
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVan der Waals heterostructures via spontaneous self-restacked assembling for enhanced water oxidation†
Rui
Wang‡
a,
Shuhui
Li‡
a,
Yang
Hu
a,
Shanshan
Wu
a,
Jiamin
Zhu
a,
Li
An
*a,
Pinxian
Xi
 *a and
Chun-Hua
Yan
ab
*a and
Chun-Hua
Yan
ab
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, Frontiers Science Center for Rare Isotopes, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: xipx@lzu.edu.cn; anli@lzu.edu.cn
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 15th May 2025
Abstract
The pursuit of sustainable energy solutions has identified water oxidation as a crucial reaction, with the oxygen evolution reaction (OER) serving as a decisive efficiency determinant in water technologies. This study presents a novel van der Waals (vdW) heterostructure catalyst, synthesized through a spontaneous self-restacking of nickel–iron-based phosphorus-sulfur compounds (NiPS3 and FePS3). Density Functional Theory (DFT) calculations underpinned the thermodynamic spontaneity of the restacking process, uncovering an electronic transition that significantly amplifies electrocatalytic functionality. The catalyst demonstrates a remarkable OER performance, achieving a low overpotential of 257 mV at 20 mA cm−2 and a Tafel slope of 49 mV dec−1 and demonstrates remarkable durability sustaining 500 mA cm−2 for 140 hours. In addition to its high performance, the material's rapid reconstruction facilitated by surface electron enrichment and the release of phosphate and sulfate during the OER underscores a dual enhancement in both activity and stability. The universality of the synthesis method is further demonstrated by extending the approach to other MPS3 materials (M = Mn, Co, Zn), establishing a generalized platform for developing high-performance OER catalysts. This work represents a significant advancement in the application of restacked vdW heterostructures as a foundation for advanced electrocatalytic materials.
Introduction
Exploring efficient energy conversion and storage technologies is critical for achieving a sustainable future.1,2 Electrocatalytic water decomposition plays a vital role in energy conversion applications, providing a clean and abundant source of hydrogen.3,4 The oxygen evolution reaction (OER), alongside the hydrogen evolution reaction (HER), constitutes a critical semi-reaction wherein water is oxidized to produce oxygen.5,6 Given its nature as a four-electron transfer process, the OER requires a significant amount of energy to overcome the kinetic barriers, thereby restricting the development of industrial water splitting technologies.7 However, the development of highly active and stable OER electrocatalysts can effectively reduce the reaction energy barrier and enhance the efficiency of water electrolysis. Constructing heterostructure electrocatalysts is a powerful approach that can improve catalytic activity and stability by modifying the electronic structure of the active site.8,9 Compared with ordinary heterostructures connected by chemical bonds, van der Waals (vdW) heterostructures offer greater potential for the development of new materials via stacking monolayers with varying crystal symmetries, structures, and lattice parameters.10 Meanwhile, with weak vdW bonding, the additional generated heterogeneous interface can achieve efficient electron–hole separation in vertically stacked adjacent layers, resulting in a more pronounced synergy effect.11The synthesis of vdW heterostructures is not only anticipated to facilitate mass production by directly synthesizing large quantities of target materials, but it can also result in diverse properties via varying the type of monolayer nanosheet unit.12 Specifically, depending on the specifics of the chemical reaction, different monolayer building blocks can be selected for stacking to create single-functional or multi-functional catalysts, thereby enhancing reaction rates, efficiency, selectivity, and stability.13 Therefore, selecting an appropriate assembly strategy for integrating diverse structural units is of critical importance. For the OER, nickel–iron materials, which exhibit excellent activity, are among the most promising types of catalysts and serve as optimal stacked monolayer nanosheet units.14 It is widely accepted that monolayer MoS2 is a high-performance electrocatalyst for the HER and NiFe-layered double hydroxide (NiFe-LDH) for the OER.15,16 Consequently, various types of MoS2 monolayer nanosheets (NSs) and NiFe-LDH monolayer NSs with opposite charges were assembled and restacked by solution-phase synthesis of the electrocatalytic water-splitting reaction.17 This assembly strategy ingeniously harnesses the mutual attraction of opposite charges to fabricate a high-efficiency bifunctional catalyst, while the introduction of hetero-charges onto material surfaces significantly enhances synthetic manoeuvrability during the loading process.
The non-metallic atoms in electrocatalysts play an important role in site regulation and catalytic performance during the OER.18 In metal phosphorus chalcogenides, the presence of sulfur and phosphorus has a synergistic effect on the surface electronic structure of the center metal atoms.19 Compared to layered materials with covalent bonds, these materials exhibit significantly stronger ionic bonds, significantly enhancing their conductivity.20 During the OER process, phosphorus and sulfur will be oxidized into phosphate and sulfate, respectively, which has been proven to be beneficial for the electrocatalytic reaction.21,22 Therefore, developing simple and efficient methods to assemble specified elements in vdW heterostructure catalysts with high OER activity and stability, while understanding the electronic structure, remains significantly challenging.
Herein, we present a method for constructing vdW heterostructures using spontaneously self-restacked NiPS3 (referred to as NPS) and FePS3 (referred to as FPS). Density functional theory (DFT) calculations predicted the Gibbs free energy required for the synthesis of these materials, demonstrating the feasibility of spontaneous restacking to create heterostructures. Meanwhile, DFT calculations were also performed to simulate the electronic structure of the restacked materials and evaluate their electrocatalytic activity. We used a straightforward process of mixing and filtering; the nanosheet units can be restacked into vertically arranged heterostructures. This synthesis method exhibits a degree of universality and can be extended to other MPS3 with similar structures. Furthermore, this special vdW heterostructure shows a completely different electronic distribution from the doped materials of the same composition. The surface of catalysts is rich in electrons, facilitating rapid reconstruction of the material during the electrocatalytic process. Additionally, cation segregation occurs during the reconstruction process. These differences significantly enhance the activity and stability of the material.
Results and discussion
Theoretical prediction and synthesis of self-restacked vdW heterostructures
The DFT calculations were performed to study the change in Gibbs free energy (ΔG) upon different restacked mole ratios of NPS NSs and FPS NSs (Fig. S1†). As the basic units of restacking, the ΔG of NPS NS and FPS NS self-assembly and combination in different proportions is negative. This indicates that their restacking is spontaneous, but the combination proportions have a tendency.23 As shown in Fig. 1a, the ΔG of self-assembly of NPS NSs is −5.30 eV, and the Gibbs free energy of self-assembly of FPS NSs is not much different, being −5.24 eV. This more negative ΔG makes it more likely that when the proportions of these two basic units are similar, the restacking process prefers self-assembly rather than forming vdW heterostructures. However, when the input mole ratio of NPS to FPS is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the ΔG of restacking to form a vdW heterostructure is −8.05 eV. This ratio is often the highest performance ratio in many nickel–iron based catalysts, and at the same time, this ratio is also the most likely to be obtained. This makes it possible to spontaneously synthesize nickel–iron vdW heterostructures using the restacking chemical potential of the material at this ratio. The density of states (DOS) reflects the distribution of electrons in different energy states which is crucial for understanding the electronic structure of materials. Fig. 1b displays DOS of pristine NPS, pristine FPS, Fe-doped NPS (Fe = 20 mol%, named DNFPS), and restacked NPS with FPS (Fe = 20 mol%, named RNFPS). NPS is a semiconductor with a bandgap of 0.82 eV, implying that it has low electrical conductivity and weak interaction with the adsorbent. The situation is similar for FPS as well.24 However, there emerge electronic states around the Fermi level (EF) in RNFPS, indicating the presence of a smaller band gap compared to the original semiconductor structure. In contrast, DNFPS remains semiconducting, although it has a small bandgap of 0.49 eV. It can be predicted through DOS calculations that the vertical restacking of RNFPS changes its electronic structure, making it beneficial for electrocatalytic reactions. Based on the predictions of the above theoretical calculations, we designed and synthesized nickel–iron nanosheet restacking materials (Fig. 1c). First, we obtained a large amount of bulk materials through solid-phase synthesis. After ultrasonic exfoliation and differential centrifugation of the bulk materials, nanosheet units were obtained (Fig. S2†). Then, the two kinds of nanosheet units were uniformly redispersed by ultrasonic treatment in the liquid phase with the proportion. The monolayers can be reconfigured under mechanical force. Pressure was applied to the basic structural units through vacuum filtration, which was proven effective in inducing structural restacking.25 In line with the predicted results, the target material RNFPS could be obtained through vacuum filtration. Inductively coupled plasma optical emission spectrometry (ICP-OES) verified that the ratio of nickel to iron elements in the obtained catalyst was consistent with the feeding ratio (Table S1†).
:1, the ΔG of restacking to form a vdW heterostructure is −8.05 eV. This ratio is often the highest performance ratio in many nickel–iron based catalysts, and at the same time, this ratio is also the most likely to be obtained. This makes it possible to spontaneously synthesize nickel–iron vdW heterostructures using the restacking chemical potential of the material at this ratio. The density of states (DOS) reflects the distribution of electrons in different energy states which is crucial for understanding the electronic structure of materials. Fig. 1b displays DOS of pristine NPS, pristine FPS, Fe-doped NPS (Fe = 20 mol%, named DNFPS), and restacked NPS with FPS (Fe = 20 mol%, named RNFPS). NPS is a semiconductor with a bandgap of 0.82 eV, implying that it has low electrical conductivity and weak interaction with the adsorbent. The situation is similar for FPS as well.24 However, there emerge electronic states around the Fermi level (EF) in RNFPS, indicating the presence of a smaller band gap compared to the original semiconductor structure. In contrast, DNFPS remains semiconducting, although it has a small bandgap of 0.49 eV. It can be predicted through DOS calculations that the vertical restacking of RNFPS changes its electronic structure, making it beneficial for electrocatalytic reactions. Based on the predictions of the above theoretical calculations, we designed and synthesized nickel–iron nanosheet restacking materials (Fig. 1c). First, we obtained a large amount of bulk materials through solid-phase synthesis. After ultrasonic exfoliation and differential centrifugation of the bulk materials, nanosheet units were obtained (Fig. S2†). Then, the two kinds of nanosheet units were uniformly redispersed by ultrasonic treatment in the liquid phase with the proportion. The monolayers can be reconfigured under mechanical force. Pressure was applied to the basic structural units through vacuum filtration, which was proven effective in inducing structural restacking.25 In line with the predicted results, the target material RNFPS could be obtained through vacuum filtration. Inductively coupled plasma optical emission spectrometry (ICP-OES) verified that the ratio of nickel to iron elements in the obtained catalyst was consistent with the feeding ratio (Table S1†).
 | ||
| Fig. 1 Theoretical computing and schematic illustration of the synthesis. (a) Density functional theory (DFT) calculated Gibbs free energy change upon different restacked mole ratios. (b) Computed DOS of NPS, FPS, RNFPS and DNFPS. (c) Schematic of the restacked layer structure. | ||
Structure and morphology characterization
The crystal structures of the RNFPS and DNFPS have obvious differences in the arrangement mode (Fig. 2a). The van der Waals heterostructures of RNFPS show horizontal homogeneity and vertical differentiation. However, DNFPS randomly substitutes nickel sites with iron atoms on the structure of NPS, showing multi-dimensional disorder. In both the structures, they are composed of the basic three trigonally distorted octahedral groups, [NiS6], [FeS6], and [P2S6]. Among them, the ratio of NiS6 to FeS6 is consistent with the mole ratio in synthesis, while the ratio of the P2S6 units is half of the total number of metals.26 The powder X-ray diffraction (XRD) techniques were selected to characterize the crystal structure of the materials (Fig. 2b). All the diffraction peaks of both NPS and FPS obtained are in good agreement with the standard XRD index cards of NiPS3 (PDF No. 00-033-0952) and FePS3 (PDF No. 00-033-0672) with the space group C2/m and no other peaks from impurities were observed.27 The DNFPS shows similar spectral signal patterns to NPS, but the peak positions have certain displacements, which is due to the doping of iron into the NPS unit cell. Unlike the above three, RNFPS shows peaks corresponding to (001), (002) and (004) planes, which is due to the large size of (001) facets in the crystal structure. The partial enlarged view of the XRD patterns further demonstrates their structural differences (Fig. 2b). For the (002) facets, the peak position of DNFPS is between that of NiPS3 and FePS3, which further indicates the success of Fe doping,28 while RNFPS shows peak splitting, which is caused by the NPS units and the FPS units respectively. As shown in Fig. 2c, Raman spectroscopy shows the differences in the valence bond composition of different materials. Normal Raman-active phonon modes have been experimentally observed in NPS and FPS.29,30 In RNFPS, the excitation wavelengths are similar to those of NPS. This is because most of the crystal structure is the same as that of NPS. It is worth noting that a special excitation wavelength is different from that of the other three materials, which is often observed in DNFPS (red cycle).28 | ||
| Fig. 2 Structure and morphological characterization studies of catalysts. (a) Crystal structures of RNFPS and DNFPS. (b) XRD patterns of RNFPS, DNFPS, NPS and FPS. (c) Raman spectrum of RNFPS, DNFPS, NPS and FPS. (d)–(f) HAADF-STEM image of the RNFPS with different resolutions. (g) EDS elemental mappings of the RNFPS. | ||
Scanning electron microscopy (SEM) characterized the morphology of these materials (Fig. S3–S6†). NPS, FPS and DNFPS are layered structures and have micron size with a smooth surface. But for RNFPS, its surface is formed by the arrangement of multiple small-diameter flakes as a whole block. This is because RNFPS is formed by the restacking of exfoliated NSs and does not have the long surface continuity of chemically synthesized restacked materials. Meanwhile, this also increases the specific surface area of the material, which is more conducive to the exposure of active sites. Furthermore, scanning electron microscope energy dispersive spectrum (SEM-EDS) elemental mapping images prove the content and distribution of elements in these materials. The transmission electron microscopy (TEM) image shows the morphology of RNFPS with irregular lamellar layers (Fig. S7†), and the corresponding EDS elemental mapping images also verified the above conclusion. The differences in composition also bring about differences in wettability.31 The wettability measurements were used to test the wettability of catalysts, which affects the interaction between the catalysts and electrolytes relating to their electrocatalysis performance.32 The contact angle of RNFPS is 84.74° decreased compared with DNFPS, NPS and FPS, which benefits from the van der Waals heterostructures (Fig. S8†). To further explore the fine morphology, aberration-corrected high angle annular dark-field scanning TEM (HAADF-STEM) with different resolution images was directly conducted to analyse the specific restacking arrangement situation (Fig. 2d–f). Obviously, the material is composed of different sizes and is restacked interlacedly. In high resolution HAADF-STEM images, the lattice fringes in different regions show different lattice spacings, which are produced by NPS and FPS. The corresponding EDS elemental mapping image further proves that these two parts belong to NPS and FPS respectively (Fig. 2g). These results indicate that the RNFPS material is a vdW heterostructure composed of NPS and FPS restacked along the vertical direction.
Electronic structure analysis
UV-vis diffuse reflectance absorption spectra are recorded to measure the band gap of the catalysts. RNFPS exhibits the smallest bandgap (1.71 eV) among the materials, demonstrating its superior electrical conductivity, which aligns with the trends predicted by prior DOS analysis (Fig. S9†). X-ray photoelectron spectroscopy (XPS) is used to determine the types of elements and the chemical environment on these materials surfaces. The XPS wide survey scan spectra clearly indicate the existence of Ni, Fe, P, and S elements in materials (Fig. S10†), which are consistent with SEM-EDS elemental mapping. Narrow scan spectra of Ni 2p3/2 show one prominent peak at about 855 eV with two satellite features as shown in Fig. 3a, which is characteristic of Ni2+ species.33 Slightly differently, compared with the main peak position of NPS at 855.2 eV, the main peak positions of DNFPS and RNFPS both shift to lower binding energies, being 855 eV and 854.6 eV. This indicates that the valence state of Ni is reduced and is proved to be caused by the introduction of Fe. Narrow scan spectra of Fe 2p show changes in binding energy of materials (Fig. 3b). The increase in the binding energy of Fe indicates the increase in the valence state and the loss of electrons proving the electron transfer between Ni and Fe.34 Based on the above conclusions, when the ratio of nickel to iron is the same, we believe that the difference in electron transfer is caused by the different synthesis methods. The charge density changes in RNFPS between the NPS layer and FPS layer shows that significant electron migration occurred in the material using a differential charge density map (Fig. 3c).35 The electrons in the FPS layer have undergone significant dispersion, increasing the electron density of the NPS layer. This also causes the electron density of the P and S elements around the metal sites to increase and the binding energy to decrease (Fig. S11†). To further verify the charge transfer in RNFPS, ultraviolet photoelectron spectroscopy (UPS) is carried out. The secondary electron cutoff edge and Fermi edge were recorded by UPS (Fig. S12†). The experimental results demonstrate that RNFPS has smaller work functions than DNFPS, which is related to the charge transfer at the NPS/FPS vdW interlayer. Notably, lower work functions strongly correlate with accelerated interfacial electron migration, which optimizes the adsorption–activation energetics of reactants and significantly improves OER performance.36 Furthermore, the work function disparity between NPS and FPS induces directional electron migration from FPS to NPS, thereby establishing an interfacial built-in electric field within the vdW heterojunction. This electronic redistribution effectively lowers the Ni oxidation state while increasing its electron density, aligning with our prior XPS and DFT analyses.37 Zeta potential measurements were conducted to further characterize the surface charge of the catalysts (Table S2†). NPS exhibits a positively charged surface, whereas FPS demonstrates negative polarity. Such complementarity in electrostatic profiles drives their spontaneous tendency to self-assemble into vdW heterostructures through coulombic attraction. In contrast, DNFPS maintains a weakly positive surface potential (2.44 mV), indicative of Fe doping-mediated charge redistribution within NPS. Notably, RNFP displays pronounced negative surface charge (−21.44 mV). The directional electron transfer from Fe to Ni elevates the Ni electron density, generating a surface electron-enriched configuration. Importantly, this electron reservoir facilitates dynamic surface reconstruction at operational potentials, as evidenced by in situ Raman spectroscopic detection of a metastable NiOOH intermediate.38 It is precisely because the vertically restacked heterostructure increases conductivity that the electron exchange of the material is enhanced. | ||
| Fig. 3 Electronic structure characterization of catalysts. (a) Ni 2p3/2 and (b) Fe 2p XPS spectra. (c) DFT calculations for the deformation charge density of RNFPS. Yellow region = electron accumulation and blue region = electron depletion. (d) Ni K-edge FT-EXAFS spectra in R space. (e) WT analysis of Ni K-edge. (f) Partial density of states (PDOS) of different Ni sites on RNFPS, DNFPS, and NPS. (g) Schematic diagram of the d orbital spin states of different Ni sites and their combination with OH. | ||
The electronic structure and coordination environment of Ni in materials were further identified by X-ray absorption near edge structure (XANES). A semi-quantitative assessment of nickel oxidation states in the synthesized catalysts was performed through half-height photon energy measurements of the Ni K-edge (Fig. S13†).9 In the clear detection diagram, the half-height absorption edge position of the obtained materials showed negligible variations. This phenomenon is due to the fact that X-ray absorption spectroscopy (XAS) is bulk analysis technology for obtaining structural information of the entire material volume, with limited surface information. The local coordination environment of nickel species was probed via extended X-ray absorption fine structure (EXAFS) spectroscopy, revealing Ni configurations within distinct structural components. (Fig. S14†).39 The Fourier transform (FT) Ni K-edge EXAFS spectrum was used to minutely investigate the coordination situation of obtained materials as shown in Fig. 3d. The first peak at around 2.0 Å corresponds to Ni–S coordination, and the second peak at around 3.1 Å is attributed to Ni–Ni/Fe coordination.9 The position of the first peak shows no obvious change, but the peak intensity of the iron-containing materials decreases. This indicates that the unsaturated coordination of nickel sites occurs. By contrast, for the second peak, the peak position of the iron-containing materials undergoes a positive shift compared to NPS, indicating the expansion of the unit cell. In RNFPS, the adjacent metal of the Ni site is Ni. Apparently, this kind of unit cell expansion is triggered by the adjacent FPS layer. In DNFPS, the adjacent sites of the Ni site are Ni/Fe. The ionic radius of Fe is larger than that of Ni, causing the unit cell expansion. Both kinds of unit cell distortions are beneficial for the reconstruction of the material and the improvement of its activity, but the enhancement of the activity of the material may not be the same.40 The wavelet transform (WT) EXAFS contour plots of RNFPS display two intensity maxima at 2.0 and 3.1 Å−1 (Fig. 3e), which also identifies the existence and change of both Ni–S and Ni–Ni/Fe bonds (Fig. 3d). To further analyze the charge state of the active Ni sites and the interaction with oxygen intermediates after Fe incorporation, we calculated the partial density of states (PDOS) of Ni (Fig. 3f). The introduction of Fe increased the unpaired electrons of Ni, which enhanced the adsorption of oxygen intermediates (Fig. 3g). Furthermore, compared with the doped material, the stacked material exhibited not only a higher number of unpaired electrons at Ni sites but also a spin-state transition from spin-up to spin-down. This spin orientation opposes the single-electron spin state of OH, potentially facilitating bonding between oxygen and metal atoms. This is demonstrated in the following electrochemical section.41,42
Electrochemical performance and surface reconstruction
To characterize the oxygen evolution reaction (OER) activity of synthesized catalysts, linear scan voltammetry (LSV) measurements were performed using a standard three electrode system at room temperature.43 As shown in Fig. 4a, RNFPS displays brilliant OER performance with an overpotential of 257 mV at a current density of 20 mA cm−2, which is better than that of other counterparts. The Tafel slope can measure the difficulty of the electron transfer steps of the material and the kinetic process in the chemical reaction.44 The Tafel slope of RNFPS is 49 mV dec−1, which is smaller than that of DNFPS (66 mV dec−1), physically mixed NPS and FPS (mole ratio = 4:1, named MNFPS, 80 mV dec−1), NiFe-LDH (86 mV dec−1), NPS (81 mV dec−1) and FPS (105 mV dec−1), indicating faster OER kinetics on RNFPS (Fig. 4b and S15†). In order to further evaluate electrocatalysis activity by comparisons, we tested the electrochemical double-layer capacitance (Cdl) to evaluate the electrochemical surface area (ECSA) of these catalysts (Fig. 4b, S16 and S17†).22 The Cdl of RNFPS is 1.46 mF cm−2, while calculated ECSA is 36.5 cm2, showing larger active surface area than contrast materials. The normalized electrocatalytic activity using the ECSA indicated that the observed differences primarily originate from electronic structure driven variations in intrinsic catalytic activity (Fig. S18†). These results also prove that RNFPS has excellent OER activity. Electrochemical stability is also one of the indicators for evaluating the performance of catalysts. Therefore, testing the electrochemical stability of the catalysts is essential. Chronopotentiometry tests were used to evaluate the long-term catalytic stability of RNFPS. The RNFPS showed great stability at different current densities (100, 200 and 500 mA cm−2) in 1 M KOH for 140 h, which is much better than that of DNFPS and NPS (Fig. 4c and S19†). We searched for and compared the representative catalysts reported with similar elemental compositions, and the RNFPS presents as the state-of-the-art alkaline OER catalyst (Fig. 4d and Table S3†).28,45–53
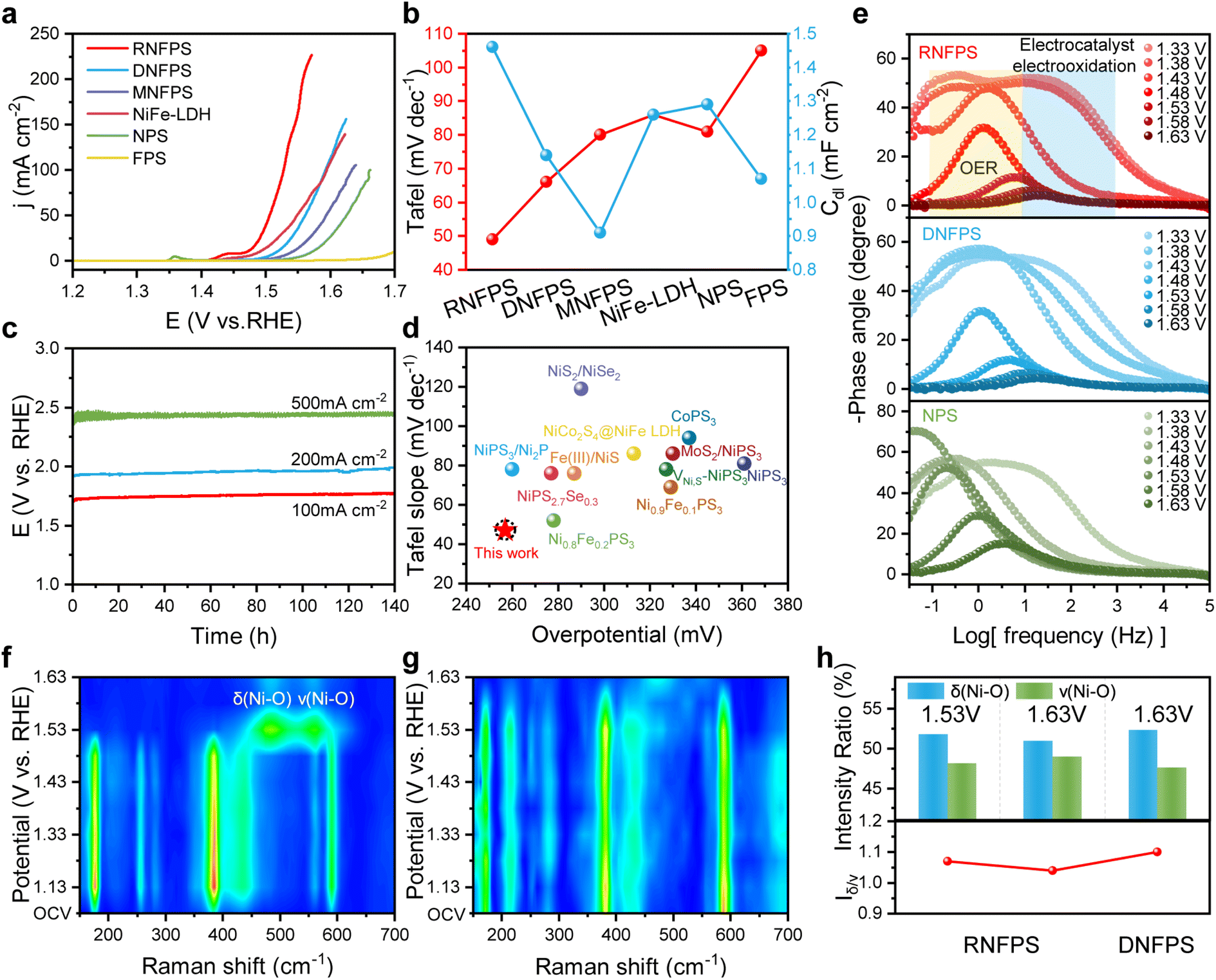 | ||
| Fig. 4 Electrochemical performance and chemical structure characterization of catalysts. (a) IR-corrected LSV curves. (b) Tafel slopes (left axis) and estimation of Cdl (right axis) by fitting linear regression at different scan rates. (c) Chronopotentiometry stability tests of RNFPS at different current densities in 1 M KOH. (d) Comparison of merit with kinetics and activity. (e) Bode phase plots of the in situ electrochemical impedance spectra of RNFPS, DNFPS and NPS. (f) Potential-dependent in situ Raman spectra of RNFPS and (g) DNFPS with heat maps. (h) Intensity ratio in part of in situ Raman spectra of RNFPS and DNFPS at different potentials. | ||
To elucidate the structural evolution mechanisms underlying electrocatalytic oxygen evolution, comprehensive investigation of dynamic surface transformations during operation becomes imperative. Notably, valence transitions in nickel-based oxides have been identified as pivotal activation pathways, wherein prior oxidation of Ni2+ to Ni3+/Ni3+δ facilitates the formation of catalytically active oxyhydroxide phases through structural reorganization.54 This metastable coordination environment has been extensively documented as essential for optimizing intermediate adsorption energetics. Furthermore, anionic constituent modifications exhibit synergistic enhancement effects, with elemental phosphorus and sulfur demonstrating progressive conversion into phosphate/sulfate species at oxidative potentials, thereby modulating electronic configurations and improving charge transfer kinetics.21,22 Such in situ compositional adjustments provide thermodynamic driving forces for maintaining structural integrity while enhancing interfacial reaction efficiency. To study the pre-oxidation of Ni in catalysts, cyclic voltammetry (CV) tests were performed (Fig. S20†). The cyclic voltammetry analysis reveals distinct charge storage characteristics between initial and subsequent cycles, with the primary scan demonstrating significantly higher capacitive behavior compared to subsequent measurements.55–57 Notably, RNFPS is activated rapidly to reach a stable state within the 5th cycle. Meanwhile, DNFPS and NiFe LDH show a continuous electrochemical corrosion phenomenon with a gradual increase in cycles. This electrochemical stabilization phenomenon indicates the occurrence of morphological evolution at the RNFPS electrode interface, wherein the catalyst undergoes permanent structural reorganization into stable metastable oxyhydroxide species. Such surface reconstruction processes ultimately yield a robust catalytic architecture with optimized coordination environments for maintaining efficient oxygen evolution functionality. To investigate the charge transfer characteristics across distinct material phases, electrochemical impedance spectroscopy (EIS) measurements were systematically performed. This technique enables quantitative assessment of interfacial processes by analyzing frequency-dependent impedance variations, thereby elucidating the kinetic limitations associated with electron transport mechanisms under varying structural conditions. The RNFPS demonstrates superior interfacial kinetics revealing a substantial reduction in charge-transfer resistance (Rct) relative to DNFPS, NPS, and FPS (Fig. S21†).58 Furthermore, in situ EIS measurements were carried out to deeply detect charge transfer at electrode/electrolyte interfaces of catalysts (Fig. 4e and S22†).59,60 In Bode plots, the high-frequency region (101–105 Hz) primarily corresponds to charge transfer processes within the catalyst electrode and potential oxidation mechanisms. Compared to DNFPS, RNFPS exhibits stronger adsorption capacity of activity sites for –OH at the same potentials, and the accumulation of –OH is more conducive to driving the overall catalytic process. This also indicates that the valence state of the active sites in the material increases. The low-frequency region (10−1–101 Hz) exhibits strong correlation with the OER. RNFPS shows an earlier decrease in the phase angle at 1.43 V and a lower phase angle at the same potential compared to other materials in the low-frequency region, indicating that RNFPS is more prone to polarization and undergoes the OER.
In addition, we utilized in situ Raman spectroscopy to observe the changes in the catalysts during the electrochemical process. For RNFPS, as the applied voltage increases from open-circuit voltage to 1.48 V, no significant changes are observed in the spectrum, which is consistent with the material under dry conditions, but the intensity slightly weakens, indicating that certain electrochemical corrosion occurs in the material, although to a minor extent. When the applied voltage is 1.53 V, the spectral peak undergoes a significant mutation (Fig. 4f). The Raman spectrum exhibits two prominent vibrational modes characteristic of Ni3+–O coordination, appearing at 483 and 560 cm−1. These spectral features are assigned to specific lattice dynamics in the layered nickel oxyhydroxide structure, with the lower-frequency peak corresponding to the Eg symmetry bending mode (δ(Ni–O)) and the higher-frequency signal arising from the A1g symmetry stretching vibration (ν(Ni–O)).61 These distinct fingerprint regions confirm the structural evolution toward catalytically active NiOOH phases under electrochemical polarization conditions. The same phenomenon can be found in DNFPS (1.63 V), but not in NPS (Fig. 4g and S23†). This indicates that the introduction of Fe is beneficial for the reconstruction of the material. In general, NiOOH contains β and γ phases, with the vibrational intensity of the ν(Ni–O) being notably weaker in γ-NiOOH compared to the β phase. This reduced intensity, reflected by a higher variation of δ(Ni–O)-to-ν(Ni–O) ratios (labelled as Iδ/ν), can be attributed to the more disordered and loosely packed structural arrangement of γ-NiOOH.54,61 We compared the Iδ/ν of RNFPS at 1.53 V and 1.63 V, observing a decrease from 1.07 to 1.04, indicating a reduction in the proportion of γ-NiOOH and its transformation into the more ordered, catalytically active, and stable β-NiOOH phase (Fig. 4h and S24†). In contrast, for DNFPS at 1.63 V, the Iδ/ν was 1.1, which is inferior to that of the restructured material of RNFPS. Therefore, we infer that the surface electron enrichment brought by this stacked van der Waals heterojunction structure can greatly increase the reducibility of the material surface, enabling the material to be rapidly, continuously and thoroughly reconstructed in a weak oxidation environment to perform catalysis with both high activity and long-term durability.
We also conducted spectroscopic and morphological studies on the RNFPS and DNFPS after long-term stability tests to investigate the differences among the materials after reconstruction. The XRD patterns of tested RNFPS (named TRNFPS) and tested DNFPS (named TDNFPS) show amorphous materials while no peak can be clearly observed (Fig. S25†). The Raman spectra of TRNFPS and TDNFPS confirm that the materials are nickel–iron-based oxyhydroxides (Fig. S26†). Specifically, the Iδ/ν ratio of TRNFPS is significantly higher than that of TDNFPS, indicating that TRNFPS exhibits a stronger tendency to form highly active and stable β-NiOOH, consistent with the results from in situ Raman characterization. Moreover, a distinct new Raman peak is observed at 677 cm−1, which we attribute to the presence of FeOOH.62 The XPS of TRNFPS shows the valence state changes of each element after the oxidation of materials (Fig. S27†). Ni 2p3/2 and Fe 2p3/2 are at 855.9 eV and 712.4 eV respectively, which is consistent with their trivalent peak positions.34 Compared to TDNFPS, TRNFPS has a higher metal valence state, which is more conducive to the progress of the OER. Meanwhile, P 2p and S 2p are completely oxidized to phosphate and sulfate respectively.28 After the OER of the material, each element has undergone oxidation, and after reconstruction, each element has dissolved to varying degrees due to the oxidation process. This phenomenon can also be found in the same situation in other comparative materials. We also utilized synchrotron radiation to observe the changes in the coordination situation of the main active center Ni site. TRNFPS has a higher valence state of Ni (Fig. S28a†), which is consistent with the XPS spectra. The peak observed at approximately 5.8 Å in the FT-EXAFS spectra of TRNFPS and TDNFPS provides insight into the alignment of the three Ni atoms in a linear configuration, suggesting a method for estimating the material's particle size.40 Notably, the particle size can be inferred to be smaller when the intensity of this peak is reduced. Based on the FT-EXAFS spectra, the peak intensity of TRNFPS at approximately 5.8 Å decreased compared to TDNFPS, suggesting a reduction in the particle size (Fig. S28b†). HAADF-STEM was used to further observe the morphology and element distribution of the material after the test. Both materials have amorphous parts and crystalline particles, while the particle size of the crystalline particles in TRNFPS is smaller (Fig. S29†). The HAADF-STEM elemental mapping images of TRNFPS and TDNFPS show differences in Fe, P and S contents (Fig. S30–S32†). This indicates that the excessive dissolution of the active elements in TDNFPS reduces the activity of the material, and this dissolution also reduces the catalytic stability. To sum up, RNFPS is more prone to reconstruction, and the reconstructed material is more conducive to the progress of the OER.
Universal expansion
Metal thiophosphates, with a general formula of MPS3 (M = Mn, Fe, Co, Ni and Zn), exist in multiple structures according to the different component metal elements.63 In addition to NPS and FPS mentioned in this paper, when the metal elements are selected as Mn, Co, and Zn, the formed MPS3 is also of a monoclinic structure and belongs to the C2/m space group. The same structure and similar cell parameters imply the possibility of their mutual stacking. So, we synthesized the other three powder materials, MnPS3 (named MPS), CoPS3 (named CPS) and ZnPS3 (named ZPS) by using the same solid-phase synthesis method and exfoliated them under the same conditions to obtain the basic nanosheet units of different elements (Fig. S33 and S34†). NPS has intermediate cell parameters among them, so we took the NPS structural unit as the main component. NPS NSs and other metal units were restacked and assembled at a mole ratio of 4:1 to test the scalability of this synthesis method (Fig. 5a). The RNMPS, RNCPS, and RNZPS are short for restacked NPS NSs with MPS NSs, restacked NPS NSs with CPS NSs, and restacked NPS NSs with ZPS NSs. The XRD patterns of different samples indicated that nickel-based restacked materials of different elements could also be synthesized (Fig. 5b). To conduct the same comparison with restacked materials, we also synthesized doped Ni0.8Mn0.2PS3 (named DNMPS), doped Ni0.8Co0.2PS3 (named DNCPS) and doped Ni0.8Zn0.2PS3 (named DNZPS) by using the solid-phase synthesis method. It could be found from the Raman spectra that they were all based on the Raman shift of NPS, and this result was close to that of previous materials (Fig. 5c and S35†). Meanwhile, TEM also proved the successful restacking of the materials from the morphology (Fig. S36†). Therefore, we tested the OER activities of different nickel-based restacked materials and doped materials. The van der Waals heterostructures composed of the restacking of the same element have different degrees of improvement in electrocatalytic activity compared to the corresponding doped materials (Fig. 5d and S37†). This indicates that this synthetic method of spontaneously restacked van der Waals heterostructures is universal, which also means that more catalysts with different functions can be synthesized.
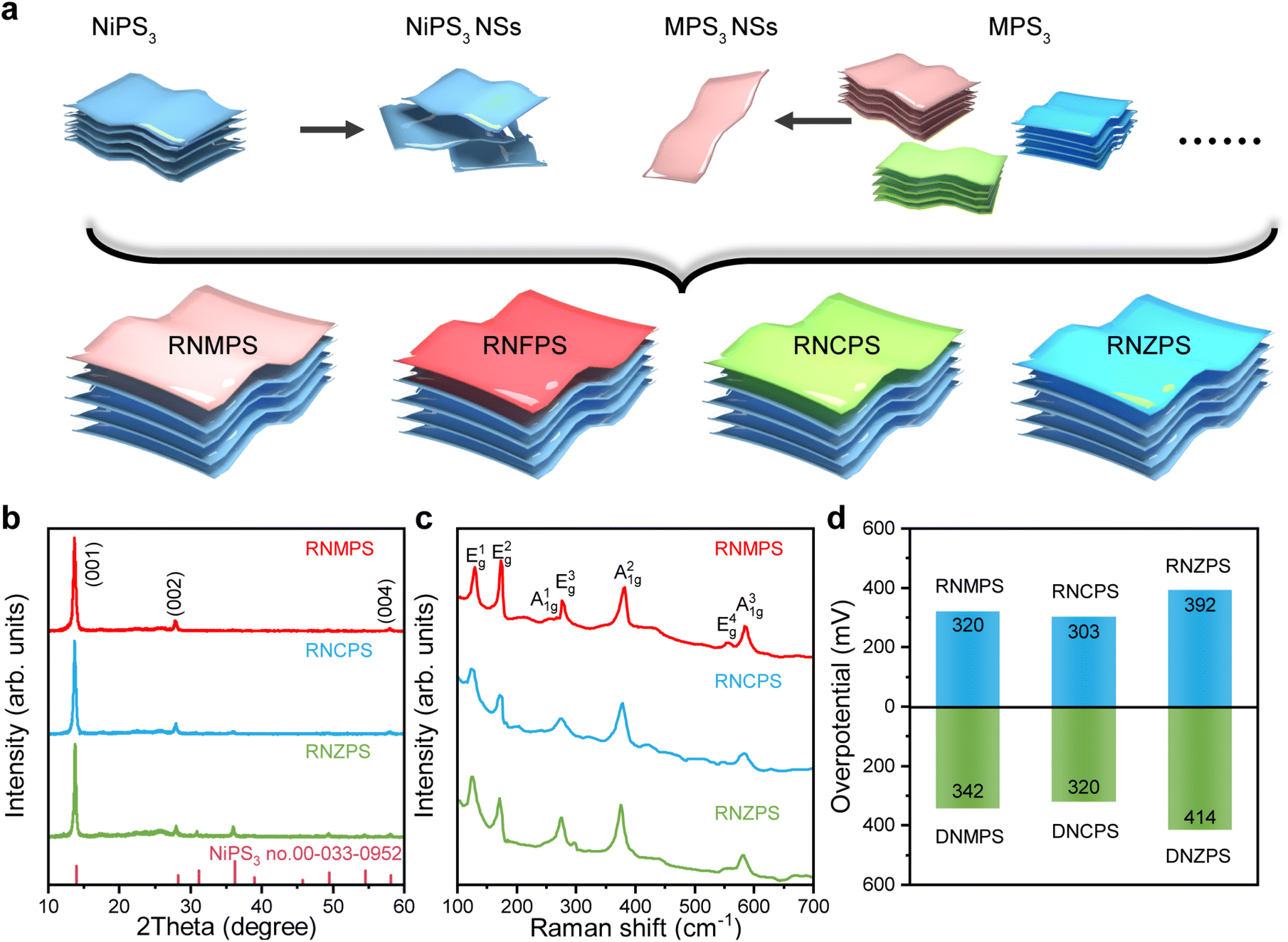 | ||
| Fig. 5 Universal expansion of RNMPS, RNCPS, and RNZPS. (a) Schematic of the restacked Ni with other transition metal layer structures. (b) XRD pattern, (c) Raman spectra and (d) overpotential contrast between restacked catalysts and doped catalysts at 10 mA cm−2. | ||
Conclusions
In this work, we predicted the material performance in advance based on DFT calculations and fabricated a high-performance OER catalyst composed of vertically restacked nickel–iron-based phosphorus–sulfur van der Waals heterostructures from bottom to top. The DFT calculations indicate that the basic units of NPS NSs and FPS NSs in a molar ratio of 4:1 exhibit the lowest restacking Gibbs free energy. This characteristic allows the basic units to spontaneously restack in a specific ratio, forming a van der Waals heterostructure. The synthesis method of this restacked heterostructure has been rarely reported in published studies. Furthermore, by calculating the DOS of the related comparison materials, it is found that this structure can transform the material from semiconductive into metallic, greatly changing the electronic structure of the material and enhancing the electrocatalytic activity of the material. This special vertically restacked van der Waals heterostructure can effectively transfer the internal electrons to the surface, endowing the surface with high reducibility, which is conducive to the rapid reconstruction of the material. This process is crucial in the OER. Meanwhile, the material releases phosphate and sulfate during the reconstruction process, further promoting the occurrence of the OER. However, the reconstructed structure can prevent the dissolution of metal ions and adjust the proportion of metal sites in the material, enhancing the stability of the material. Specifically, RNFPS shows excellent alkaline OER performance with a fast kinetic process, low η and long-term stability, such as a Tafel slope of 49 mV dec−1, 257 mV at j = 10 mA cm−2 and 140 h at j = 500 mA cm−2, much better than recently reported noble metal free catalysts. We have also extended this synthesis method. We found that by taking NPS as the basic component and combining different basic units (MPS3, M = Mn, Fe, Co and Zn), multiple van der Waals heterostructures can also be obtained. This work provides a promising way to prepare vital restacked van der Waals heterostructure catalysts with boosted OER activity and stability in alkaline media.
Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
P. X. and L. A. provided guidance and advice for this work. R. W. completed the main part of the experiments and the writing of the paper. S. L. provided support on the theoretical calculation. Y. H. conducted the HADDF-STEM tests. S. W. and J. Z. helped with the analysis of the data. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Key R&D Program of China (2021YFA1501101), the National Natural Science Foundation of China (No. 22425105, 22221001, 22271124, 22471103, and 22201111), China Northern Rare Earth (Group) High-Tech Co., Ltd (No. BFXT-2022-D-0078), the 111 Project (B20027), and the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001). We also acknowledge support from the Science and Technology Major Plan of Gansu Province (24ZD13GA015, 23ZDGA012, and 23ZDKA014) and the Natural Science Foundation key project of Gansu Province (24JRRA394).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- L. Magnier, G. Cossard, V. Martin, C. Pascal, V. Roche, E. Sibert, I. Shchedrina, R. Bousquet, V. Parry and M. Chatenet, Nat. Mater., 2024, 23, 252–261 CrossRef CAS PubMed.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 Search PubMed.
- R. Wang, Y. Wei, L. An, R. Yang, L. Guo, Z. Weng, P. Da, W. Chen, J. Jin, J. Li and P. Xi, Chin. J. Chem., 2020, 38, 772–786 CrossRef CAS.
- N. Zhang, Y. Hu, L. An, Q. Li, J. Yin, J. Li, R. Yang, M. Lu, S. Zhang, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2022, 61, e202207217 CrossRef CAS PubMed.
- A. Castellanos-Gomez, X. Duan, Z. Fei, H. R. Gutierrez, Y. Huang, X. Huang, J. Quereda, Q. Qian, E. Sutter and P. Sutter, Nat. Rev. Methods Primers, 2022, 2, 58 Search PubMed.
- Q. Zheng, W. A. Saidi, Y. Xie, Z. Lan, O. V. Prezhdo, H. Petek and J. Zhao, Nano Lett., 2017, 17, 6435–6442 CrossRef CAS PubMed.
- J. Li, X. Yang, Y. Liu, B. Huang, R. Wu, Z. Zhang, B. Zhao, H. Ma, W. Dang, Z. Wei, K. Wang, Z. Lin, X. Yan, M. Sun, B. Li, X. Pan, J. Luo, G. Zhang, Y. Liu, Y. Huang, X. Duan and X. Duan, Nature, 2020, 579, 368–374 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- L. Wu, M. Ning, X. Xing, Y. Wang, F. Zhang, G. Gao, S. Song, D. Wang, C. Yuan, L. Yu, J. Bao, S. Chen and Z. Ren, Adv. Mater., 2023, 35, 2306097 CrossRef CAS PubMed.
- J. Hu, C. Zhang, L. Jiang, H. Lin, Y. An, D. Zhou, M. K. H. Leung and S. Yang, Joule, 2017, 1, 383–393 CrossRef CAS.
- Z. Zhu, H. Yin, C. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H. Yang, Z. Tang, D. Wang, X. Chen and H. Zhao, Adv. Mater., 2018, 30, 1801171 Search PubMed.
- P. Xiong, X. Zhang, H. Wan, S. Wang, Y. Zhao, J. Zhang, D. Zhou, W. Gao, R. Ma, T. Sasaki and G. Wang, Nano Lett., 2019, 19, 4518–4526 CrossRef CAS PubMed.
- Q. Kang, D. Lai, W. Tang, Q. Lu and F. Gao, Chem. Sci., 2021, 12, 3818–3835 RSC.
- C. Du, K. N. Dinh, Q. Liang, Y. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127 CrossRef.
- B. Konkena, J. Masa, A. J. R. Botz, I. Sinev, W. Xia, J. Koßmann, R. Drautz, M. Muhler and W. Schuhmann, ACS Catal., 2017, 7, 229–237 CrossRef CAS.
- A. Dutta, S. Mutyala, A. K. Samantara, S. Bera, B. K. Jena and N. Pradhan, ACS Energy Lett., 2018, 3, 141–148 CrossRef CAS.
- J. Jin, J. Yin, Y. Hu, Y. Zheng, H. Liu, X. Wang, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2024, 63, e202313185 CrossRef CAS PubMed.
- X. C. Liu, S. Zhao, X. Sun, L. Deng, X. Zou, Y. Hu, Y.-X. Wang, C.-W. Chu, J. Li, J. Wu, F.-S. Ke and P. M. Ajayan, Sci. Adv., 2020, 6, eaay4092 CrossRef CAS PubMed.
- J. Wang, X. Li, B. Wei, R. Sun, W. Yu, H. Y. Hoh, H. Xu, J. Li, X. Ge, Z. Chen, C. Su and Z. Wang, Adv. Funct. Mater., 2020, 30, 1908708 CrossRef CAS.
- J. Pan, C. Guo, C. Song, X. Lai, H. Li, W. Zhao, H. Zhang, G. Mu, K. Bu, T. Lin, X. Xie, M. Chen and F. Huang, J. Am. Chem. Soc., 2017, 139, 4623–4626 Search PubMed.
- Q. Liang, L. Zhong, C. Du, Y. Luo, Y. Zheng, S. Li and Q. Yan, Nano Energy, 2018, 47, 257–265 CrossRef CAS.
- C. C. Mayorga-Martinez, Z. Sofer, D. Sedmidubský, Š. Huber, A. Y. S. Eng and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 12563–12573 Search PubMed.
- B. Kirubasankar, Y. S. Won, S. H. Choi, J. W. Kim, L. A. Adofo, S. M. Kim and K. K. Kim, Chem. Commun., 2023, 59, 9247–9250 Search PubMed.
- S. Yan, C. Wang, Q. Xie, L. Chen, W. Wang and X. Ai, Solid State Commun., 2022, 348–349, 114764 CrossRef CAS.
- I. Kerrache, Solid State Ionics, 1996, 92, 37–43 Search PubMed.
- M. Kim, K. L. Firestein, J. F. S. Fernando, X. Xu, H. Lim, D. V. Golberg, J. Na, J. Kim, H. Nara, J. Tang and Y. Yamauchi, Chem. Sci., 2022, 13, 10836–10845 Search PubMed.
- S. Zi, J. Zhu, Y. Zhai, Y. Hu, N. Zhang, S. Li, L. Liu, L. An, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2025, 64, e202413348 Search PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434 Search PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 Search PubMed.
- L. An, Y. Hu, J. Li, J. Zhu, M. Sun, B. Huang, P. Xi and C. Yan, Adv. Mater., 2022, 34, 2202874 CrossRef CAS PubMed.
- Y. Zhou, P. Guan, F. Chen, Z. Feng, H. Jia, T. Liang, M. Li, T. Wan, R. Tian, Z. Han and D. Chu, J. Colloid Interface Sci., 2023, 642, 23–28 Search PubMed.
- D. Chen, R. Lu, R. Yu, Y. Dai, H. Zhao, D. Wu, P. Wang, J. Zhu, Z. Pu, L. Chen, J. Yu and S. Mu, Angew. Chem., Int. Ed., 2022, 61, e202208642 CrossRef CAS PubMed.
- Y. H. Lee, S. Park, K. Lee, M. Y. Lee, K. H. Cho, S. J. Kim and K. T. Nam, ChemCatChem, 2019, 11, 1665–1672 CrossRef CAS.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y.-W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702–708 CrossRef CAS PubMed.
- H. Zhong, X. Wang, G. Sun, Y. Tang, S. Tan, Q. He, J. Zhang, T. Xiong, C. Diao, Z. Yu, S. Xi, W. S. V. Lee and J. Xue, Energy Environ. Sci., 2023, 16, 641–652 RSC.
- X. Zhang, H. Zhong, Q. Zhang, Q. Zhang, C. Wu, J. Yu, Y. Ma, H. An, H. Wang, Y. Zou, C. Diao, J. Chen, Z. G. Yu, S. Xi, X. Wang and J. Xue, Nat. Commun., 2024, 15, 1383 CrossRef CAS PubMed.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 CrossRef CAS PubMed.
- J. Zhang, G. Chen, D. Sun, Y. Tang, W. Xing, H. Sun and X. Feng, Chem. Sci., 2024, 15, 17900–17911 RSC.
- J. Zhu, S. Zi, N. Zhang, Y. Hu, L. An and P. Xi, Small, 2023, 19, 2301762 CrossRef CAS PubMed.
- Y. Liu, Y. Chen, Y. Tian, T. Sakthivel, H. Liu, S. Guo, H. Zeng and Z. Dai, Adv. Mater., 2022, 34, 2203615 CrossRef CAS PubMed.
- Y. Tong, P. Chen, L. Chen and X. Cui, ChemSusChem, 2021, 14, 2576–2584 CrossRef CAS PubMed.
- Y. Yang, Y. Kang, H. Zhao, X. Dai, M. Cui, X. Luan, X. Zhang, F. Nie, Z. Ren and W. Song, Small, 2020, 16, 1905083 Search PubMed.
- Q. Liang, L. Zhong, C. Du, Y. Luo, J. Zhao, Y. Zheng, J. Xu, J. Ma, C. Liu, S. Li and Q. Yan, ACS Nano, 2019, 13, 7975–7984 CrossRef CAS PubMed.
- B. Song, K. Li, Y. Yin, T. Wu, L. Dang, M. Cabán-Acevedo, J. Han, T. Gao, X. Wang, Z. Zhang, J. R. Schmidt, P. Xu and S. Jin, ACS Catal., 2017, 7, 8549–8557 CrossRef CAS.
- J. Song, S. Qiu, F. Hu, Y. Ding, S. Han, L. Li, H. Chen, X. Han, C. Sun and S. Peng, Adv. Funct. Mater., 2021, 31, 2100618 Search PubMed.
- F. M. Oliveira, J. Paštika, V. Mazánek, M. Melle-Franco, Z. Sofer and R. Gusmão, ACS Appl. Mater. Interfaces, 2021, 13, 23638–23646 Search PubMed.
- X. Feng, Q. Jiao, W. Chen, Y. Dang, Z. Dai, S. L. Suib, J. Zhang, Y. Zhao, H. Li and C. Feng, Appl. Catal., B, 2021, 286, 119869 Search PubMed.
- M. Zhong, W. Li, J. Chen, S. Ren, R. Qi, C. Wang and X. Lu, Sep. Purif. Technol., 2023, 310, 123164 Search PubMed.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 Search PubMed.
- X. Ren, C. Wei, Y. Sun, X. Liu, F. Meng, X. Meng, S. Sun, S. Xi, Y. Du, Z. Bi, G. Shang, A. C. Fisher, L. Gu and Z. J. Xu, Adv. Mater., 2020, 32, 2001292 Search PubMed.
- L. An, H. Zhang, J. Zhu, S. Xi, B. Huang, M. Sun, Y. Peng, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2023, 62, e202214600 CrossRef CAS PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 Search PubMed.
- Y. Wei, Y. Hu, P. Da, Z. Weng, P. Xi and C.-H. Yan, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312224120 Search PubMed.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. Liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- Z. Xiao, Y.-C. Huang, C.-L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 Search PubMed.
- L. Bai, S. Lee and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 3095–3103 Search PubMed.
- H. Zhang, T. Wei, Y. Qiu, S. Zhang, Q. Liu, G. Hu, J. Luo and X. Liu, Small, 2023, 19, 2207249 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02417j |
| ‡ Rui Wang and Shuhui Li have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |