DOI:
10.1039/D5SC00932D
(Review Article)
Chem. Sci., 2025,
16, 9092-9108
Advances in layer manganese dioxide for energy conversion and storage: mechanisms, strategies and prospects
Received
5th February 2025
, Accepted 17th April 2025
First published on 17th April 2025
Abstract
Layer manganese dioxide with its special structure, low price and large theoretical specific capacitance/capacity is considered a competitive candidate for various energy conversion and storage devices, such as supercapacitors and batteries (Li-ion, Na-ion, and Zn-ion) However, challenges such as low electronic/ionic conductivity, sluggish diffusion kinetics, and structural collapse during cycling are still the main factors limiting its practical application. A solid understanding of the correlation between structure and performance will greatly promote the performance and the further application of layer manganese dioxide. In this review, the energy storage mechanism of layer manganese dioxide in different energy storage devices is discussed in detail. Additionally, considering the current difficulties and challenges, recent advances in strategies for electrochemical performance improvement are systematically summarized, including synthetic methods, structure design, and interlayer engineering. Finally, suggestions for the future directions and developments in preparing layer manganese dioxide cathodes with high electrochemical performance are put forward.
1. Introduction
With the development of renewable energy as an alternative to fossil fuels, the demand for high-efficiency energy storage/conversion devices is increasing dramatically in order to improve the efficiency of hydroelectricity, achieve the load levelling of electric power grids and improve the reliability of renewable energy, and so on.1,2 Until now, supercapacitors and rechargeable batteries have been considered the most representative candidates for energy conversion/storage, which can achieve reversible conversion of electrical and chemical energies.3 In terms of the cost, safety and environmental friendliness, MnO2-based materials have attracted intensive attention as electrode materials in supercapacitors and different kinds of batteries, such as Li-ion, Na-ion, Mg-ion and Zn-ion batteries. In addition, MnO2 has high theoretical pseudocapacitance (≈1370 F g−1) and a wide positive potential window (1.0 V) compared with most of the other transition metal oxides (e.g., NiO and Co3O4 are 0.5 and 0.45 V, respectively), thereby offering high energy density.4–9 However, based on various arrangements of the basic MnO6 octahedra units, there are six polymorphs of MnO2, namely, α-MnO2, β-MnO2, R-MnO2, γ-MnO2, δ-MnO2, and λ-MnO2,10 as shown in Fig. 1. Among these polymorphs, α-MnO2 is in a 2 × 2 tunnel or hollandite structure with a tunnel size of ≈0.46 nm and δ-MnO2 is in a two-dimensional (2D) layered or birnessite structure with a large interlayer separation of ≈0.7 nm, which are suitable for intercalation/deintercalation of the majority charge carrier. The narrow tunnels in λ-MnO2 and β-MnO2 give rise to a lower performance and phase transition during the initial discharge process. The structure of γ-MnO2 consists of an intergrowth between 1 × 1 and 2 × 1 tunnels that can facilitate proton intercalation in alkaline batteries.11–14 Obviously, MnO2 with different crystallographic polymorphs exhibit completely disparate redox reaction kinetics when they are used as electrode materials.
 |
| | Fig. 1 Crystal structures of MnO2 polymorphs (Mn: magenta and O: red). Water molecules and guest cations are omitted for clarity.10 Reproduced with permission from ref. 10. Copyright 2019, Sage. | |
Among them, layer manganese dioxide, also known as birnessite-type MnO2 or δ-MnO2, is the most widely used electrode material because of its adjustable interlayer spacing that could provide reasonable transport channels for insertion of alkaline ions (from monovalent to trivalent, e.g., Li+, Na+, K+, Zn2+, Mg2+, Ca2+, and Al3+) and abound chemically active sites for redox reaction. It is widely acknowledged that layer manganese dioxide (usually represented as AxMnO2·yH2O (A is Na, K, or Ca)) is not a simple manganese oxide but a hydrous manganese oxide constructed with a layer of edge-sharing MnO6 octahedra, with various combinations of cationic species and H2O molecules depending upon the environment of formation.15–18 Even though it has been used as an electrode material for a long time, some intrinsic issues still limit its electrochemical performance, such as low electronic/ionic conductivity, structural instability upon cycling resulting from Jahn–Teller distortion and sluggish diffusion kinetics. Besides, the valence of Mn is based on the combination of Mn2+, Mn3+ and Mn4+, which raises the possibility of redox reactions involving ion exchange between MnOx and electrolyte. Initially, some strategies have been introduced to improve electrochemical performance on the macro level. For example, the preparation of a carbon-containing composite,19,20 and the design of a nanostructure from 0D to 3D, reducing the dead mass of layer manganese dioxide.21–23 These pathways can increase electronic conductivity, enhance electrochemically active sites and promote material utilization, achieving 15–25% of theoretical capacitance. To further improve electrochemical performance, many studies focus on the crystal structure level, such as interlayer pre-intercalation, element doping, defect engineering, and so on, which can effectively improve the intrinsic electronic/ionic conductivity, expand the interlayer spacing and stimulate reaction kinetics. The major strategies used on layer manganese dioxide electrode materials are illustrated in Fig. 2. Nevertheless, there is still room for further improvement in electrochemical performance of layer manganese dioxide through other strategies. Herein, we reveal the intrinsic mechanisms of layer manganese dioxide in various energy conversion and storage devices and summarize the previously reported solutions. Furthermore, bottlenecks in electrochemical performance of layer manganese dioxide electrode materials and the corresponding solutions are proposed to guide future research.
 |
| | Fig. 2 Some strategies applied on layer manganese dioxide electrode materials to improve their electrochemical performance. | |
2. Energy storage mechanism of layer manganese dioxide
2.1 Surface absorption and desorption mechanism
Two effects typically occur when the electrolyte ions are in contact with the electrode interface: one is the adsorption behavior of fully solvated ions on the electrode interface through electrostatic interaction, resulting in a non-Faraday electric double layer (EDL) capacitance, and the other is the specific adsorption of desolvated or partially solvated ions on the electrode interface through chemical bonding, resulting in a Faraday pseudocapacitance. In fact, EDL capacitance exists in nearly all electrodes, similar to a parallel-plate capacitor. In some studies, an almost rectangular profile was delivered in cyclic voltammetry (CV) curves of layer manganese dioxide in neutral electrolyte, analogous to non-faradaic energy storage mechanisms.24 Researchers have investigated whether EDL capacitance is proportional to the specific surface area available for the active material,25–27 according to C = Aεrε0/d, where A is the specific surface area of the electrode accessible to the electrolyte ions, εr is the electrolyte dielectric constant, ε0 is the permittivity of a vacuum, and d is the effective thickness of the EDL. The specific surface area is a factor that must be considered in the process of material preparation.
2.2 Surface redox reaction mechanism
Studies have shown that layer manganese dioxide exhibits characteristics related to pseudocapacitance, such as changes in structure, local bonding and Mn oxidation state, which was first described in acidic electrolytes.28,29 Subsequently, a similar mechanism was found in mild electrolyte.30 This is mainly due to the adsorption of alkaline cations (Li+, Na+, K+, etc.) in the electrolyte with the opposite electrical properties of O in the MnO6 octahedron. Then, the charge is transferred to the adjacent Mn atom, its valence state is changed from +4 to +3, and the reaction is described below:
| (MnO2)surface + C+ + e− → (MnOOH)surface |
where C+ = Li+, Na+, and K+. It should be noted that the reaction only occurs on the surface atom or in a thin layer of layer manganese dioxide. Therefore, constructing an ultra-thin layer is an effective strategy to improve the capacitance. The redox reaction is very fast and it is difficult to directly distinguish from EDL capacitance, but it involves a chemical reaction resulting in higher capacitance. Additionally, the choice of potential window will cause a difference in the energy storage mechanism. For example, Xia et al. discussed the electrochemical behavior of the birnessite Na0.5MnO2 in different potential windows, finding that the reversible redox reaction of Mn3+/Mn4+ was accompanied by the insertion/extraction of Na+ in a window of 0–1.0 V, the deintercalation and intercalation of Na+ results in the large potential window of 0–1.3 V.23 As reported in the literature,4,31,32 when the upper cutoff potential is set below 0.8 or 1 V, Mn3+/Mn4+ redox reaction would not be not complete, and the energy storage of layer MnO2 would be dominated by pseudocapacitive resulting from rapid surface Faraday reactions. When the potential window is widened to 1.0–1.4 V, a hybrid energy storage mechanism with synergistic pseudocapacitance and ion intercalation/deintercalation occurs. Oxygen vacancies usually reduce the Jahn–Teller distortion by reducing the coordination symmetry of MnO6 octahedron and inhibiting manganese dissolution and structural collapse at high potentials. Besides, element doping can reduce the band gap of MnO2 and reduce polarization, resulting in the layered structure working under a wide potential window. Therefore, the influence of various factors on the energy storage mechanism should be considered comprehensively.
2.3 Cation intercalation/deintercalation mechanism
In recent studies, another important energy storage mechanism in layer manganese dioxide is the (de)intercalation of alkaline cations in the bulk of the material. The reaction involved in the charge/discharge process is shown below:
where C+ = H+, Na+, K+, and Li+. This process is more like a battery behaviour than a capacitive behaviour, which is limited by ion diffusion within the crystalline framework of active material. Currently, a CV investigation proposed by Dunn is employed to distinguish diffusion-controlled contribution and surface capacitive contribution,33 as shown in the following formula:
where i is the current (A), ν is the sweep rate (V s−1), a and b are adjustable parameters, and b value has special significance. In particular, b = 0.5 means that the redox reaction is controlled by ion diffusion, while b = 1 means a typical capacitive behavior. The capacitive contribution includes EDL capacitance and pseudocapacitance. To enhance the electrochemical performance of layer manganese dioxide, pre-intercalated MnO2 (cation or polymer) has been widely studied by researchers, and the corresponding energy storage mechanism has been investigated. Teng et al. have explored the sodium storage mechanism of sodium rich disordered birnessite (Na0.27MnO2).34In situ X-ray diffraction illustrated that sodium ion and water co-deintercalated in the bulk of Na0.27MnO2 during high voltage discharge (Fig. 3a), which prevented excessive increase of layer spacing and stabilized the layer structure, benefiting in a large voltage (2.5 V). Lin et al. proposed a simple “hydrothermal insertion of potassium” strategy that effectively increased the potassium content in δ-MnO2.35In situ X-ray diffraction (XRD) and theoretical calculations exploring the charge storage mechanism shows that the multi-stage charging and discharging platforms appears with increasing potassium content, proving K+ intercalation/deintercalation behavior in the layered matrix. Moreover, the remaining K+ between the layers can inhibit the collapse of the layered structure in the charging state, ensuring a long cycle life. Zhang et al. reported the charge storage mechanisms of 2D cation intercalated manganese oxide in neutral electrolyte (NaSO4).36 It showed surface-controlled pseudocapacitive behaviour at low potential (0–0.8 V), while intercalation pseudocapacitive behaviour became dominant when the potential was higher than 0.8 V (Fig. 3b).
 |
| | Fig. 3 (a) Schematic of the motion of Na-ions and water during charging and discharging processes.34 Reproduced with permission from ref. 34. Copyright 2019, Springer Nature. (b) Movement tendency of ions and electrons during charging and discharging processes, and the whole charge storage of NMO in Na2SO4 solution.36 Reproduced with permission from ref. 36. Copyright 2019, Wiley-VCH. (c) Schematic of the degree of ion interaction with the electrode surface.37 Reproduced with permission from ref. 37. Copyright 2021, Springer Nature. (d) Schematic of the displacement/intercalation reaction mechanism in the first cycle, and the insertion/extraction mechanism of zinc ions in subsequent electrochemical discharge/charge processes.38 Reproduced with permission from ref. 38. Copyright 2020, Springer Nature. (e) Illustration of the reaction process of δ-MnO2 electrode in AZIBs.44 Reproduced with permission from ref. 44. Copyright 2024, Wiley-VCH. | |
To elucidate the relevant capacitance mechanism in layer manganese dioxide, plenty of experimental and computational studies were carried out by the Augustyn group.37 It has been found that high efficiency charge storage in neutral electrolyte can be achieved through the combination of water molecules and confined alkaline cations. The weight change before and after charge and discharge measured by electrochemical quartz crystal microbalance (EQCM) showed that the storage of capacitive/charge in birnessite was controlled by interlayer cation (de)intercalation (Fig. 3c). At the same time, the distance between the intercalated cation and birnessite increased due to the presence of nanoconfined interlayer structure water, resulting in reduced interaction and exhibiting capacitive properties which mediated the interaction between the intercalated cation and the main host of birnessite and led to minimal structural changes. Zhai et al. successfully synthesized the layered Na0.55Mn2O4·0.57H2O (NMOH) co-intercalated with sodium ion and crystal water and explored the zinc ion storage mechanism.38 In this research, a displacement/intercalation mechanism was confirmed for the first time. In detail, Zn2+ replaced some Na+ and remained in the interlayer, which played a supporting role to stabilize the layered structure and promoted the subsequent Zn2+/H+ (de)intercalation process in the first electrochemical process (Fig. 3d) so that the battery obtained a high specific capacity and excellent cycle stability. In addition, Na+ removed from NMOH in the first cycle will be re-adsorbed to the anode surface during the discharge process, generating pseudocapacitance to increase the specific capacity of the electrode material. The same energy storage mechanism was also confirmed in the literature.16,39–41 A cation (de)intercalation mechanism was also illustrated in a Mg/Al-pre-intercalated MnO2 battery.42,43 However, Cui et al. suggested that Zn2+ initially irreversibly inserted to replace the interlayer K+ without capacity contribution, and the charge storage was mainly dominated by the electrochemical H+ intercalation/extraction, the electrodissolution of δ-MnO2 and the electrodissolution-electrodeposition of vernadite rather than Zn2+/H+ (de)intercalation process (Fig. 3e).44 These two distinct conclusions should be further probed.
3. Strategies to enhance electrochemical performance
Electronic conductivity, active site and specific surface area of electrode materials are closely related to electrochemical performances of energy storage/conversion devices, which mainly determine the charge transfer ability and diffusion of electrolyte ions at the electrode/electrolyte interface during the charge/discharge process. Given this, some strategies were attempted to alleviate the above situation, such as the conductive species composite, pre-intercalation, transition metal-doping, defect engineering, and so on, and these will be reviewed in detail.
3.1 Layer manganese dioxide/conductive species composite
Graphene, carbon nanotubes (CNTs), and porous carbon are commonly used conductive backbone or matrix compounded with layer dioxide, creating a high specific surface area abound electrochemically active sites, and high electronic conductivity.45–48 The formation of layer manganese dioxide/carbon composite is mainly based on in situ redox deposition in acid solution.49,50 The redox reaction of KMnO4 and carbon is expressed as:
| 4KMnO4 + 3C + 2H2SO4 → 4MnO2 + 3CO2 + 2MnSO4 + 2H2O |
In the process, carbon acts as a reducing agent and substrate, and electrons transfer from carbon to MnO4−, forming a thin, uniform and continuous MnO2 coating. It should be noted that carbon materials need to have some defects to facilitate the growth of MnO2. Therefore, a hydrothermal process, microwave-assisted irradiation, and/or a refluxing technique are usually carried out to synthesize layer manganese dioxide/carbon composite. Liu et al. reported δ-MnO2/graphene flower-like microspheres using thermally-exfoliated graphite.51 SEM images (Fig. 4a) showed that the resulting MnO2 nanosheets arrays dispersed on the graphene surface exhibited a honeycomb-like structure. The composite delivered a maximum specific surface area of 252.3 m2 g−1, resulting in excellent performances such as cycle stability and capacitance retention. Peng et al. prepared birnessite/graphene composite with a layer-by-layer structure to assemble planar supercapacitors (Fig. 4b).52 The novel hybrid nanostructure with uniform MnO2 distribution (Fig. 4c) introduced more electrochemically active surfaces for electrolyte ions and extra interfaces to facilitate charge transport. Additionally, a graphene film with good flexibility can adapt to various bending and deformation and is commonly used for the preparation of flexible electrochemical devices. Based on the high length–diameter ratio of CNTs, Li et al. synthesized MnO2/CNTs composite with uniformly cross-linked MnO2 nanoflakes anchored on CNTs through a modified one-pot reaction process.50 Wu et al. used CNTs prepared by the chemical vapor deposition (CVD) method as a conductive substrate material to support δ-MnO2.53 Then, flexible MnO2@CNTs/CNTs film was prepared by vacuum filtration with the addition of CNTs (Fig. 4d). As shown in Fig. 4e, δ-MnO2 grew uniformly on the surface of carbon nanotubes. The porous self-supported MnO2@CNTs/CNTs film electrode showed excellent pseudocapacitance behaviour, including high volume capacitance of about 177.5 F cm−3 at 0.2 A g−1 and good stability of 90% capacitance retention after 5000 cycles at 50 mV s−1. Meanwhile, there was almost no performance fading at different bending sates, illustrating a high flexibility. Porous carbon has a large specific surface area and abundant pore structure, which facilitates the flow of electrolyte ions while increasing the specific surface area. An et al. used carbonized conjugated microporous polymer hydrogels as conductive substrate and matrix to support manganese dioxide to obtain C-CMPAs@MnO2 composite as a binder-free cathode for a zinc-ion battery.54 The special structure increased the contact area of the electrode/electrolyte, provided abundant active sites, and effectively shortened the zinc ion insertion/extraction path. The test results showed that C-CMPAs@MnO2 composite exhibited a high specific capacity (670.7 mA h g−1 at 0.1 A g−1) and an excellent rate performance (149.2 mA h g−1 at 3 A g−1). To accelerate the electron migration and transfer rate of Zn2+/H+, carbon cloth (CC) was employed as a collector to support tailoring hierarchical MnO2 (MNSMO@CC) resulting from leveraging the nanomicellar properties of cetyltrimethylammonium bromide (CTAB) (Fig. 4f(i–iii)).55 The mass loading increased to 6.1 mg cm−2, and the areal capacitance reached 0.64 mA h cm−2 at 0.1 A g−1 with a good reversibility. Additionally, a long cycle lifespan was achieved, with a capacity retention of 81.8% at 0.8 A g−1 after 1300 cycles, owing to a buffer of CC against δ-MnO2 structural strains during ion intercalation. Besides carbon substrate, nickel foam,56,57 stainless steel,58 Pt,59 Au,60–62 and conductive polymer (e.g., polyaniline, polypyrrole, polydopamine)21,63–67 are used as substrates for supporting layer manganese dioxide to improve the electrochemical performance. The preparation of layer manganese dioxide/conductive species composite mainly improves the electronic conductivity, while the introduction of carbon materials inevitably increases the quality of electrode, which is not conducive to the improvement of specific capacity, making it particularly important to modify/design the structure of layer manganese dioxide itself.
 |
| | Fig. 4 (a) SEM image of MnO2/graphene flower.51 Reproduced with permission from ref. 51. Copyright 2022, Frontiers Media Sa. (b) Schematic of the ultra-flexible planar supercapacitor. (c) TEM image of the 2D hybrid structure with δ-MnO2 nanosheets integrated on graphene surfaces.52 Reproduced with permission from ref. 52. Copyright 2013, the American Chemical Society. (d) Schematics of the stacked films' preparation process. (e) SEM image of MnO2@CNT composites.53 Reproduced with permission from ref. 53. Copyright 2016, the American Chemical Society. (f) Synthesis process and mechanism of MNSMO@CC. (g) Rate performance with different mass loadings.55 Reproduced with permission from ref. 55. Copyright 2024, the Royal Society of Chemistry. | |
3.2 Novel nano-structural design
The energy storage mechanism in layer manganese dioxide involves EDL and pseudocapacitance behavior on the surface, and the interlayer intercalation–deintercalation behavior of alkaline ion. Therefore, it is an effective strategy to reduce the dead mass in the bulk of layer manganese dioxide to enhance the electrochemical performance. Nano-structural design can enlarge the electrode/electrolyte contact area and shorten the diffusion path of electrolyte ions, which will increase the utilization of Mn in the interlayer and eliminate the low intrinsic conductivity of layer manganese dioxide caused by a high content of crystal water, sparse atoms and poor crystalline degree. Layer manganese dioxide are easily fabricated with various nanostructures through different synthetic methods and processing parameters, such as 0D nanoparticles (nanospheres) and dots, 1D nanowires and nanorods, 2D nanosheets and nanobelts, and 3D nanoflowers self-assembled from the above low-dimensional nanostructured building blocks and porous nanostructures.68–72 In this section, we provide a detailed review on the effect of the nanostructure of layer manganese dioxide on its electrochemical properties.
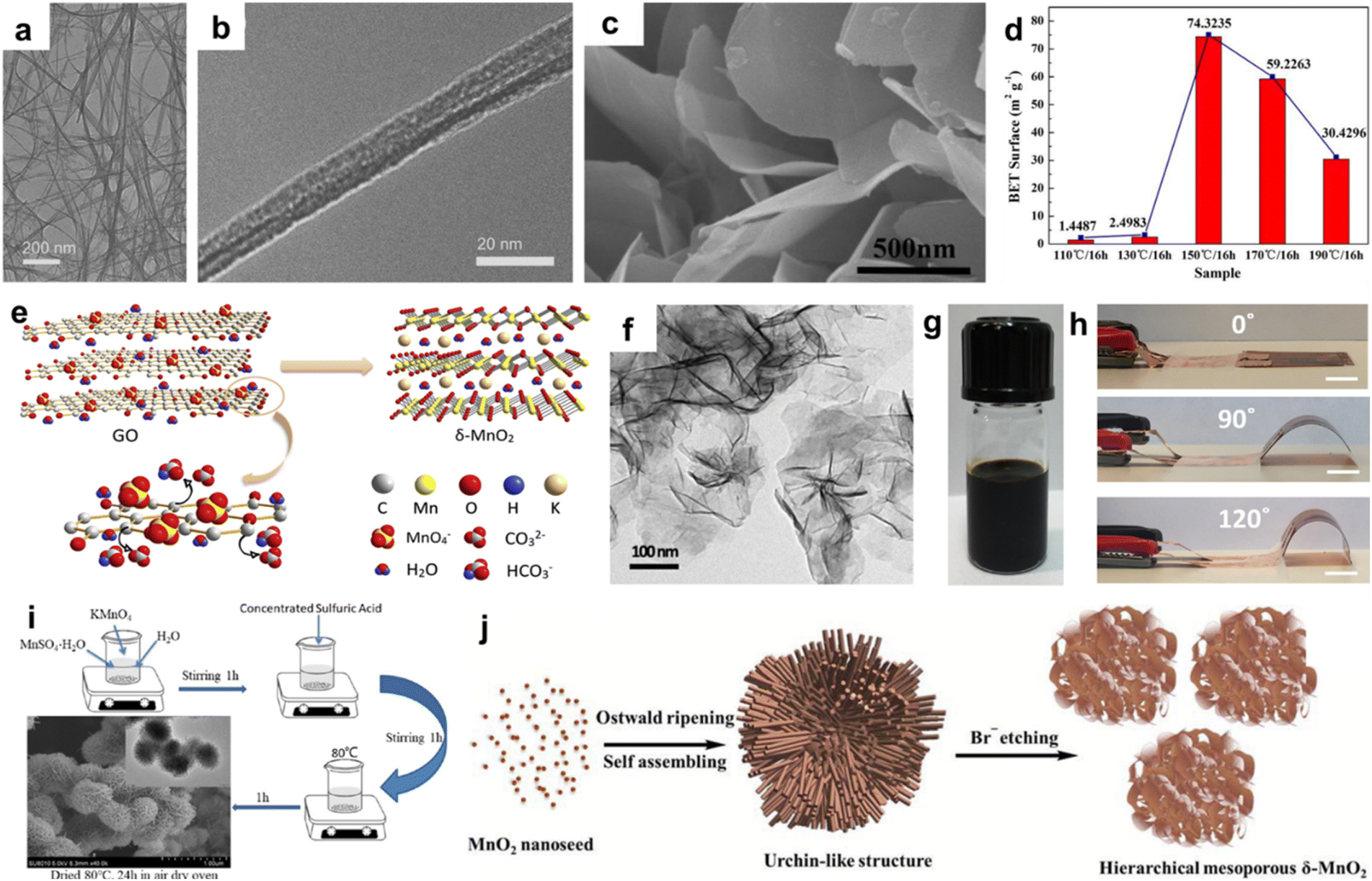
0D nanoparticles are conducive to a good distribution on conductive matrix, which can improve the conductivity and the utilization of electrode material. Xie et al. reported graphite-like carbon-decorated δ-MnO2 composite by a water bath method that was applied to zinc-ion batteries (Fig. 5a).73 The size of δ-MnO2 nanoparticles was about 25 nm (Fig. 5b), with abundant active sites and a short ionic diffusion path, facilitating high specific capacity and excellent cyclic stability. Yun et al. coated MnO2 nanoparticles on porous carbon nanofiber (CNF) matrix (Fig. 5c).74 This strategy maximized the synergistic effect of the pseudocapacitance of MnO2 and the electric double-layer capacity induced by the highly porous surface. Tang et al. synthesized novel MnO2 nanodots/reduced graphene oxide (MnO2 NDs/rGO) composite via the hydrothermal approach followed by ultrasonic treatment (Fig. 5d).75 The synergistic effect of small-sized MnO2 nanodots and conductive rGO accelerated the diffusion kinetics and charge transfer ability of ZIBs, resulting in high rate performance (124 mA h g−1 at 2.0 A g−1) and cycling stability (90.1% capacity retention after 1000 cycles at 1.0 A g−1) (Fig. 5e). Xiao et al. prepared hierarchical mesoporous δ-MnO2 hollow microspheres (δ-MnO2 HMS) using a hydrothermal method (Fig. 5f).76 The hierarchical mesoporous shell (Fig. 5g) with a large specific surface area provided abundant electroactive sites and accessibility for electrolyte ions, guaranteeing a high utilization of active materials. Furthermore, interior cavities within δ-MnO2 HMS regarded as ion reservoirs can shorten the diffusion path of the electrolyte and provide sufficient ions. Based on this special feature, δ-MnO2 HMS delivered a long cycle life (only 8.8% capacitance fading after 3000 cycles at 5 A g−1) (Fig. 5h). It needs to be stressed that the small particle radius of nanoparticles leads to low thermodynamic stability and easy agglomeration, making them not conducive to synthesis, preparation and application.
 |
| | Fig. 5 (a) Schematic illustration of the preparation of GC-δ-MnO2 nanoparticles. (b) FETEM image of GC-δ-MnO2-2.73 Reproduced with permission from ref. 73. Copyright 2022, Springer. (c) HR-TEM image of MnO2 nanoparticles/carbon nanofiber composite.74 Reproduced with permission from ref. 74. Copyright 2022, the American Chemical Society. (d) TEM image of MnO2 NDs/rGO. (e) Rate performance at varying current densities. (f) Cyclic performance and coulombic efficiency at 1.0 A g−1 of MnO2 and MnO2 NDs/rGO.75 Reproduced with permission from ref. 75. Copyright 2023, Elsevier. (g) TEM image of δ-MnO2 HMS. (h) Cyclic performance of δ-MnO2 HMS at 5 A g−1.76 Reproduced with permission from ref. 76. Copyright 2018, Elsevier. | |
1D layer manganese dioxide such as nanowires and nanorods have high length–diameter ratios and nanoscale dimensions that can realize a short ion diffusion distance, so it is favored by researchers.77–80 The most common method for preparing 1D layer manganese dioxide is the hydrothermal method. Liu et al. employed a simple hydrothermal method to prepare a 1D-3D interconnected δ-MnO2 nanowire network (KMO NNT).69 KMO NNT shows 1D–3D network hybrid architecture (Fig. 6a and b), consisting of the interconnected nanowires with average estimated diameters of 10–70 nm. KMO NNT acts as cathode material for Zn-ion batteries, exhibiting a reversible capacity of 342 mA h g−1 at 0.162C, 150 mA h g−1 at 6.494C, illustrating a good rate performance, which benefited from an enhanced contact surface and short ion diffusion pathway. Like nanoparticles, 1D nanowires and nanofibers can be combined with conductive substrates to enhance the performance.81,82 Nanofilms and nanoflakes are the most popular morphology types due to the flexibility and the large surface areas that improve abundant redox-active sites compared with 1D and 2D layer manganese dioxide such as nanosheets.83–86 Xie's group employed the hydrothermal method at different temperatures to prepare δ-MnO2 with various microstructures.87 Porous δ-MnO2 nanosheets were obtained when the hydrothermal process was carried out at 150 °C for 16 h (Fig. 6c). The BET results showed that δ-MnO2 nanosheets delivered the largest specific surface area (Fig. 6d). Guo et al. reported ultrathin δ-MnO2 nanosheets with a thickness of about 2–4 nm via in situ reduction using graphene oxide as a reductant and self-sacrificing template (Fig. 6e and f).88 The electrochemical performance of Zn-ion battery was better than δ-MnO2 microspheres due to favourability of the ultrathin nanosheets for fast diffusion during cycling. In addition, a δ-MnO2 nanosheet flexible electrode was fabricated by Wang et al. using inkjet printing, which can be used as binder-free electrode to assemble all-solid-state symmetrical micro-supercapacitors with highly mechanical flexibility (Fig. 6g and h).89 The major advantage of hierarchical 3D materials is that they have a uniform pore size, high surface area and facile transport of electrolyte ions. Zhang et al. displayed spherical δ-MnO2 nanoflowers with a high specific surface area (228.0 m2 g−1) and abundant mesopores (primarily around 4 and 50 nm, Fig. 6i).90δ-MnO2 serving as an electrode material for the supercapacitor with a nanoflower structure can achieve higher bulk phase utilization and fast kinetics according to the linear potential sweep technique. Furthermore, Bag and Raj synthesized hierarchical mesoporous δ-MnO2 using the thermodynamically favourable redox reaction (Fig. 6j).91 The novel structure gave δ-MnO2 a high specific surface area of 238 m2 g−1 and an average pore size of 3.6 nm, which was beneficial for charge transfer/ion diffusion during charge storage. A specific capacitance of 364 F g−1 at a current density of 1 A g−1 was obtained, which could be further enhanced by reducing the size of δ-MnO2 clusters to increase the redox-active sites. Apart from the above methods, 3D δ-MnO2 microflowers can be prepared by the microwave and hydrothermal methods.92,93
 |
| | Fig. 6 (a and b) TEM images of KMO NNT.69 Reproduced with permission from ref. 69. Copyright 2021, Elsevier. (c) SEM image of δ-MnO2 at 150 °C, (d) Specific surface area of δ-MnO2 at various temperatures.87 Reproduced with permission from ref. 87. Copyright 2016, Elsevier. (e) Illustration of the formation of δ-MnO2 nanosheets. (f) TEM image of the as-prepared δ-MnO2 nanosheets.88 Reproduced with permission from ref. 88. Copyright 2019, Elsevier. (g) Image of the formulated δ-MnO2 nanosheet ink. (h) Optical images of MSC bent under different angles.89 Reproduced with permission from ref. 89. Copyright 2018, Elsevier. (i) Schematic for the preparation of spherical δ-MnO2 nanoflowers.90 Reproduced with permission from ref. 90. Copyright 2024, the American Chemical Society. (j) Plausible mechanism involved in the growth of mesoporous δ-MnO2 nanostructures.91 Reproduced with permission from ref. 91. Copyright 2016, the Royal Society of Chemistry. | |
According to the above discussions on structural designs for layer manganese dioxide nanomaterials, a perfect structure should include the following features: (1) The size of nanostructure unit (diameter of nanodots/nanofiber, thickness of nanosheet/nanofilm) should be refined to 2–4 nm as much as possible to provide accessible surface area and redox active sites; (2) a conductive matrix should be used so that the layer manganese dioxide based nanomaterials grow better, ensuring high electric conductivity; (3) reasonable pore size and distribution should be maintained, providing a smooth diffusion path for electrolyte ions.
3.3 Defect engineering
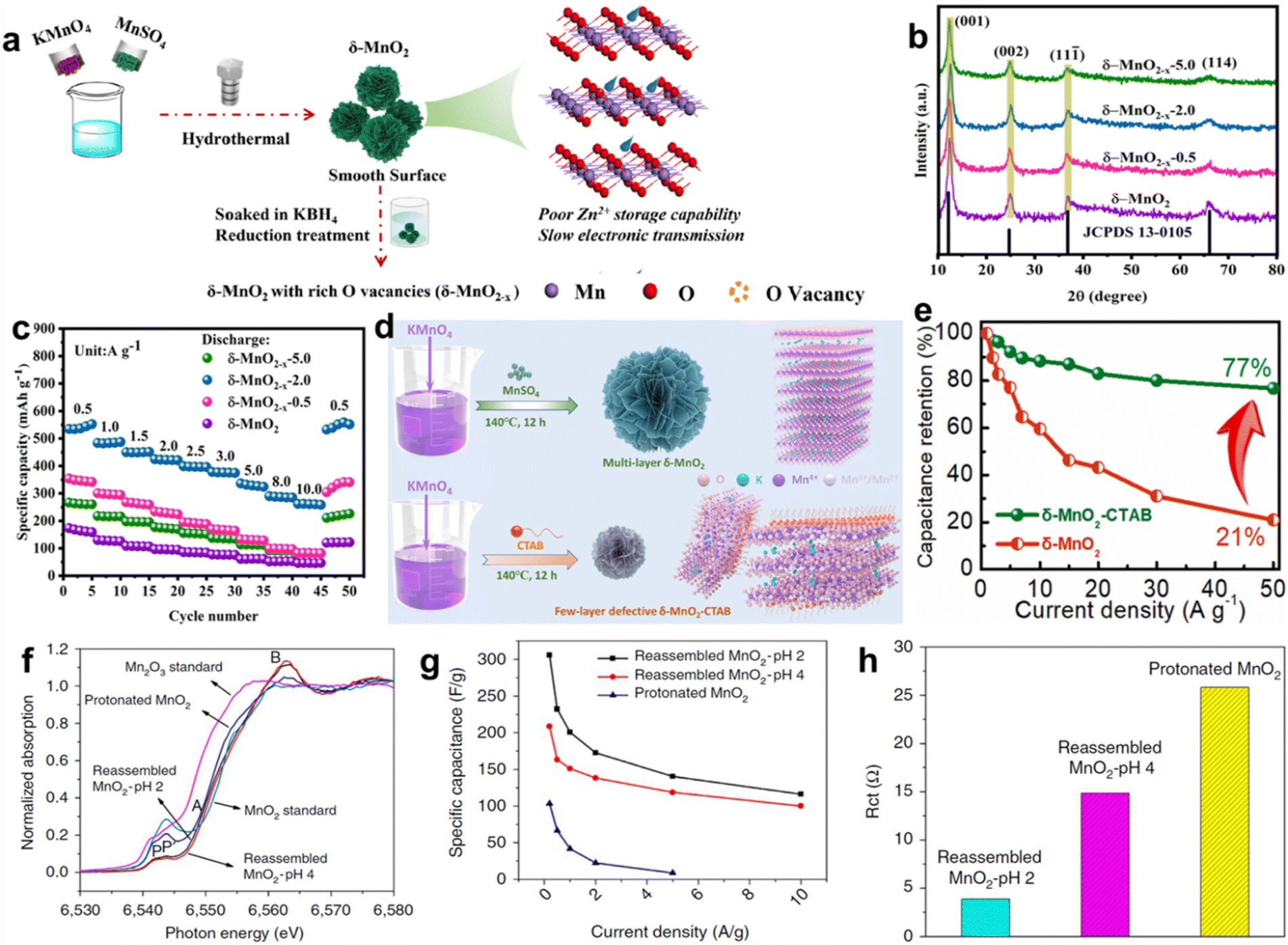
The reported specific capacitance/capacity of layer manganese dioxide are much lower than the theoretical value and the rate capability is poor at high current density, which are mainly caused by intrinsic inferior electronic/ionic conductivity and sluggish reaction kinetics. Among various strategies, defect engineering including anion and cation vacancies is an outstanding approach to boost the electrochemical performance. Oxygen vacancy is the most widely explored anion vacancy in transition metal oxides because of its low energy and easy fabrication. The existence of oxygen vacancies may have a great influence on the physical and chemical properties of layer manganese dioxide, such as the change of the oxidation states of Mn ions (Mn2+, Mn3+, and Mn4+), the modulation of the band gap, and the variation of the charge carrier density, fundamentally improving the electrical/ionic conductivity.94–96 Additionally, the introduction of oxygen vacancies can produce more electrochemically active sites for the accession of electrolyte to active materials, attributed to the enhancement of capacitance/capacity. In fact, the concentration of the oxygen vacancy has a double-edged sword effect on the performance of the MnO2 electrode. For example, a moderate vacancy can significantly improve the electrochemical activity and conductivity, but excessive vacancy will damage the structural stability. Li's group fabricated flower-like δ-MnO2 nanostructures with different oxygen vacancy concentrations controlled by the reduction treatment time in KBH4 solution (Fig. 7a).97 The wider and weaker diffraction peak intensity in XRD patterns illustrated typical disordered structures owing to the introduction of vacancy defects (Fig. 7b). Although the Mn3+/Mn4+ ratio increased with higher oxygen vacancy concentrations to provide abundant active sites, Mn4+ balanced the introduced oxygen vacancies, resulting in structural instability. Additionally, extremely robust H+/Zn2+ binding strength with oxygen vacancy leads to more difficulties in completing the charge reaction. Therefore, in the case of zinc-ion battery cathode, the research findings stated that δ-MnO2−x-2.0 (the reduction time is 2 min) delivered the highest diffusion coefficient and the lowest reaction resistance, resulting in the most remarkable electrochemical performance (large specific capacity of 551.8 mA h g−1 at 0.5 A g−1, high-rate capability of 262.2 mA h g−1 at 10 A g−1) among all the samples, including δ-MnO2, δ-MnO2−x-0.5, δ-MnO2−x-2.0, δ-MnO2−x-5.0, as shown in Fig. 7c. Peng et al. regulated the Mn atomic coordination environment through electrochemical deposition and chemical reduction strategies.98 The optimized oxygen vacancy coordination environment can improve the reactivity, form a localized electric field, accelerate the ion/electron mobility, and realize an extremely high specific capacitance of 4.8 F cm−2/402.6 F g−1. It is expected that the coexistence of exfoliation and oxygen vacancies in δ-MnO2 would enlarge its electrochemical performance due to the large surface area and high electric/ion conductivity. Tang et al. proposed few-layer defective δ-MnO2 nanosheets (named δ-MnO2-CTAB) by using a redox reaction between cetyltrimethylammonium bromide (CTAB) and KMnO4.99 The exchange of CTA+ and K+ in the interlayer caused exfoliation of δ-MnO2 to obtain the ultrathin nanosheet structure (Fig. 7d). Meanwhile, the spatial steric effect of CTA+ weakened the van der Waals force between the layers, introducing many oxygen vacancies, which was confirmed by X-ray photoelectron spectroscopy (XPS) results. The synergistic effect of exfoliation and oxygen vacancies led to a larger specific surface area than pure δ-MnO2, providing more active sites, channels, and space for ion storage and transport. An ultra-high rate-capability of 77% was achieved as the current density was increased 50-fold (1 to 50 A g−1) (Fig. 7e). The intercalation of doping ions in δ-MnO2 will inevitably lead to the expansion or contraction of the lattice, resulting in lattice distortion and defects because of the different ionic radius or valence state, which is also called doping type vacancies. The synergy of ion pre-intercalation and oxygen vacancies in δ-MnO2 can also tremendously improve its electrochemical performance because of the enhanced structural stability and the fast charge transfer capability.100,101
 |
| | Fig. 7 (a) Schematic of the preparation procedure for δ-MnO2 with rich oxygen vacancies (δ-MnO2−x). (b) XRD patterns of all samples. (c) Rate performance of δ-MnO2−x-2.0.97 Reproduced with permission from ref. 97. Copyright 2023, Springer. (d) Preparation scheme of δ-MnO2 and δ-MnO2-CTAB samples. (e) Specific capacitance values of δ-MnO2-CTAB and δ-MnO2 (different current densities).99 Reproduced with permission from ref. 99. Copyright 2023, Elsevier. (f) XANES spectra of protonated MnO2, pH = 2 and 4 treated reassembled MnO2. (g) Comparison of specific capacitance of the samples as a function of current density. (h) Comparison of the charge transfer resistance among the three samples.103 Reproduced with permission from ref. 103. Copyright 2016, Springer Nature. | |
Unlike the anion vacancy, the formation energy for cationic vacancy is higher, making its construction more difficult. The similarity is that the cationic vacancy in metal oxides can significantly regulate the electronic structure, increase the density of states near the Fermi level, and improve their electrical conductivity. Moreover, the relatively low atomic escape energy of ultra-thin two-dimensional materials makes it easier to construct cationic vacancies. Manceau et al. confirmed that Mn atoms migrate steadily from the interlayer position (above or below the Mn layer vacancy), and the remaining intralayer Mn atoms shift toward the vacancy at lower pH, leading to Mn vacancies in the nanosheets.102 On the basis of this research, Gao and coworkers investigated the amount of Mn vacancies by reassembling the as-exfoliated birnessite at different pH values.103 This observation in X-ray absorption near edge structure (XANES) spectra (Fig. 7f) confirmed the existence of mixed oxidation states of Mn3+/Mn4+ in the samples. The Mn surface Frenkel defect content was controlled to a value of 26.5% and 19.9% at pH = 2 and 4, respectively, which was in direct proportion to capacitance due to increase of the Mn3+/Mn4+ ratio and the reduced charge transfer resistance in 1 mol L−1 Na2SO4 electrolyte (Fig. 7g and h). Besides the defects mentioned above, many lattice defects can also significantly change the physical and chemical properties, such as lattice dislocation, expansion, and deformation. However, there is still a lot of room for exploration, and many difficulties need to be overcome.
3.4 Element doping
Recently, researchers attempted to improve the electrochemical performance of layer manganese dioxide by using element doping, such as V, Mo, Fe, Ag, Co, Ca, etc.104–107 Element doping can improve the conductivity of layer manganese oxide electrode material and enhance the electron transport ability inside the material. At the same time, doping can introduce additional redox reaction center sites to enhance the energy storage capacity of the material. In addition, element doping can also improve the interface reaction performance of the material, slow down the side reaction between the electrode material and the electrolyte, and improve the electrochemical stability.108 Liu and coworkers prepared V-doped birnessite with different contents of vanadium atoms and investigated the supercapacitance performance (Fig. 8a).109 With an increase of V/Mn molar, the crystallinity of birnessite decreased (Fig. 8b), and the relative proportion of Mn3+ first increased and then decreased, while the relative proportion of Mn4+ first decreased and then increased; that is, a high doping concentration will eventually lead to a decreased Mn3+/Mn4+ redox couple, which could result in a decrease in the specific capacitance. Therefore, the highest specific capacitance of 245 F g−1 was obtained for doped birnessite with a V/Mn molar ratio of 0.14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (HB-V15%), with excellent cyclic stability (Fig. 8c). Xia's group also investigated the electrochemical properties of Cr-doped δ-MnO2. The specific capacitance first increased and then decreased with a higher doping concentration.110 Li et al. proposed group VIII metal (Fe, Co and Ni) doped δ-MnO2 using the hydrothermal method for aqueous Zn-ion batteries.111 XRD results stated that Fe-doping (FMO) might introduce lattice disorders or defects (Fig. 8d), with no change in the nanoflower morphology. The zinc ion diffusion path in pure δ-MnO2 (PMO) and FMO was modeled (Fig. 8e and f), and the lower energy diffusion barrier of FMO compared with PMO is expected to result in faster diffusion kinetics (Fig. 8g). In situ XRD and ex situ TEM elucidated a highly reversible phase conversion (δ-MnO2 → ZnxMnO2 → ZnMn2O4/Mn3O4 → ZnxMnO2 → δ-MnO2), giving rise to a high specific capacity of 338.2 mA h g−1 at 1 A g−1 with a capacity retention of 86.3% after 200 cycles, like that of Co and Ni-doping MnO2 electrode. However, the capacity of δ-MnO2 is mainly limited by the one-electron reaction of Mn4+/Mn3+ redox, which could be boosted through reduction of Mn oxygen state. Therefore, Xia et al. reported MnO2 based cathode with two-electron redox reaction through Mo doped δ-MnO2 (Mo–MnO2).112 Mo doping had no effect on the crystal structure and interlayer spacing (Fig. 8h), illustrating that the Mo atom partially replaced the Mn atom in the lamellar. The XPS result stated the existence of mixed valence states of Mn4+, Mn3+ and Mn2+ due to the introduction of Mo5+ dopant (Fig. 8i). Density functional theory (DFT) calculations stated that the narrowed band gap created excellent electric conductivity. The zinc ion storage mechanism of Mo–MnO2 (Fig. 8j) was based on a reversible two-step two-electron redox deduced by ex-site XRD, XPS and SEM images. As the cathode of the zinc-ion battery, the capacity of 652 mA h g−1 at 0.2 A g−1 and rate capability was better than that of pure MnO2 (Fig. 8k). Additionally, Ye et al. reported Se-dopant layer manganese dioxide (Se–MnO2) as a Zn-ion battery. The doping content of Se can control the ratio of H+ intercalation in MnO2 and inhibit the formation of ZnMn2O4 by-products, further creating excellent long-term cycle stability.113 Notably, element doping can increase the specific capacity and improve the stability, mainly because it greatly inhibits the elongations of Mn–O bonds along the z-axis to reduce Jahn–Teller distortion. Recently, Yang's group confirmed the role of Mg substitution in layered K0.5MnO2. Mg2+ substitution could promote the formation of Mn3+ ions with electron configurations eg(1)t2g(3) and t2g(4), effectively alleviating the structural deformation usually caused by Jahn–Teller distortion.114 Beyond that, Zhao et al. doped sulfur (S) anion and introduced oxygen vacancies into the layered manganese dioxide lattice by defect engineering. The preparation process and atomic structure model of S–MnO2 are shown Fig. 8l and m, respectively.115 S doping increased the intrinsic electron conductivity, and enhanced the diffusion kinetics of Zn ions, achieving good rate capability and excellent endurance cycle capacity of over 1000 cycles at a current density of 3 A g−1 (Fig. 8n). Nevertheless, the doping process may introduce new impurities, which may affect the purity and stability of the material, and doped elements may migrate or fall off at high temperature or humidity. Therefore, the doping conditions and element types need to be further studied.
:1 (HB-V15%), with excellent cyclic stability (Fig. 8c). Xia's group also investigated the electrochemical properties of Cr-doped δ-MnO2. The specific capacitance first increased and then decreased with a higher doping concentration.110 Li et al. proposed group VIII metal (Fe, Co and Ni) doped δ-MnO2 using the hydrothermal method for aqueous Zn-ion batteries.111 XRD results stated that Fe-doping (FMO) might introduce lattice disorders or defects (Fig. 8d), with no change in the nanoflower morphology. The zinc ion diffusion path in pure δ-MnO2 (PMO) and FMO was modeled (Fig. 8e and f), and the lower energy diffusion barrier of FMO compared with PMO is expected to result in faster diffusion kinetics (Fig. 8g). In situ XRD and ex situ TEM elucidated a highly reversible phase conversion (δ-MnO2 → ZnxMnO2 → ZnMn2O4/Mn3O4 → ZnxMnO2 → δ-MnO2), giving rise to a high specific capacity of 338.2 mA h g−1 at 1 A g−1 with a capacity retention of 86.3% after 200 cycles, like that of Co and Ni-doping MnO2 electrode. However, the capacity of δ-MnO2 is mainly limited by the one-electron reaction of Mn4+/Mn3+ redox, which could be boosted through reduction of Mn oxygen state. Therefore, Xia et al. reported MnO2 based cathode with two-electron redox reaction through Mo doped δ-MnO2 (Mo–MnO2).112 Mo doping had no effect on the crystal structure and interlayer spacing (Fig. 8h), illustrating that the Mo atom partially replaced the Mn atom in the lamellar. The XPS result stated the existence of mixed valence states of Mn4+, Mn3+ and Mn2+ due to the introduction of Mo5+ dopant (Fig. 8i). Density functional theory (DFT) calculations stated that the narrowed band gap created excellent electric conductivity. The zinc ion storage mechanism of Mo–MnO2 (Fig. 8j) was based on a reversible two-step two-electron redox deduced by ex-site XRD, XPS and SEM images. As the cathode of the zinc-ion battery, the capacity of 652 mA h g−1 at 0.2 A g−1 and rate capability was better than that of pure MnO2 (Fig. 8k). Additionally, Ye et al. reported Se-dopant layer manganese dioxide (Se–MnO2) as a Zn-ion battery. The doping content of Se can control the ratio of H+ intercalation in MnO2 and inhibit the formation of ZnMn2O4 by-products, further creating excellent long-term cycle stability.113 Notably, element doping can increase the specific capacity and improve the stability, mainly because it greatly inhibits the elongations of Mn–O bonds along the z-axis to reduce Jahn–Teller distortion. Recently, Yang's group confirmed the role of Mg substitution in layered K0.5MnO2. Mg2+ substitution could promote the formation of Mn3+ ions with electron configurations eg(1)t2g(3) and t2g(4), effectively alleviating the structural deformation usually caused by Jahn–Teller distortion.114 Beyond that, Zhao et al. doped sulfur (S) anion and introduced oxygen vacancies into the layered manganese dioxide lattice by defect engineering. The preparation process and atomic structure model of S–MnO2 are shown Fig. 8l and m, respectively.115 S doping increased the intrinsic electron conductivity, and enhanced the diffusion kinetics of Zn ions, achieving good rate capability and excellent endurance cycle capacity of over 1000 cycles at a current density of 3 A g−1 (Fig. 8n). Nevertheless, the doping process may introduce new impurities, which may affect the purity and stability of the material, and doped elements may migrate or fall off at high temperature or humidity. Therefore, the doping conditions and element types need to be further studied.
 |
| | Fig. 8 (a) Schematic diagram of V-doped birnessite. (b) XRD patterns of birnessites. (c) Charge/discharge profiles of HB, HB-V5%, HB-V10%, HB-V15%, and HB-V20% at 100 mA g−1.109 Reproduced with permission from ref. 109. Copyright 2015, Elsevier. (d) XRD spectra of PMO and FMO. (e and f) Zn ion diffusion path in PMO and FMO, respectively. (g) Lowest energy barrier for the diffusion of Zn ions through the FMO and PMO.111 Reproduced with permission from ref. 111. Copyright 2022, Elsevier. (h) TEM image of Mo–MnO2. (i) XPS of Mn atom. (j) Schematic of the mechanism for Zn//Mo–MnO2 during the charging/discharging process. (k) Rate test of MnO2 and Mo–MnO2.112 Reproduced with permission from ref. 112. Copyright 2022, Springer. (l) Illustration of the preparation process, (m) atomic structure model, and (n) cycling performance of S–MnO2.115 Reproduced with permission from ref. 115. Copyright 2022, Elsevier. | |
3.5 Expanding interlayer spacing
The capacitance of layer manganese dioxide acting as the electrode material of the supercapacitor is mainly composed of surface EDL capacitance, surface redox pseudocapacitance and intercalated pseudocapacitance in the bulk. Meanwhile, the capacity is dominated by the (de)intercalation of charge carriers as the electrode material of battery. Therefore, a reasonable interlayer distance is one of the most indispensable parameters to obtain high electrochemical performance. A comprehensive review of the literature stated that the pre-intercalation strategy involving the insertion of water molecules,116 guest ions or polymers into the layer manganese dioxide host structure before cycling is an advanced way to boost interlayer distance.117–119 Specially, Zhao et al. reviewed the interlayer pre-intercalation strategy and results in manganese oxides.120 The interlayer pre-intercalation can simultaneously improve the electronic conductivity, activate more active sites, promote diffusion kinetics, stabilize structure integrity, and depress the phase transition of MnO2 cathode materials.
3.5.1 Guest ion pre-intercalation.
Intercalated guest ions including single valent (Li+, Na+, and K+) and multivalent metal ions (Ca2+, Zn2+, La3+, and Ce3+) could reduce the bandgap between the valence and conductive bonds, contributing to higher intrinsic electronic/ionic conductivity. Beyond that, the functionality of pre-intercalation has been proposed, such as creating active sites, facilitating ionic diffusion kinetics and stabilizing the crystal structure during the charge/discharge process.121–123 Ma et al. synthesized Li+-intercalated vis a one-pot process and studied the electrochemical behavior in the sulfate-based electrolyte (Li2SO4, Na2SO4, K2SO4, Rb2SO4, and Cs2SO4).124 Li-manganese dioxide displayed the highest charge storage capacity in 0.5 mol L−1 Na2SO4, which mainly depended on the synergistic effect of the hydrated ion diameter of the electrolytic cation, the active site utilization of Mn and the change in the oxidation state (Fig. 9a). The charge storage mechanism of the layered manganese dioxide nanosheet is shown in Fig. 9b. Additionally, single valent alkali ions pre-intercalated layered MnO2 was prepared to use as a cathode in zinc ion batteries. Potassium ion pre-intercalated layered MnO2 nanosheet (K0.27MnO2·0.54H2O, known as KMO) was prepared by Simon's group via a rapid molten salt method (Fig. 9c).125 The XPS spectrum illustrated that the interlayer water molecules formed chemical bonds with the MnO2 layers (Fig. 9d). The pillaring effect of K+ and water crystals benefited for the good structure stability of KMO and a rapid diffusion of cations in the KMO structure, contributing to high power capability (90 mA h g−1 at 10C) and good cycling stability (91% after 1000 cycles), as shown in Fig. 9e. However, the univalent ion pre-intercalation has a limitation on the expansion of layer spacing. Multivalent metal ions (e.g., Co2+, Pb2+, and Y3+) intercalated layer manganese dioxides improve the electrochemical characteristics.14,126,127 However, it is a pity that these intercalants are toxic and/or expensive. Therefore, Long et al. produced multivalent metal ions (Cu2+ and Bi3+) intercalated manganese dioxide (CuMO and BiMO) using the hydrothermal method for aqueous zinc ion batteries.128 The XRD pattern indicated the co-intercalation of copper ion, bismuth ion and water molecules (Fig. 9f). CuMO displayed the highest capacity of 493.3 mA h g−1 at 100 mA g−1 because of the additional capacity contribution based on the transformation reaction between CuMO and metallic Cu. BiMO delivered the longest cycle life with a capacity retention of 98.6% after 1100 cycles at 1 A g−1 due to the pillar role of pre-intercalated Bi3+ and water molecules, which were superior to that of pure δ-MnO2 (Fig. 9g). Moreover, lower overpotential of CuMO and BiMO manifested faster reaction kinetics compared with δ-MnO2. Yadav et al. reported Cu2+ intercalated Bi-birnessite (Bi–δ-MnO2) cathode using metallic Cu as an additive for the zinc battery to integrate the excellent electrochemical performances of the electrode material.129 During charging, Cu transferred to Cu2+ to intercalate in the interlayer regions of Bi-δ-MnO2 at the redox potential, and was reduced to Cu0 with the Mn(OH)2-layered material during discharge (Fig. 9h). Cu addition contributed towards the stability and electronic/ionic conductivity of Bi-δ-MnO2, resulting in an excellent areal capacity and cycle life. This wonderful idea can be applied to layered materials in other fields. Although the intercalation of metal ion in layered MnO2 can effectively enhance the stability and electronic/ionic conductivity, the expanded layer spacing is still less than 0.8 nm, which seriously prevents the movement of large carriers, such as Ca2+, Mg2+ and Al3+. It is particularly important to further expand the layer spacing and maintain stability.
 |
| | Fig. 9 (a) Specific capacitance as a function of current densities for various electrolytes. (b) Schematic diagram of the layer manganese oxide charge storage mechanism via electrochemical intercalation/deintercalation of alkali cation.124 Reproduced with permission from ref. 124. Copyright 2020, Elsevier. (c) Low resolution TEM image of K0.27MnO2·0.54H2O. (d) XPS spectrum of O 1s. (e) Discharge–charge curves at various current rates.125 Reproduced with permission from ref. 125. Copyright 2021, Wiley-VCH. (f) XRD pattern of as-synthesized CuMO, BiMO, and δ-MnO2. (g) Charge–discharge curves of CuMO, BiMO, and δ-MnO2 at 0.1 A g−1 (in the second cycle).128 Reproduced with permission from ref. 128. Copyright 2022, Elsevier. (h) Electrochemical reactions for the regeneration cycle of Cu2+-intercalated Bi-birnessite.129 Reproduced with permission from ref. 129. Copyright 2017, Springer Nature. | |
3.5.2 Organic ions/polymer pre-intercalation.
The larger size of organic ions or polymer compared with metal ions can effectively extend the layer spacing of layered manganese dioxide to 0.9 nm, and even 1.3 nm. In general, organic ions/polymers are difficult to intercalate in layered manganese dioxide by the one-step hydrothermal method and calcination method because they tend to form composites rather than intercalation. Therefore, ion-exchange and interface reactions are the most commonly used methods. Based on the swelling and delamination behaviors of the protonated birnessite in tetrabutylammonium hydroxide (TMAOH) solutions, TMA+ intercalated birnessite (TMA-birnessite) with large layer spacing was prepared via ion-exchange reactions for electrochemical energy storage.33,43,130 Zhao et al. also used the ion exchange method to prepare TMA-birnessite with different molar ratio of TMA+/H+ for pseudocapacitors (Fig. 10a).131 When TMA+/H+ increased up to 1000, H+ is almost totally replaced by TMA+, resulting in an enlarged interlayer spacing of 0.96 nm. The ion diffusion tunnels were enlarged thanks to the expansion of interlayer spacing, and the layer interaction and resistance of charge diffusion decreased. Therefore, TMA+/H+ = 1000 displayed the largest specific capacitance of 585 F g−1 and the best rate capability of 86% from 1 to 20 A g−1 (Fig. 10b). Wang's group reported TMA-birnessite through a two-step ion-exchange reaction from K-birnessite to H-birnessite to TMA-birnessite for bivalent Mg-ion storage.132 The exchange of K+ and TMA+ in the interlayer made the interlayer spacing expand from 0.70 to 0.97 nm, which was beneficial to a faster diffusion of Mg2+ and lower overpotential. TMA-birnessite delivered twice as much specific capacity as K-birnessite with excellent rate performance. Polymer-intercalated layered manganese dioxide have been intensively investigated to further enhance layered spacing. Zhang and co-workers proposed polyvinylpyrrolidone (PVP) pre-intercalation δ-MnO2 (PVP–MnO2) for Zn-ion batteries (Fig. 10c).133 The as-prepared PVP-MnO2 exhibited a terrace-shape hybrid superlattice structure and an expanded interlayer spacing of 1.14 nm more than TMA-birnessite reported previously, which was beneficial to Zn2+/H+ co-insertion. Obviously pronounced redox intensity and reduced potential hysteresis in CV profiles (Fig. 10d) illustrated the improved electrochemical reactivity and diffusion kinetics due to PVP pre-intercalation. PVP-MnO2 delivered impressive electrochemical performance, such as a high specific capacity (317.2 mA h g−1 at 0.125 A g−1), improved rate performance (106.1 mA h g−1 at 12.5 A g−1) and excellent cycle stability (capacity retention of almost 100% after 20000 cycles at 10 A g−1). Polyquaternium-2 (PQN2) was intercalated in layered manganese oxide through a delamination/reassembling process, resulting in an expanded interlayer spacing of 0.94 nm.134 Beyond that, Xia's group designed polyaniline-intercalated layered manganese dioxide using a simple one-step inorganic/organic interface reaction.135,136 The formation of PANI restricted the growth of 2D MnO2, eventually resulting in a mesoporous structure (Fig. 10e).136 A high specific capacity of 298 mA h g−1 was obtained after subsequent cycles thanks to the typical nanosize (∼10 nm), expanded interlayer space (∼1.0 nm), uniform mesostructure (∼4 nm) and polymer-reinforced layered structure, which was barely the theoretical specific capacity of 308 mA h g−1. Meanwhile, an amazing rate performance (Fig. 10f) and cycle stability was observed without phase transformation and collapse of the layered structure. However, the yield of the interface reaction is low, and polyaniline intercalation and coating coexist. This causes difficulties to quantify the insertion amount of polyaniline. Zuo et al. developed MnO2-PANI hybrid cathode (MnO2–P) with PANI intercalation and coating for Ca-ion batteries.137 The presence of PANI improves the conductivity and enlarges the interlayer spacing, facilitating electron transport and Ca2+ diffusion. Moreover, the formed Mn–N bond based on the PANI coating can enhance structural stability (Fig. 10g). The above characteristics gave excellent electrochemical performances to the MnO2-PANI hybrid cathode, including high capacity, outstanding rate performance (Fig. 10h) and a long-term cycle. These results indicate that the interlayer spacing is efficiently expanded in favour of ion transport by the organic ions/polymer pre-intercalation strategy. However, the current research is limited to expanding the layer spacing, and the correlation between layer spacing and electrochemical properties of aluminum has not been studied. In addition, there are still some questions that need to be further clarified, such as whether the intercalation amount has a negative effect on the ion transport rate and mechanical structure stability, and how to achieve accurate regulation of the intercalation amount.
 |
| | Fig. 10 (a) Schematic illustration of the ion-exchange process. (b) Comparison of specific capacitance at different current densities.131 Reproduced with permission from ref. 131. Copyright 2017, Elsevier. (c) Schematic of PVP intercalation in δ-MnO2. (d) CV profiles of MnO2 and PVP-MnO2.133 Reproduced with permission from ref. 133. Copyright 2023, Wiley-VCH. (e) Schematic of the expanded intercalated structure of PANI-intercalated MnO2 nanolayers. (f) Rate performance of PANI-intercalated MnO2.136 Reproduced with permission from ref. 136. Copyright 2018, Springer Nature. (g) Schematic of the synthesis of MnO2–P organic–inorganic nanosheets. (h) Rate performance of MnO2 and MnO2–P.137 Reproduced with permission from ref. 137. Copyright 2023, Wiley-VCH. | |
4. Summary and outlook
Layer manganese dioxide is still one of the preferred materials for supercapacitors and various ion batteries. Researchers have proposed a variety of strategies on the macro and micro levels to improve the electrochemical performance. In this review, we summarized the latest advances in the study of charge-storage mechanisms and related improvement strategies, including preparing conductive composite, novel nano-structural design, defect engineering, element doping and expanding interlayer spacing. These strategies improve electrical/ionic conductivity, increase electrochemically active sites and stabilize the layer structure, while providing the foundation and inspiration for future research. However, there is a long way to go for attaining the theoretical electrochemical performance levels for the available practical electrodes, for which advanced strategies should be employed.
4.1 Porous layer-by-layer assembly
The low utilization of layer manganese dioxide is a challenge to obtain high capacitance/capacity. However, it is feasible to maximize the specific surface area by assembling the manganese dioxide layer with other conductive two-dimensional material to enhance the utilization of layer manganese dioxide as an electrode. Furthermore, creating pores on the layers can achieve a desirable ionic conductivity. This idea deserves more experiments.
4.2 Special element doping
It has been confirmed that element doping (metal or non-metal elements) is an effective way to improve the performance of layer manganese dioxide. It is challenging to uncover the relationship between doping concentration and electrochemical properties. In addition, element doping technology can be combined with other modification strategies to further improve the performance of manganese oxide electrode materials, such as nanostructure design, layer engineering, etc.
4.3 Coating and pre-intercalation integrated design
The pre-intercalation of polymer in layer manganese dioxide can expand the layer spacing to about 1.0 nm, facilitate the fast transfer of electrolyte ion, and accelerate the kinetic process. It will be interesting to use conductive polymer to pre-intercalate manganese dioxide and to achieve coating at the same time, which may facilitate the simultaneous transfer of electrons and ions. The interaction between layer spacing and electrochemical properties needs further investigation.
4.4 Research on energy storage mechanism
Layer manganese dioxide exhibits different energy storage mechanism as an electrode material of various kinds of batteries, which urgently needs to be understood and explored using various spectral approaches. Meanwhile, the energy storage mechanism of manganese dioxide may be different within different potential windows, which can be studied according to the high voltage characteristics of high concentration aqueous electrolytes.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
Y. Z., H. X., X. H. Z., Z. G. and B. B. X. developed the concept and performed literature scoping and review with M. E., X. Z. The copyright of literatures were acquired by H. X., X. Z. and K. J. Figures were processed by Y. Z., X. Z. and K. J. All authors were involved in drafting the manuscript with the special assistances from M. E. and K. J. to adjust the format of manuscripts. X. H. Z., Z. G. and B. B. X. critically reviewed the context. The manuscript was proofread by M. E., X. H. Z. and Z. G. Y. Z., Z. G. and B. B. X. provided the funding, administration and supervision to support the project. The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2025-80-01”.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the Gansu Provincial Youth Science and Technology Fund Projects (No. 23JRRA1371), the National College Students’ Innovation and Entrepreneurship Training Program (No. 202316209003). B. B. X. is grateful for the support from UK Engineering and Physical Sciences Research Council (EPSRC, UK) RiR Grant RIR18221018-1. The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA for funding this research work through the project number “NBU-FFR-2025-80-01”.
Notes and references
- H. Zhao, G. Wilde and Y. Lei, Small Methods, 2019, 3, 1900283 CrossRef.
- R. Sharma, H. Kumar, G. Kumar, S. Sharma, R. Aneja, A. K. Sharma, R. Kumar and P. Kumar, Chem. Eng. J., 2023, 468, 143706 CrossRef CAS.
- M. Das, ES Energy Environ., 2024, 24, 1128 CAS.
- N. Jabeen, Q. Xia, S. V. Savilov, S. M. Aldoshin, Y. Yu and H. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 33732–33740 CrossRef CAS PubMed.
- S. Vyas, A. Shuklaand, S. Shivhare, R. Das and R. Venkatesh, ES Mater Manuf., 2024, 23, 1002 CAS.
- Y. Liao, C. Yang, J. Bai, Q. He, H. Wang, H. Chen, Q. Zhang and L. Chen, Chem. Sci., 2024, 15, 7441–7473 RSC.
- Y. Guo, C. Guo, P. Huang, Q. Han, F. Wang, H. Zhang, H. Liu, Y.-C. Cao, Y. Yao and Y. Huang, eScience, 2023, 3, 100091 CrossRef.
- S. Guo, L. Qin, J. Wu, Z. Liu, Y. Huang, Y. Xie, G. Fang and S. Liang, Natl. Sci. Rev., 2024, 11, nwae181 CrossRef CAS.
- S. Naeem, S. Av, S. Up and P. Av, ES Energy Environ., 2023, 21, 915 CAS.
- J. Shin, J. K. Seo, R. Yaylian, A. Huang and Y. S. Meng, Int. Mater. Rev., 2019, 65, 356–387 CrossRef.
- N. D. Ingale, J. W. Gallaway, M. Nyce, A. Couzis and S. Banerjee, J. Power Sources, 2015, 276, 7–18 CrossRef CAS.
- C. Julien, M. Massot, S. Rangan, M. Lemal and D. Guyomard, J. Raman Spectroscopy, 2002, 33, 223–228 CrossRef CAS.
- L. Zhang, Z.-H. Liu, X. Tang, J. Wang and K. Ooi, Mater. Res. Bull., 2007, 42, 1432–1439 CrossRef CAS.
- Q. Zhang, J. Zhao, X. Chen, R. Yang, T. Ying, C. Cheng, B. Liu, J. Fan, S. Li and Z. Zeng, Adv. Funct. Mater., 2024, 34, 2306652 CrossRef CAS.
- T. Xiong, T. L. Tan, L. Lu, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2018, 8, 1702630 CrossRef.
- D. Wang, L. Wang, G. Liang, H. Li, Z. Liu, Z. Tang, J. Liang and C. Zhi, ACS Nano, 2019, 13, 10643–10652 CrossRef CAS PubMed.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330–2339 RSC.
- N. Naresh, S. Eom, S. J. Lee, S. H. Jeong, J.-W. Jung, Y. H. Jung and J.-H. Kim, Energy Environ. Mater., 2024, 7, e12640 CrossRef CAS.
- A. R. Akbar, A. Saleem, A. Rauf, R. Iqbal, M. Tahir, G. Q. Peng, A. S. Khan, A. Hussain, M. Ahmad, M. Akhtar, M. Ali, C. X. Xiong, Q. L. Yang, G. Ali and F. D. Liu, J. Power Sources, 2023, 579, 233181 CrossRef CAS.
- G. Wang, Y. Wang, B. Guan, J. Liu, Y. Zhang, X. Shi, C. Tang, G. Li, Y. Li, X. Wang and L. Li, Small, 2021, 17, 2104557 CrossRef CAS PubMed.
- Z. Zhuang, W. Wang, Y. Wei, T. Li, M. Ma and Y. Ma, Adv. Compos. Hybrid Mater., 2021, 4, 938–945 CrossRef CAS.
- Y. Liu, Y. Qiao, X. Lou, X. Zhang, W. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 14564–14571 CrossRef CAS.
- N. Jabeen, A. Hussain, Q. Xia, S. Sun, J. Zhu and H. Xia, Adv. Mater., 2017, 29, 1700804 CrossRef PubMed.
- M.-W. Xu, W. Jia, S.-J. Bao, Z. Su and B. Dong, Electrochim. Acta, 2010, 55, 5117–5122 CrossRef CAS.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- J. Chen and P. S. Lee, Adv. Energy Mater., 2020, 11, 2003311 CrossRef.
- J. Cao, X. Li, Y. Wang, F. C. Walsh, J.-H. Ouyang, D. Jia and Y. Zhou, J. Power Sources, 2015, 293, 657–674 CrossRef CAS.
- C. C. Hu and T. W. Tsou, Electrochim. Acta, 2002, 47, 3523–3532 CrossRef CAS.
- S. C. Pang, M. A. Anderson and T. W. Chapman, J. Electrochem. Soc., 2000, 147, 444 CrossRef CAS.
- C. Xu, B. Li, H. Du, F. Kang and Y. Zeng, J. Power Sources, 2008, 184, 691–694 CrossRef CAS.
- Y. Zhang, Y. An, J. Jiang, S. Dong, L. Wu, R. Fu, H. Dou and X. Zhang, Energy Technol., 2018, 6, 2146–2153 CrossRef CAS.
- Y. Zhao, X. Zhu, Q. Zhang, L. Gu, Z. Shi, C. Qiu, T. Chen, M. Ni, Y. Zhuang, S. V. Savilov, S. M. Aldoshin and H. Xia, ACS Nano, 2024, 18, 32556–32568 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS.
- X. Shan, F. Guo, D. S. Charles, Z. Lebens-Higgins, S. Abdel Razek, J. Wu, W. Xu, W. Yang, K. L. Page, J. C. Neuefeind, M. Feygenson, L. F. J. Piper and X. Teng, Nat. Commun., 2019, 10, 4975 CrossRef.
- B. Lin, X. Zhu, L. Fang, X. Liu, S. Li, T. Zhai, L. Xue, Q. Guo, J. Xu and H. Xia, Adv. Mater., 2019, 31, 1900060 CrossRef PubMed.
- Q. Zhang, M. D. Levi, Q. Dou, Y. Lu, Y. Chai, S. Lei, H. Ji, B. Liu, X. Bu, P. Ma and X. Yan, Adv. Energy Mater., 2018, 9, 1802707 CrossRef.
- S. Boyd, K. Ganeshan, W.-Y. Tsai, T. Wu, S. Saeed, D.-e. Jiang, N. Balke, A. C. T. van Duin and V. Augustyn, Nat. Mater., 2021, 20, 1689–1694 CrossRef CAS.
- X.-Z. Zhai, J. Qu, S.-M. Hao, Y.-Q. Jing, W. Chang, J. Wang, W. Li, Y. Abdelkrim, H. Yuan and Z.-Z. Yu, Nano-Micro Lett., 2020, 12, 1–15 CrossRef PubMed.
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, Small, 2020, 16, 2001228 CrossRef CAS.
- Z. Liu, M. Qin, B. Fu, M. Li, S. Liang and G. Fang, Angew. Chem., Int. Ed., 2025, 64, e202417049 CrossRef CAS.
- C. Luo, H. Lei, Y. Xiao, X. Nie, Y. Li, Q. Wang, W. Cai, C. Dai, M. Yao, Y. Zhang and D. Yuan, Energy Mater, 2024, 4, 400036 CAS.
- Z. Xu, R. Ren, H. Ren, J. Zhang, J. Yang, J. Qiu, Y. Zhang, G. Zhu, L. Huang and S. Dong, Front. Optoelectron., 2023, 16, 39 CrossRef PubMed.
- Y.-R. Tsai, T.-Y. Chen, Z.-Q. Yang, C.-C. Tsai, Y.-T. Huang, K. S. Anuratha, T.-W. Lin, Z. Sofer and J.-Y. Lin, 2D Mater., 2023, 10, 024001 CrossRef.
- S. Cui, D. Zhang and Y. Gan, Adv. Energy Mater., 2024, 14, 2302655 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Chem. Sci., 2019, 10, 5656–5666 RSC.
- T. Serikov, E. Seliverstova, A. Sadykova, N. Ibrayev and N. Nuraje, Eng. Sci., 2024, 32, 1327 Search PubMed.
- H. Luo, L. Wang, P. Ren, J. Jian, X. Liu, C. Niu and D. Chao, Nano Res., 2022, 15, 8603–8612 CrossRef CAS.
- Z. T. Gossage, D. Igarashi, Y. Fujii, M. Kawaguchi, R. Tatara, K. Nakamoto and S. Komaba, Chem. Sci., 2024, 15, 18272–18294 RSC.
- Y. Chen, Y. Zhang, D. Geng, R. Li, H. Hong, J. Chen and X. Sun, Carbon, 2011, 49, 4434–4442 CrossRef CAS.
- L. Li, Z. A. Hu, N. An, Y. Y. Yang, Z. M. Li and H. Y. Wu, J. Phy. Chem. C, 2014, 118, 22865–22872 CrossRef CAS.
- X. Liu, B. Liang, X. Hong and J. Long, Front. Chem., 2022, 10, 870541 CrossRef CAS PubMed.
- L. Peng, X. Peng, B. Liu, C. Wu, Y. Xie and G. Yu, Nano Lett., 2013, 13, 2151–2157 CrossRef CAS PubMed.
- P. Wu, S. Cheng, L. Yang, Z. Lin, X. Gui, X. Ou, J. Zhou, M. Yao, M. Wang, Y. Zhu and M. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 23721–23728 CrossRef CAS.
- N. An, J. Xin, W. Li, Z. Guo, L. Shang, Y. He, L. Lv, D. Sun, Y. Zhang and Z. Hu, Appl. Surface Sci., 2022, 599, 153881 CrossRef CAS.
- W. Zheng, Z. Cui, C. Liu, L. Yuan, S. Li, L. Lin, T. Meng, L. Yang, Y. Tong and D. Shu, Chem. Sci., 2024, 15, 20303–20314 RSC.
- X. Zhai, Z. Mao, G. Zhao, D. Rooney, N. Zhang and K. Sun, J. Power Sources, 2018, 402, 373–380 CrossRef CAS.
- Y. Yang, Y. Yang, X. Qi, T. Zeng, Y. Wang and S. Yuan, J. Electroanal. Chem., 2024, 972, 118610 CrossRef CAS.
- A. K. Sundramoorthy, Y.-C. Wang and S. Gunasekaran, Nano Res., 2015, 8, 3430–3445 CrossRef CAS.
- Y.-J. Lee, G.-H. An and H.-J. Ahn, J. Nanosci. Nanotechnol., 2015, 15, 8931–8936 CrossRef CAS.
- H. Hu, Z. Pei, H. Fan and C. Ye, Small, 2016, 12, 3059–3069 CrossRef CAS PubMed.
- Y. Dai, S. Tang, X. Wang, X. Huang, C. Zhu, Z. Hang and X. Meng, Chem. Lett., 2014, 43, 122–124 CrossRef CAS.
- Y. Han and H. V. Huynh, Dalton Trans., 2011, 40, 2141–2147 RSC.
- H. Jiang, J. Ma and C. Li, J. Mater. Chem., 2012, 22, 16939–16942 RSC.
- A. Bahloul, B. Nessark, E. Briot, H. Groult, A. Mauger, K. Zaghib and C. M. Julien, J. Power Sources, 2013, 240, 267–272 CrossRef CAS.
- S. Grover, S. Shekhar, R. K. Sharma and G. Singh, Electrochim. Acta, 2014, 116, 137–145 CrossRef CAS.
- G. Zhang, J. Zhu, L. Lin, Y. Liu, S. Li, Q. Li, X.-X. Liu and X. Sun, Chem. Sci., 2024, 15, 3545–3551 RSC.
- S. Vyasa, A. Shukla, S. Shivhare, R. Das and V. R, ES Mater. Manuf., 2024, 23, 1002 Search PubMed.
- H. Tang, W. Chen, N. Li, Z. Hu, L. Xiao, Y. Xie, L. Xi, L. Ni and Y. Zhu, Energy Storage Mater., 2022, 48, 335–343 CrossRef.
- D.-S. Liu, Y. Mai, S. Chen, S. Liu, E. H. Ang, M. Ye, Y. Yang, Y. Zhang, H. Geng and C. C. Li, Electrochim. Acta, 2021, 370, 137740 Search PubMed.
- Z. Jia, J. Wang, Y. Wang, B. Li, B. Wang, T. Qi and X. Wang, J. Mater. Sci. Technol., 2016, 32, 147–152 Search PubMed.
- Y. Y. Rivera-Lugo, R. M. Félix-Navarro, B. Trujillo-Navarrete, E. A. Reynoso-Soto, C. Silva-Carrillo, C. A. Cruz-Gutiérrez, E. Quiroga-González and J. C. Calva-Yáñez, Fuel, 2021, 287, 19463 Search PubMed.
- C. Tang, Y. Zhang, S. Lu, Y. Zeng, Q. Hua and Y. Zhang, ACS Appl. Nano Mater., 2025, 8, 1568–1576 CrossRef CAS.
- Q. Xie, L. Huang, Z. Liang, S. Tang, W. Ling, Q. Huang, Z. Zhou, X. Su, T. Xue and G. Cheng, J. Electron. Mater., 2022, 52, 41–49 CrossRef.
- S. I. Yun, J.-W. Song and B.-H. Kim, ACS Appl. Nano Mater., 2022, 5, 5688–5698 CrossRef CAS.
- H. Tang, C. Liu, R. Zhou, T. Cai, C. Guo, X. Liu and Y. Zhu, J. Power Sources, 2023, 566, 232915 CrossRef CAS.
- W. Xiao, W. Zhou, H. Yu, Y. Pu, Y. Zhang and C. Hu, Electrochim. Acta, 2018, 264, 1–11 Search PubMed.
- A. Ramakrishnan, S. Swaminathan, J. Mayandi and D. K G, Nano Express, 2024, 5, 015024 CrossRef CAS.
- X. Fu, X. Wang, Y. Chen, W. Huo, X. Liu, K. Chen, F. Dong, H.-C. Yao and Y. Zhang, J. Colloid. Interfaces Sci., 2019, 557, 168–173 CrossRef CAS.
- R. Zhang, P. Liang, H. Yang, H. Min, M. Niu, S. Jin, Y. Jiang, Z. Pan, J. Yan, X. Shen and J. Wang, Chem. Eng. J., 2022, 433, 133687 CrossRef CAS.
- C. Tanggarnjanavalukul, N. Phattharasupakun, K. Kongpatpanich and M. Sawangphruk, Nanoscale, 2017, 9, 13630–13639 Search PubMed.
- L. Wang, M. Huang, S. Chen, L. Kang, X. He, Z. Lei, F. Shi, H. Xu and Z.-H. Liu, J. Mater. Chem. A, 2017, 5, 19107–19115 Search PubMed.
- X. Li, J. Mao and F. Cheng, ACS Appl. Energy Mater., 2024, 7, 7808–7817 CrossRef CAS.
- Y. Hu, J. Wang, X. Jiang, Y. Zheng and Z. Chen, Appl. Surf. Sci., 2013, 271, 193–201 CrossRef CAS.
- M. H. Alfaruqi, J. Gim, S. Kim, J. Song, D. T. Pham, J. Jo, Z. Xiu, V. Mathew and J. Kim, Electrochem. Commun., 2015, 60, 121–125 CrossRef CAS.
- M. Pang, G. Long, S. Jiang, Y. Ji, W. Han, B. Wang, X. Liu and Y. Xi, Electrochim. Acta, 2015, 161, 297–304 CrossRef CAS.
- H. Xu, Y. Du, A. Emin, X. Long, Y. Fu, Y. Li, J. Li, D. Liu and D. He, J. Electrochem. Soc., 2021, 168, 030540 CrossRef CAS.
- A. Xia, W. Yu, J. Yi, G. Tan, H. Ren and C. Liu, J. Electroanal. Chem., 2019, 839, 25–31 CrossRef CAS.
- Q. Wei, Y. Fu, G. Zhang, D. Yang, G. Meng and S. Sun, Nano Energy, 2019, 55, 234–259 CrossRef CAS.
- Y. Wang, Y.-Z. Zhang, D. Dubbink and J. E. ten Elshof, Nano Energy, 2018, 49, 481–488 CrossRef CAS.
- D. Zhang, J. Dai, J. Zhang, Y. Zhang, H. Liu, Y. Xu, J. Wu and P. Li, ACS Omega, 2024, 9, 18032–18045 CrossRef CAS.
- S. Bag and C. R. Raj, J. Mater. Chem. A, 2016, 4, 8384–8394 RSC.
- S. Khamsanga, R. Pornprasertsuk, T. Yonezawa, A. A. Mohamad and S. Kheawhom, Sci. Rep., 2019, 9, 8441 CrossRef.
- J. Wu, J. Chen, X. Chen, Y. Liu, Z. Hu, F. Lou, S. Chou and Y. Qiao, Chem. Sci., 2024, 15, 9591–9598 Search PubMed.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. N. Waterhouse, W. Huang and T. Zhang, Adv. Energy Mater., 2017, 7, 1700005 CrossRef.
- Y. Zheng, Z. Yao, Z. Shadike, M. Lei, J. Liu and C. Li, Adv. Funct. Mater., 2021, 32, 2107060 CrossRef.
- R. Samal, M. Kandasamy, B. Chakraborty and C. S. Rout, Int. J. Hydrogen Energy, 2021, 46, 28028–28042 CrossRef CAS.
- Y. Wang, Y. Zhang, G. Gao, Y. Fan, R. Wang, J. Feng, L. Yang, A. Meng, J. Zhao and Z. Li, Nano-Micro Lett., 2023, 15, 219 CrossRef CAS.
- Z. Peng, Y. Huang, A. G. Bannov, S. Li, L. Tang, L. Tan and Y. Chen, Energy Environ. Sci., 2024, 17, 3384–3395 RSC.
- C. Tang, X. Wang, M. Ma, Z. Wang, Y. Li, H. Li, B. Li, Y. Zhang and X. Zhu, Chem. Eng. J., 2023, 471, 144784 CrossRef CAS.
- Y. Wang, H. Quan, Q. Zhang, B. Tan, W. Chen and D. Chen, ACS Appl. Nano Mater., 2024, 7, 27988–27997 CrossRef CAS.
- W. Lv, J. Meng, Y. Li, W. Yang, Y. Tian, X. Lyu, C. Duan, X. Ma and Y. Wu, Nano Energy, 2022, 98, 107274 CrossRef CAS.
- A. Manceau, M. A. Marcus, S. Grangeon, M. Lanson, B. Lanson, A.-C. Gaillot, S. Skanthakumar and L. Soderholm, J. Appl. Crystallogr., 2013, 46, 193–209 CrossRef CAS.
- P. Gao, P. Metz, T. Hey, Y. Gong, D. Liu, D. D. Edwards, J. Y. Howe, R. Huang and S. T. Misture, Nat. Commun., 2017, 8, 14559 CrossRef CAS.
- Q. Gao, J. Wang, B. Ke, J. Wang and Y. Li, Ceram. Int., 2018, 44, 18770–18775 CrossRef CAS.
- S. Yang, F. Li, P. Fu, C. Zhen, J. Wu, Y. Feng, H. Lu and Z. Sheng, J. Power Sources, 2024, 611, 234767 CrossRef CAS.
- X. Pu, X. Li, L. Wang, H. Maleki Kheimeh Sari, J. Li, Y. Xi, H. Shan, J. Wang, W. Li, X. Liu, S. Wang, J. Zhang and Y. Wu, ACS Appl. Mater. Interfaces, 2022, 14, 21159–21172 CrossRef CAS PubMed.
- K. Ma, G. Liang, Q. Jiao, H. Jin, Y. Su, N. Li, J. Li, Z. Xiong, C. Feng and Y. Zhao, J. Mater. Chem. C, 2025, 13, 4727–4736 RSC.
- S. Wei, Y. Wang, S. Chen and L. Song, Chem. Sci., 2024, 15, 7848–7869 RSC.
- L. Liu, M. Min, F. Liu, H. Yin, Y. Zhang and G. Qiu, J. Power Sources, 2015, 277, 26–35 CrossRef CAS.
- Y. Zhao, Q. Fang, X. Zhu, L. Xue, M. Ni, C. Qiu, H. Huang, S. Sun, S. Li and H. Xia, J. Mater. Chem. A, 2020, 8, 8969–8978 RSC.
- H. Li, Z. Huang, B. Chen, Y. Jiang, C. Li, W. Xiao and X. Yan, J. Power Sources, 2022, 527, 231198 CrossRef CAS.
- X. Xia, Y. Zhao, Y. Zhao, M. Xu, W. Liu and X. Sun, Nano Res., 2022, 16, 2511–2518 CrossRef.
- J. J. Ye, P. H. Li, Z. Hou, W. Zhang, W. Zhu, S. Jin and H. Ji, Angew. Chem., Int. Ed., 2024, 136, e202410900 CrossRef.
- Z. Hou, Y. Liu, S. Yao, S. Wang, Y. Ji, W. Fu, J. Xie, Y.-M. Yan and Z. Yang, Mater. Horiz., 2024, 11, 5674–5683 RSC.
- Y. Zhao, P. Zhang, J. Liang, X. Xia, L. Ren, L. Song, W. Liu and X. Sun, Energy Storage Mater., 2022, 47, 424–433 CrossRef.
- K. W. Nam, H. Kim, J. H. Choi and J. W. Choi, Energy Environ. Sci., 2019, 12, 1999–2009 RSC.
- C.-Y. Tsai, T.-S. Su, T.-C. Wei and M.-S. Wu, Electrochim. Acta, 2022, 430, 141107 CrossRef CAS.
- L. L. Cao, B. Z. Yu, T. Cheng, X. L. Zheng, X. H. Li, W. L. Li, Z. Y. Ren and H. M. Fan, Ceram. Int., 2017, 43, 14897–14904 CrossRef CAS.
- N. Bensalah and Y. De Luna, Energy Technol., 2021, 9, 2100011 CrossRef CAS.
- Q. Zhao, A. Song, S. Ding, R. Qin, Y. Cui, S. Li and F. Pan, Adv. Mater., 2020, 32, 2002450 CrossRef CAS.
- X. Yao, Y. Zhao, F. A. Castro and L. Mai, ACS Energy Lett., 2019, 4, 771–778 CrossRef CAS.
- L. Chen, C. Hao, Y. Zhang, Y. Wei, L. Dai, J. Cheng, H. Zhang and L. Ci, J. Energy Chem., 2021, 60, 480–493 CrossRef CAS.
- D. Xie, Y. Wang, L. Tian, H. Huang, J. Sun, D. W. Kim, J. Zhao and J. Mao, Adv. Funct. Mater., 2024, 35, 2413993 CrossRef.
- N. Ma, S. Kosasang, P. Chomkhuntod, S. Duangdangchote, N. Phattharasupakun, W. Klysubun and M. Sawangphruk, J. Power Sources, 2020, 455, 22796 CrossRef.
- L. Liu, Y. C. Wu, L. Huang, K. Liu, B. Duployer, P. Rozier, P. L. Taberna and P. Simon, Adv. Energy Mater., 2021, 11, 210128 Search PubMed.
- T.-H. Wu, Y.-Q. Lin and J.-X. Huang, Nanoscale, 2023, 15, 1869–1879 RSC.
- J. Ji, H. Wan, B. Zhang, C. Wang, Y. Gan, Q. Tan, N. Wang, J. Yao, Z. Zheng, P. Liang, J. Zhang, H. Wang, L. Tao, Y. Wang, D. Chao and H. Wang, Adv. Energy Mater., 2020, 11, 2003203 CrossRef.
- F. Long, Y. Xiang, S. Yang, Y. Li, H. Du, Y. Liu, X. Wu and X. Wu, J. Colloid Interface Sci., 2022, 616, 101–109 CrossRef CAS.
- G. G. Yadav, J. W. Gallaway, D. E. Turney, M. Nyce, J. Huang, X. Wei and S. Banerjee, Nat. Commun., 2017, 8, 14424 CrossRef.
- Y. Wang, A. Yuan and X. Wang, J. Solid State Electrochem., 2007, 12, 1101–1107 CrossRef.
- P. Xiong, R. Ma, N. Sakai, X. Bai, S. Li and T. Sasaki, ACS Appl. Mater. Interfaces, 2017, 9, 6282–6291 CrossRef CAS.
- R. Zhao, L. Zhang, C. Wang and L. Yin, J. Power Sources, 2017, 353, 77–84 CrossRef CAS.
- A. Zhang, R. Zhao, Y. Wang, J. Yue, J. Yang, X. Wang, C. Wu and Y. Bai, Angew. Chem., Int. Ed., 2023, 62, e202313163 CrossRef CAS.
- Z.-H. Liu, L. Kang, Z. Yang, Z. Wang and K. Ooi, J. Mater. Res., 2006, 21, 1718–1725 CrossRef CAS.
- Y. G. Wang, W. Wu, L. Cheng, P. He, C. X. Wang and Y. Y. Xia, Adv. Mater., 2008, 20, 2166–2170 CrossRef CAS.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2960 CrossRef.
- C. Zuo, F. Chao, M. Li, Y. Dai, J. Wang, F. Xiong, Y. Jiang and Q. An, Adv. Energy Mater., 2023, 13, 2301014 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  d,
Zhanhu
Guo
d,
Zhanhu
Guo