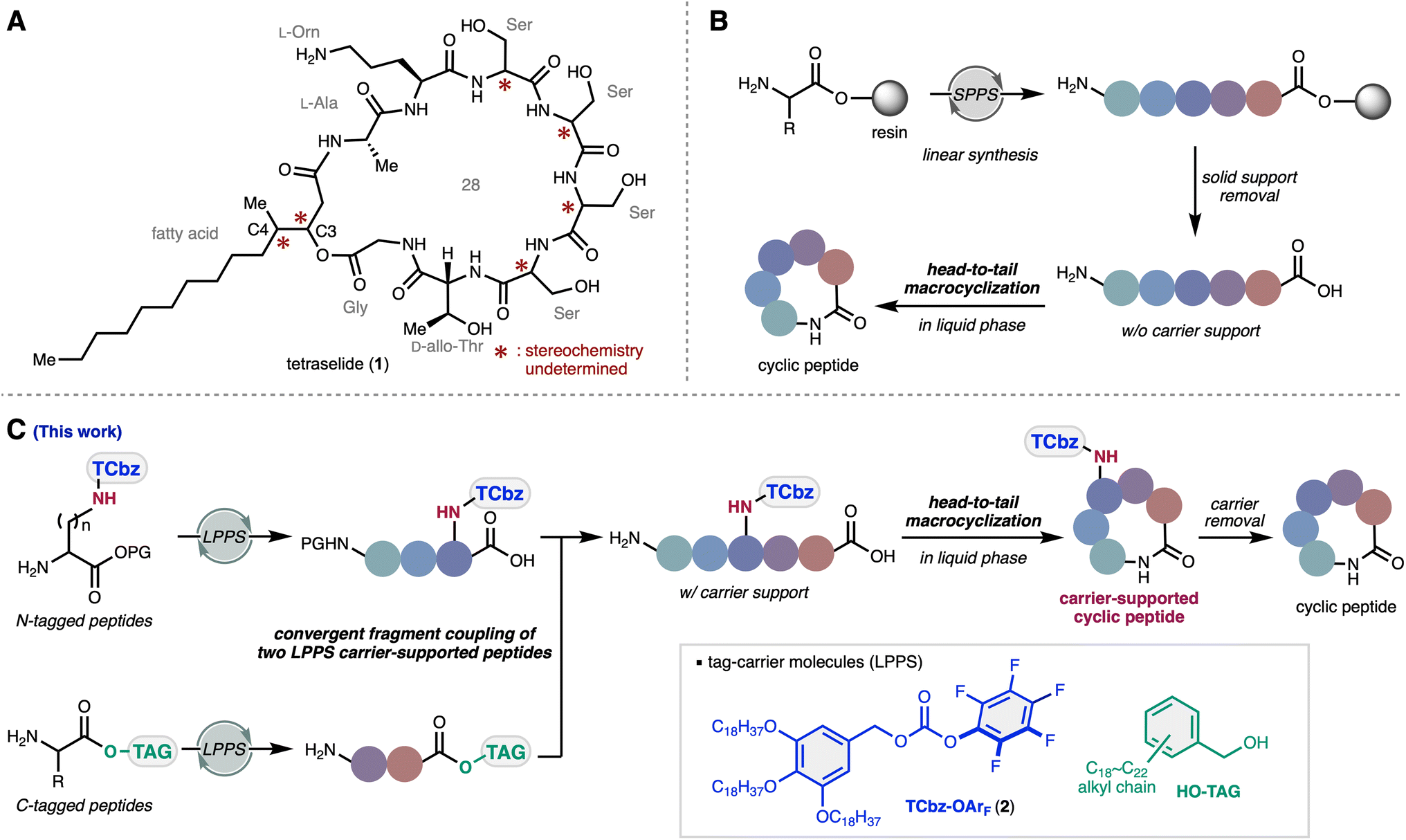 | ||
| Fig. 1 Natural product tetraselide and peptide synthesis strategy. (A) The structure of tetraselide. (B) Typical SPPS strategy for cyclic peptides. (C) Our convergent strategy to synthesize macrocyclic peptides. SPPS: solid-phase peptide synthesis. LPPS: liquid-phase peptide synthesis. PG: protecting group. | ||
Since the invention of the Merrifield resin,12 solid-phase peptide synthesis (SPPS) has become one of the most common methods to elaborate peptide compounds (Fig. 1B).13 Although SPPS serves as a robust and reliable method, excess amounts of reagents and coupling partners are often required to achieve sufficient reactivity due to the heterogeneous nature of the reaction. As such, the convergent synthetic approach, which requires the preparation of peptide fragments through their isolation/purification over several steps, is generally considered to be less efficient than the more commonly employed SPPS strategies.14 Despite this, several scalable approaches for peptide fragment coupling in solution have been developed.15 Additionally, head-to-tail macrocyclization on the solid-phase requires side chain anchoring strategies or removal from the solid-phase carrier necessitates a solution phase cyclization.16 The solubility and polarity of peptides without the carrier support sometimes result in troublesome handling, particularly with amphiphilic and zwitterionic peptides.17
As an alternative approach to SPPS, our group has been interested in liquid-phase peptide synthesis (LPPS)18 using soluble hydrophobic tags.19 We applied this method to the total syntheses of argifin,20 kozupeptin,21 verticilide,22 and emodepside derivatives.23 Recently, we also developed a carbonate-type tag reagent TCbz-OArF (2) that enabled attachment of the tag-carrier to the amine group of amino acids (Fig. 1C).24 Orthogonal to the typical approach that relies on installation of the carrier molecule to the C-terminus, we thought to develop a de novo synthetic strategy using the tag-reagent 2. Herein, we report the convergent total synthesis and structure determination of tetraselide (1) via LPPS utilizing two tag-carrier molecules. The homogeneous reaction conditions of LPPS enabled convergent fragment coupling of two carrier-supported peptides in equimolar amounts. Because the TCbz group was installed on the amine group of the Orn side chain, we successfully synthesized head-to-tail macrocyclic peptides supported on a carrier molecule. This strategy facilitated systematic convergent syntheses of all plausible isomers of the four structurally ambiguous consecutive Ser moieties and allowed us to determine the structure of 1.
Results and discussion
Biosynthetic analysis, chemical degradation and chiral pool synthesis to elucidate stereochemistry
As described earlier, our preliminary efforts for the structural elucidation of tetraselide (1) suggested that there were 16 possible diastereomers—four of the consecutive Ser moieties and four of the fatty acid moiety. Our failure to clarify the structure of 1 using Edman degradation25 and X-ray crystallographic analysis led us to implement biosynthetic analysis to narrow down the structural candidates of 1. First, from the draft genome sequence of the FKJ-0225 strain, we found five genes (ttsA to ttsE, ESI Fig. S12A,† accession number: LC847109) associated with the biosynthesis of 1 by mining a candidate biosynthetic gene cluster using antiSMASH 7.0 (ref. 26) and 2ndfind.27 This indicated that the β-hydroxy-γ-methyl fatty acid moiety produced by the polyketide synthase (PKS; ttsA) is transferred to the non-ribosomal peptide synthase (NRPS; ttsE). Subsequent peptide elongation from L-Ala to Gly, followed by macrolactonization at the C-terminus of Gly with the hydroxy group at C3, furnishes tetraselide (ESI Fig. S12B†). To predict the position of the D-Ser residue, Clabofold28 and AutoDock29 were used to conduct docking simulations of the adenylation (A) domains in the modules that construct the consecutive Ser moiety. Consequently, all A domains are likely responsible for the recognition of only L-Ser (ESI Fig. S13†) and the corresponding epimerization (E) domains are not involved, suggesting that L-Ser might be epimerized at a condensation (C) domain in the modules.30 As a result of phylogenic analysis of the C domains (ESI Fig. S14†), we speculated that the Ser residue next to Thr might be the D-form, although the mechanism of epimerization is unclear at this stage (see the ESI† for details).We expected that the stereocenters of the β-hydroxy-γ-methyl fatty acid moiety in 1 would be predictable because the absolute stereochemistry of compounds produced by PKSs relies on the amino acid sequence of each PKS domain. As a result of the bioinformatic analysis (ESI Fig. S15†), we found that the keto reductase (KR) domain in the PKS was classified as the B-type, which generally reduces the ketone group to a D-hydroxy group.31 In addition, we identified that the Tyr residue—one of the proton donors in the enoyl reductase (ER) domain—is substituted with Leu, suggesting that the methyl group would likely be in the D-configuration.32 These results indicated that the β-hydroxy-γ-methyl fatty acid moiety in 1 could be in the (3S)-hydroxy and (4R)-methyl configurations. To support this hypothesis, we next attempted a chemical degradation study of the naturally occurring 1 and synthesis of the degraded fragment using a chiral pool approach.
Our degradation experiments of 1 began with hydrolysis of the ester moiety by treatment with 2 M NaOH, yielding linear peptide 3 (Scheme 1A). Peptide 3 was subjected to 6 M HCl at 100 °C to hydrolyze the amide bonds, affording fatty acid 4 as the major component after acid extraction. In order to facilitate UV detection for purification by reverse-phase HPLC, selective benzylation of the acid moiety in 4 provided ester 5 in 10% overall yield over four steps from 1. With successfully degraded alcohol 5 in hand, we next chemically synthesized fatty acid derivatives to confirm the absolute stereochemistry.
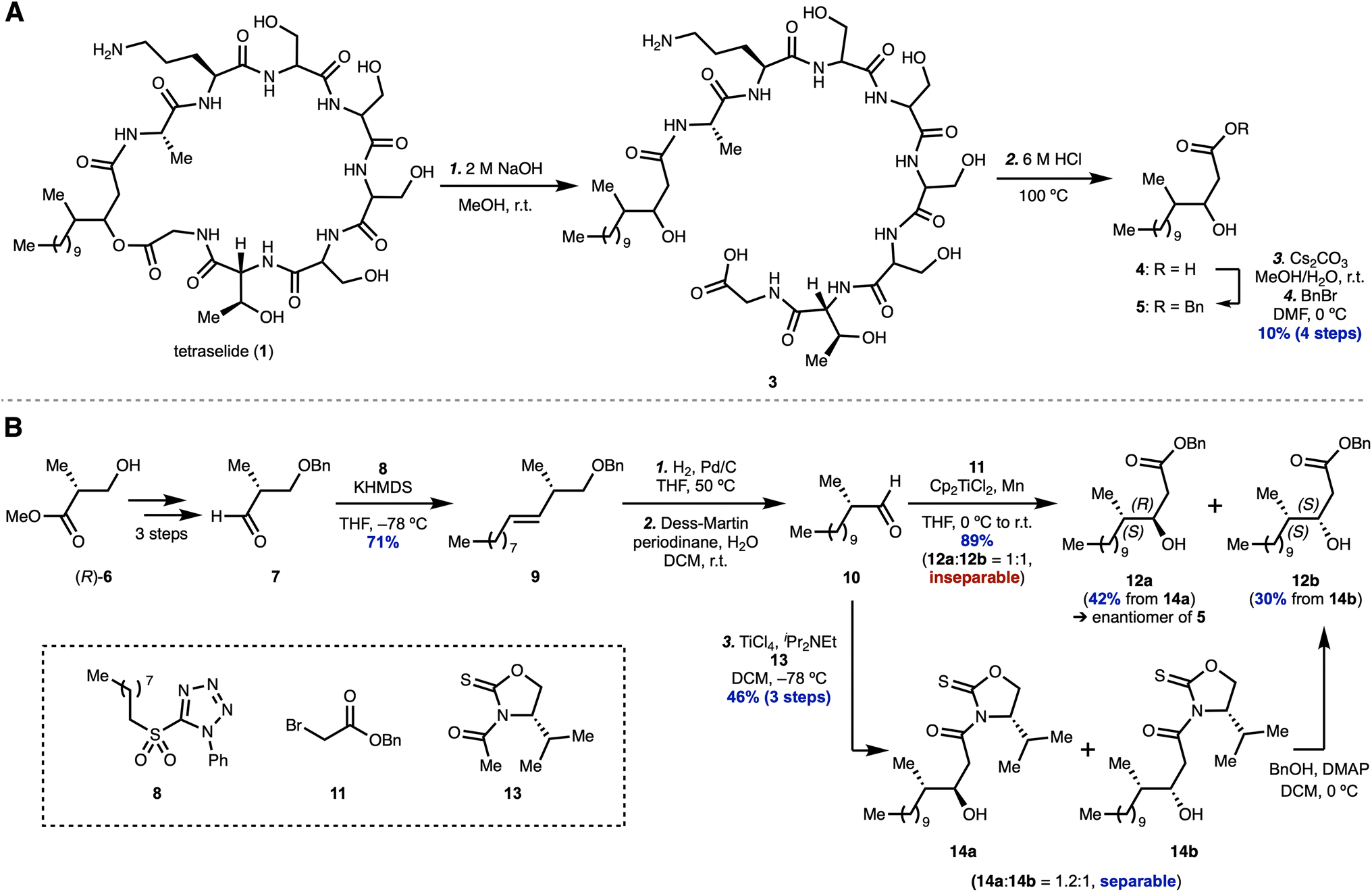 | ||
| Scheme 1 Initial experimentations to support our hypothesis through bioinformatic analysis. (A) Chemical degradation of the naturally occurring product. (B) Chiral pool synthesis of fatty acids. | ||
In order to implement enantiospecific synthesis for the structural determination of 5, we employed a chiral pool starting material. We began our investigation with Roche ester (R)-6 because the (S)-enantiomer is more expensive and less available (Scheme 1B). Following the known three-step sequence to produce aldehyde 7,33 Julia–Kocienski olefination34 of 7 using PT-sulfone 8 afforded alkene 9 in 71% yield as a single isomer. Hydrogenation of the double bond and simultaneous hydrogenolysis of the benzyl group in 9, which was followed by Dess–Martin oxidation, produced aldehyde 10. Reformatsky reaction of 10 using bromoacetate 11 gave rise to an inseparable 1:1 mixture of diastereomers (12a and 12b) in 89% yield over three steps. Fortunately, during our investigation to achieve a diastereoselective aldol reaction, we found that treatment of 10 with chiral auxiliary 13 using TiCl4 in the presence of iPr2NEt35 afforded the desired adducts 14a and 14b, which were easily separable by column chromatography. After separating the diastereomers, benzyl esterification of 14a and 14b using BnOH and DMAP provided alcohols 12a (42% yield) and 12b (30% yield), respectively. The stereochemistry of the hydroxy groups in 12a and 12b was determined by the modified Mosher method (ESI Fig. S18†).36
A comparison of the 1H NMR spectra for the synthesized alcohols 12a and 12b with degraded 5 revealed that the methyl and hydroxy groups in the natural form are in the anti-configuration. Because the optical rotation of 12a was opposite to that of 5, we assigned the absolute configuration of the β-hydroxy-γ-methyl fatty acid moiety in tetraselide (1) as (3S, 4R). This result is consistent with our bioinformatic analysis of the PKS of 1. Nevertheless, we narrowed down the plausible structure of natural product 1 to four isomers in the consecutive Ser residues. With this invaluable information, we commenced the total synthesis of all four isomers.
Synthetic strategy
To determine the structure of tetraselide (1), we envisioned a convergent route that could efficiently synthesize all four plausible isomers that differed in the position of the D-isomer in the four consecutive Ser moieties (Scheme 2). In this regard, natural product 1 could be divided into two carrier-supported peptide fragments (17 and 18) by disconnection at the C-terminus of Orn and the N-terminus of Thr. In the forward sense, head-to-tail macrolactamization of 16 at the Thr–Ser moiety after cleavage of the C-terminal tag would forge the 28-membered macrocycle in 15. We expected that the side chain bound TCbz group would allow us to assemble the macrocyclic peptides in a concise fashion by solidification, followed by global deprotection to furnish tetraselide and its constitutional isomers.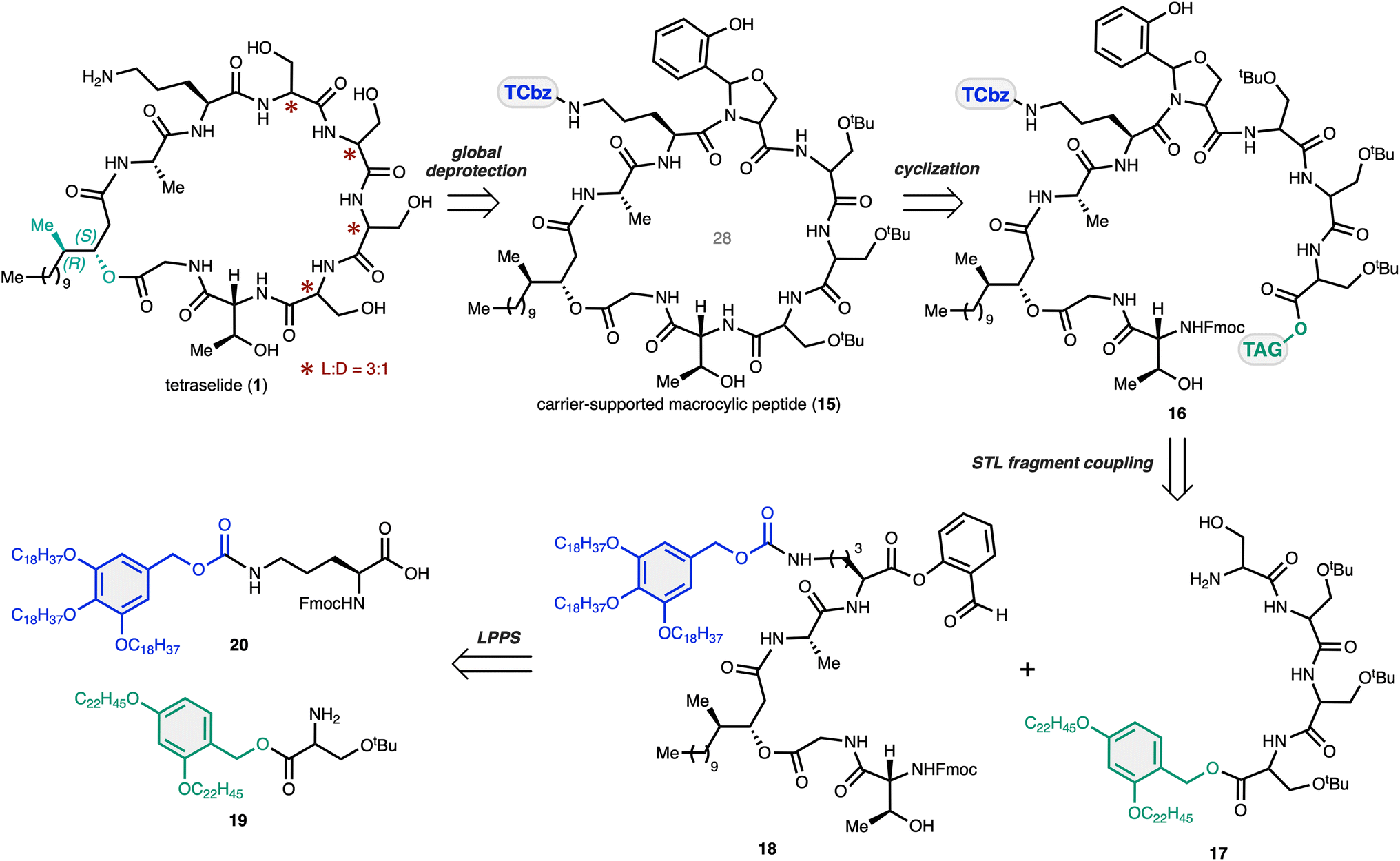 | ||
| Scheme 2 Retrosynthesis of tetraselide using two carrier supported peptides for convergent assembly in LPPS. | ||
To suppress problematic epimerization via oxazolone formation at the C-terminus for convergent fragment coupling37 and to promote facile macrocyclization, we thought to synthesize the macrocyclization precursor 16 using the serine/threonine ligation (STL) reaction38 between the N-terminus of the four consecutive Ser fragment 17 and salicylaldehyde ester 18. In this way, Ser fragment 17 could be prepared without protection of the hydroxy group at the N-terminal side chain through one-pot LPPS23 using tag-supported Ser 19. The western fragment 18 could be synthesized by peptide elongation from TCbz-supported Orn 20, which we developed previously.24
Western fragment synthesis
The western fragment synthesis commenced with diastereoselective elaboration of the fatty acid moiety in the desired enantioenriched form (Scheme 3A). The aldol reaction of the known ketone 22,39 prepared from (R)-lactate 21 over three steps, with aldehyde 23 under the conditions reported by Paterson40 gave rise to the desired anti-diastereomer 24 in excellent yield and selectivity (d.r. = 20:1). Protecting group manipulations and oxidative cleavage afforded aldehyde 25, which was subjected to the Julia–Kocienski olefination condition using PT-sulfone 8, providing alkene 26 in 80% yield as a single isomer. Simultaneous hydrogenation of the double bond and hydrogenolysis of the benzyl group in 26 gave diol 27 in 89% yield, followed by selective oxidation of the primary alcohol in 27 to the corresponding carboxylic acid, affording 28 in >99% yield. In this way, the desired fatty acid 28 was prepared in 10 steps from commercially available 21.
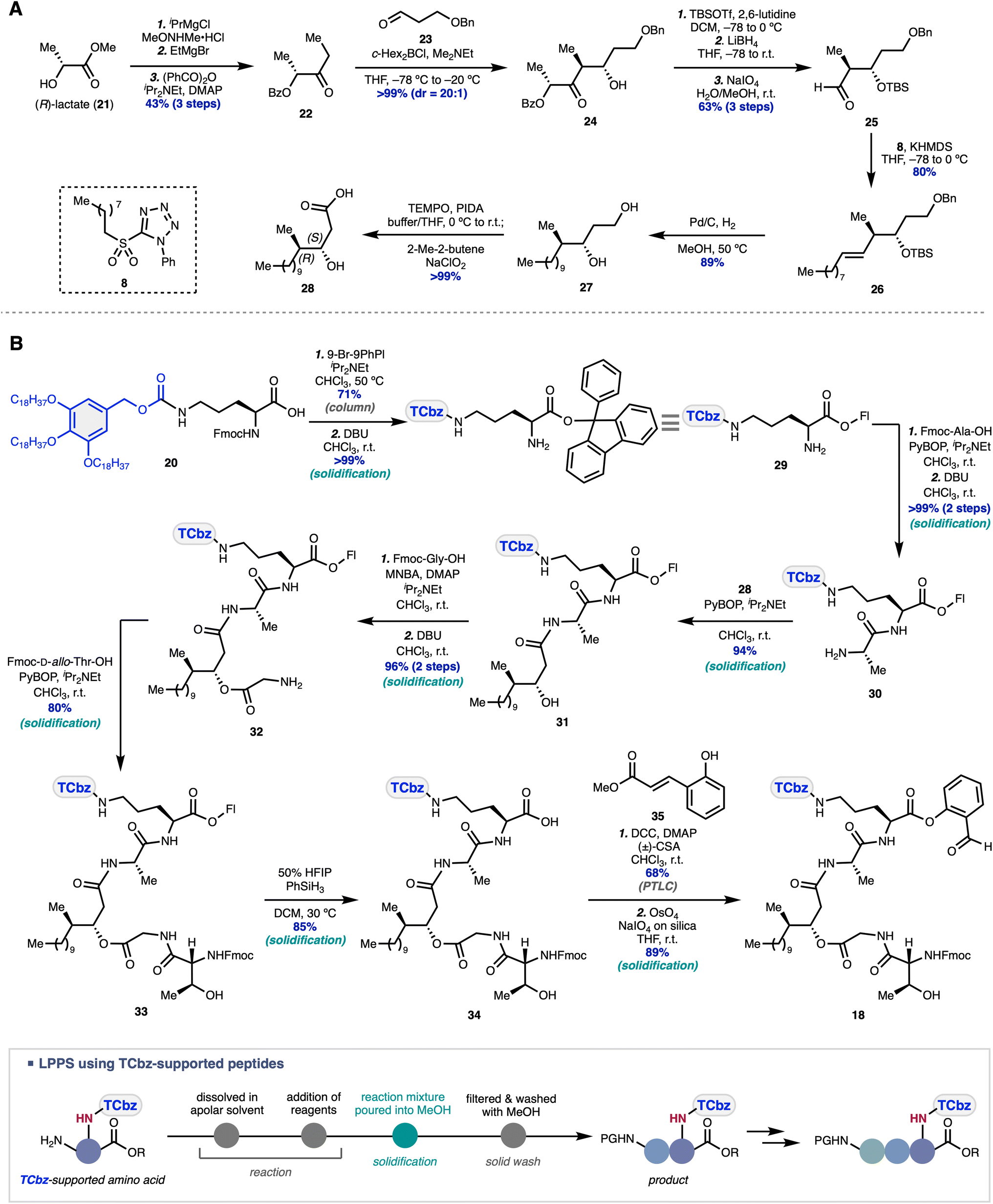 | ||
| Scheme 3 Western fragment synthesis. (A) Diastereoselective synthesis of the fatty acid fragment in an enantiospecific fashion. (B) TCbz group-enabled LPPS to elaborate the desired western fragment. The typical procedure for our LPPS is shown in the gray box. | ||
With fatty acid 28 in hand, we then undertook peptide elongation by investigation of the protecting group at the C-terminus of Fmoc-Orn(TCbz)-OH (20) (see the ESI† for details). Consequently, the 9-phenylfluorenyl (Fl) group was installed on the acid moiety of 20 in 71% yield, followed by removal of the Fmoc group, to provide amine 29 quantitatively (Scheme 3B). Condensation of 29 with Fmoc-Ala-OH and subsequent removal of the Fmoc group produced 30 in quantitative yield over two steps. Dipeptide 30 was condensed with fatty acid 28 (94% yield), followed by esterification of alcohol 31 with Fmoc-Gly-OH using MNBA (2-methyl-6-nitrobenzoic anhydride) and deprotection of the amine group, providing 32 in 96% yield over two steps. Subjection of 32 to the condensation conditions with Fmoc-D-allo-Thr-OH to produce 33 (80% yield) and subsequent deprotection of the acid group under mild conditions provided 34 in 85% yield. Finally, salicylaldehyde ester 18 was synthesized by esterification of 34 with phenol 35 (68% yield) and oxidative cleavage of the corresponding unsaturated ester (89% yield). Thus, the western fragment 18 was prepared in a total of 11 steps from Fmoc-Orn(TCbz)-OH (20).
Consecutive Ser fragment synthesis
We began our investigation of the Ser fragment synthesis with a one-pot protocol similar to that we previously developed in the total synthesis of emodepsides.23 Considering the orthogonality to the TCbz group in the downstream deprotection step, we chose tag-carrier 36 reported by Chiba and coworkers41 which could be selectively removed at the late-stage under mild conditions. Esterification of 36 with Fmoc-D-Ser(tBu)-OH using DCC and DMAP, followed by trapping of the excess activated ester reagent residue with propylamine and cleavage of the Fmoc group using DBU, furnished the corresponding amine D-19.Unexpectedly, the typical purification procedure of LPPS using the tag-carrier by solidification with MeOH led to gelation of the mixture and diminished the yield (Scheme 4A). To our delight, we found that solidification using MeOH containing HOBt as a moderately acidic additive, which we previously employed to prevent the undesired cross condensation in the total synthesis of argifin,19 successfully suppressed the problematic gelation. The addition of HOBt may affect the intermolecular hydrogen bonding network of the carrier-supported Ser derivative in the MeOH solution.
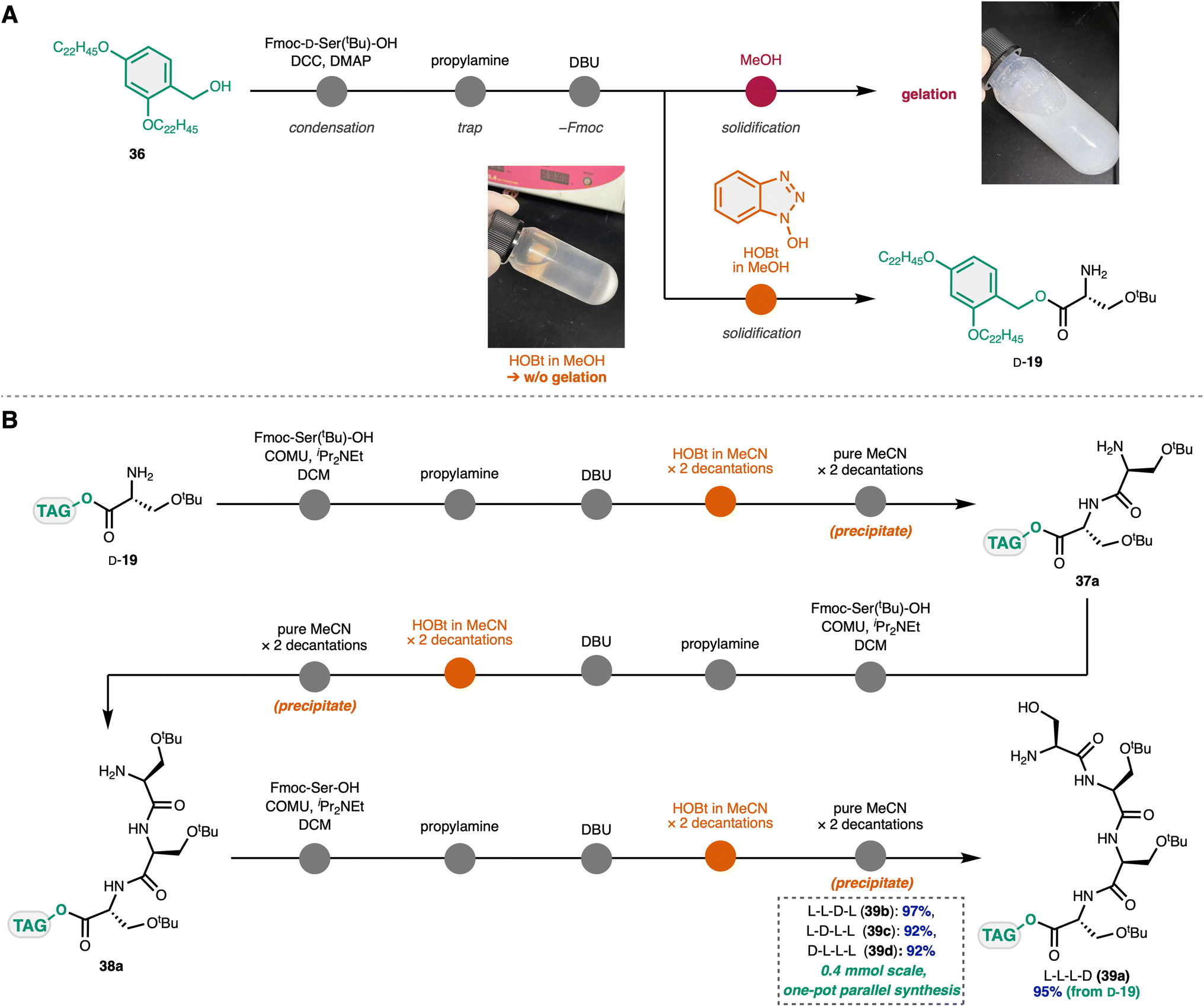 | ||
| Scheme 4 One-pot Ser fragment synthesis. (A) Synthesis of tag-supported Ser 19. (B) Rapid peptide elongation using a one-pot protocol. | ||
With this simple and practical procedure to handle the carrier-supported polar peptide, we attempted the one-pot synthesis of the consecutive Ser fragment (Scheme 4B). Carrier-supported Ser D-19 was subjected to the condensation conditions using COMU with Fmoc-Ser(tBu)-OH. Subsequent treatment with propylamine and DBU removed the Fmoc group. A solution of HOBt in MeCN was used for solidification, in which the precipitate was washed once with MeCN containing HOBt, followed by addition of pure MeCN twice to provide dipeptide 37a. This procedure was iterated using 37a and Fmoc-Ser(tBu)-OH to afford tripeptide 38a, and then analogously with Fmoc-Ser-OH to lead to tetrapeptide 39a without incident. Similarly, all four plausible isomers of the Ser fragments (39b–d) were synthesized in a range of 92–97% yield. This method could be performed on a 0.4 mmol scale, demonstrating a one-pot parallel synthesis to rapidly access the four fragments at scale.
Total synthesis and the absolute structure determination of the natural product
With both fragments in hand, we investigated fragment coupling by the STL reaction (Scheme 5). After a survey of buffered reaction conditions that could promote imine formation while maintaining the solubility of the carrier-supported peptide fragments,42 we found that treatment of 18 and 39a with 5% AcOH/pyridine (1:1 mol mol−1) in DCM (0.03 M) gave rise to the desired coupled product 40a in quantitative yield as a diastereomeric mixture of the N,O-acetal moiety.43 Remarkably, the STL reaction proceeded efficiently with only a slight excess (1.05 eq.) of 39a without epimerization of the peptide backbone. This highlights the power of convergent LPPS strategy under the homogeneous reaction conditions.
 | ||
| Scheme 5 Fragment coupling using the STL reaction, and complete synthesis of tetraselide and its constitutional isomers through systematic elaboration of the carrier-supported cyclic peptides. | ||
After cleavage of the Fmoc group in 40a under basic conditions, the carrier molecule at the C-terminus in 41a was selectively removed under mild conditions using HFIP to furnish amino acid 42a. Macrolactamization of 42a using HATU in the presence of iPr2NEt afforded head-to-tail macrocyclic peptide 43a. Finally, global deprotection of the amino acids in 43a including the TCbz group was accomplished by treatment with TFA in HFIP/CHCl3. At this stage, the solid residue derived from the carrier molecules was removed by filtration and washed with MeCN. The crude filtrate was purified by reverse-phase HPLC, affording the desired peptide 44a in 39% yield over four steps. Analogously, we prepared the other three isomers 44b–d in four steps (17–23% yield). All spectral data of synthesized 44a were consistent with those of naturally occurring tetraselide (1), thus determining the order of the four consecutive Ser moieties in the natural product 1 as L-L-L-D. Consequently, our rapid systematic syntheses of all plausible isomers of tetraselide through the assembly of the carrier-supported macrocyclic peptides allowed us to reveal the absolute structure of the natural product.
Conclusions
We achieved the first total synthesis of tetraselide to determine its absolute structure. Motivated by our group's long-standing interest in exploring new natural products, this macrocyclic depsipeptide was isolated as a potent antifungal compound from a marine-derived filamentous fungus. We implemented bioinformatic analysis to predict two ambiguous stereocenters in the fatty acid moiety, as well as chemical degradation and chiral pool synthesis to elucidate the stereochemistry. With this information, a convergent synthetic strategy using two carrier-supported peptide fragments was developed to synthesize the four plausible isomers of the natural product.The western fragment was synthesized via unconventional LPPS based on our previous work using the TCbz group. We also established the one-pot parallel synthesis of the four Ser fragments. The challenging peptide fragment coupling was accomplished utilizing the serine/threonine ligation reaction. Head-to-tail macrocyclization of the carrier-supported precursors using the TCbz auxiliary allowed us to handle the macrocyclic peptides in a systematic way.
Overall, our work highlights the power of LPPS to enable practical convergent syntheses of macrocyclic peptides. The synthesized isomers were evaluated for in vitro antifungal activities that were used for our screening method to explore the natural product (see the ESI† for details). Further investigations for antifungal properties as well as structure–activity relationship studies are currently underway at our institute.
Data availability
All data associated with this publication are provided in the ESI.†Author contributions
T. H. and T. S. supervised the project. The design of this work was conceptualized by G. S. and T. H. with input from H. N. and H. T. H. N., H. A., and Y. W. carried out the experimental work. H. T. performed bioinformatic analyses and docking simulations. The experimental data were recorded by H. N., H. A., and Y. W. The ESI† was written by H. N., G. S., H. A., H. T. and Y. W. and reviewed by I. M. and T. H. The manuscript was written by H. N., G. S. and H. T. and reviewed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Distinguished Emeritus Professor Satoshi Ōmura (Kitasato University) for his helpful support and suggestions. We thank Drs Yuriko Nagano (Japan Agency for Marine-Earth Science and Technology) and Kenichi Nonaka (Teikyo University of Science) for the deposition of the fungal strain. This work was supported by a Grant-in-Aid for Transformative Research Areas (A) “Latent Chemical Space” [JP24H01789] for G. S. from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This study was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant No. 24K18256 (G. S.), 23K06053 (Y. N.), 21K05293 (Y. W.), 22K06535 (T. H.), and Research Support Project for Life Sciences Research and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED (Grant No. JP24ama121035). The authors thank Dr Kenichiro Nagai, Ms Noriko Sato, and Ms Reiko Seki (School of Pharmacy, Kitasato University) for spectroscopic assistance.Notes and references
- (a) A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 Search PubMed; (b) C. Morrison, Nat. Rev. Drug Discovery, 2018, 17, 531–533 Search PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 Search PubMed.
- (a) P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 Search PubMed; (b) A. Ohta, M. Tanada, S. Shinohara, Y. Morita, K. Nakano, Y. Yamagishi, R. Takano, S. Kariyuki, T. Iida, A. Matsuo, K. Ozeki, T. Emura, Y. Sakurai, K. Takano, A. Higashida, M. Kojima, T. Muraoka, R. Takeyama, T. Kato, K. Kimura, K. Ogawa, K. Ohara, S. Tanaka, Y. Kikuchi, N. Hisada, R. Hayashi, Y. Nishimura, K. Nomura, T. Tachibana, M. Irie, H. Kawada, T. Torizawa, N. Murao, T. Kotake, M. Tanaka, S. Ishikawa, T. Miyake, M. Tamiya, M. Arai, A. Chiyoda, S. Akai, H. Sase, S. Kuramoto, T. Ito, T. Shiraishi, T. Kojima and H. Iikura, J. Am. Chem. Soc., 2023, 145, 24035–24051 Search PubMed.
- (a) E. Biron, J. Chatterjee, O. Ovadia, D. Langenegger, J. Brueggen, D. Hoyer, H. A. Schmid, R. Jelinek, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2008, 47, 2595–2599 Search PubMed; (b) T. R. White, C. M. Renzelman, A. C. Rand, T. Rezai, C. M. McEwen, V. M. Gelev, R. A. Turner, R. G. Linington, S. S. F. Leung, A. S. Kalgutkar, J. N. Bauman, Y. Zhang, S. Liras, D. A. Price, A. M. Mathiowetz, M. P. Jacobson and R. S. Lokey, Nat. Chem. Biol., 2011, 7, 810–817 Search PubMed; (c) C. K. Wang, C. K. S. E. Northfield, B. Colless, S. Chaousis, I. Hamernig, R.-J. Lohman, D. S. Nielsen, C. I. Schroeder, S. Liras, D. A. Price, D. P. Fairlie and D. J. Craik, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 17504–17509 Search PubMed.
- (a) S. Sivanathan and J. Scherkenbeck, Molecules, 2014, 19, 12368–12420 Search PubMed; (b) S.-X. Liu, S.-Y. Ou-Yang, Y.-F. Lu, C.-L. Guo, S.-Y. Dai, C. Li, T.-Y. Yu and Y.-H. Pei, Front. Microbiol., 2023, 14, 1276928 Search PubMed.
- (a) M. Weinmüller, F. Rechenmacher, U. Kiran Marelli, F. Reichart, T. G. Kapp, A. F. B. Räder, F. S. Di Leva, L. Marinelli, E. Novellino, J. M. Muñoz-Félix, K. Hodivala-Dilke, A. Schumacher, J. Fanous, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2017, 56, 16405–16409 Search PubMed; (b) A. Schumacher-Klinger, J. Fanous, S. Merzbach, M. Weinmüller, F. Reichart, A. F. B. Räder, A. Gitlin-Domagalska, C. Gilon, H. Kessler and A. Hoffman, Mol. Pharmaceutics, 2018, 15, 3468–3477 Search PubMed; (c) Y. Hosono, S. Uchida, M. Shinkai, C. E. Townsend, C. N. Kelly, M. R. Naylor, H.-W. Lee, K. Kanamitsu, M. Ishii, R. Ueki, T. Ueda, K. Takeuchi, M. Sugita, Y. Akiyama, S. R. Lokey, J. Morimoto and S. Sando, Nat. Commun., 2023, 14, 1416 Search PubMed; (d) L. Cao, Z. Xu, T. Shang, C. Zhang, X. Wu, Y. Wu, S. Zhai, Z. Zhan and H. Duan, J. Med. Chem., 2024, 67, 1888–1899 Search PubMed.
- S. Ōmura, Angew. Chem., Int. Ed., 2016, 55, 10190–10209 Search PubMed.
- (a) Y. Watanabe, T. Suga, S. Narusawa, M. Iwatsuki, K. Nonaka, T. Nakashima, Y. Shinohara, T. Shiotsuki, N. Ichimaru, H. Miyoshi, Y. Asami, S. Ōmura and K. Shiomi, J. Antibiot., 2017, 70, 395–399 Search PubMed; (b) Y. Watanabe, Y. Asami, S. Narusawa, S. Hashimoto, M. Iwatsuki, K. Nonaka, Y. Shinohara, T. Shiotsuki, N. Ichimaru, H. Miyoshi, S. Ōmura and K. Shiomi, J. Antibiot., 2018, 71, 146–148 Search PubMed; (c) Y. Watanabe, Y. Yoshida, T. Tokiwa, M. Higo, S. Ban, A. Ikeda, Y. Noguchi, T. Hirose, T. Sunazuka, K. Nonaka, T. Yaguchi and M. Iwatsuki, J. Gen. Appl. Microbiol., 2022, 68, 200–206 Search PubMed; (d) Y. Watanabe, S. Takahashi, S. Ito, T. Tokiwa, Y. Noguchi, H. Azami, H. K. M. Higo, S. Ban, K. Nagai, T. Hirose, T. Sunazuka, T. Yaguchi, K. Nonaka and M. Iwatsuki, Org. Biomol. Chem., 2023, 21, 2320–2330 Search PubMed; (e) H. Azami, Y. Watanabe, K. Sakai, H. Nakahara, H. Kojima, T. Tokiwa, K. Nonaka, Y. Noguchi, Y. Nagano, T. Hirose, T. Sunazuka, H. Matsui, N. Arima, K. Abe, H. Hanaki and M. Iwatsuki, J. Antibiot., 2024, 77, 156–162 Search PubMed.
- Of note, the same planar structure has been reported in the patent (JP 2004-168685). However, the reported NMR spectra did not match with those of tetraselide (1).
- F. Rosconi, D. Davyt, V. Martínez, M. Martínez, J. A. Abin-Carriquiry, H. Zane, A. Butler, E. M. De Souza and E. Fabiano, Environ. Microbiol., 2013, 15, 916–927 Search PubMed.
- K. Harada, K. Fujii, K. Hayashi, M. Suzuki, Y. Ikai and H. Oka, Tetrahedron Lett., 1996, 37, 3001–3004 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 Search PubMed.
- (a) D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 Search PubMed; (b) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 Search PubMed; (c) H. Liu, L. Bai and X. Jiang, Tetrahedron Lett., 2024, 151, 155314 Search PubMed.
- V. Martin, P. H. G. Egelund, H. Johansson, S. Thordal Le Quement, F. Wojcik and D. Sejer Pedersen, RSC Adv., 2020, 10, 42457–42492 Search PubMed.
- B. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 Search PubMed.
- (a) A. Thakkar, T. B. Trinh and D. Pei, ACS Comb. Sci., 2013, 15, 120–129 Search PubMed; (b) L. Digal, S. C. Samson, M. A. Stevens, A. Ghorai, H. Kim, M. C. Mifflin, K. R. Carney, D. L. Williamson, S. Um, G. Nagy, D.-C. Oh, M. C. Mendoza and A. G. Roberts, ACS Chem. Biol., 2024, 19, 81–88 Search PubMed.
- (a) A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 Search PubMed; (b) L. K. Mueller, A. C. Baumruck, H. Zhdanova and A. A. Tietze, Front. Bioeng. Biotechnol., 2020, 8, 162 Search PubMed.
- (a) E. Bayer and M. Mutter, Nature, 1972, 237, 512–513 Search PubMed; (b) M. Mutter and E. Bayer, Angew. Chem., Int. Ed., 1974, 13, 88–89 Search PubMed.
- H. Tamiaki, T. Obata, Y. Azefu and K. Toma, Bull. Chem. Soc. Jpn., 2001, 74, 733–738 Search PubMed.
- T. Hirose, T. Kasai, T. Akimoto, A. Endo, A. Sugawara, K. Nagasawa, K. Shiomi, S. Ōmura and T. Sunazuka, Tetrahedron, 2011, 67, 6633–6643 Search PubMed.
- (a) Y. Hayashi, W. Fukasawa, T. Hirose, M. Iwatsuki, R. Hokari, A. Ishiyama, M. Kanaida, K. Nonaka, A. Také, K. Otoguro, S. Ōmura, K. Shiomi and T. Sunazuka, Org. Lett., 2019, 21, 2180–2184 Search PubMed; (b) Y. Hayashi, T. Hirose, M. Iwatsuki, S. Ōmura and T. Sunazuka, Org. Lett., 2019, 21, 8229–8233 Search PubMed.
- A. Watanabe, Y. Noguchi, T. Hirose, S. Monma, Y. Satake, T. Arai, K. Masuda, N. Murashima, K. Shiomi, S. Ōmura and T. Sunazuka, Tetrahedron Lett., 2020, 61, 151699 Search PubMed.
- Y. Noguchi, S. Sekikawa, Y. Nogaki, Y. Satake, N. Murashima, T. Kirisawa, G. Schiffer, J. Köebberling, T. Hirose and T. Sunazuka, Tetrahedron, 2022, 128, 133100 Search PubMed.
- H. Nakahara, G. Sennari, Y. Noguchi, T. Hirose and T. Sunazuka, Chem. Sci., 2023, 14, 6882–6889 Search PubMed.
- P. Edman, Acta Chem. Scand., 1950, 4, 283–293 Search PubMed.
- K. Blin, S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann, M. Alanjary, A. Fetter, B. R. Terlouw, W. W. Metcalf, E. J. N. Helfrich, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 51, W46–W50 Search PubMed.
- A Web-Based Support Tool to Find Secondary Metabolite Biosynthetic Gene Cluster, https://biosyn.nih.go.jp/2ndfind/.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 Search PubMed.
- (a) O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 Search PubMed; (b) J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 Search PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. H. Huson, BMC Evol. Biol., 2007, 7, 78 Search PubMed.
- (a) A. T. Keatinge-Clay, Chem. Biol., 2007, 14, 898–908 Search PubMed; (b) A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 Search PubMed.
- K. J. Weissman, J. Org. Chem., 2017, 13, 348–371 Search PubMed.
- R. D. C. Pullin, A. H. Rathi, E. Y. Melikhova, C. Winter, A. L. Thompson and T. J. Donohoe, Org. Lett., 2013, 15, 5492–5495 Search PubMed.
- P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 23, 2563–2585 Search PubMed.
- M. T. Crimmins and J. She, Synlett, 2004, 8, 1371–1374 Search PubMed.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 Search PubMed.
- L. A. Carpino and A. El-Faham, Tetrahedron, 1999, 55, 6813–6830 Search PubMed.
- X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Org. Lett., 2010, 12, 1724–1727 Search PubMed.
- L. C. Dias and E. C. Polo, J. Org. Chem., 2017, 82, 4072–4112 Search PubMed.
- (a) I. Paterson, D. J. Wallace and S. M. Velázquez, Tetrahedron Lett., 1994, 35, 9083–9086 Search PubMed; (b) I. Paterson and D. J. Wallace, Tetrahedron Lett., 1994, 35, 9087–9090 Search PubMed; (c) N. Y. S. Lam, T. P. Stockdale, M. J. Anketell and I. Paterson, Chem. Commun., 2021, 57, 3171–3189 Search PubMed.
- G. Tana, S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Chem. Commun., 2010, 46, 8219 Search PubMed.
- H. Wu, Z. Sun and X. Li, Angew. Chem., Int. Ed., 2023, 62, e202310624 Search PubMed.
- (a) H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. T. Wong, C. Xu and X. Li, J. Am. Chem. Soc., 2013, 135, 6272–6279 Search PubMed; (b) Z. Sun, Z. Shang, N. Forelli, K. H. L. Po, S. Chen, S. F. Brady and X. Li, Angew. Chem., Int. Ed., 2020, 59, 19868–19872 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00566c |
| This journal is © The Royal Society of Chemistry 2025 |