DOI:
10.1039/D4SC08795J
(Edge Article)
Chem. Sci., 2025, Advance Article
Light-induced twisting, untwisting, and retwisting of aromatic polyamides: an interplay between the induced chirality and the co-facial π-stacking interactions†
Received
30th December 2024
, Accepted 28th April 2025
First published on 7th May 2025
Abstract
The ability of proteins to undergo conformational changes in response to varying environmental conditions has inspired chemists to devise smart materials that can achieve comparable functions. Oligopeptides, which are simplified versions of proteins, have demonstrated the ability to undergo conformational changes in response to stimuli, transitioning between two ordered structures: helix and sheet. In contrast, such conformational transitions in non-peptidic synthetic polymers are generally limited to order-disorder transitions, specifically shifting between helix and coil states. This report presents a novel approach in which we designed a periodically functionalized aromatic polyamide that exhibits the conformational dynamicity between two order structures (helix ↔ pleated-sheet ↔ helix). The enantiopure pendants of this aromatic polyamide induce a helical structure into the achiral polyamide backbones. At the same time, incorporating the guest molecule enhances the co-facial π-stacking and mediates a conformational transition from a helix to a pleated sheet-like structure. Subsequently, we employed photoresponsive merocyanine as the planar guest molecule, which served as a reversible conformational switch for this aromatic polyamide. The planer merocyanine induces the host–guest complex with this polymer and transforms the helical structure of polyamides into a pleated sheet-like structure. When exposed to visible light, the planar merocyanine changes into a non-planar spyropyran, which breaks apart the host–guest complex and effectively restores the helical structure of aromatic polyamides. Therefore, we present an intriguing demonstration of the twisting, untwisting, and retwisting of aromatic polyamides by balancing two key interactions, such as co-facial π-stacking along the aromatic polyamide backbone and the helical induction from the grafted enantiopure residues.
Introduction
The secondary structure of polypeptides, including α-helices, β-sheets, and turns, comprises amino acid building blocks crucial for achieving the functional 3D structure of proteins.1–4 It is important to note that changes in these secondary structures, particularly in response to environmental factors, play a significant role in various biological processes.5–7 Beyond the essential biological functions for survival, improper conformational transitions of peptide segments can disrupt natural processes, leading to malfunction. For instance, neurodegenerative diseases such as Alzheimer's, Creutzfeldt-Jakob, and others are linked to protein misfolding, where there is an irreversible transition from α-helix to β-sheet structures.8–12 Understanding these processes is vital not just for biological insights but also because the responsiveness of secondary structures makes them appealing for developing smart materials.13–18 Synthetic peptides are the most popular choice for study, as their backbone is similar to natural proteins.14,19–21 Researchers have successfully induced conformational changes in synthetic polypeptides, switching between α-helices and β-sheets by manipulating non-covalent interactions among side chains or applying external stimuli like temperature, pH changes, salt concentrations, redox potential, light exposure, and metal ion binding, among others.9,22–26 However, most of these peptides undergo an irreversible transition from α-helices to β-sheets.9,22,26–29 Along with polypeptides, other synthetic polymer chains have also been engineered to exhibit a stimuli-responsive secondary structure.30–35 Yet, the conformational changes in these macromolecules are generally limited to an order-disorder (helix ↔ coil) transition.
Herein, we report the design of periodically grafted aromatic polyamides (non-peptide polymer) exhibiting the conformational dynamicity between two order structures (helix ↔ pleated-sheet). This is achieved by carefully adjusting two crucial interactions: the induced chirality, which stabilizes the helical form, and the co-facial π-stacking interactions, which support the stability of the intrachain sheet-like structure. The chirality introduced by the grafted phenylalanine residues promotes the twisting of aromatic polyamide backbones, effectively suppressing the co-facial π-stacking interactions among the pyrene amides. Furthermore, incorporating planar guest molecules into these helical aromatic polyamides improves the co-facial π-stacking, inducing the pleated sheet-like arrangement. Lastly, to facilitate reversible conformational transitions, we propose using an aromatic guest molecule capable of shifting its structure from planar to non-planar in response to external stimuli. We hypothesized that the planar guest molecule effectively forms a host–guest complex and enhances the co-facial π-stacking interactions to promote an intrachain sheet-like arrangement of the aromatic polyamides. In contrast, the host–guest complex breaks apart when the planer guest transitions to a non-planar conformation, restoring the aromatic polyamides' twisted structure. As a result, the stimuli-responsive conformational change of the guest molecule can be utilized to tune the secondary structure of these aromatic polyamides.
Result and discussion
A series of periodically functionalized (enantiopure- and racemic-phenyl alanine derivative) aromatic polyamides have been synthesized from a common precursor polyamide (P1 in Scheme 1) via post-polymerization modifications using azide–alkyne click chemistry.36 The precursor polymers' (P1) pendant propargyl group served as a reactive handle to transform the achiral aromatic polyamides into enantiopure or racemic polymers using azide–alkyne click reactions (Schemes 1 and S2†). Moreover, we have also prepared the related model compound (Scheme S3†) to understand the chiroptical properties of these aromatic polyamides. The experimental section has described the detailed synthesis and characterization of the azido acid units, functionalized polymer chains, and the model compounds (Schemes S1–S3; also see Fig. S1–S5 and S7–S9†). The azide–alkyne click transformation was nearly quantitative as assessed by comparing the NMR (1H and 13C) spectra of the post-polymerization modified polymer (P1-Phe(L) or P1-Phe(D) or P1-Phe(D/L)) with the precursor polymer (Fig. 1 and S2–S5†). The characteristic 1H NMR signal of the alkyne proton (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH at ∼3.78 ppm) in the precursor polymer (P1) disappeared (Fig. 1, S2, S3 and S5;† highlighted in the purple shade) in the click-modified final polymers (P1-Phe(L) or P1-Phe(D) or P1-Phe(D/L)). Additionally, a downfield shift (∼0.28 ppm) of the CH2 propargyl group (at 5.25 ppm, c′ to c″ in Fig. 1, S2, S3 and S5†) signifies that post-polymerization modifications underwent quantitatively. Next, the molecular weights of those periodically functionalized polymers were estimated using size exclusion chromatography (SEC) analysis with DMF as the eluent (Fig. S6, also see Table S1†).
CH at ∼3.78 ppm) in the precursor polymer (P1) disappeared (Fig. 1, S2, S3 and S5;† highlighted in the purple shade) in the click-modified final polymers (P1-Phe(L) or P1-Phe(D) or P1-Phe(D/L)). Additionally, a downfield shift (∼0.28 ppm) of the CH2 propargyl group (at 5.25 ppm, c′ to c″ in Fig. 1, S2, S3 and S5†) signifies that post-polymerization modifications underwent quantitatively. Next, the molecular weights of those periodically functionalized polymers were estimated using size exclusion chromatography (SEC) analysis with DMF as the eluent (Fig. S6, also see Table S1†).
 |
| | Scheme 1 Design and synthesis of azido acid functionalized aromatic polyamides. | |
 |
| | Fig. 1 Stack plot 1H NMR spectra of azido acid (Phe(D)-N3) (bottom), precursor polymer (P1), and post-polymerization modified aromatic polyamide (P1-Phe(D)) (top). Inset showing the expanded region (5.05–5.70 ppm) of P1 and P1-Phe(D). It showcases the downfield shift of – CH2 propargyl protons (c′ to c″) after post-polymerization modifications (P1-Phe(D)), indicating completion of the click reaction. [All the NMR spectra were recorded in a mixture of DMSO-d6 and trifluoroacetic acid-d (TFA-d) at room temperature. A zoomed NMR spectra (8.2–12 ppm) of Phe(D)-N3, showcasing the peak for TFA, has been depicted as the inset]. | |
Helical induction and single-handedness (twisting)
The secondary structure of the periodically functionalized aromatic polyamides was studied in a DMSO (dimethyl sulfoxide) and triethylamine (99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5) mixture using absorption and CD spectroscopy. The details of sample preparations have been described in the ESI file.† While the precursor polymer (P1) dissolves well in DMSO, azido acid-functionalized polymers do not exhibit a similar solubility in pure DMSO. However, adding less than 1% of organic acids or bases in DMSO improves polymer solubility significantly. Fig. 2a (also see Fig. S11†) displays typical absorption and CD spectra of the functionalized aromatic polyamides in a dilute solution (50 µM) of DMSO–Et3N (99.5:0.5). The absorption spectrum of the precursor polymer exhibits a broad, featureless peak with λmax = 366 nm (see Fig. S11b†), corresponding to the pyrene diamide residues. After functionalization with azido acid, the absorption maximum of the pyrene diamides shifts to a higher wavelength (∼385 nm) (Fig. S11b†). This change suggests a weakening of the co-facial π-stacking (H-aggregation) interactions along the aromatic polyamide backbone, likely due to the twisting pyrene amides induced by the enantiopure pendant units. Next, we investigated the chiroptical properties of aromatic polyamides using CD spectroscopy. Polymers with enantiopure pendants, P1-Phe(D) and P1-Phe(L), exhibited bisignated cotton effects corresponding to pyrene diamide backbone in the CD spectra, which were mirror images of each other (Fig. 2a). In contrast, the precursor (P1) and the polymer with racemic pendants do not exhibit a CD signal (Fig. S11a and 2a†). Additionally, the arrangement of electric transition dipole moments in adjacent pyrene diamides caused exciton-coupled circular dichroism,37 leading to the observed couplet CD patterns (Fig. 2d). To gain insights into the secondary structure of aromatic polyamides, we compared them with related model compounds (Fig. 2e; M1-Phe(L) and P1-Phe(L), as well as M1-Phe(D) and P1-Phe(D), while maintaining a pyrene amide concentration of 50 µM). The absorption spectrum of M1-Phe(L) (λmax = 345 nm) was about 40 nm blue-shifted compared to P1-Phe(L) (λmax = 385 nm), indicating significant co-facial π-stacking in the model compound (Fig. 2b (bottom) and S12a†).38,39 While comparing the model compound's and polymer's emission spectra (Fig. 2c), the quenching of monomeric emission followed by red-shifted broad excimer band at 500 nm in the model compound reconfirmed the predominant intramolecular π-stacking interactions.40,41 Notably, the polymer exhibited higher CD intensity at similar concentrations than the model compounds. The lower CD intensity of the model compound could be due to the stronger co-facial π-stacking in the model compound outmatched the chiral influence of the pendant chain, resulting in a weak CD signal (Fig. 2b (top)). Moreover, the inherent structural differences between the polymer chain and the model compound may lead to varying π-stacking distances and electronic coupling among the consecutive pyrene amides. For instance, unlike the model compound, a pyridine spacer separates the pyrene diamides of the polymer chains at both ends, resulting in lengthier π-stacking distances than the model compound, which could explain the observed difference in their photophysical and CD studies. Next, we visualized the enantiopure aromatic polyamides through high-resolution transmission electron microscopy (Fig. S13†).
:0.5) mixture using absorption and CD spectroscopy. The details of sample preparations have been described in the ESI file.† While the precursor polymer (P1) dissolves well in DMSO, azido acid-functionalized polymers do not exhibit a similar solubility in pure DMSO. However, adding less than 1% of organic acids or bases in DMSO improves polymer solubility significantly. Fig. 2a (also see Fig. S11†) displays typical absorption and CD spectra of the functionalized aromatic polyamides in a dilute solution (50 µM) of DMSO–Et3N (99.5:0.5). The absorption spectrum of the precursor polymer exhibits a broad, featureless peak with λmax = 366 nm (see Fig. S11b†), corresponding to the pyrene diamide residues. After functionalization with azido acid, the absorption maximum of the pyrene diamides shifts to a higher wavelength (∼385 nm) (Fig. S11b†). This change suggests a weakening of the co-facial π-stacking (H-aggregation) interactions along the aromatic polyamide backbone, likely due to the twisting pyrene amides induced by the enantiopure pendant units. Next, we investigated the chiroptical properties of aromatic polyamides using CD spectroscopy. Polymers with enantiopure pendants, P1-Phe(D) and P1-Phe(L), exhibited bisignated cotton effects corresponding to pyrene diamide backbone in the CD spectra, which were mirror images of each other (Fig. 2a). In contrast, the precursor (P1) and the polymer with racemic pendants do not exhibit a CD signal (Fig. S11a and 2a†). Additionally, the arrangement of electric transition dipole moments in adjacent pyrene diamides caused exciton-coupled circular dichroism,37 leading to the observed couplet CD patterns (Fig. 2d). To gain insights into the secondary structure of aromatic polyamides, we compared them with related model compounds (Fig. 2e; M1-Phe(L) and P1-Phe(L), as well as M1-Phe(D) and P1-Phe(D), while maintaining a pyrene amide concentration of 50 µM). The absorption spectrum of M1-Phe(L) (λmax = 345 nm) was about 40 nm blue-shifted compared to P1-Phe(L) (λmax = 385 nm), indicating significant co-facial π-stacking in the model compound (Fig. 2b (bottom) and S12a†).38,39 While comparing the model compound's and polymer's emission spectra (Fig. 2c), the quenching of monomeric emission followed by red-shifted broad excimer band at 500 nm in the model compound reconfirmed the predominant intramolecular π-stacking interactions.40,41 Notably, the polymer exhibited higher CD intensity at similar concentrations than the model compounds. The lower CD intensity of the model compound could be due to the stronger co-facial π-stacking in the model compound outmatched the chiral influence of the pendant chain, resulting in a weak CD signal (Fig. 2b (top)). Moreover, the inherent structural differences between the polymer chain and the model compound may lead to varying π-stacking distances and electronic coupling among the consecutive pyrene amides. For instance, unlike the model compound, a pyridine spacer separates the pyrene diamides of the polymer chains at both ends, resulting in lengthier π-stacking distances than the model compound, which could explain the observed difference in their photophysical and CD studies. Next, we visualized the enantiopure aromatic polyamides through high-resolution transmission electron microscopy (Fig. S13†).
 |
| | Fig. 2 (a) CD spectra of P1-Phe(L), P1-Phe(D), P1-Phe(D/L) (top) and UV-vis of P1-Phe(L), P1-Phe(D), P1-Phe(D/L) (bottom); (b) CD spectra of P1-Phe(L) and the model compound (M1-Phe(L)) (top), normalized UV-vis spectra of P1-Phe(L) and the model compound (M1-Phe(L)) (bottom); (c) normalized fluorescence spectra of P1-Phe(L) and the model compound (M1-Phe(L)) [all the solutions were prepared in DMSO + triethylamine (TEA) (99.5:0.5) at 50 µM and the arrows (a and b) indicate the corresponding y-axes for CD (left) and UV-vis (right) spectra]; (d) structural comparison of the enantiopure azido acid clicked polymers (P1-Phe(D and P1-Phe(L))) depicting chirality induction from pendant units (enantiopure) to aromatic polyamide backbone and the orientations of the electronic transition moments (the assignment of helical handedness depicted in this figure is purely illustrative); (e) structural comparison of the model compounds. | |
Effect of the guest molecule on helical folding (twisting → untwisting)
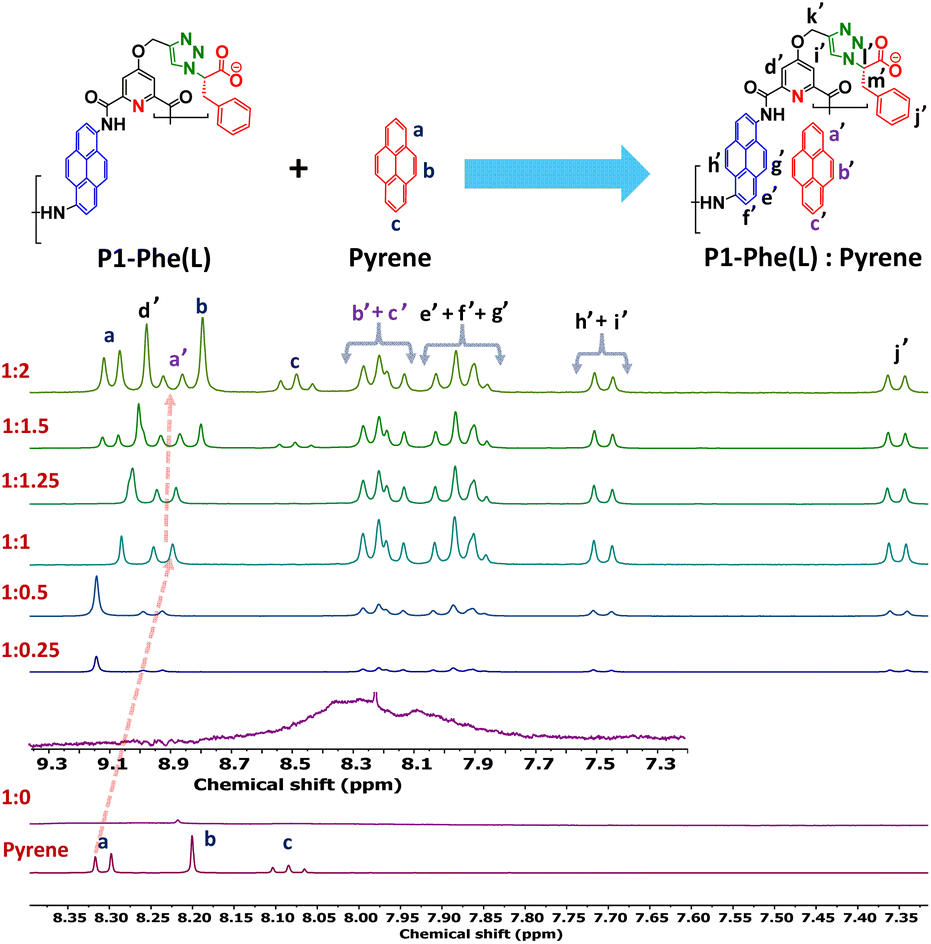
In the previous section, we noted that chiral induction from the pendant groups (ICD) outperformed the co-facial π-stacking interactions between adjacent pyrene diamides, leading to a twisted structure in the aromatic polyamides. We then explored the possibility of altering the secondary structure of aromatic polyamides by enhancing π-stacking interactions. This approach aims to counterbalance the predominant chiral induction and promote an intrachain sheet-like arrangement of pyrene diamides in the aromatic polyamide backbone. A rigid pyridine spacer between the pyrene diamide units likely limits robust co-facial π-stacking interactions along the polymer backbones. Therefore, a suitable planar guest molecule fitting into the free space between the consecutive pyrene diamides might enhance the co-facial π-stacking interactions. However, two scenarios could arise when an aromatic guest enters into the twisted pockets of the polyamides (P1-Phe(L), P1-Phe(D)): (a) the guest may adopt a twisted orientation, allowing the ICD continued to dominate, or (b) the planar π-surface could strengthen the co-facial π-stacking along the polyamide backbone, overshadowing the ICD and resulting in a π-stacked pleated structure (Scheme S4†). We investigated the host–guest complexation and related structural changes using various spectroscopic techniques. The 1H NMR spectra of P1-Phe(L) in DMSO-d6 and triethylamine (99.5:0.5) solvent showed broad signals (depicted as an inset in Fig. 3), likely due to nonspecific intermolecular interactions involving several H-bond donors and acceptors (polyamide backbone) at the NMR concentration (1 mM, much higher than spectroscopic studies carried out before), leading to diverse rotational conformers (Fig. 3, 1:0). Notably, as pyrene (guest) is added to the polymer solution, the 1H NMR signals remarkably sharpen, suggesting the formation of a discrete conformer through successful host–guest complexation as illustrated in Fig. 3 (P1-Phe(L):pyrene; also see Fig. S14–S16†).42 An upfield shift of pyrene diamides (host) and pyrene (guest) proton signals indicates possible co-facial π-stacking interactions between the polymer and the guest molecules.43–45 Moreover, the upfield shift of the pyrene proton (Fig. 3; at ∼8.32 ppm, marked a′) increases with the guest-to-host ratio, leveling off at a ∼1:1 ratio, indicating the stoichiometric nature of the host–guest complex (see Fig. S16 and S17†). Furthermore, the partial NOESY spectra of the 1:1 host–guest complex at room temperature have been depicted in Fig. S18.† Strong NOE was observed between pyrene amides (host) and the guest molecule (pyrene). For example, the pyrene (guest) protons were correlated with the e′, f′, g′, and h′ protons of the pyrene amides (host polymer).
 |
| | Fig. 3 A stacked plot of the 1H NMR spectra (aromatic region) of pyrene, P1-Phe(L) (1:0), and P1-Phe(L):pyrene (host–guest) complex, demonstrating the spectral changes upon addition of an increasing amount of pyrene to the polymer scaffold; a zoomed NMR spectra of the uncomplexed host polymer (1:0) has been depicted as the inset; [1H NMR spectra were recorded in a mixture of DMSO-d6 and triethylamine (TEA) ((99.5:0.5)) at 1 mM]. | |
Next, we investigated the chiroptical properties of host–guest complexes using CD spectroscopy. In a titration experiment involving a polymer solution at a concentration of 50 µM, we observed that as the amount of guest molecules increased, the intensity of the circular dichroism (CD) signal from pyrene amides significantly decreased. This effect became particularly pronounced at a guest-to-host ratio of 1:1, where the CD signal nearly disappeared (refer to Fig. 4a, c and S20†). Beyond this point, additional pyrene had minimal effect on the ICD signal (Fig. 4b and S20†). These findings suggest that host–guest complexation results in the planarization of aromatic polyamide chains. Notably, pyrene plays a significant role in reinforcing co-facial π-stacking by bridging gaps between pyrene diamide units, converting the twisted structure of aromatic polyamides into a pleated sheet-like structure (Fig. 4e). Fluorescence studies also support the proposed structural change, showing a shift in emission from 425 nm in the host polymer to a structureless broad emission band at 489 nm in the host–guest complex (Fig. 4d and S21†). This shift indicates the formation of an exciplex between the pyrene diamide (host) and the pyrene (guest), suggesting the likelihood of a co-facially stacked H-aggregated conformation of the host–guest complex.46 Overall, the NMR, CD, and fluorescence spectroscopic data indicate that the planar aromatic π-surface of pyrene enhances the π-stacking interactions within the host–guest complex, outweighing the chiral induction (ICD) and leads to a conformational change from a one-handed helix to a pleated sheet-like (untwisted) structure (see Fig. 4e).
 |
| | Fig. 4 (a) CD spectra of P1-Phe(L):pyrene (host–guest) complex, demonstrating the spectral changes upon addition of increasing amount of pyrene to the aromatic polyamide scaffold; (b) a titration plot of CD intensity (mdeg) at 428 nm of the P1-Phe(L):pyrene (host–guest) complex as a function of added equivalents of pyrene (the solid line represents the calculated fitting curve for the experimental data point series); (c) CD spectra of P1-Phe(D):pyrene (host–guest) complex, demonstrating the spectral changes upon addition of increasing amount of pyrene to the host polymer; (d) normalized emission spectra of P1-Phe(L) (1:0) and P1-Phe(L):pyrene (host–guest, 1:1) complex [all the solutions were prepared in DMSO + triethylamine (TEA) (99.5:0.5) at 50 µM]; (e) a schematic representation illustrating the untwisting of the helically folded aromatic polyamide into a planar sheet upon complexation with pyrene (guest). | |
Twisting → untwisting → retwisting of aromatic polyamides
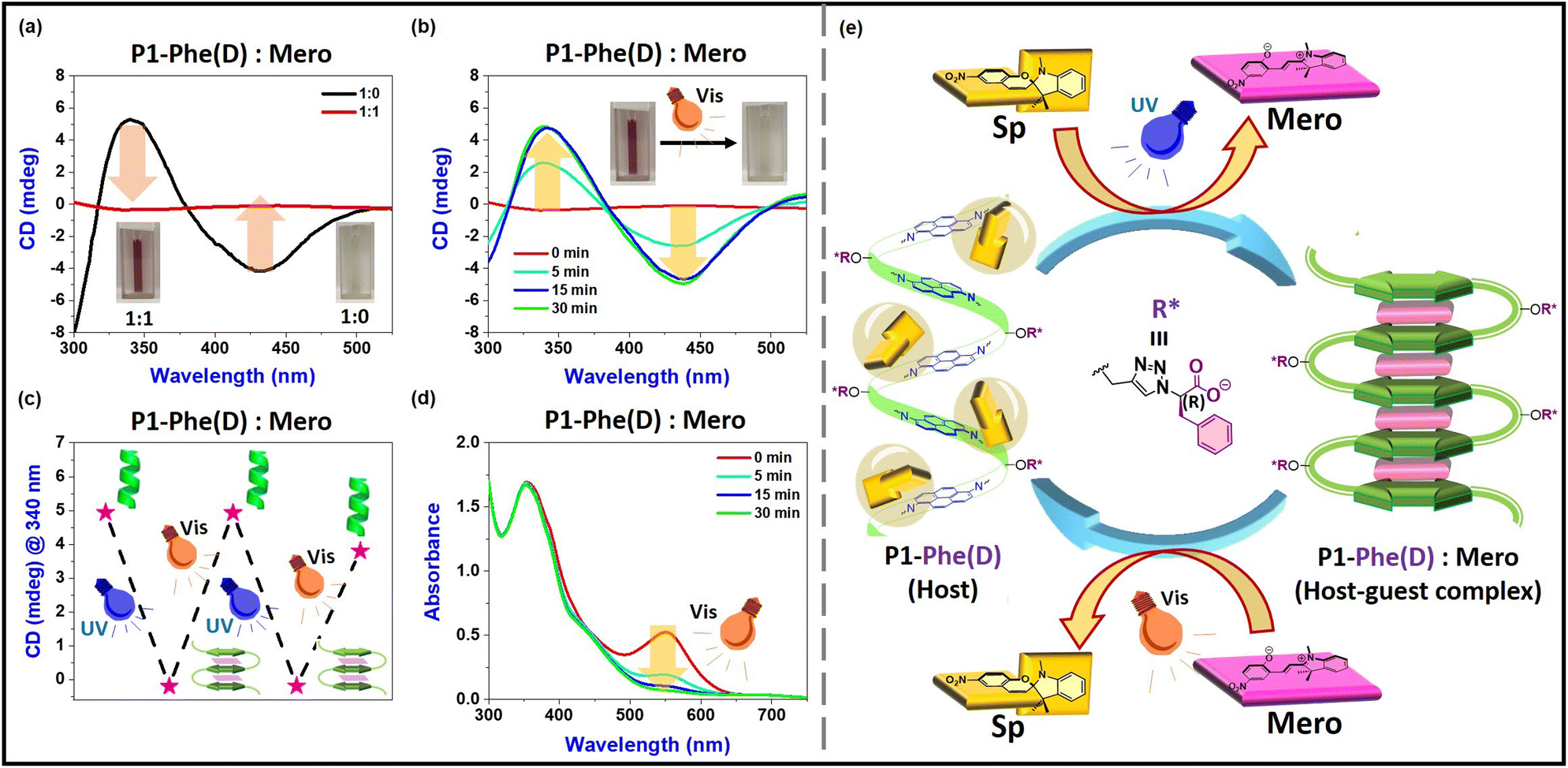
Our next objective was to fine-tune the conformational changes of these aromatic polyamides, enabling a reversible transformation that would dynamically enhance their properties. As previously demonstrated, a guest molecule featuring a planar aromatic π-surface can mediate the conformational transition of this polymer from helix to a pleated sheet-like structure. Ideally, upon removing the guest, the polyamides would return to their twisted state. However, selective removal of pyrene (the guest) from this host–guest complex is challenging. We propose using an aromatic guest that induces structural changes from planner to nonplanner in response to external stimuli. The planar guest molecule effectively forms a host–guest complex, which is expected to promote a sheet-like arrangement of the pyrene diamides along the polymer chain. However, this complex disassembles when the guest shifts to a non-planar conformation, leading to reforming the aromatic polyamides' twisted structure. We selected spiropyran as a conformational switch, which undergoes structural isomerization when exposed to stimuli.47–49 Spiropyran (Sp) transforms its planar merocyanine (Mero) form when exposed to UV light (λ = 365 nm), characterized by a distinct absorption peak at 557 nm (see Fig. S22 and S23a†). Additionally, the merocyanine reverts to spiropyran under visible light (557 nm), demonstrating reversible photoswitches to serve our purposes.47–49 The reverse isomerization of merocyanine, monitored by its absorbance at 557 nm over time under visible light, displays first-order kinetics with a conversion rate of 0.228 min−1 (Fig. S23b and S23c†).
Similar to pyrene, merocyanine can act as a planar guest molecule to induce the transition of helical aromatic polyamides into a planar sheet-like structure. In contrast to pyrene, the light-induced structural change (planner ↔ nonplanner) between merocyanine and spiropyran acts as a photoswitch, promoting the reversible conformational changes (helix ↔ sheet ↔ helix) of aromatic polyamides. At the beginning of our experiments, spiropyran was converted to planer merocyanine via UV-light irradiation (λ = 365 nm). We then mixed one equivalent of merocyanine with a P1-Phe(D) polymer solution (50 µM, DMSO:TEA = 99.5:0.5) and stirred in the dark to promote host–guest complex formation. The guest molecule-induced conformational change of the host–guest complex was monitored using UV-vis and circular dichroism (CD) spectroscopy. We observed a shift in the absorption peak of merocyanine from 557 nm to 550 nm upon host–guest complexations, changing the solution color from pink to brown (Fig. S24†). The induced CD (ICD) intensity of P1-Phe(D) dropped to zero after complexation with merocyanine (1:1 ratio), indicating the polymer's secondary structure was untwisted (Fig. 5a and e). Upon exposing the host–guest (P1-Phe(D)-Mero) complex to visible light, the ICD signal gradually recovered, restoring the original CD pattern and reducing the 550 nm absorption peak (Fig. 5b and d). This indicated that merocyanine was photo-isomerized back to spiropyran, led to its exclusion from the well-defined pockets of the polymer scaffold (P1-Phe(D)), allowing the twisted aromatic polyamide structure to re-emerge (Fig. 5c and e). Thus, we demonstrated the twisting, untwisting, and retwisting of aromatic polyamides by utilizing the photoisomerization between merocyanine and spiropyran as a conformational switch. This approach holds great promise for the development of responsive materials. The kinetics of merocyanine's photoisomerization to spiropyran within a host polymer were studied by measuring absorbance at 550 nm over time. In contrast with the photoisomerization of the uncomplexed guest molecule (k[uncomplexed guest] = 0.228 min−1), the photoisomerization of the host–guest complex followed second-order kinetics (k[host–guest] = 0.06 L mol−1 min−1) (Fig. 5d and S24†). Moreover, the stability of the merocyanine is remarkably enhanced when it is inside the host–guest complex.
 |
| | Fig. 5 (a) CD spectra P1-Phe(D) (1:0, colourless), P1-Phe(D) and merocyanine (P1-Phe(D):Mero, 1:1; brown) host–guest complex; (b) CD spectral changes of P1-Phe(L):Mero (host–guest) complex upon irradiation with visible light; (c) CD intensities (at 336 nm) of P1-Phe(D) and spiropyran (Sp) mixture upon alternative UV and visible light irradiation; (d) UV-vis spectra of the P1-Phe(L):Mero (host–guest) complex at different time intervals upon irradiation with visible light [all the solutions were prepared in DMSO + triethylamine (TEA) (99.5:0.5) at 50 µM]; (e) schematic representation of twisting → untwisting → retwisting of aromatic polyamide via host–guest complexation using photo-responsive merocyanine (Mero) as guest. | |
Conclusion
Our research has revealed the design of aromatic polyamides with a tuneable secondary structure. By strategically manipulating two fundamental interactions—co-facial π-stacking and chiral induction from the periodically functionalized pendant units—we tuned the secondary structure of the aromatic polyamide backbone to adopt either helical or planar sheet-like conformations reversibly. While the extent of chirality induced by the pendant chain remained consistent, we varied the π-stacking interactions using controlled host–guest complexation to fine-tune the secondary structure of these aromatic polyamides. The synthesis of functionalized aromatic polyamides from precursor polymer (P1) was accomplished through precise post-polymerization modifications with suitable azido acids (enantiopure or racemic). In these periodically functionalized aromatic polyamides, the chirality of pendant units dominated over π-stacking, leading to helix-sense-selective twisting of the polyamide backbone. Planar aromatic guest molecules could revert the twisted helical structure to a pleated sheet-like conformation due to enhanced co-facial π-stacking from host–guest interactions. We confirmed these findings with detailed 1H NMR, photophysical, and chiroptical studies. Finally, we introduced photoresponsive guest molecules, such as merocyanine, enabling dynamic control over the polymer's secondary structure. Upon complexation with planner merocyanine, the twisted aromatic polyamide transformed into a pleated sheet-like structure. The subsequent photoisomerization of merocyanine to nonplanner spyropyran triggered its expulsion from the host polymer, resulting in the retwisting of the aromatic polyamide backbone. Therefore, merocyanine served as a molecular switch to tune these aromatic polyamides' secondary structure (helix ↔ sheet ↔ helix). This innovative approach can open new avenues in designing conformationally dynamic smart materials.
Data availability
The data supporting this article have been included as part of the ESI† and are also available upon reasonable request to the corresponding author.
Author contributions
The manuscript was written with contributions from both authors, and the authors have approved the final version.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
R. K. R. dedicates this work to Professor Eiji Yashima in celebration of his 65th birthday and to honor his remarkable contributions to the field of helical polymers and foldamers. R. K. R. thanks IISER Mohali for the infrastructure and financial support. S. S. thanks IISER Mohali for his fellowship.
Notes and references
-
A. Lesk, Introduction to Protein Science: Architecture, function and genomics, Oxford University Press, 2010 Search PubMed.
- E. J. Stollar and D. P. Smith, Essays Biochem., 2020, 64, 649–680 CrossRef CAS PubMed.
-
P. W. B. Alberts, A. Johnson, J. Lewis, M. Raff and K. Roberts, 'The shape and structure of proteins', Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- N. Koga, R. Tatsumi-Koga, G. Liu, R. Xiao, T. B. Acton, G. T. Montelione and D. Baker, Nature, 2012, 491, 222–227 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- M. Garcia-Viloca, J. Gao, M. Karplus and D. G. Truhlar, Science, 2004, 303, 186–195 CrossRef CAS PubMed.
- J. Deisenhofer, O. Epp, K. Miki, R. Huber and H. Michel, Nature, 1985, 318, 618–624 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- Y. Takahashi, A. Ueno and H. Mihara, Chem.–Eur. J., 1998, 4, 2475–2484 CrossRef CAS.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- J. P. Taylor, J. Hardy and K. H. Fischbeck, Science, 2002, 296, 1991–1995 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2002, 418, 729–730 CrossRef CAS PubMed.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed.
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC.
- C. Ge, J. Zhu, H. Ye, Y. Wei, Y. Lei, R. Zhou, Z. Song and L. Yin, J. Am. Chem. Soc., 2023, 145, 11206–11214 CrossRef CAS PubMed.
- Z. Song, H. Fu, R. Wang, L. A. Pacheco, X. Wang, Y. Lin and J. Cheng, Chem. Soc. Rev., 2018, 47, 7401–7425 RSC.
- Z. Song, R. A. Mansbach, H. He, K.-C. Shih, R. Baumgartner, N. Zheng, X. Ba, Y. Huang, D. Mani, Y. Liu, Y. Lin, M.-P. Nieh, A. L. Ferguson, L. Yin and J. Cheng, Nat. Commun., 2017, 8, 92 CrossRef PubMed.
- M. Kumar, P. Brocorens, C. Tonnelé, D. Beljonne, M. Surin and S. J. George, Nat. Commun., 2014, 5, 5793 CrossRef CAS PubMed.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS PubMed.
- K.-S. Krannig and H. Schlaad, J. Am. Chem. Soc., 2012, 134, 18542–18545 CrossRef CAS PubMed.
- P. Ghosh, J. Torner, P. S. Arora and G. Maayan, Chem.–Eur. J., 2021, 27, 8956–8959 CrossRef CAS PubMed.
- X. Wang, I. Bergenfeld, P. S. Arora and J. W. Canary, Angew. Chem., 2012, 124, 12265–12267 CrossRef.
- K. Pagel, T. Vagt, T. Kohajda and B. Koksch, Org. Biomol. Chem., 2005, 3, 2500–2502 RSC.
- H. Liu, Y. Zhou, Y. Liu, Z. Wang, Y. Zheng, C. Peng, M. Tian, Q. Zhang, J. Li, H. Tan, Q. Fu and M. Ding, Angew. Chem., Int. Ed., 2023, 62, e202213000 CrossRef CAS PubMed.
- H. Dong and J. D. Hartgerink, Biomacromolecules, 2007, 8, 617–623 CrossRef CAS PubMed.
- K. Pagel and B. Koksch, Curr. Opin. Chem. Biol., 2008, 12, 730–739 CrossRef CAS PubMed.
- Y. Takahashi, A. Ueno and H. Mihara, Bioorg. Med. Chem., 1999, 7, 177–185 CrossRef CAS PubMed.
- M. Nguyen, J.-L. Stigliani, G. Pratviel and C. Bonduelle, Chem. Commun., 2017, 53, 7501–7504 RSC.
- M. Lago-Silva, M. Fernández-Míguez, R. Rodríguez, E. Quiñoá and F. Freire, Chem. Soc. Rev., 2024, 53, 793–852 RSC.
- X. Lu, L. Ren, X. Zhang, A. K. Whittaker, W. Li and A. Zhang, Macromolecules, 2024, 57, 5915–5928 CrossRef CAS.
- E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef CAS.
- K. Matsumura, K. Kinjo, K. Tateno, K. Ono, Y. Tsuchido and H. Kawai, J. Am. Chem. Soc., 2024, 146, 21078–21088 CrossRef CAS PubMed.
- R. Rodríguez, E. Suárez‐Picado, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 8616–8622 CrossRef PubMed.
- N. Ousaka, K. Shimizu, Y. Suzuki, T. Iwata, M. Itakura, D. Taura, H. Iida, Y. Furusho, T. Mori and E. Yashima, J. Am. Chem. Soc., 2018, 140, 17027–17039 CrossRef CAS PubMed.
- S. Samanta, D. Mallick and R. K. Roy, Polym. Chem., 2022, 13, 3284–3293 RSC.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- M. Carini, M. P. Ruiz, I. Usabiaga, J. A. Fernández, E. J. Cocinero, M. Melle-Franco, I. Diez-Perez and A. Mateo-Alonso, Nat. Commun., 2017, 8, 15195 CrossRef CAS PubMed.
- I. O. Aparin, G. V. Proskurin, A. V. Golovin, A. V. Ustinov, A. A. Formanovsky, T. S. Zatsepin and V. A. Korshun, J. Org. Chem., 2017, 82, 10015–10024 CrossRef CAS PubMed.
- V. Berl, M. J. Krische, I. Huc, J.-M. Lehn and M. Schmutz, Chem.–Eur. J., 2000, 6, 1938–1946 CrossRef CAS PubMed.
- C. Zhang, H. Wang, J. Zhong, Y. Lei, R. Du, Y. Zhang, L. Shen, T. Jiao, Y. Zhu, H. Zhu, H. Li and H. Li, Sci. Adv., 2019, 5, eaax6707 CrossRef CAS PubMed.
- P. Mondal, S. Banerjee and S. P. Rath, Eur. J. Inorg. Chem., 2019, 2019, 3629–3637 CrossRef CAS.
- V. R. Naidu, M. C. Kim, J. Suk, H.-J. Kim, M. Lee, E. Sim and K.-S. Jeong, Org. Lett., 2008, 10, 5373–5376 CrossRef CAS PubMed.
- X. Tang, L. Lin, F. Liu, X. Yue, B. Gong, M. Li and L. He, Tetrahedron, 2013, 69, 8487–8493 CrossRef CAS.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*