
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand non-innocence and an unusual σ-bond metathesis step enables catalytic borylation using 9-borabicyclo-[3.3.1]-nonane†
Milan Kumar
Bisai‡
 a,
Justyna
Łosiewicz‡
a,
Gary S.
Nichol
a,
Andrew P.
Dominey
b,
Stephen P.
Thomas
a,
Stuart A.
Macgregor
*c and
Michael J.
Ingleson
*a
a,
Justyna
Łosiewicz‡
a,
Gary S.
Nichol
a,
Andrew P.
Dominey
b,
Stephen P.
Thomas
a,
Stuart A.
Macgregor
*c and
Michael J.
Ingleson
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: mingleso@ed.ac.uk
bGSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK
cEaStCHEM School of Chemistry, University of St Andrews, KY16 9ST, UK
First published on 15th April 2025
Abstract
The metal-catalyzed intermolecular C–H borylation of arenes is an extremely powerful C–H functionalization methodology. However, to date it is effectively restricted to forming organo-boronate esters (Aryl–B(OR)2) with its application to form other organoboranes rarely explored. Herein, we report a catalytic intermolecular heteroarene C–H borylation method using the commercial hydroborane 9-borabicyclo-[3.3.1]-nonane, (H–BBN)2. This process is effective for mono- and di-borylation to form a range of heteroaryl–BBN compounds using either NacNacAl or NacNacZn (NacNac = {(2,6-iPr2C6H3)N(CH3)C}2CH) based catalysts. Notably, mechanistic studies indicated a highly unusual σ-bond metathesis process between NacNacZn–Aryl and the dimeric hydroborane, with first order kinetics in the hydroborane dimer ((H–BBN)2). Our calculated metathesis pathway involves ligand non-innocence and addition of both H–BBN units in (H–BBN)2 to the NacNacZn–heteroaryl complex. This is in contrast to the conventional σ-bond metathesis mechanism using other hydroboranes which invariably proceeds by reaction of one equivalent of a monomeric hydroborane (e.g., H–B(OR)2) with a M–C unit. Overall, this work demonstrates the potential of extending catalytic arene C–H borylation beyond boronate esters, while highlighting that the σ-bond metathesis reaction can be mechanistically more complex when utilizing dimeric hydroboranes such as (H–BBN)2.
Introduction
The catalytic borylation of arenes is well established as an extremely useful C–H functionalization methodology.1–3 However, catalytic intermolecular C–H borylation processes are almost exclusively limited to forming heteroaryl- and aryl-boronate esters (collectively termed herein Ar–B(OR)2), particularly pinacol derivatives (Ar–BPin, Fig. 1A).1–3 This is due to the commercially available and bench-stable precursors HBPin/B2Pin2 reacting appropriately with transition metal complexes to enable catalysis to form Ar–BPin products that have considerable synthetic utility. In contrast, catalytic intermolecular borylation to access other aryl boranes (i.e., non Ar–B(OR)2) is extremely rare despite the fact that many of these boranes display distinct reactivity profiles compared to the organo-boronate ester analogues. In this area a notable exception are the reports using H–BDan (1,8-naphthalenediaminatoborane) in catalytic C–H borylation.4–6 However, the Ar–BDan products and Aryl–BPin compounds are both weakly Lewis acidic at boron. In contrast, B-Ar-9-borabicyclo-[3.3.1]-nonane compounds (Ar–BBN) are more Lewis acidic at boron. While this results in lower bench stability, it enables these organoboranes to effect transformations that are not possible using Ar–B(OR)2/Ar–BDan.7–9 Furthermore, the hydroborane, 9-borabicyclo-[3.3.1]-nonane, (H–BBN)2, is commercially available, inexpensive, and widely used in synthesis.10 However, to date the use of (H–BBN)2 in catalytic intermolecular C–H borylation to form a range of Ar–BBN compounds is not reported to our knowledge (Fig. 1A),11–13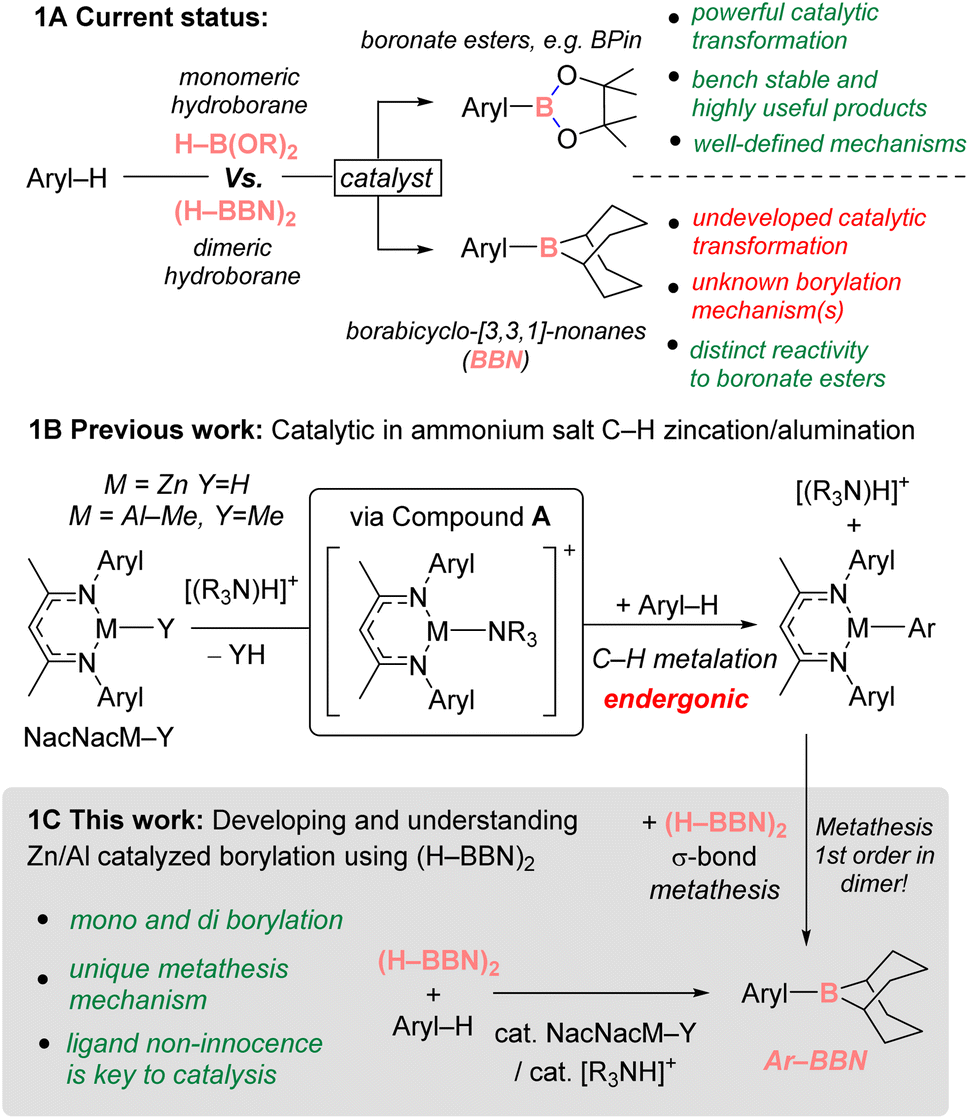 | ||
| Fig. 1 (A): Comparison of the current status of catalytic C–H borylation using monomeric H–B(OR)2 and dimeric (H–BBN)2. (B): Previous catalyzed C–H zincation and alumination. (C): This work. | ||
Previously, we and others have reported zinc-catalyzed C–H borylations using the monomeric boranes H–BPin, H–BCat, (Cat = o-C6H4O22−) and H–BDan.5,14–20 The coordination of these monomeric boranes through O/N to zinc electrophiles was proposed to be key for borylation.14,15 This is consistent with catalytic borylation not proceeding in these systems when using (H–BBN)2 as this borane does not contain a Lewis basic O or N unit.15 Furthermore, (H–BBN)2 is dimeric and thus often reacts differently to the monomeric dioxaborolanes. More recently, we have used NacNacM–Y (M = Zn, Y![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H or M = Al–Me, Y = Me) and sub-stoichiometric ammonium salts to form compound A that effects the C–H zincation and C–H alumination of heteroarenes (Fig. 1B, NacNac = {(2,6-iPr2C6H3)N(CH3)C}2CH).21 Given that NacNacM–R complexes have been reported that undergo σ-bond metathesis with monomeric hydroboranes (e.g. H–BPin) to form organoboranes and NacNacM–H,17,19,22–24 we wondered if performing arene C–H zincation/alumination in the presence of (H–BBN)2 could enable catalytic C–H borylation to form Ar–BBN compounds. This would require the product from C–H metalation, NacNacM–Ar, to undergo σ-bond metathesis with (H–BBN)2 dimer to produce Aryl–BBN and NacNacM–H. NacNacM–H would then react rapidly with [(R3N)H]+ salts to reform compound A.21 However, the feasibility of this catalytic borylation process is contingent upon (i) the dimeric (H–BBN)2 undergoing σ-bond metathesis with NacNacM–Aryl via a low barrier process, and (ii) the Lewis acidic Y–BBN species (YH or Ar) not quenching any on-cycle species. A low barrier metathesis step is essential given the endergonic nature of the first step in the putative catalytic cycle, C–H metalation using compound A. This results in an increase in the effective transition state energies for all steps after the C–H metalation. Notably, there are no previously reported studies into the mechanism and associated barriers of the σ-bond metathesis between dimeric hydroboranes, such as (H–BBN)2, and (main group element)–Y species (YR, OR, NR2).22,25–29 Note, the dissociation of (H–BBN)2 into two equivalents of monomer is significantly endergonic,26b therefore it will react in a distinct way to the monomeric boranes widely used to date in catalytic borylation, e.g. HBPin/HBCat/HBDan, which could significantly impact the mechanism of the σ-bond metathesis, and other, step(s) in the catalytic cycle.
H or M = Al–Me, Y = Me) and sub-stoichiometric ammonium salts to form compound A that effects the C–H zincation and C–H alumination of heteroarenes (Fig. 1B, NacNac = {(2,6-iPr2C6H3)N(CH3)C}2CH).21 Given that NacNacM–R complexes have been reported that undergo σ-bond metathesis with monomeric hydroboranes (e.g. H–BPin) to form organoboranes and NacNacM–H,17,19,22–24 we wondered if performing arene C–H zincation/alumination in the presence of (H–BBN)2 could enable catalytic C–H borylation to form Ar–BBN compounds. This would require the product from C–H metalation, NacNacM–Ar, to undergo σ-bond metathesis with (H–BBN)2 dimer to produce Aryl–BBN and NacNacM–H. NacNacM–H would then react rapidly with [(R3N)H]+ salts to reform compound A.21 However, the feasibility of this catalytic borylation process is contingent upon (i) the dimeric (H–BBN)2 undergoing σ-bond metathesis with NacNacM–Aryl via a low barrier process, and (ii) the Lewis acidic Y–BBN species (YH or Ar) not quenching any on-cycle species. A low barrier metathesis step is essential given the endergonic nature of the first step in the putative catalytic cycle, C–H metalation using compound A. This results in an increase in the effective transition state energies for all steps after the C–H metalation. Notably, there are no previously reported studies into the mechanism and associated barriers of the σ-bond metathesis between dimeric hydroboranes, such as (H–BBN)2, and (main group element)–Y species (YR, OR, NR2).22,25–29 Note, the dissociation of (H–BBN)2 into two equivalents of monomer is significantly endergonic,26b therefore it will react in a distinct way to the monomeric boranes widely used to date in catalytic borylation, e.g. HBPin/HBCat/HBDan, which could significantly impact the mechanism of the σ-bond metathesis, and other, step(s) in the catalytic cycle.
Herein, we report the Zn/Al-catalyzed C–H borylation of a range of heteroarenes using (H–BBN)2. Notably, mechanistic studies indicate a highly unusual σ-bond metathesis process proceeding by the addition of both H–BBN units in dimeric (H–BBN)2 to the metal complex which is enabled by NacNac ligand non-innocence.
Results and discussion
C–H borylation studies
In our ammonium salt catalyzed aryl C–H zincation,21 the highest yields were observed using [(Et3N)H]+ containing salts and after that [(DMT)H]+ (DMT = N,N-dimethyl-p-toluidine). Therefore, these salts were explored in combination with NacNacZnH, 1, for the C–H borylation of 2-methyl-thiophene using (H–BBN)2 (Table 1). From this it was found that [(DMT)H][B(C6F5)4] significantly outperformed the [(Et3N)H]+ congener (entries 1–2), suggesting that a stronger Brønsted acid is vital (pKa [(DMT)H]+ = 10.8, [(Et3N)H]+ = 18.5).30 This is distinct to performance in C–H zincation. An anion effect also was observed, with [B(C6F5)4]− being more effective than the more coordinating anion [OTf]− (entry 3). In the absence of 1 or the ammonium salt no C–H borylation was observed (entries 4–5). Ultimately, 5 mol% of 1 and [(DMT)H][B(C6F5)4] was identified as optimal (entry 7, for full optimization see Table S1†).|
|
|||||
|---|---|---|---|---|---|
| Brønsted acid | x/y mol% | T (°C) | t (h) | Yielda (%) | |
| a Yield relative to (H–BBN)2 using an internal standard. b Using 1.15 equiv. 2a. | |||||
| 1 | [(Et3N)H][B(C6F5)4] | 10/10 | 60 | 18 | <5 |
| 2 | [(DMT)H][B(C6F5)4] | 10/10 | 60 | 18 | 55 |
| 3 | [(DMT)H][OTf] | 10/10 | 60 | 18 | < 5 |
| 4 | [(DMT)H][B(C6F5)4] | 0/10 | 60 | 18 | 0 |
| 5 | — | 10/0 | 60 | 18 | 0 |
| 6 | [(DMT)H][B(C6F5)4] | 10/10 | 80 | 24 | 98 |
| 7 | [(DMT)H][B(C 6 F 5 ) 4 ] | 5/5 | 80 | 24 | 94 |
| 8 | [(DMT)H][B(C6F5)4] | 2.5/2.5 | 80 | 24 | 84 |

With optimized conditions in hand the scope and limitations of the C–H borylation were explored (Chart 1). Other C2 substituted thiophenes were amenable and produced 3b and 3c in good yield, while the less nucleophilic heteroarene benzothiophene was converted to 3d in 81% yield. 3-Me-thiophene was functionalized predominantly at the C5 position to form 3e (with only a minor amount of C2-functionalized product formed), while thiophene and bithiophene also were borylated to form 3f and 3g, respectively, in good yield. Other heteroarenes, such as furan and N–Me-indole, also were amenable to C–H borylation to form 3h and 3i, respectively, in moderate yield. Note, the products from these reactions are all consistent with SEAr type selectivity, while less activated heteroaromatics (e.g., benzofuran) and activated arenes such as N–Me-carbazole, anisole and anthracene did not undergo borylation, analogous to the outcome from the C–H zincation (see ESI, Section 3.4†). Furthermore, attempts using a chiral dimeric R2BH borane, diisopinocampheylborane, under optimized conditions resulted in no C–H borylation of 2-methylthiophene (after 24 h at 80 °C).
 | ||
| Chart 1 Substrate scope for zinc-catalyzed borylation. a Yield relative to (H–BBN)2 using an internal standard. b at 100 °C. | ||
Next, we assessed if aluminium-catalyzed borylation was possible. It was found that 1 could be replaced with NacNacAlMe24 in the catalytic borylation using (H–BBN)2 (Chart 2). Aluminium-catalyzed borylation also was applied to N–Me-pyrrole to form diborylated 3j (attempts to form mono-borylated N–Me-pyrrole resulted in mono and diborylated products).
 | ||
| Chart 2 Aluminium catalyzed C–H borylation. a Yield versus an internal standard. b 1.5 equiv. (H–BBN)2. | ||
From the reactions producing 3i and 3j a small quantity of crystals suitable for X-ray diffraction analysis were formed. Notably, this analysis showed these were not 3i/3j but instead were the H–BBN adducts of 3i and 3j, termed 3i-(H–BBN) and 3j-(H–BBN) (Fig. 2). In situ analysis of the Zn/Al catalyzed reactions that form 3i/3j revealed that while the major resonance is due to 3i/3j (δ11B ≈ 72), an additional minor 11B resonance at ca. 5 ppm was present consistent with 3i/3j-(H–BBN).31 Indeed, dissolution of crystals of 3i-(H–BBN) or 3j-(H–BBN) led to formation of 3i/3j and (H–BBN)2 as the major product (by NMR spectroscopy), indicating a solution equilibrium favoring 3i/3j and (H–BBN)2. Note, no resonances for thienyl analogues of 3i-/3j-(H–BBN) were observed in any of the reactions, presumably due to the lower nucleophilicity of thiophenes relative to indoles/pyrroles32 and consistent with the computed trend observed in our DFT calculations (see Table S5†). The formation of 3i/3j-(H–BBN) is related to the reaction of Et2N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–BBN with (H–BBN)2 which forms a compound also containing a cyclic CB2H unit (Fig. 2 inset, compound B).31
C–BBN with (H–BBN)2 which forms a compound also containing a cyclic CB2H unit (Fig. 2 inset, compound B).31
 | ||
| Fig. 2 Reaction of 3i/3j with (H–BBN)2. Bottom, solid-state structures of 3i/3j-(H–BBN), ellipsoids at 50% probability and most hydrogens omitted for clarity. Inset right the formation of compound B also containing a CB2H core. | ||
With the mono-borylation scope assessed, our attention turned to the diborylation of thiophenes given the importance of diborylated precursors in accessing organic materials.33 Note, to date the catalytic C–H diborylation of thiophenes via an SEAr type process (i.e. transition metal free) is limited to only the most highly nucleophilic thiophenes, such as 3,4-dialkoxy-substituted thiophenes.34 Diborylated thienyl products 5a–5f proved accessible through zinc catalysis simply by increasing the equivalents of (H–BBN)2 used (Chart 3). Notably, most of the diborylated products were poorly soluble in the reaction solvent, chlorobenzene, facilitating their facile isolation in good yield. X-ray diffraction studies on a number confirmed their formation (inset Chart 3 for 5c, for 5d and 5e see ESI†).
 | ||
| Chart 3 Zinc catalyzed diborylation of thiophenes. a Yield versus an internal standard. b Isolated yield. | ||
Synthetic utility
With a range of mono- and di-borylated products accessible their utility was explored. Given the importance of thienyl–C(H)(OH)Ph units in active pharmaceutical ingredients, e.g., tiemonium salts,35 a one-pot route to these motifs from a thiophene precursor was targeted. The reaction of crude (i.e., made in situ and taken forward with no purification) Br–thienylBBN (3c) with benzaldehyde proceeded to form 6a in 70% yield (Scheme 1 top) with the alcohol 6b then formed using the previously reported O–BBN hydrolysis conditions (see Fig. S76†).8 Importantly, thienyl–BPin compounds do not react with benzaldehyde under identical conditions, presumably due to their lower Lewis acidity at boron. This demonstrates that this catalytic C–H borylation approach can be harnessed in tandem with the distinct reactivity profile of Ar–BBN derivatives to access value-added products in one-pot from the starting heteroarene. The crude products from zinc-catalyzed C–H borylation also can be telescoped into a Suzuki–Miyaura cross-coupling reaction (Scheme 1, middle), which led to compound 7 in excellent yield for a multi-step process. Bench stable borylated derivatives also were prepared by trans-borylation of thienyl–BBN species with HBPin to generate the Aryl–Bpin congeners (e.g., to form 8).36 | ||
| Scheme 1 Utilization of thienyl–BBN products. a Borylation conditions from Table 1. | ||
Mechanistic studies
With the scope and utility of the process assessed our attention turned to the mechanism. For this catalytic borylation a NacNacM–Y complex (e.g.1), and [(DMT)H][B(C6F5)4] were both required. This is consistent with the observation that (H–BBN)2 and [(DMT)H][B(C6F5)4] do not react (even on heating to 80 °C) to form a [(DMT)–BBN]+ salt (with related salts known to be effective borylating agents,37 including in catalytic processes when using [CatB(amine)]+ derivatives).38,39 To further preclude the possibility of [(DMT)–BBN]+ mediated borylation occurring in this process, the borylation of 2,5-N-trimethylpyrrole was attempted under the optimized aluminium-catalyzed conditions. This substrate was chosen as it undergoes C–H borylation using stoichiometric [(amine)–BBN]+ in high yield at room temperature,37 while it has a sterically hindered borylation position located alpha to a methyl (C–H alumination is highly sensitive to steric environment).21 Under our optimized aluminium-catalyzed conditions < 5% C–H borylation of 2,5-N-trimethylpyrrole was observed at 100 °C, further disfavoring a [(DMT)–BBN]+ mediated catalytic borylation process (Scheme 2). Furthermore, DFT calculations (vide infra and Fig. S115†) disfavored a process proceeding through the borenium equivalent [NacNacZn–(μ-H)–BBN]+. Therefore, our mechanistic studies focused on the hypothesis that heteroarene C–H metalation followed by σ-bond metathesis leads to Aryl–BBN. | ||
| Scheme 2 Disparate outcomes from the borylation of 2,5-N-trimethylpyrrole. | ||
Given the greater substrate scope of the zinc catalyzed metalation/borylation21 all subsequent work focused on this system. The zinc-catalyzed reactions use NacNacZnH (1) and [(DMT)H][B(C6F5)4]; and these react to form [NacNacZn(DMT)][B(C6F5)4] ([9][B(C6F5)]4) rapidly through a low-energy transition state.21 It was proposed that C–H borylation then would proceed by: (i) [9]+ reacting with a heteroarene to form NacNacZn–Aryl and [(DMT)H]+; (ii) the Zn–Aryl species would then undergo σ-bond metathesis with (H–BBN)2 to form the Ar–BBN product, and regenerate the zinc-hydride 1; (iii) 1 would then react with [(DMT)H]+ to form [9]+ and H2. At this point it is important to highlight that 2-methyl-thiophene C–H zincation using [9]+ is endergonic by +15 kcal mol−1.21 Therefore, to have a feasible ΔG‡span for this catalytic C–H borylation cycle the σ-bond metathesis step involving dimeric (H–BBN)2 has to proceed via a low barrier process.
To probe the metathesis process NacNacZn(thienyl) complex 10 was synthesized and reacted with 0.5 equivalent of (H–BBN)2. While the metathesis did proceed at room temperature to form thienyl–BBN, 3a, only 50% of 10 was consumed to form a single new NacNacZn product that was not compound 1. Instead, the zinc complex displayed NMR data consistent with formation of NacNacZn–(μ-H)2–BBN, 11 (Fig. 3). This included a 11B NMR resonance (at −13.5 ppm) comparable with other M–(μ-H)2–BBN complexes.25,26a,40 The use of 1 equiv. of (H–BBN)2 resulted in the full conversion of 10 into 3a and 11. The formation of 11 was confirmed by single crystal X-ray diffraction studies (inset Fig. 3). The structure of 11 contains a Zn⋯B distance of 2.179 Å consistent with a borohydride unit bound to zinc via two bridging hydrogens, with 11 having comparable metrics to NacNacZn–(μ-H)2–BH2.41 Complex 11 also can be synthesized from NacNacZnH 1 by addition of 0.5 equivalent of (H–BBN)2. Note, while the boron center in 11 is structurally similar to the active boron electrophile in Wang's borocation-mediated catalytic C–H borylation,42 complex 11 is neutral and thus is a weak electrophile at boron which is not active in C–H borylation (Table 1, entry 5).
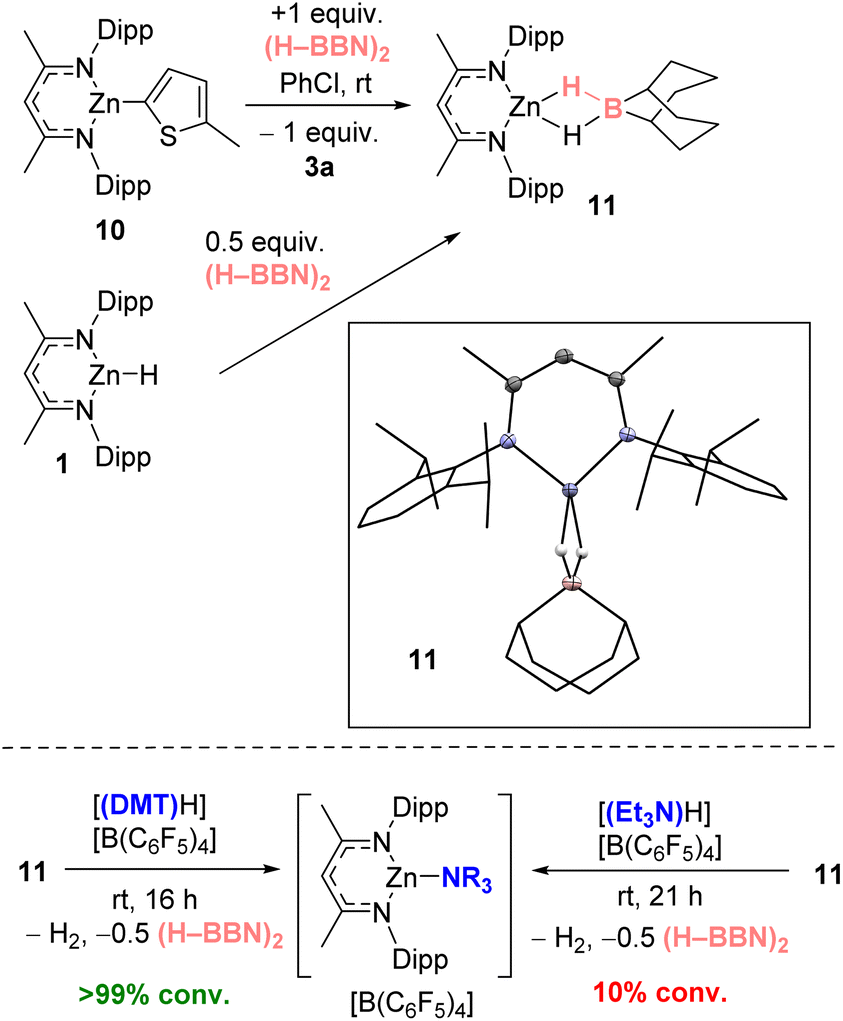 | ||
| Fig. 3 Top, the formation of 11 by metathesis of 10 with (H–BBN)2 (also forming 3a) or by direct addition of 0.5 equiv. (H–BBN)2 to 1. Inset, the solid-state structure of 11. Bottom, the disparate outcome from protonolysis of 11. | ||
The formation of 11 instead of 1 is significant as it demonstrates a key difference in reactivity using (H–BBN)2versus H–BPin.17 Furthermore, to close the C–H borylation cycle the Brønsted acid, [(DMT)H]+, produced during the arene C–H zincation step has to react with a hydridic species to reform 9 (and concomitantly H2). A protonolysis step starting from 11 is distinct to the protonolysis of NacNacZnH 1, which is facile with a range of [(R3N)H]+ salts.21 Therefore, the reaction of [(DMT)H][B(C6F5)4] and 11 was investigated. This revealed a rapid reaction at room temperature to form [9][B(C6F5)4], H2 and 0.5 equiv. (H–BBN)2 (Fig. 3, bottom). In contrast, the reaction between [(Et3N)H][B(C6F5)4] and 11 proceeds very slowly at room temperature indicating a higher barrier protonolysis step starting from 11 than starting from 1 (Fig. 3, bottom). This may explain the poor outcomes using [(Et3N)H][B(C6F5)4] in the catalytic borylation compared to that using [(DMT)H][B(C6F5)4].
Overall, the proposed cycle is outlined in Fig. 4, proceeding via 1. Arene C–H zincation, 2. σ-bond metathesis and 3. Protonolysis. The latter could potentially occur directly from 11 or by endergonic conversion of 11 into 1 which would then undergo protonolysis. While the stoichiometric experiments confirmed that both the σ-bond metathesis and protonolysis of 11 using [(DMT)H][B(C6F5)4] proceed at room temperature, how they proceed remained a key question. The σ-bond metathesis process was of particular interest, as the C–H borylation reactions used (H–BBN)2 which strongly favors its dimeric form in contrast to the monomeric hydroboranes (e.g., H–BPin) previously studied.
 | ||
| Fig. 4 Proposed catalytic cycle for C–H borylation. | ||
Cognizant that hidden catalysis can facilitate transformations in organoboron chemistry,43 several species that could be present at low concentration during the catalysis were tested for their effect on the metathesis step. The addition of 5 mol% of DMT, [(DMT)H][B(C6F5)4] and Me2S–BH3 separately to the reaction between Zn–Aryl 10 and (H–BBN)2 led to no change in the rate of the metathesis reaction, indicating that these species do not catalyze this metathesis reaction. With the positive effect of catalyst loading on borylation rate determined (Table 1 and Fig. S107 and S108†), variable time normalization analysis (VTNA)44 was used to determine the order with respect to (H–BBN)2. This analysis gave data that was consistent with a first order in (H–BBN)2 dimer for the metathesis step (Fig. S100†). Some alkene hydroboration reactions using (H–BBN)2 are first order with respect to the dimer,45 which indicates the dissociation of the (H–BBN)2 dimer into two equivalents of (H–BBN) monomer is rate limiting. In these cases, the hydroboration reactions are zero order with respect to the other component(s). The situation here is different, as the rate is also affected by the concentration of the zinc complex and the thienyl substrate (see Tables 1, S1 and Fig. S107 and S108†). This precludes (H–BBN)2 dimer dissociation into two monomers being the turnover limiting step in this zinc catalyzed arene borylation. The standard metathesis mechanism involves a monomeric hydroborane species reacting via a four membered transition state (e.g., involving M–C and H–B). However, such a process is not consistent with the above data as when one equivalent of monomeric H–BBN reacts with a substrate in the rate limiting step the reaction is 0.5 order with respect to (H–BBN)2 dimer.45 Therefore, computational analysis was performed to provide further insight into the mechanism.
Computational studies
All calculations discussed herein were performed at the B3PW91(def2-TZVP, D3(BJ), PhCl)//B3PW91(Zn: SDD; S: SDD(d); other atoms: 6-31G**) level of theory which was chosen based on its performance in our work on the related catalyzed C–H zincation.21 Note, in the solvent used for the borylation reactions, PhCl, H–BBN exists as a dimer, (H–BBN)2, and at this level of theory +9.9 kcal mol−1 (ΔG) per monomer unit is required for dissociation of the dimer. Therefore, calculations involving association/dissociation of H–BBN use 0.5 equiv. of (H–BBN)2 for determining the energy change.As mentioned above a borylation mechanism involving borenium equivalents e.g., INT-1B+ or INT-2B+ was disfavored based on DFT calculations (Scheme 3). Specifically, the displacement of DMT by H–BBN proceeds via formation of INT-1B+ which is formed in a significantly endergonic step involving a high energy transition state (ΔG‡ = +37.0 kcal mol−1) which precludes this mechanism. Notably, the formation of INT-2B+ from [9]+ is much more endergonic than the displacement of DMT in [9]+ by HBCat (which coordinates through an oxo group to zinc).14 This significant difference presumably is why there is no C–H borylation using (H–BBN)2 when catalyzed by zinc electrophiles that do not effect C–H metalation.15
 | ||
| Scheme 3 Calculated free energies (kcal mol−1) to form the borenium equivalent INT-2B+. | ||
A range of mechanisms then were explored for the catalytic borylation of 2-methyl-thiophene that proceed via C–H zincation, σ-bond metathesis and protonolysis and a computed profile that is consistent with the VTNA analysis (1st order in (H–BBN)2 dimer) is shown in Fig. 5(a). Here [NacNacZn(DMT)]+ ([9]+) and 2-methyl-thiophene are taken as the starting point of the cycle. Overall, the reaction proceeds as postulated, with an endergonic C–H zincation to form 10 (+14.8 kcal mol−1) proceeding as outlined in our previous report (via a transition state at +21.7 kcal mol−1).21 This is followed by a σ-bond metathesis phase that is effectively thermoneutral (+0.9 kcal mol−1) with the overall process driven forward by exergonic dehydrocoupling to reform [9]+ with loss of H2 (ΔG = −16.9 kcal mol−1). The overall process is therefore exergonic (ΔG = −1.2 kcal mol−1).
 | ||
| Fig. 5 (a) Computed free energy reaction profile (kcal mol−1) for the catalytic C–H borylation of 2-methyl-thiophene focusing on the σ-bond metathesis phase. (Method: B3PW91(def2-TZVP, D3(BJ), PhCl)//B3PW91(Zn: SDD; S: SDD (d); other atoms: 6-31G**)). (b) Details of key intermediates in the σ-bond metathesis process (distances in Å; inset defines the metallacycle folding angle, ϕ;21 NacNac, BBN and 2-methyl-thiophene substituents shown in wireframe for clarity). | ||
The σ-bond metathesis phase proceeds through addition of the (H–BBN)2 dimer to 10 to form INT-1Zn-B at +20.4 kcal mol−1. This involves the Cγ position of the NacNac ligand acting as a Lewis base towards one H–BBN unit, with the other H–BBN unit interacting with the Zn–thienyl moiety.46INT-1Zn-B therefore contains two B–H⋯Zn interactions, with that bridging the Zn–thienyl unit being stronger (Zn⋯H1–B1 = 1.69 Å; B1–C1 = 1.64 Å cf. Zn⋯H2–B2 = 2.15 Å; Cγ–B2 = 1.77 Å, see Fig. 5(b)). This also induces significant elongation of the Zn–C1 bond (2.41 Å vs. 1.93 Å in 10) such that its cleavage has a barrier of only 1 kcal mol−1viaTS1Zn-B. This forms INT-2Zn-B in which the Zn–H1⋯B1 moiety approaches linearity (165°) and from which dissociation of the thienyl–BBN product proceeds viaTS2Zn-B with concomitant contraction of the B2–H2⋯Zn distance to 1.83 Å. Thus, the non-innocence of the NacNac ligand47 not only assists the cleavage of the (H–BBN)2 dimer by Cγ–B bond formation, but the resultant Cγ-bound hydroborane provides flexible ligation of the zinc centre during the metathesis phase.
This is also reflected in the variation of the Zn–NacNac metallacycle folding angle, ϕ, as previously discussed (inset Fig. 5(b))21 and is also seen in INT-3Zn-B, formed after product dissociation (B2–H2⋯Zn = 1.93 Å; ϕ = 80.7°). To complete the σ-bond metathesis phase, dissociation of H–BBN occurs viaTS3Zn-B at +27.6 kcal mol−1.
Details of the final phase, protonolysis, are shown in Scheme 4. No pathway involving direct protonolysis of 11 by [(DMT)H]+ to form [9]+ was found with feasible barriers (see Fig. S111†). However, dissociation of 0.5 equiv. of (H–BBN)2 from 11 to form NacNacZnH, 1, is endergonic by only 7.4 kcal mol−1. Complex 1 can then react with [(DMT)H]+via the mechanism described in our previous work which proceeds by protonation of the Cγ position of NacNac.21 Note, the highest-lying transition state for this process lies at +26.9 kcal mol−1 and so is competitive with TS3Zn-B at +27.6 kcal mol−1 in the σ-bond metathesis phase. However, we disfavor this protonolysis step being rate limiting as that scenario would result in an inverse dependence on [(H–BBN)]2,26 not the first order dependence observed. Finally, as with the σ-bond metathesis phase, we highlight that NacNac ligand non-innocence again appears essential for accessing sufficiently low barrier mechanisms for the protonolysis phase.
 | ||
| Scheme 4 Free energies (kcal mol−1) for the protonolysis phase that proceeds via1 with only the highest transition state energy shown. Energies are in kcal mol−1 relative to the zero energy defined in Fig. 5. | ||
Conclusions
A catalytic intermolecular arene C–H borylation process using (H–BBN)2 to make a range of mono- and di-borylated heteroarenes is reported for the first time to our knowledge. Due to the endergonic nature of the C–H metalation step, for this C–H borylation process to be energetically feasible it requires low barrier σ-bond metathesis and protonolysis steps. Kinetic studies revealed that the σ-bond metathesis process proceeds via addition of both H–BBN units in the (H–BBN)2 dimer to the NacNacZn–Aryl complex – i.e., the order of reaction with respect to (H–BBN)2 was found to be 1. This is distinct to conventional σ-bond metatheses which involve one equivalent of a monomeric hydroborane reacting with a M–Y unit of a metal complex. The involvement of both H–BBN units in the turnover limiting process is crucial as this offsets the significant energetic cost of cleaving dimeric (H–BBN)2. This highlights the importance of considering the correct speciation of the hydroborane when designing borylation processes using (H–BBN)2, as this has a considerable impact on the energy profile for a catalytic cycle. In this case, the ability of the metal complex to interact with two H–BBN units is enabled by ligand non-innocence, specifically interaction of the NacNac Cγ position with one of the H–BBN units derived from (H–BBN)2. The second H–BBN unit then effects the σ-bond metathesis with the M–Aryl unit. Thus, NacNac ligand non-innocence is essential for low barrier σ-bond metathesis and protonolysis phases, which is crucial for enabling borylation catalysis given the endergonic nature of the C–H metalation phase. Thus, this study showcases the benefits of ligating main group metals with non-innocent NacNac ligands, the dramatic impact (H–BBN)2 has on the mechanism relative to monomeric boronate esters (e.g., HBPin), and the utility of extending catalytic C–H borylation beyond the synthesis of the aryl-boronate esters that currently dominate this field.Data availability
The data supporting this article has been uploaded as part of the ESI.† This includes NMR spectra for all new compounds, in situ NMR spectra for catalytic and mechanistic reactions and Cartesian coordinates for all calculated structures.Author contributions
MKB, JL, SPT, MI and SAM conceived the research concept and aims and analyzed all data. MKB and JL performed the majority of the synthetic work and the majority of the analytical components of this project. GN collected and solved all the crystal structures. JL performed all the calculations. Combined, all authors drafted, reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the EPSRC (EP/V03829X/1 and the Program Grant “Boron: Beyond the Reagent” (EP/W007517/1)). We thank the Mass Spectrometry facility (SIRCAMS) at the University of Edinburgh (UoE) for carrying out MS analysis. S. A. M thanks the University of St. Andrews for support. Prof. M. R. Crimmin is thanked for useful discussions.References
- I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed.
- S. Guria, M. M. M. Hassan and B. Chattopadhyay, Org. Chem. Front., 2024, 11, 929–953 RSC.
- M. M. M. Hassan, S. Guria, S. Dey, J. Das and B. Chattopadhyay, Sci. Adv., 2023, 9, eadg3311 CrossRef CAS PubMed.
- S. Kamio and H. Yoshida, Adv. Synth. Catal., 2021, 363, 2310–2324 CrossRef CAS.
- T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Adv. Synth. Catal., 2015, 357, 77–82 CrossRef CAS.
- N. Iwadate and M. Suginome, J. Organomet. Chem., 2009, 694, 1713–1717 CrossRef CAS.
- M. J. O'Donnell, J. T. Cooper and M. M. Mader, J. Am. Chem. Soc., 2003, 125, 2370–2371 CrossRef PubMed.
- P. I. Jacob and H. C. Brown, J. Org. Chem., 1977, 42, 579–580 CrossRef CAS.
- S. Jin, H. T. Dang, G. C. Haug, V. D. Nguyen, H. D. Arman and O. V. Larionov, Chem. Sci., 2020, 11, 9101–9108 RSC.
- H. C. Brown, Organic syntheses via boranes, New York : Wiley, 1975 Search PubMed.
- The following reference contains only one example of catalytic borylation using (H–BBN)2, specifically of the highly activated heteroarene N-Me-pyrrole. M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed.
- The following reference contains only one example of catalytic borylation using (H–BBN)2, specifically of the highly activated heteroarene N-Me-indole. T. Stahl, K. Müther, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed.
- Note, to our knowledge all other studies reported to date using (H–BBN)2 in catalytic C–H borylation proceed by Lewis base directed C–H borylation. These are thus intramolecular borylation reactions and form four-coordinate at boron products (e.g., base→BBN(Ar)) in which the presence of a dative bond significantly reduces the utility of these products in subsequent transformations. See (and references therein): L. Britton, A. D. Bage, S. L. McOnie and S. P. Thomas, Angew. Chem., Int. Ed., 2024, e202423929 Search PubMed.
- M. E. Grundy, L. Sotorrios, M. K. Bisai, K. Yuan, S. A. Macgregor and M. J. Ingleson, ACS Catal., 2023, 13, 2286–2294 CrossRef CAS PubMed.
- M. E. Grundy, K. Yuan, G. S. Nichol and M. J. Ingleson, Chem. Sci., 2021, 12, 8190–8198 RSC.
- R. J. Procter, M. Uzelac, J. Cid, P. J. Rushworth and M. J. Ingleson, ACS Catal., 2019, 9, 5760–5771 CrossRef CAS.
- M. Uzelac, K. Yuan and M. J. Ingleson, Organometallics, 2020, 39, 1332–1338 CrossRef CAS.
- R. Zhou, W. Wang, J. Zhang, Q. Wang, Z. Chen, Y. Shao and F. Zhang, Org. Chem. Front., 2025, 12, 906–911 RSC.
- (a) R. K. Sahoo, A. G. Patro, N. Sarkar and S. Nembenna, ACS Omega, 2023, 8, 3452–3460 CrossRef CAS PubMed; (b) R. K. Sahoo, S. Rajput, A. G. Patro and S. Nembenna, Dalton Trans., 2022, 51, 16009–16016 RSC.
- B. Zhang, Y. Zou, L. Wang and H. Zhang, Chem. Commun., 2021, 57, 11185–11188 RSC.
- M. K. Bisai, J. Łosiewicz, L. Sotorrios, G. S. Nichol, A. P. Dominey, M. J. Cowley, S. P. Thomas, S. A. Macgregor and M. J. Ingleson, Angew. Chem., Int. Ed., 2024, 63, e202404848 CrossRef PubMed.
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS PubMed.
- J. Li, M. Luo, X. Sheng, H. Hua, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- M. Arrowsmith, T. J. Hadlington, M. S. Hill and G. Kociok-Köhn, Chem. Commun., 2012, 48, 4567–4569 RSC.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 4076–4079 CrossRef CAS.
- (a) Note, this reference reported significant differences between the σ-bond metathesis involving a NacNacMg–NR2 complex with H–BPin Vs. (H–BBN)2 based on kinetic analysis. D. J. Liptrot, M. S. Hill, M. F. Mahon and A. S. S. Wilson, Angew. Chem., Int. Ed., 2015, 54, 13362–13365 CrossRef CAS PubMed; (b) This reference, focused on trans-borylation, reports an energy change of +15 kcal/mol for the dissociation of (H–BBN)2 into two monomers. D. R. Willcox, E. Cocco, G. S. Nichol, A. Carlone and S. P. Thomas, Angew. Chem., Int. Ed., 2024, e202401737 CAS.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2020, 59, 19282–19291 CrossRef CAS PubMed.
- A. Kobayashi, K. Suzuki, R. Kitamura and M. Yamashita, Organometallics, 2020, 39, 383–387 CrossRef CAS.
- Mayr's Database Of Reactivity Parameters - Start page, https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/, (accessed January 8, 2025).
- See (and references therein) H. Xiong, Q. Lin, Y. Lu, D. Zheng, Y. Li, S. Wang, W. Xie, C. Li, X. Zhang, Y. Lin, Z.-X. Wang, Q. Shi, T. J. Marks and H. Huang, Nat. Mater., 2024, 23, 695–702 CrossRef CAS PubMed.
- Note, transition metal free catalysts reported to date result in (at best) low yields of diborylated products for less activated thiophenes even when using two equivalents of HBCat e.g. 5% diborylation of thienothiophene was reported for one of the most active systems see: V. Desrosiers, A. Gaudy, K.-A. Giroux and F.-G. Fontaine, Z. Anorg. Allg. Chem., 2023, 649, e202300006 CrossRef CAS.
- See (and references therein) W. K. O. Teixeira, D. Y. de Albuquerque, S. Narayanaperumal and R. S. Schwab, Synthesis, 2020, 52, 1855–1873 CrossRef CAS.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, Synthesis, 2022, 55, 62–74 Search PubMed.
- A. Prokofjevs, J. W. Kampf and E. Vedejs, Angew. Chem., Int. Ed., 2011, 50, 2098–2101 CrossRef CAS PubMed.
- Q. Yin, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 3712–3717 CrossRef CAS PubMed.
- For recent reviews of this area see: (a) X. Tan and H. Wang, ChemCatChem, 2023, 15, e202300734 CrossRef CAS; (b) S. Hazra, S. Mahato, K. K. Das and S. Panda, Chem.–Eur. J., 2022, 28, e202200556 CrossRef CAS PubMed.
- A. Caise, E. L. Kolychev, J. Hicks, M. Á. Fuentes, J. M. Goicoechea and S. Aldridge, Dalton Trans., 2019, 48, 10845–10852 RSC.
- J. Prust, H. Hohmeister, A. Stasch, H. W. Roesky, J. Magull, E. Alexopoulos, I. Usón, H.-G. Schmidt and M. Noltemeyer, Eur. J. Inorg. Chem., 2002, 2002, 2156–2162 CrossRef.
- X. Tan, X. Wang, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2022, 144, 23286–23291 CrossRef CAS PubMed.
- (a) A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed; (b) A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
- C. D.-T. Nielsen and J. Burés, Chem. Sci., 2019, 10, 348–353 RSC.
- H. C. Brown, K. K. Wang and C. G. Scouten, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 698–702 CrossRef CAS PubMed.
- Several pathways for the formation of INT-1Zn-B were characterized computationally, but these all suffered from a high entropic cost of adding the (H–BBN)2 dimer to 10. Our favored mechanism involves the stepwise addition of two H–BBN monomers, first at Cγ and then across the Zn–thienyl unit. See section S7 of the supporting information† for full details..
- For a review on NacNac non-innocence see: (a) C. Camp and J. Arnold, Dalton Trans., 2016, 45, 14462–14498 RSC; (b) for a paper showing binding of BH3 to the γ position of NacNac see: D. D. L. Jones, A. J. R. Matthews and C. Jones, Dalton Trans., 2019, 48, 5785–5792 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2417816–2417821. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02085a |
| ‡ These authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |