
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombined effect of high voltage and large Li-ion flux on decomposition of Li6PS5Cl†
Deye Sun‡
abcd,
Wenru Li‡abc,
Yantao Wang‡abc,
Jiangwei Juabc,
Pengxian Hanabc,
Shanmu Dong*abc,
Jun Ma *abc and
Guanglei Cui*abcd
*abc and
Guanglei Cui*abcd
aQingdao Industrial Energy Storage Research Institute, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: dongsm@qibebt.ac.cn; majun@qibebt.ac.cn; cuigl@qibebt.ac.cn
bShandong Energy Institute, Qingdao 266101, China
cQingdao New Energy Shandong Laboratory, Qingdao 266101, China
dCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 5th May 2025
Abstract
High voltage has been considered the primary factor causing electrolyte decomposition in all-solid-state lithium batteries. However, whether high voltage is the only decisive factor in sulfide electrolyte decomposition is an open question. Herein, we redefined the decomposition conditions of sulfide electrolytes under the combined effect of high voltage (≥5 V vs. Li+/Li) and large Li+ flux by recording the decomposition process of Li6PS5Cl via in situ Raman spectroscopy during cyclic voltammetry measurement. The result shows that under the combined action of high voltage and large Li+ flux, PS43− anions of Li6PS5Cl undergo much more severe deformation and decomposition than they do only at high voltage or large Li+ flux. At the same time, it also suggests that the much severe decomposition of Li6PS5Cl located near the surface of cathode particles compared to that in the bulk region of Li6PS5Cl may be the combined result of electrochemical/chemical reactions and high voltage/large Li+ flux. Furthermore, the effect of large Li+ flux on the irreversible decomposition of Li6PS5Cl supplements our understanding of the electrochemical stability of solid electrolytes. This work redefines the stability of sulfide electrolyte and gives directions to design highly stable bulk and interfacial structures of sulfide electrolytes at high voltage and high current density.
Introduction
The solid-state electrolytes (SSEs) represented by the sulfide Li6PS5Cl (LPSCl) have ultra-high ionic conductivity, but their application has been hampered by structural instability.1–3 The irreversible decomposition of LPSCl is hardly avoidable when it is paired with high-voltage cathodes (>4 V vs. Li+/Li) to assemble solid-state batteries. Specifically, the extensive deintercalation of Li+ causes charge compensation of lattice oxygen as the layered oxide material-based cathode is charged to high voltage, generating highly reactive oxygen species. These released oxygen species exhibiting high oxidizing properties easily react with reductive LPSCl, leading to the accumulation of poorly conductive byproducts, such as S, SOx−, and POx−, at the electrolyte/electrode interfaces. This decelerates the Li+ transport kinetics through the interphase and aggravates the electrochemical performance of ASSLBs.4,5 Therefore, the stability of LPSCl at the cathode surface is one of the most concerning issues for researchers. Surface modification of cathode particles by using materials with lithium conductivity and electrochemically inert properties could reduce the decomposition of LPSCl.6,7 However, whether high voltage is the only decisive factor in sulfide electrolyte decomposition is an open question. How the stability of LPSCl changes from the surface area to the bulk area in more extreme environments is still unknown in the field.Apart from the factor of high voltage, Li+ flux would play an important role in the LPSCl decomposition in the composite cathode. According to reports, the high ionic conductivity of LPSCl takes root in the significant dynamic coupling between the translational motion of Li+ and the rotational motion of the anion PS43−.5 Therefore, the high Li+ flux during the high current density charge/discharge process inevitably causes deformation in the crystal structure of LPSCl, owing to the coordination bonding.8 Generally, there is a correlation between ion migration, lattice distortions, and stability.9 Therefore, it is reasonably deduced that Li+ flux should also be another factor affecting the decomposition of LPSCl. This decomposition behavior is clearly different from the decomposition of LPSCl caused by electrochemical/chemical reactions. Investigating the effect of the Li+ flux on the decomposition of LPSCl may be a breakthrough in discovering the failure mechanism and critically stable condition of SSEs at high operating voltage, which is a milestone for deep insight into kinetically driven sulfide electrolyte decomposition theory.
Herein, the experimental variables, including working voltage and Li+ flux, were controlled by changing testing parameters and the components of composite cathodes. To observe the decomposition behaviors of LPSCl under varying voltage and current density conditions, the chemical bond-sensitive in situ Raman spectroscopy technique combined with cyclic voltammetry measurement is employed to monitor the structural evolution of the electrolyte in real time during the electrochemical process.10,11 Through continuous observation of the variations in the Raman signatures of PS43− anions under diverse voltage and current conditions, we can accurately and precisely capture the onset and progression of the decomposition process.
The research results indicate that increasing voltage or ion flux alone cannot be the direct cause of significant electrolyte decomposition. Nevertheless, their combined effect greatly alters the structural stability of PS43− anions and ultimately leads to the violent decomposition of LPSCl. At the same time, we also observed a faster decomposition phenomenon of LPSCl at the cathode/electrolyte interface. This phenomenon indicates that the decomposition of LPSCl in high-voltage ASSLBs should be caused by a combination of electrochemical reactions and large Li+ flux. This work effectively supplements our understanding of the electrochemical behavior of LPSCl in ASSLBs, providing support for the design of high-voltage ASSLBs in future studies.
Results and discussion
Battery construction with different electrical properties
First of all, the LPSCl SSEs are prepared through a high-temperature sintering method. The crystal structure and phase identification of the prepared LPSCl powder are examined with XRD. The primary diffraction peaks of the produced material match well with the LPSCl phase, indicating that the pure target material with the argyrodite phase is effectively synthesized (Fig. S1a†).4,12 The Raman spectrum of pristine LPSCl is shown in Fig. S1b and Table S1.† The peaks located at 196, 573, and 600 cm−1 are attributed to vibrational modes of PS43− anions within LPSCl, while those at 266 cm−1 and 423 cm−1 originate from the δdef. (S–P–S) and us. (PS43−) in PS43− anions.13–17 The impedance spectra of ball-milled LPSCl powder show that the ionic conductivity is as high as ∼2 × 10−3 S cm−1 at 30 °C (Fig. S1c†). The above measurements confirm that LPSCl materials with good purity and high conductivity have been synthesized. And argyrodite-structured sulfide solid electrolytes are currently the primary solid electrolytes used in all-solid-state batteries. Research on their conductivity is extensive, with site disorder and changes in carrier concentration being the reasons for their high conductivity. Notably, variations in carrier concentration can significantly induce changes in the lattice,18,19 which may indirectly affect the stability of anion groups.20,21 These changes can cause shifts in the position, intensity, or width of the Raman characteristic peaks.To observe whether the LPSCl structure changes during Li+ transfer, the Raman spectra of LPSCl in a LiIn symmetric cell before and after cycling 10 times at a current density of 0.1 mA cm−2 were collected. As shown in Fig. S1e,† compared to the initial state of LPSCl, the peak positions of PS43− in both the interior and surface region of cycled LPSCl electrolyte exhibit slight shifts to high wavenumber. Moreover, the Raman shift in the interior region of LPSCl is much higher than that in the surface region of LPSCl. Since the interior region of LPSCl does not contact with LiIn, the interface side reactions can be excluded. Therefore, we attribute the distortions of PS43− in the interior LPSCl more likely to Li+ transfer. The effect of Li+ transfer on PS43− is reflected not only in the Raman shifts but also in the abnormal enhancement of Raman intensity in the SSE layer region.
Next, by changing the test parameters and the components of the composite cathodes, four kinds of all-solid-state batteries with different cutoff voltages and Li+ flux were assembled. The battery compositions and electrical properties are summarized in Table 1. First, the LPSCl-VGCF composite cathode was designed to study the electrochemical behaviors of LPSCl under a small Li+ flux and high working voltage with the absence of cathode active materials (Fig. S2a†). To eliminate the influence of oxygen generated by the cathode at high voltage on the electrolyte decomposition behaviour, LiNi0.5Mn1.5O4 (LNMO) with a three-dimensional Li+ diffusion pathway was selected as the cathode research object. Unlike active materials with layered structures, LNMO can retain structural stability at high voltage (5 V vs. Li).22 However, the galvanostatic charge–discharge curves shown in Fig. S3a† displayed a quite low discharge capacity of 13 mA h g−1 at a rate of 0.1C at 30 °C, which may be caused by the high interface impedance at the cathode/electrolyte interface of the LNMO-LPSCl-VGCF composite cathode (Fig. S2b†). To optimize the Li+ transport through the cathode/electrolyte interface, the Li2Ta2(P2O7)3 (LTaP) layer with a wide electrochemical window (5.44 V) is coated on the surface of LNMO particles. As demonstrated by the typical SEM images and EDS mappings (Fig. S4†), LTaP grows in an irregular island-like form on the smooth surface of the LNMO particles. After the LTaP coating, XRD and Raman spectra of LNMO display negligible changes, indicating that LNMO remains in its original crystal structure and chemical state (Fig. S5 and Table S2†). Compared with bare LNMO, LNMO modified with LTaP can provide a discharge capacity of 121.4 mA h g−1, which is 10 times higher than the former. Notably, after coating with LTaP, the Ri of the LNMO battery decreased from ∼240 Ω to ∼160 Ω, indicating a decrease in the transport barrier of Li+ at the electrode/electrolyte interface (Fig. S2c†). This result confirms that after LTaP coating, the cathode/electrolyte interface has good Li+ transport kinetics, in accordance with the galvanostatic charge–discharge test results. Consequently, the LTaP@LNMO-LPSCl-VGCF (Fig. S5f†) composite was selected to achieve both high voltage and large Li+ flux conditions. Besides, due to the low interface impedance of 110 Ω and high charge–discharge capacities (Fig. S2d and S3c†), the LCO-LPSCl-VGCF composite cathode was prepared to create an environment with large Li+ flux at low working voltage.
| Samples cathode|SSEs|anode | Composite cathode (wt%) | Li+ flux (mol cm−2 h−1) | Cutoff voltage (V vs. LiIn) | |
|---|---|---|---|---|
| 1 | LPSCl-VGCF|LPSCl|LiIn | LPSCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VGCF = 70:30 :VGCF = 70:30 |
<0.01 | 4.5 |
| 2 | Uncoated LNMO-LPSCl-VGCF|LPSCl|LiIn | LNMO:LPSCl:VGCF = 40:60:3 |
0.01–0.247 | 4.5 |
| 3 | LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn | LTaP@LNMO:LPSCl:VGCF = 40:60:3 |
>0.247 | 4.5 |
| 4 | LCO-LPSCl-VGCF|LPSCl|LiIn | LCO:LPSCl:VGCF = 70:30:3 |
>0.247 | 3.7 |
The CV test is carried out to demonstrate the improvement in Li+ flux for the prepared composite cathodes of LPSCl-VGCF, LNMO-LPSCl-VGCF, and LTaP@LNMO-LPSCl-VGCF, respectively. As shown in Fig. 1a–c, the area enclosed by the CV curves of LPSCl-VGCF|LPSCl|LiIn, LNMO-LPSCl-VGCF|LPSCl|LiIn, and LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn cells increases sequentially, implying the capacities of Li+ in these composite cathodes are successively enhanced. Among them, the LPSCl-VGCF|LPSCl|LiIn cell has the minimum amount of Li+ (the peak current is 0.009 mA and the maximal Li+ flux is 0.004 mol cm−2 h−1) runs away from LPSCl. The CV curves of the LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn cell have distinct anodic and cathodic peaks, indicating that the addition of cathode particles provides more lithium sources than that of the LPSCl-VGCF|LPSCl|LiIn cell. Benefiting from the modification of the LTaP coating layer, the peak current of the LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn cell (0.09 mA, 0.04 mol cm−2 h−1) is larger than that of the LNMO-LPSCl-VGCF|LPSCl|LiIn cell (0.02 mA, 0.01 mol cm−2 h−1). This suggests that LPSCl obtained small, medium, and large Li+ fluxes, respectively, by changing the components of the composite cathodes. And the CV result of the LCO-LPSCl-VGCF|LPSCl|LiIn cell shows significant redox peaks and a large peak current (0.518 mA) within the voltage range of 2.4–3.7 V (Fig. 1d), indicating larger Li+ flux (0.247 mol cm−2 h−1) in the LCO-LPSCl-VGCF composite cathode than that of the LTaP@LNMO-LPSCl-VGCF composite cathode.
 | ||
| Fig. 1 The CV curves of (a) LPSCl-VGCF|LPSCl|LiIn, (b) uncoated LNMO-LPSCl-VGCF|LPSCl|LiIn, (c) LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn and (d) LCO-LPSCl-VGCF|LPSCl|LiIn cells, respectively. The battery used for measurement is newly assembled. | ||
Decomposition conditions of LPSCl
To exclude the decomposition reaction between the LTaP coating layer and LPSCl, we first studied the possibility of LPSCl decomposition caused by contact with LTaP. Raman measurement was carried out to test the difference between two kinds of LTaP@LNMO-LPSCl composite cathodes; one freshly mixed without any thermal treatment, and the other subjected to heat-treatment at 60 °C for 24 h. As shown in Fig. S6a,† the characteristic peaks of PS43− anions of both samples are observed at 420 cm−1, and they show unnoticeable differences, indicating that the LTaP coating layer is stable when contacted with LPSCl. This is also confirmed by the XPS results in Fig. S6 b and c.†During the battery operation, if the crystal structure of LPSCl changes dynamically, the electron cloud of PS43− anions will be deformed in this process due to the limitations of the lattice, which could be detected through Raman tests. Thereby, in situ Raman spectroscopy is employed to characterize the structural change of LPSCl with the variation of voltage and current density by monitoring the intensity changes of Raman signals of PS43− anions at 420 cm−1. In in situ experiments, the Raman spectroscopy testing was carried out based on the CV tests (Fig. 1) instead of the galvanostatic charge–discharge tests. The voltage range is 2.4–4.5 V (vs. LiIn), and the scanning rate is 0.05 mV s−1. Under these conditions, the changes in the Raman signal are related to the voltage and current, instead of the testing time. In the experiment, the electrolyte far away from the cathode active particle/LPSCl particle interface in the composite cathode was selected for Raman testing. The size of the testing area is 2 µm × 2 µm. Fig. 2a shows that the position of the characterization peaks of PS43− anions in the LPSCl-VGCF composite cathode has no apparent change. But a slight decrease happened in intensity during the charging/discharging process, which indicates that LPSCl undergoes mild decomposition in high-voltage and small Li+ flux environments (Fig. 2b and S7a†). In the LNMO-LPSCl-VGCF composite cathode, the intensity of the Raman characteristic peak of PS43− anions is basically stable during the charging/discharging process, except for a slight and step-like decrease in the middle and later stages of the charging process (Fig. 2c, d, and S7b†). The structural fluctuation in PS43− anions could be related to the increased Li+ flux during charging at high voltage. For the LTaP@LNMO-LPSCl-VGCF composite cathode with the large Li+ flux, when the voltage reaches 4.5 V (vs. LiIn) and the current is about 0.33 mA, the Raman signals representing PS43− units that appear at 196, 266, and 420 cm−1 disappear simultaneously (Fig. 2e, f, and S7d†). Among them, the peak at 226 cm−1 corresponding to the distortion vibration of σdef.(S–P–S) in PS43− evolves into a gradually increasing broadened peak. This confirms the decomposition of LPSCl (Fig. S8a†). As shown in Fig. S8b,† new characteristic peaks appear at 147, 217, and 471 cm−1 corresponding to the bending vibration and the stretching vibration of S, respectively, illustrating the generation of by-products at about 3.4 V (vs. LiIn).23,24 From this, we can basically conclude that the adequate Li+ flux is one of the necessary conditions for the decomposition of LPSCl.
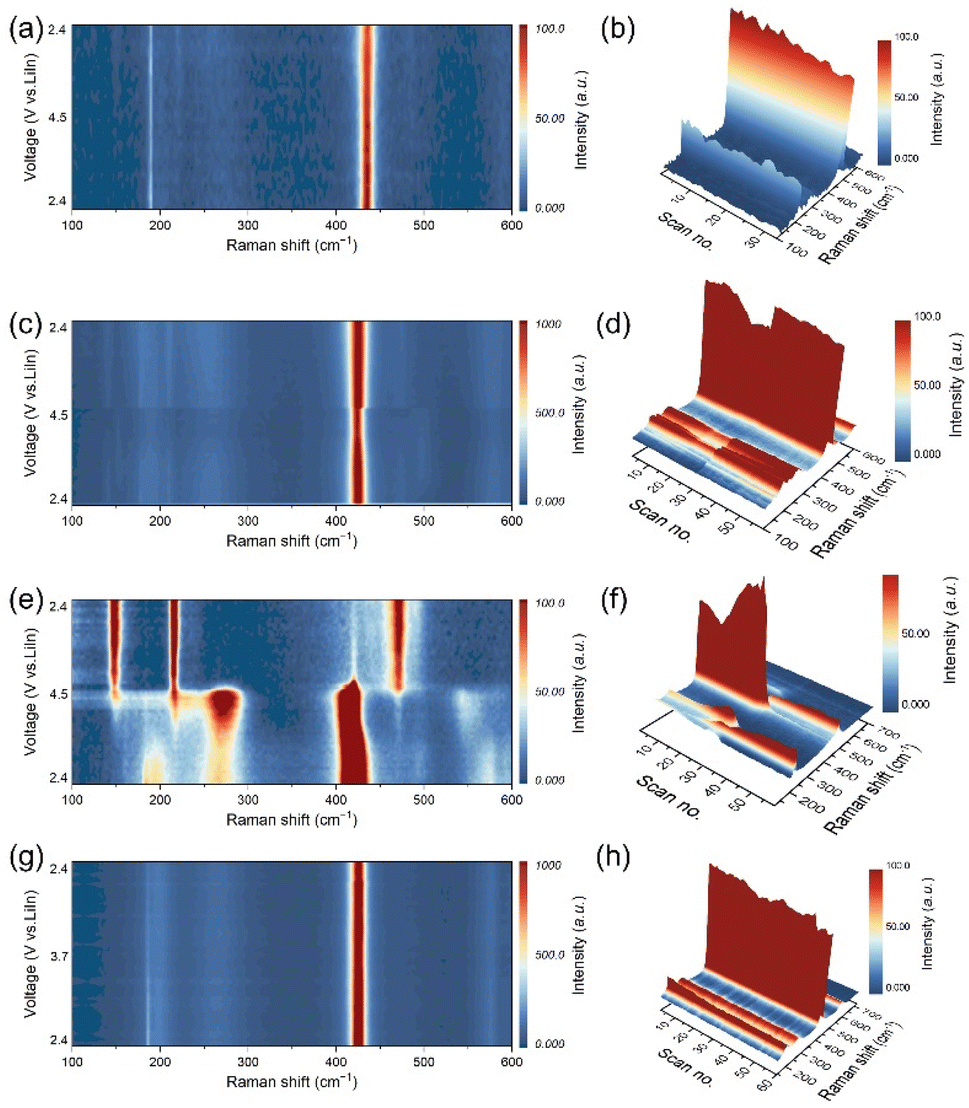 | ||
| Fig. 2 The in situ Raman spectra of (a and b) LPSCl-VGCF|LPSCl|LiIn, (c and d) uncoated LNMO-LPSCl-VGCF|LPSCl|LiIn, (e and f) LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn, and (g and h) LCO-LPSCl-VGCF|LPSCl|LiIn cells from two-dimensional and three-dimensional views, respectively. The positions for Raman signal collection are the LPSCl-rich area far from the cathode active particle/LPSCl particle interface in the composite cathode. | ||
Subsequently, we discussed the effect of voltage on the decomposition of LPSCl under the condition of large Li+ flux. The in situ Raman measurement was conducted for the LCO ASSLBs with a voltage window of 2.4–3.7 V (vs. LiIn), the upper cutting-off voltage of which is lower than that of the LNMO ASSLBs. It should be noted that although LCO has a high electronic conductivity. But for the sake of consistency in experimental conditions, we also mixed VGCF into the LCO composite cathode. The in situ Raman test results show no apparent change in PS43− characterizing peaks but a slight decrease in the intensity (Fig. 2g, h, and S7c†). Therefore, it can be determined that the decomposition of LPSCl is slight at low voltage and large Li+ flux. Combined with the previous experimental results, we conclude that high voltage and large Li+ flux are two key factors for the complete decomposition of LPSCl. The decomposition of LPSCl far away from the cathode surface indicates that the stability of LPSCl not only depends on its voltage window, but also on the intensity of the ion flux. This kinetic instability is mainly determined by the material characteristics of sulfide electrolytes, which is different from the decomposition caused by electrochemical/chemical reactions.
Decomposition behaviour of LPSCl at different positions in the composite cathode
After understanding the high-voltage decomposition mechanism in the bulk phase, the decomposition behaviour of LPSCl in the near-surface region of the cathode was also studied. In the experiment, we characterized two adjacent positions, i.e., the bulk phase (position 1) and near the cathode surface (position 2), in the LTaP@LNMO-LPSCl-VGCF composite cathode through in situ Raman spectroscopy to find out the decomposition behaviour of LPSCl (Fig. 3a). As shown in Fig. 3b and c, the content distribution of PS43− anions (420 cm−1) and Ni2+–O (490 cm−1) in the composite cathode is measured to display the distribution of LPSCl and LNMO, respectively. The warmer the color, the higher the content of the corresponding molecular group/chemical bond. Conversely, the colder the color, the lower the content. From this, the LPSCl content at position 2 is much lower than that of LNMO, indicating that position 2 is near the surface of the cathode particles (Fig. 3d and S7b†). The in situ Raman characterization of PS43− anions (420 cm−1) at position 2 shows that LPSCl completely decomposes as soon as the charging process begins (Fig. 3e, S7d, e and Table S3†). Therefore, this result indicates that the decomposition of LPSCl is more severe in the near-surface region of the cathode surface (Position 2) than in the bulk phase (Position 1). Although we have confirmed that the LTaP@LNMO cathode has good chemical compatibility with LPSCl, it is still difficult to experimentally verify the electrochemical stability between the two materials. Based on the existing results, we can make a reasonable inference that the decomposition of LPSCl in the near-surface region of the cathode should be a combined effect of electrochemical decomposition and high voltage/large Li+ flux.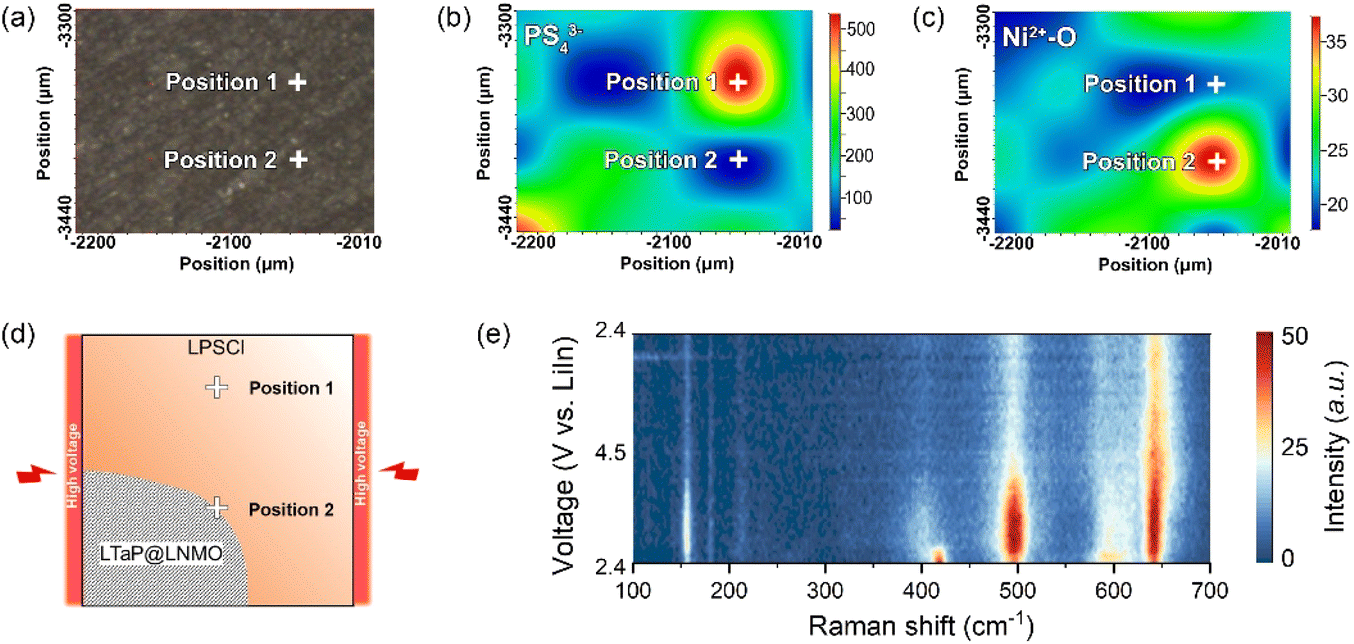 | ||
| Fig. 3 (a) Micrograph of the in situ Raman surface scanning mode and selected points in the LTaP@LNMO-LPSCl-VGCF|LPSCl|LiIn cell. (b and c) Distribution of the cathode and sulfide electrolyte, distinguished by observing the local content distribution of PS43− (420 cm−1) and Ni2+–O (490 cm−1) in the composite cathode, respectively. (d) Schematic diagram of position 1 and position 2 studied in the in situ Raman experiments. (e) The in situ Raman spectra of the LTaP@LNMO-LPSCl-VGCF composite cathode at position 2 from a two-dimensional view. | ||
Decomposition process of LPSCl
To have a comprehensive understanding of the decomposition mechanism of LPSCl, it is necessary to study the types and changes of decomposition products. For this concern, XPS is performed to characterize the changes in S and P elements of the LTaP@LNMO-LPSCl-VGCF composite cathode during different charging/discharging cycles (Fig. S9 and Table S4†). In the spectrum of S element, there are two new peaks generated at 169.1 and 170.5 eV, which are the characteristic peaks of SO42− and SO32− (Fig. S9a†).25 The contents of sulfur/polysulfide species (163.9/165.2 eV) increase during the charging process, but decrease during the discharging process. Meanwhile, the signal of SO42−/SO32− disappeared after the first cycle. After 50 cycles, the XPS spectrum shows that the intensity and area of each peak are basically consistent with those after the first cycle of charging, indicating that the sulfide by-products are unstable at high voltage and generate and decompose repeatedly. From the XPS spectrum of the P element, the POx− species by-products are generated (133.8/134.7–134.8/135.6 eV) after the first cycle and have an obvious cumulative effect as the cycle progresses (Fig. S9b†). It should be noted that the binding energy of POx− species has a slight shift between the charged and discharged states. This result suggests reversible changes in the different types of POx− species during the charging and discharging processes.25 The XRD patterns also show that the presence of POx− species is evident in the LTaP@LNMO-LPSCl-VGCF composite cathode after 50 cycles (Fig. S10†). This result suggests that the POx− species by-products are stable during the high-voltage operation of the battery.At this point, we have a clear understanding of the decomposition of LPSCl at high voltage and large Li+ flux. LPSCl decomposes to produce by-products mainly composed of SOx− and POx−. Among them, SOx− species by-products are unstable at high voltage, resulting in the unstable structure of the cathode–electrolyte interface and the continuous consumption of LPSCl during battery cycling. While the by-products of POx− species are stable at high voltage and accumulate continuously with the cycling of the battery. However, the POx− species have a low ionic conductivity of ∼3 × 10−7 S cm−1, which badly limits the charge transports.26 As a result, the capacity of LTaP@LNMO ASSLBs rapidly decreases with battery cycling (Fig. S3b†). Furthermore, this high impedance by-product results in the aggregation of Li+ ions at the electrolyte decomposition layer, particularly at large Li+ flux, forming interfacial electric fields and Li+ ion concentration gradients between LNMO and LPSCl. As a result, the extent of lattice local distortion and the Raman intensity of LPSCl would decrease gradually from the LNMO/LPSCl interface to the bulk of LPSCl.27,28
Conclusions
To sum up, the decomposition of bulk phase LPSCl is mainly caused by the combined action of high voltage and large Li+ flux, which directly aggravates the distortion of PS43− anions, and eventually leads to the destruction of the molecular structure. In addition, LPSCl decomposition near the cathode surface is more severe than that in the bulk region. During the decomposition, SOx− by-products continuously decompose after formation due to their inability to withstand high voltage, resulting in the unstable structure of the cathode–electrolyte interface. While high-voltage-resistant POx− by-products continue to accumulate, causing an increase in interface resistance and deteriorating battery performance. As far as we are concerned, future research can start with designing high-voltage-stable sulfide electrolytes from aspects such as decomposition kinetics, material components, polarizability, bond energy of anions, etc., which is expected to accelerate the commercial application of all-solid-state batteries.Experimental
Materials and methods
Synthesis of Li6PS5Cl (LPSCl). The raw materials Li2S (Alfa, 99.9%), P2S5 (Alfa, 99.9%), and LiCl (Alfa, 99.9%) were first mixed in the stoichiometric ratio by ball milling with ZrO2 balls at 600 rpm for 1 h. Next, the mixture adhered to the wall of the ball milling tank was scraped off, and the same ball milling process was repeated 10 times. Then, the mixture was pressed into pellets and calcined in an argon atmosphere at 500 °C for 5 h. After calcination, the pellets were thoroughly ground for later use.
Synthesis of lithium tantalum pyrophosphate (Li2Ta2(P2O7)3) coated LiNi0.5Mn1.5O4 (LTaP@LNMO). First, tantalum ethoxide (Aladdin, 99.98%) and lithium nitrate (Aladdin, 99.99%) were weighed in a stoichiometric ratio and completely dissolved in anhydrous ethanol by ultrasonic treatment. Next, phosphoric acid (Sinopharm, 85%) was added to the above solution with continued ultrasonic treatment. The molar ratio of tantalum ethoxide
:lithium nitrate:phosphoric acid was 2:2:6. Meanwhile, LiNi0.5Mn1.5O4 (LNMO) (Beijing MREDA Technology Co., Ltd) powder with a weight ratio of LTaP:LNMO = 0.02:1 was dispersed in anhydrous ethanol was treated by ultrasonic treatment. Then, the above two solutions were mixed by continuously stirring for 6 h. After that, the obtained mixture was dried at 60 °C overnight in an air environment. Finally, dried materials were pre-calcined in a muffle furnace at 250 °C for 1.5 h and subsequently calcined them at 400 °C for 7 h before naturally cooling to room temperature.
Cell assembly
The composite cathodes of batteries for the in situ Raman test29,30 were prepared as described in Table 1. In sample 1, LPSCl and vapor-grown carbon fibers (VGCFs) (ShenZhen KeJing Star Technology Company) were mixed evenly in a mortar. And in samples 2–4, the cathode active material and Li6PS5Cl were uniformly mixed in a mortar to prepare the composite cathodes, which are also mixed with 3 wt% VGCF to construct a good electronic conductive network. And the electrolyte pellet was prepared by pressing 80 mg of electrolyte powder at 200 MPa for 2 min inside a PEEK mold (diameter of 10 mm). Then, 10 mg of the cathode composites were dispersed on the surface of the as-prepared electrolyte pellet and pressed at 350 MPa for 2 min. Finally, one piece of indium foil (0.1 mm thickness, 8 mm diameter) was attached on the other side of the electrolyte pellet and pressed at 100 MPa for 1 min. The prepared battery was placed in a mold equipped with SiO2 glass windows for in situ Raman testing.A steel-to-steel symmetrical battery was utilized to measure the ionic conductivity of a solid electrolyte. 50 mg of solid electrolyte powder was added to a polyether ether ketone (PEEK) mold with a diameter of 10 mm, along with two steel columns as current collectors. The mixture was pressed under a pressure of 375 MPa using a hydraulic press for 2 min.
Materials characterization
X-ray diffraction (XRD) measurements were conducted in the 2θ range of 10–80° using an X-ray diffractometer (Bruker-AXS Micro-diffractometer, D8 ADVANCE) equipped with a copper target (λ1 = 1.54060 Å, λ2 = 1.54439 Å) to characterize the crystal structure of materials. Due to the air and moisture sensitivity, the sulfide electrolyte powder was sealed in a sample holder covered with a polyimide film in a glove box before XRD measurement. The surface morphology of materials was characterized using a scanning electron microscope (SEM, Hitachi S-4800 field) equipped with energy dispersive spectroscopy (EDS). The surface structure of materials was characterized using a scanning transmission electron microscope (STEM, JEOL-JEM 2100 F) with an acceleration voltage of 200 kV. The X-ray photoelectron spectroscopy technology (XPS, Thermo SCIENTIFIC ESCALAB 250Xi) was used for surface composition analysis. The measured XPS data were calibrated with reference to the standard carbon spectrum (C 1s, 284.8 eV). Raman spectra are acquired using a micro-Raman spectrometer (Renishaw, in Via) equipped with a 532 nm Ar ion laser. The laser beam with a laser spot size of ≈2 µm was focused on each sample, and the acquisition time for each spectrum was 20 s.Electrochemical measurements
Electrochemical impedance spectroscopy (EIS) measurement was conducted with a multi-channel electrochemical workstation (Zahner Zennium Electrochemical Workstation) at 30 °C, with an amplitude of 10 mV in the range of 7 MHz to 0.1 Hz.The ionic conductivity of the prepared sulfide electrolyte was calculated using the following formula:

The galvanostatic cycling tests of the assembled all-solid-state lithium batteries were conducted using a Land battery test system (Land CT2001A, Wuhan Land Electronic Co. Ltd) at 30 °C. The voltage ranges of LNMO and LiCoO2 (LCO) based ASSLBs were 2.4–4.5 V (vs. LiIn) and 2.4–3.7 V (vs. LiIn), respectively. The current rate was 0.1C (1C = 140 mA g−1). The cyclic voltammetry (CV) test31 was conducted via an electrochemical working station (Zahner Zennium Electrochemical Workstation) in the voltage range of 2.4–4.5 V (vs. LiIn) and at a scanning rate of 0.05 mV s−1. And the Li+ flux was calculated using the following formula:

Based on our experimental data, we define “high lithium-ion flux” as a lithium-ion flux greater than 0.247 mol cm−2 h−1, which is the lithium-ion flux value obtained from the LCO-LPSCl-VGCF composite cathode in our experiments. This value represents a condition where the impact of the lithium-ion flux on the decomposition of LPSCl is significant.
For “medium lithium-ion flux”, we define it as a range between 0.01 mol cm−2 h−1 and 0.247 mol cm−2 h−1. The lower value, 0.01 mol cm−2 h−1, is derived from the LNMO-LPSCl-VGCF composite cathode data, which shows a relatively less intense effect on LPSCl decomposition compared to the higher values. “Small lithium-ion flux” is defined as less than 0.01 mol cm−2 h−1, as exemplified by the LPSCl-VGCF|LPSCl|LiIn cell in our study.
Data availability
The data supporting this study are available within the article and its ESI.† The raw data are available from the corresponding authors upon reasonable request.Author contributions
Research design and funding acquisition: Guanglei Cui; writing-original draft: Deye Sun; data collection and experiment: Deye Sun and Yantao Wang; data collection and analysis: Deye Sun, Jun Ma, and Wenru Li; review and editing: Guanglei Cui, Jiangwei Ju, Pengxian Han, Shanmu Dong, and Jun Ma; refining the ideas and finalizing this paper: Guanglei Cui, Jun Ma, and Deye Sun.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Sun, W. Li, and Y. Wang contributed equally to this work. This work was financially supported by the Key Scientific and Technological Innovation Project of Shandong (No. 2023CXGC010302), the National Natural Science Foundation of China (No. 22379155, No. 52202326, No. 52372245, No. 22139001 and No. 52037006), the Shandong Provincial Natural Science Foundation (No. ZR2022QB180 and No. ZR2023ME027), the Shandong Postdoctoral Innovation Project (No. SDCX-ZG-202202037), the Science & Technology Cooperation Program of Shandong (2024KJHZ018), the Youth Innovation Promotion Association of CAS (No. 2021210), the Qingdao New Energy Shandong Laboratory (No. QIBEBT/SEI/QNESLS202304), the Taishan Scholars Program (No. tsqn202306308 and No. tsqn202408286), and the Emerging Industry Cultivation Plan of the Qingdao Future Industry Cultivation Project (No. 24-1-4-xxgg-7-gx).References
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- D. Cao, Y. Zhang, A. M. Nolan, X. Sun, C. Liu, J. Sheng, Y. Mo, Y. Wang and H. Zhu, Nano Lett., 2020, 20, 1483–1490 CrossRef CAS.
- C. Wang, S. Hwang, M. Jiang, J. Liang, Y. Sun, K. Adair, M. Zheng, S. Mukherjee, X. Li, R. Li, H. Huang, S. Zhao, L. Zhang, S. Lu, J. Wang, C. V. Singh, D. Su and X. Sun, Adv. Energy Mater., 2021, 11, 2100210 CrossRef CAS.
- R. Schlenker, A.-L. Hansen, A. Senyshyn, T. Zinkevich, M. Knapp and T. Hupfer, Chem. Mater., 2020, 32, 8420–8430 CrossRef CAS.
- J. G. Smith and D. J. Siegel, Nat. Commun., 2020, 11, 1483 CrossRef CAS.
- W. Li, S. Zhang, W. Zheng, J. Ma, L. Li, Y. Zheng, D. Sun, Z. Wen, Z. Liu, Y. Wang, G. Zhang and G. Cui, Adv. Funct. Mater., 2023, 33, 2300791 CrossRef CAS.
- X. Sun, L. Wang, J. Ma, X. Yu, S. Zhang, X. Zhou and G. Cui, ACS Appl. Mater. Interfaces, 2022, 14, 17674–17681 CrossRef CAS PubMed.
- Z. Zhang and L. F. Nazar, Nat. Rev. Mater., 2022, 7, 389–405 CrossRef.
- K. Kim and D. J. Siegel, J. Mater. Chem. A, 2019, 7, 3216–3227 RSC.
- C. Gao, J. Zhang, C. He, Y. Fu, T. Zhou, X. Li, S. Kang, L. Tan, Q. Jiao, S. Dai, Y. Yue and C. Lin, Adv. Energy Mater., 2023, 13, 2204386 CrossRef CAS.
- X. Zhang and R. Frech, J. Electrochem. Soc., 1998, 145, 847 CrossRef CAS.
- S. Boulineau, M. Courty, J.-M. Tarascon and V. Viallet, Solid State Ionics, 2012, 221, 1–5 CrossRef CAS.
- S. Yubuchi, S. Teragawa, K. Aso, K. Tadanaga, A. Hayashi and M. Tatsumisago, J. Power Sources, 2015, 293, 941–945 CrossRef CAS.
- J. Zhang, C. Zheng, L. Li, Y. Xia, H. Huang, Y. Gan, C. Liang, X. He, X. Tao and W. Zhang, Adv. Energy Mater., 2020, 10, 1903311 CrossRef CAS.
- Y. Zhou, C. Doerrer, J. Kasemchainan, P. G. Bruce, M. Pasta and L. J. Hardwick, Batteries Supercaps, 2020, 3, 647–652 CrossRef CAS.
- L. Sang, R. T. Haasch, A. A. Gewirth and R. G. Nuzzo, Chem. Mater., 2017, 29, 3029–3037 CrossRef CAS.
- K. Ohara, A. Mitsui, M. Mori, Y. Onodera, S. Shiotani, Y. Koyama, Y. Orikasa, M. Murakami, K. Shimoda, K. Mori, T. Fukunaga, H. Arai, Y. Uchimoto and Z. Ogumi, Sci. Rep., 2016, 6, 21302 CrossRef CAS.
- L. Zhou, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, J. Am. Chem. Soc., 2019, 141, 19002–19013 CrossRef CAS.
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, J. Power Sources, 2022, 520, 230849 CrossRef CAS.
- K. Seol, C. Kaliyaperumal, A. Uthayakumar, I. Yoon, G. Lee and D. Shin, Electrochim. Acta, 2023, 441, 141757 CrossRef CAS.
- D. Li, X. Liu, Y. Li, X. Zhao, M. Wu, X. Qi, L. Gao and L.-Z. Fan, Adv. Energy Mater., 2024, 14, 2402929 CrossRef CAS.
- G. Liu, Y. Lu, H. Wan, W. Weng, L. Cai, Z. Li, X. Que, H. Liu and X. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 28083–28090 CrossRef CAS.
- J. Hassoun, R. Verrelli, P. Reale, S. Panero, G. Mariotto, S. Greenbaum and B. Scrosati, J. Power Sources, 2013, 229, 117–122 CrossRef CAS.
- M. Ishii, K. Shibata and H. Nozaki, J. Solid State Chem., 1993, 105, 504–511 CrossRef CAS.
- J. Auvergniot, A. Cassel, J.-B. Ledeuil, V. Viallet, V. Seznec and R. Dedryvère, Chem. Mater., 2017, 29, 3883–3890 CrossRef CAS.
- N. I. P. Ayu, E. Kartini, L. D. Prayogi, M. Faisal and Supardi, Ionics, 2016, 22, 1051–1057 CrossRef CAS.
- S. N. Clafton, D. A. Beattie, A. Mierczynska-Vasilev, R. G. Acres, A. C. Morgan and T. W. Kee, Langmuir, 2010, 26, 17785–17789 CrossRef CAS PubMed.
- X.-Y. Liu, Y. Cui, J.-P. Deng, Y.-Y. Liu, X.-F. Ma, Y.-X. Hou, J.-Y. Wei, Z.-Q. Li and Z.-W. Wang, J. Phys. Chem. Lett., 2022, 13, 8858–8863 CrossRef CAS PubMed.
- R. Zhang, Y. Wu, Z. Chen, Y. Wang, J. Zhu and X. Zhuang, J. Mater. Chem. A, 2023, 11, 19195–19209 RSC.
- F. Nicolson, L. E. Jamieson, S. Mabbott, K. Plakas, N. C. Shand, M. R. Detty, D. Graham and K. Faulds, Chem. Sci., 2018, 9, 3788–3792 RSC.
- W. Brown, Y. Li, R. Yang, D. Wang, M. Kvetny, H. Zheng and G. Wang, Chem. Sci., 2020, 11, 5950–5958 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02018b |
| ‡ D. Sun, W. Li, and Y. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |