
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring hyperfine coupling in molecular qubits†
Joan Cardona a,
Àlex Soléab,
Pablo Mellac,
Daniel Aravena*c,
Javier Ruiz-Hidalgo*b,
Silvia Gómez-Coca*a and
Eliseo Ruiz*a
a,
Àlex Soléab,
Pablo Mellac,
Daniel Aravena*c,
Javier Ruiz-Hidalgo*b,
Silvia Gómez-Coca*a and
Eliseo Ruiz*a
aDepartament de Química Inorgànica i Orgànica, Institut de Recerca de Química Teòrica i Computacional, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain. E-mail: eliseo.ruiz at qi.ub.edu
bDepartment of Image Processing Group – Signal Theory and Communications Department, Universitat Politècnica de Catalunya, Jordi Girona 1–3, 08034 Barcelona, Spain
cDepartamento de Química de los Materiales, Facultad de Química y Biología, Universidad de Santiago de Chile, Casilla 40, Correo 33, Santiago, Chile
First published on 23rd May 2025
Abstract
Molecular qubits represent a promising avenue for advancing quantum sensing and computing technologies, yet significant challenges remain in optimising their performance. Hyperfine coupling has a critical influence on molecular qubit properties. While previous studies have exhaustively investigated this phenomenon, a comprehensive understanding of the underlying mechanisms across different systems remains elusive. A benchmark test was performed using DFT to assess which methodology worked best to accurately predict hyperfine coupling constants in molecular qubits predominantly composed of VIV and CuII. We systematically analysed the decomposition of hyperfine coupling and examined how variations in coordination sphere and molecular geometry impact dipolar, isotropic and spin–orbit contributions. By modelling diverse systems, we demonstrate how molecular design can fine-tune hyperfine coupling contributions, either minimising overall interaction or enhancing coupling along specific axes. This study provides useful insights into the structure–property relationships governing hyperfine coupling mechanisms and assesses the accuracy of different choices of density functional, basis sets and relativistic corrections in the prediction of hyperfine coupling constants.
Introduction
Magnetic molecules play a prominent role in quantum devices due to their electronic spin behaviour, making them potential candidates as qubits and quantum sensors.1–4 A variety of two-state systems have been explored as potential platforms for qubit realisation, benefiting from different physical implementations such as photons,5 electronic spins,6 trapped ions,7 ultracold atoms,8 superconductor Josephson junctions,9 and quantum dots.10 Electronic spins can directly behave as qubits, and controlling their direction is a fundamental element in achieving sufficiently long coherence times for technological applications.2,11 Currently, diamond NV centres are being implemented in technological devices, mainly for quantum sensing. These systems are employed in scanning probe microscopy or single-photon emitters despite the difficulty of controlling the concentration of spin centres and the behaviour of such defects. In this way, magnetic molecules present a great potential to be competitive with NV centres if coherence times can be improved, as molecular crystals can be designed to yield an ordered array of spin centres, avoiding the problems associated with qubits based on randomly distributed defects.12,13Quantum coherence represents a critical parameter for qubit performance, with coherence times necessarily exceeding gate operation timescales (0.1–1 ms) to facilitate fault-tolerant quantum computation.14 Measuring coherence time is experimentally carried out using pulsed Electron Paramagnetic Resonance (EPR), which is a highly specific technique that involves a Hahn echo pulse15 to study the dephasing of electron spins. Furthermore, the level of complexity and resources required to obtain most qubits is very high; hence, molecular spin-based qubits are an attractive alternative. They offer great tunability,16,17 homogeneity in comparison with the defects of solid-state spin qubits, and easy synthetic approaches. However, they still face the same challenge of maintaining coherence over spin–lattice (T1) and spin–spin (T2) relaxation mechanisms. The former involves vibrational relaxation of the lattice, while the latter centres on interactions between spins, both contributing to dephasing.18 At lower temperatures, the limiting factor is T2 due to reduced molecular vibrations from insufficient thermal energy; on the other hand, when higher temperatures are reached, T1 decreases exponentially, and it becomes the main hindrance due to efficient spin–phonon coupling.19
Hyperfine coupling (HFC) between electron and nuclear spins introduces additional spin interactions that can significantly play a role in such effects in qudits or quantum sensing.3,18,20–26 This interaction arises from the magnetic coupling between the electron spin and the nuclear spin(s) of the same atom (hyperfine) or nearby atoms (superhyperfine).27 The spin Hamiltonian approach is often employed to study these interactions systematically. In this formulation, the energy spectrum is projected to the small, low-energy manifold, which is important to the magnetic properties of the system, as expressed in eqn (1).27,28
 | (1) |
Here, the first term is attributed to the Zero-Field Splitting (ZFS) which describes the interactions resulting from the presence of more than one unpaired electron, where Ŝ is the operator for the electron spin and D describes the spin–spin interaction between two or more unpaired spins. The second term represents the electronic Zeeman effect (where μB is Bohr magneton, g is the electron g-factor and B is the applied magnetic field). For a free electron, the g-factor is approximately 2.0023. However, in chemical systems, the g-factor can vary significantly due to spin–orbit coupling and other interactions. The magnetic anisotropy described by the g-factor offers detailed information about the spatial arrangement and geometry of the metal complex. In anisotropic systems, such as those with low symmetry or strong ligand fields, the g-factor becomes directionally dependent, reflecting the interaction between the unpaired electron magnetic moment and the local environment. Finally, the third term represents the HFC (where A(A) is the hyperfine tensor and Î(A) is the nuclear spin for a nucleus A).
Recent molecular qubit studies19,29 seem to point towards mononuclear complexes based on S = 1/2 systems as potential candidates for molecular systems with long coherence time. We have chosen first-row transition metals, VIV and CuII, as they present d1 and d9 electronic configurations and reported the best coherence times.4,6,16,30,31 Longer coherence times have also been achieved through ligand design by favouring spin-free nuclei, reducing electronic-nuclear spin interactions.32 Beyond minimising spin–spin interactions, a ligand should also possess rigidity to minimise vibrational perturbations, thereby reducing spin–phonon coupling.3,33,34
In recent advances within quantum computing materials, understanding the fundamental principles governing molecular spin qubits has become increasingly crucial.35 The gap we address is to translate a simple explanation of the different contributions of the hyperfine coupling and how to modify them experimentally. This work aims to understand how to modulate HFC in copper and vanadium compounds to develop systems for their application in quantum devices. This investigation led us to evaluate various computational methods to identify a precise approach for calculating the hyperfine coupling constant A. Through this methodology, we analysed the geometric and electronic factors that determine the HFC constants, ultimately providing insights into how this parameter can be effectively modulated for optimal qubit performance.
Hyperfine coupling contributions
The theoretical framework presented in this section draws on established equations from the literature.27,28 The HFC interaction (A(A)μν) can be decomposed by considering the isotropic, also called Fermi contact, (A(A)iso), dipolar (A(A;dip)μν and the spin–orbit contributions (A(A;SOC)μν). This term is treated as a 3 × 3 tensor where μ, ν = x, y, z and δμν is a Kronecker delta such as| A(A)μν = A(A)isoδμν + A(A;dip)μν + A(A;SOC1)μν + A(A;SOC2)μν | (2) |
Since the isotropic contribution is based on the electronic spin density within the nucleus, transition metal atoms should not exhibit such contribution as the unpaired electron resides in d orbitals, which present a node at the nucleus position. However, the exchange terms for a d1 system between the d orbital bearing the unpaired alpha electron and the paired core electrons differ for alpha and beta orbitals because such contributions are non-zero for electrons with the same spin. Thus, a spin-dependent interaction occurs within the atom, resulting in a spin polarisation of the inner shell s electrons. This exchange energy leads to a non-uniform distribution of the α and β electrons near the nucleus, consequently inducing a spin density in the internal orbitals.36 Consequently, s orbitals, with their spherical symmetry, dominate the isotropic term in eqn (2) and require theoretical methods incorporating relativistic approaches to describe this term properly.37 For a nucleus A, we use the following expression:
 | (3) |
 | (4) |
 | (5) |
 is the position vector of the electron relative to the nucleus ({ϕ} is the set of basis functions). Since the dipolar interaction exhibits dependence on rA−3, the integral can be simplified by retaining solely one-centre contributions. When introducing ligand-field theory, cAi relates to the covalency of the metal–ligand bonding and inclusion of the one-centre reduced field gradient integrals fμν, states that dipole contributions are proportional to the expectation value of rA−3 over the rather compact d-orbitals and the value PA that can be substantial for some metal nuclei.28
is the position vector of the electron relative to the nucleus ({ϕ} is the set of basis functions). Since the dipolar interaction exhibits dependence on rA−3, the integral can be simplified by retaining solely one-centre contributions. When introducing ligand-field theory, cAi relates to the covalency of the metal–ligand bonding and inclusion of the one-centre reduced field gradient integrals fμν, states that dipole contributions are proportional to the expectation value of rA−3 over the rather compact d-orbitals and the value PA that can be substantial for some metal nuclei.28
 | (6) |
| Pd = PA〈rA−3〉d | (7) |
The last two terms from eqn (2) are attributed to the spin–orbit coupling, which describes the second-order interaction between the electronic spin-magnetic moment and the orbit magnetic moment of the same and other electrons.
 | (8) |
 | (9) |
| L3μij = Im(〈ψi|lμArA−3|ψj〉) | (10) |
And:
 | (11) |
The effective nuclear charges (ZeffA), used in the spin–orbit coupling expression are semi-empirical parameters. This expression considers two types of excitations: Type I involves an electronic promotion from a doubly occupied molecular orbital (i) to a singly occupied molecular orbital (oj). Type II, on the other hand, entails a promotion from a singly occupied molecular orbital (oi) to an empty orbital (a). Since only states with identical spin multiplicities contribute to the second-order HFC term, we restrict our focus to these states. This equation exhibits significant similarities to the expression for the second-order g-tensor (gμν). This connection leads to:
| A(A;SOC1)μν = PdΔgμν | (12) |
The second spin–orbit coupling contribution comes from a cross-term between the electron-nuclear dipole–dipole Hamiltonian and the spin–orbit coupling. It is written as:
 | (13) |
| Fμν;Aij(〈ψi|rA−5{δμνrA2 − 3rA,μrA,μ}|ψj〉) | (14) |
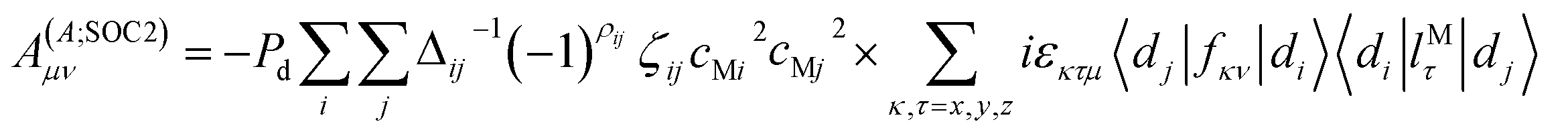 | (15) |
Computational details
These effective Hamiltonian terms can be estimated using modern electronic structure methodologies. A key aspect for HFC calculation is a precise description of relativistic effects.38,39 Dirac's one-electron Hamiltonian containing a 4-spinor vector has been used as a four-component wavefunction. It describes the state of an electron, where these components are split as two-component spinor vectors. Although the four-component methodology exhibits remarkable accuracy as a potential candidate for describing quantum mechanical systems, it incurs a significantly higher computational demand compared to Schrödinger-based methods.40The major shortcoming surfaces from the positronic states (negative energy states) are coupled by the off-diagonal terms of Pauli's spin matrices.41 As we are interested in electronic states, Hamiltonian corrections, which eliminate the negative energy components from the Dirac equation, presented very efficient results in practice, where zeroth-order regular approximation (ZORA)42–45 stands out as a popular choice using a lower-order approximation. The exact decoupling of the upper and lower components of the spinor was achieved by Barysz and Sadlej,46 leading to a unique transformation technique, the Douglas–Kroll–Hess (DKH) method.47–49 Although presenting an interesting alternative to calculate fully relativistic quantum mechanics and properties, it also exhibits certain challenges, such as introducing spin–orbit and many-electron terms in the transformation of the many-electron Hamiltonian (in higher orders) and the calculation of molecular properties,50 where picture change effects must be dealt with.48 The eXact Two-Component (x2c) method offers an alternative to DKH for performing relativistic quantum chemistry calculations.51–54 Unlike DKH, x2c utilises a non-iterative construction process. This means it directly obtains a matrix operator from the electronic solutions of the Fock–Roothaan equation. Additionally, x2c achieves a one-step solution by expanding the components of the 4-spinor in a one-electron basis set.53 This bypasses the requirement for additional unitary transformations that contribute to the computational cost, as in DKH. As a result, x2c offers a streamlined approach that reduces computational effort compared to higher order DKH41 which has been shown to yield notably accurate HFC and g-tensor calculations for a range of small 3d(1), 4d(1), and 5d(1) complexes, including larger 5d(7) Ir and Pt complexes.55
Basis and exchange–correlation functional assessment calculations were performed with Turbomole 7.7 package, using ridft module for x2c calculations, where the default density-fitted operator has been replaced by the exact operator.56 All x2c calculations of EPR parameters were computed for three distinct orthogonal directions of the spin-magnetization.57 A radial ultrafine/most dense grid (5a) was used for numerical quadrature. A 10−9 (hartree) energy threshold was chosen unless stated otherwise. Computation of HFC-tensors was achieved using the x2c transformation applying the finite nucleus approximation.58 The modified Scaled Nuclear Spin–Orbit (mSNSO) approximation was used to calculate HFC constants as suggested by Wodynski and Kaupp.59 In troublesome cases, maximum and minimum damping settings were set to 1.5–0.5, and orbital shifting was added (0.2 eV). ZORA calculations were performed using the ORCA 5.0.4 package,60,61 employing B3LYP functional with an uncontracted version of ANO-DK3 basis set, adjusting to a 10−8 hartree convergence criteria, and including picture change effects and finite nucleus approximation, unless stated otherwise. The hybrid functionals selected for benchmarking were B3LYP,62 PBE0 (ref. 63) and a PBE40, as suggested by Wodynski and Kaupp for heavy metal complexes,59 a PBE-based functional with 40% exact-type exchange. A long-range corrected functional was also analysed in the context of this study (ωB97X).64 A series of basis sets, mainly relativistic, were experimented upon, including x2c-TZVPall,56 x2c-TZVPPall,56 x2c-QZVPPall,65 x2c-QZVPPall-s,65 def2-QZVPP,66 UGBS,67 ANO-DK3.68 IGLO-III69 was employed for light atoms in conjunction with ANO-DK3 and x2c-TZVPPall used for the metal.
The benchmark set of molecules comprised of a series of mononuclear first-row metal transition complexes showing large reported coherence times, consisting of [Cu(mnt)2]2− (1),29 [V(dmit)3]2− (2),19 [V(C8S8)3]2− (3),6 [VO(dmit)2]2− (4),19 and a TiIII complex, [CpTi(cot)] (5).70 For validation purposes we added Cu(Pc) (6),71 [V(C6Br4O2)3]2− (7),31 [V(C6H4O2)3]2− (8),72 [Cu(acacen)] (9),3 [Cu(tmtaa)] (10),3 [Cu(C6H4S2)2]2− (11),16 and [Cu(C6H4Se2)2]2− (12).16 The molecular structures determined by X-ray diffraction have been employed in the calculations (see Fig. 1). However, it is important to keep in mind that experimentally, the EPR measurements for molecular qubits are carried out in frozen solution and magnetically diluted samples employing a non-magnetic metal cation that forms an isostructural complex (further details regarding the computational implementation and its impact on the results can be found in Section 1 of the ESI†).
 | ||
| Fig. 1 Molecular structure of the 12 studied qubit molecules. [Cu(mnt)2]2− (1), [V(dmit)3]2− (2), [V(C8S8)3]2− (3), [VO(dmit)2]2− (4), CpTi(cot) (5), Cu(Pc) (6), [V(C6Br4O2)3]2− (7), [V(C6H4O2)3]2− (8), [Cu(acacen)] (9), [Cu(tmtaa)] (10), [Cu(C6H4S2)2]2− (11) and [Cu(C6H4Se2)2]2− (12). Mnt = maleonitriledithiolate, dmit = 1,3-dithiole-2-thione-4,5-dithiolate, Cp = cyclopentadienyl, cot = 1,3,5,7-cyclooctatetraene, Pc = phthalocyanine, acacen = bis(acetylacetone)ethylenediamine, tmtaa = tetramethyltetraazaannulene. Hydrogens are hidden for clarity purposes. Orange, grey, and purple spheres represent the metal atoms, copper, vanadium, and titanium, respectively. Lighter elements are represented with sticks using grey, blue, red, yellow, and orange colours for carbon, nitrogen, oxygen, sulphur and selenium, respectively. | ||
Additionally, model complexes with idealised symmetry have been generated using Shape 2.1 package73,74 employing VIV as the main metal centre and NH3 as ligands. The V–N bond distances were changed from the average of non-ideal DFT-optimized structures. The same code was employed to perform continuous shape measurements to determine the degree of distortion of the metal complexes in comparison with ideal coordination polyhedra.
To quantify the accuracy in the prediction of A parameters, the Mean Absolute Percentage Logarithmic Error (MAPLE) was chosen as a metric for each approach according to:
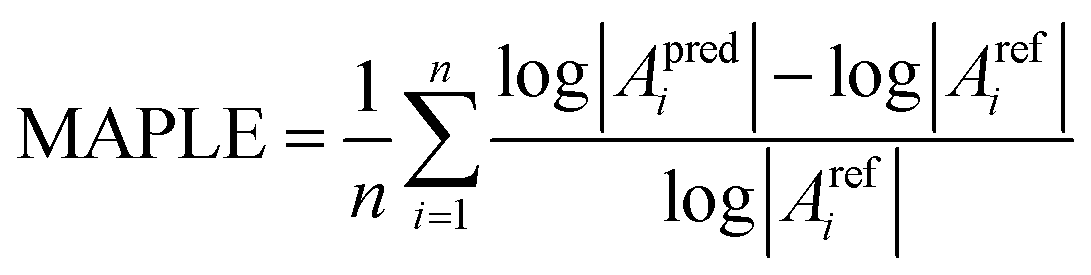 | (16) |
Results and discussion
Benchmark assessment for EPR parameters prediction
To evaluate the effect of the basis set and density functional in the calculation of HFC parameters, we selected a representative set of transition metal complexes proposed as molecular qubit candidates: [V(dmit)3]2−, [Cu(mnt)2]2−, [V(C8S8)3]2−, [CpTi(cot)] and [VO(dmit)2]2− (compounds 1–5). As illustrated in Fig. 2, basis sets containing IGLO-III for lighter atoms in conjunction with ANO-DK3 and x2c-TZVPPall for the metal centre underperformed, proving inadequate for accurate HFC prediction in these systems. Triple-ξ basis sets yielded relatively high errors, between 25–28%, while quadruple-ξ basis sets performed significantly better with MAPLEs in the 10–14% range (excluding ωB97X results). A full basis set decontraction significantly improved predictions (e.g. ANO-DK3 MAPLE error reduced from 41% to 5% with the decontraction when used with B3LYP). To isolate the influence of a more detailed s orbital description on HFC constants, we modified the ANO-DK3 basis set by decontracting only the s functions. This reduced the error to 18%, suggesting that a more accurate description of higher angular momentum orbitals, even those not directly involved in bonding, can have a significant impact on HFC values. This observation aligns with eqn (3), indicating that a better description of the spin density ρα−β(RA) using heavier s-weighted function basis sets and adding s-primitives with large exponents improves the calculation of isotropic terms. NMR-shielded x2c-type basis sets (x2c-QZVPPall-s) were also evaluated but performed slightly worse than their non-shielded counterparts, showing approximately a 4% increase in MAPLE. For more numerically detailed results, check Tables S2–S6 in ESI.† | ||
| Fig. 2 MAPLE error of computed HFC components employing various methodologies from compounds 1–5. NREL stands for non-relativistic treatment. See Table S7 of the ESI† for the numerical details. | ||
The evaluation of exchange–correlation functionals for HFC calculations revealed several notable trends. We focused our investigation on hybrid functionals based on previous research by Kossmann et al.,76 which demonstrated their superior performance over-generalized gradient approximation (GGA) functionals and highlighted their versatility across diverse compound classes compared to computationally more demanding double-hybrid approaches. The impact of the functional choice demonstrated significantly less influential on HFC predictions compared to basis set selection. Nevertheless, when evaluating functionals with the best-performing basis set (uncontracted ANO-DK3), B3LYP emerged as the superior option with a remarkably low MAPLE of 5%, outperforming both PBE0 and PBE40, which showed MAPLEs of 14% and 13%, respectively. Surprisingly, the long-range-separated hybrid functional ωB97X yielded the least accurate results among all tested functionals, on average, and showed no significant improvement when changing basis sets (Fig. 2).
To evaluate the importance of relativistic effects in first-row transition metal complexes, we compared ZORA, x2c, and non-relativistic (NREL) treatments (Fig. 2). ZORA demonstrated comparable accuracy to the x2c methodology, confirming its suitability for these systems. In contrast, non-relativistic calculations yielded a MAPLE of 17%, underscoring the significant influence of relativistic effects, even for first-row transition metals. This suggests that the simplified ZORA approach may be preferable in some cases, offering a good balance between accuracy and computational efficiency. The impact of relativistic effects appeared less pronounced when dealing with lighter transition metal atoms compared to the effects of basis set choice and basis set contraction. However, for systems containing heavier elements significantly affected by spin–orbit coupling, higher Hartree–Fock exchange might be beneficial to account for multiple two-electron terms and spin-other-orbit (2eSOO) contributions, which tend to dominate in such cases.77–79
In addition to HFC constants, g-values were also calculated for compounds 1–5, which agree well with experimental reference values (see Fig. 3 and Table S13†). Regarding the choice of the density functional, B3LYP, ωB97X, and PBE0 performed similarly well, while PBE40 delivered significantly worse results. The basis set choice had a smaller impact, where we identified the contracted ANO-DK3/IGLO-III basis set as the one providing the highest error. ZORA, in conjunction with the uncontracted basis set, was revealed to outperform every other methodology, proving superior in g-value prediction within these systems (see Tables S8–S12†).
 | ||
| Fig. 3 Average computed g-factor MAPE error for compounds 1–5. NREL stands for non-relativistic treatment. Numerical details are shown in Section 3 of ESI.† | ||
Based on these findings, we strongly recommend employing an uncontracted basis set, coupled with the B3LYP hybrid functional, demonstrating superior performance for these compounds. For relativistic treatment, ZORA provided an excellent balance between computational efficiency and accuracy, performing comparably to the x2c approach while reducing computational demands. To assess the comparison between this computational procedure and experimental data, EPR parameters from the best-performing methods were used to simulate continuous-wave EPR (cw-EPR) and were compared with experimental EPR spectra for compound 2, as shown in Fig. 4. Experimental values reported in the literature were given as absolute magnitudes for most compounds as cw-EPR cannot determine the sign of HFC constants. To address this limitation, triple ENDOR (three-pulse) techniques80,81 enable the unambiguous sign assignment of HFC constants. However, almost no experimental sign measurements are reported due to the complexity of conducting such techniques. Simulations using EasySpin82 were carried out to demonstrate no discernible impact of the HFC sign on EPR interpretation, as detailed in Section 4 of the ESI.† Compound 3 (Fig. S3†) raised significant discrepancies between the predicted and experimentally determined HFC constants, particularly for the perpendicular axes. This fact can be attributed to the experimental EPR fitting, which employed a single value for both perpendicular components.
 | ||
| Fig. 4 Simulated cw-EPR spectra of [V(dmit)3]2− (2) using EasySpin version 6.0. (A) Comparison between DFT computed and experimental hyperfine coupling constants setting experimental g-values. (B) Comparison between computed and experimental EPR parameters. Simulated cw-EPR for the rest of the compounds can be found in Section 4 of ESI.† | ||
An additional set of reported first-row transition metal molecular qubits consisting of [Cu(Pc)], [V(C6Br4O2)3]2−, [V(C6H4O2)3]2−, [Cu(acacen)], [Cu(tmtaa)], [Cu(C6H4S2)2]2− and [Cu(C6H4Se2)2]2− (compounds 6–12) were selected to check how our computational proposal performed on a different set of molecules (Fig. 5). For this second test, the B3LYP density functional was employed with various choices of relativistic approximation (x2c and ZORA) and basis set (x2c-QZVPPall and ANO-DK3, uncontracted). EPR parameters for compounds 6–12 were also computed and gathered in Tables S13 and S14.† Overall, ZORA managed to capture HFC arguably close to x2c results and exhibited superior accuracy for g-factor calculations. For compound 6, the observed error primarily stemmed from overestimating the perpendicular HFC constants. Thus, we conclude that the ZORA approach remains accurate for the extended molecule set.
 | ||
| Fig. 5 MAPLEs for computed HFC constants of compounds 6–12. Numerical details are provided in Table S14.† | ||
Hyperfine coupling term decomposition
While the isotropic term often dominates discussions due to its significant contribution in many systems, the spin–dipole and spin–orbit coupling terms can be of comparable magnitude under specific conditions. In systems with unpaired electrons primarily in p or d orbitals, the spin–dipole interaction becomes equally crucial. In molecules containing heavy atoms or those with significant orbital angular momentum, the spin–orbit coupling term, though sometimes overlooked, can contribute to hyperfine splitting of several MHz, particularly affecting g-tensor anisotropy, and zero-field splitting parameters.The simultaneous effect of the three hyperfine coupling contributions necessitates a comprehensive approach to hyperfine engineering; any attempt to precisely control spin dynamics or interpret spectroscopic data must account for the modulation of all three hyperfine components. Neglecting any single term would result in significant quantitative and sometimes qualitative errors in predicting molecular behaviour. In this paper, we propose a decomposition of HFC for compounds 1–12, enabling an in-depth evaluation of how structural changes in these systems correspond to shifts for the three contributions.
 | ||
| Fig. 6 Correlation between isotropic term (Fermi contact) and nuclear spin density for compounds 1–12 (except compound 5). Squares represent valence shell contributions, triangles represent core shell contributions and circles show the total contribution. Calculations were carried out employing B3LYP/ANO-DK3 and using ZORA relativistic treatment. Numerical details can be found in Table S15.† For clarity purposes, to reduce the limits of the Fermi contact values, the TiIII complex has not been included but can be checked in Fig. S6.† | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 relation between axes, which agreed with ab initio results.
:1:1 relation between axes, which agreed with ab initio results.
 | ||
| Fig. 7 Spin–dipolar term and SOMO visualisation for compounds 1–5. Each colour bar represents an axis; blue, orange, and green display x, y and z, respectively. MOs were represented against the first coordination sphere. Compounds 2 and 3, which present Jahn–Teller distortions, do not follow the axiality ratio in Table S16.† | ||
Compounds 2 and 3 present pseudo-octahedral geometries with a VIV(d1) and non-axial HFC components (see Tables S3 and S4†), which is likely a result of a symmetry-breaking shift induced by a Jahn–Teller distortion. We validated this theory via distortion analysis, which revealed that both compounds display a trigonal distortion, shifting from a regular octahedral (OC) geometry to a trigonal prism (TPR) as continuous shape measurement S(OC) values of 2.222 and 2.767 were obtained in contrast to S(TPR) values of 7.767 and 8.330. This distortion leads to a more stabilised dz2 orbital and shifts the ratio in function of the degree of distortion. Further analysis shows that in compounds 2 and 3, the SOMO is mostly a 3dyz orbital, which should show a −2:1:1 spin–dipolar contributions according to Table S16.† However, Jahn–Teller structural distortion in these compounds induces significant mixing of the d orbitals, departing from the expected trend. Compound 4 presented a square-base pyramid geometry with the metal out-of-plane and exhibited a similar trend as compound 1, as its SOMO orbital is 3dxy, which shows in the same pattern than complex 1 SOMO (3dx2–y2) (Table S16†). This difference in SOMO between VIV compounds, pseudo-octahedral 2 and 3 and vanadyl 4 complexes, explains the experimental and calculated values of the three HFC components (see Tables S3–S5†), which are opposite between these two families of systems. The first ones present larger perpendicular HFC components, while the vanadyl axial component is the largest.
As mentioned earlier, compound 5 counts with a TiIII(d1) metal centre (which has a negative g(A)N) and an antiprism-like geometry, which portrayed a 3dz2 SOMO, revealing an ideal relation in the spin–dipolar contributions, but with reversed signs. This leads to an almost vanishing contribution for this compound. Thus, we establish a clear relation between the pattern observed of the axial spin–dipolar terms and the nature of the orbital bearing the unpaired electron, following the coefficients of the electric field gradient matrix elements (Table S16† and eqn (6)) for the systems without important mixing of d orbitals in the SOMO.
 | ||
| Fig. 8 Computed Δgμν against total ASOCμν (where μ = ν) for compounds 1–12. All calculations were performed using a fully uncontracted ANO-DK3 basis set and B3LYP employing a ZORA relativistic treatment. | ||
Fig. 8 shows compounds 1–12, which fall into two distinct categories: d1 complexes (containing VIV and TiIII) and d9 complexes (containing CuII). Compounds with less than half-filled d-orbitals exhibited negative g-factor shifts and, consequently, negative spin–orbit coupling terms. In contrast, compounds with more than half-filled d-orbitals displayed positive g-shifts and positive spin–orbit coupling terms. Notably, CuII (d9) compounds (1, 6, 9–12) exhibited positive g shifts, which lead to positive spin–orbit coupling contribution on all axes. Conversely, VIV compounds (2–4, 7, 8) showed small negative g-shift values for every component and their ASOCμν values are either negative or small. As mentioned earlier, despite expecting negative spin–orbit coupling values for compound 5, positive contributions were obtained due to having a negative g nuclear constant (g(A)N). The compounds in Fig. 8 demonstrate how two of the three g-shift components are positioned much closer together, revealing a general axiality pattern. Our findings indicate that distortions, such as those exhibited by compounds 2 and 3, disrupting this axiality. These distortions alter the spin–orbit coupling by changing the molecular anisotropy, resulting in more widely dispersed g values that reflect a more rhombic environment. Compounds 6 and 12 presented some deviation from the expected trend, possibly presenting a higher ASOC2μν value as opposed to the positive ASOC1μν term. Metal centres also dictated the magnitude of the spin–orbit coupling term, since changes to the nucleus impact on the ξ(riA) function (see eqn (11)), revealing higher spin–orbit coupling constants for heavier metals in Fig. 8.
Based on the detailed analysis of HFC components, we can propose a strategy for modulating HFC by targeting its three main contributions. Manipulation of the coordination environment and metal–ligand interactions are key. For the isotropic term, the choice of donor atoms can control the degree of covalency in metal–ligand bonds, affecting the s shell contributions due to the subtle balance between spin delocalisation and polarisation mechanisms. Modifying the symmetry of the coordination sphere through distortions (like Jahn–Teller effects) can significantly impact the spin–dipolar term by altering the SOMO character and the electric field gradient around the metal centre. For example, introducing trigonal distortions in octahedral complexes can break the orbital occupancy pattern from the ideal 1:1:−2 axial ratio. For the spin–orbit coupling contribution, selection of the metal centre is crucial, as heavier metals exhibit larger spin–orbit coupling constants. Additionally, the electron configuration of the metal (d1 vs. d9) determines whether the g-shift and consequent spin–orbit coupling contribution will be negative or positive. Fine-tuning of the axiality or rhombicity in the three HFC parameters can be further modulated through the spin–orbit coupling term.
The role of metal coordination on the hyperfine coupling
Variations in HFC depending on geometrical differences posed a question regarding the coordination environment and its implications. To explore this hypothesis further, we performed a computational study on VIV homoleptic models coordinated with NH3. The choice of nitrogen donor atoms instead of sulphur-based ligands as those analysed in the previous sections intends to avoid convergence problems due to highly negatively charged structures.To visualize a general picture of computed HFC constants across the model structures, we generated a heatmap showing the magnitude and distribution of HFC constants for each geometric configuration (Fig. 9). The highest Fermi contact terms were observed in trigonal planar, vacant tetrahedra, and square planar geometries. In low coordination systems (angular and linear), spin contamination aberrantly overestimated Fermi contact values, significantly degrading the accuracy of the results, which were discarded. While lower coordination geometries generally showed higher values, coordination number alone does not fully explain the trend, as evidenced by the low Fermi contact terms in trigonal bipyramidal and trigonal prismatic structures. Regardless, a common feature of the highest-yielding Fermi contact term compounds is their planar geometry. This term was further analysed in detail in Fig. 10. Again, as we have previously seen in Fig. 6, there is a linear correlation between the isotropic contribution and the spin density in the metal nuclei centre. Inner-shell contributions comprise the sum of occupied core orbital (1s, 2s and 3s) contributions and consistently exhibited values found between −0.2 and −0.3 a.u.−3 and remained relatively similar throughout all geometries, with no relevant trends observed. The valence shell effects can be predominantly attributed to the electronic contributions of the coordinating ligands. Notably, as metal–ligand distances increase, reduced molecular orbital overlap results in net positive contributions.86 What is remarkable is that although this term is smaller than the contributions of the inner shells, it is, in fact, the one that makes the difference between the different modes of coordination as it is the only one that varies considerably from one system to another (see Fig. 10).
 | ||
| Fig. 9 Heatmap of computed VIV HFC components for [V(NH3)x]4+ (where x = 3–7) geometric model complexes. All calculations were performed using B3LYP/ANO-DK3 approach and using ZORA relativistic treatment. | ||
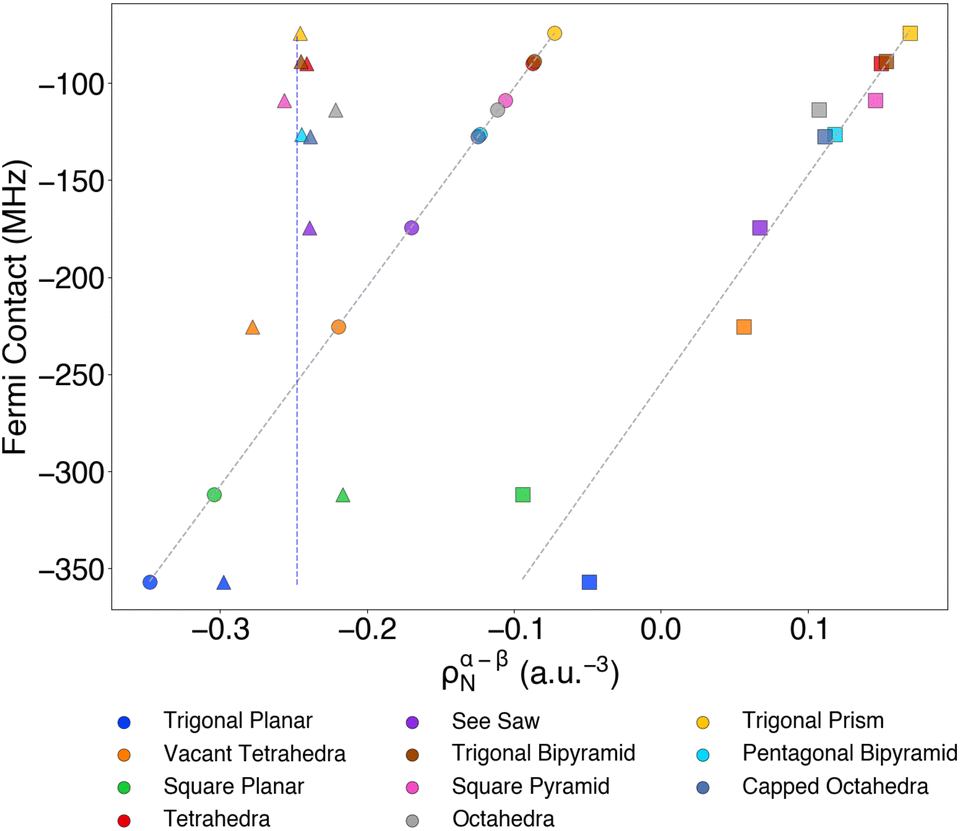 | ||
| Fig. 10 Correlation between Fermi contact contribution and total nuclear spin density for [V(NH3)x]4+ (where x = 3–7) compounds. Squares, triangles, and circles represent the valence shell, core–shell and total contributions, respectively. Numerical details can be found in Table S17.† | ||
Analysis of the metal spin populations revealed values ranging from 1.08 to 1.31, with low-coordination compounds on the higher end. VIV is a d1 electronic configuration metal centre, and its SOMO has mainly non-bonding character, spin polarisation dominates over delocalisation, and there is no mixing with ligand orbitals. This α spin metal density could induce opposite spin density onto ligands, subsequently generating additional α spin density on the metal centre through a spin polarisation mechanism.
All systems showed relatively consistent spin–dipolar values, revealing axial ratios (1:1:2) between axis components in each system (as illustrated in Fig. 9). Positive axial HFC values only were exhibited by the tetrahedra and trigonal prism compounds with a dz2 SOMO, as expected from the one-centre reduced field integral values in Table S16.† While Fermi contact interaction is isotropic, allowing only magnitude adjustments, the spin–dipolar interaction could selectively nullify one component while enhancing the others based on the SOMO configuration, which is determined by molecular geometry. Finally, the spin–orbit coupling component for all model compounds follows the same rule expressed in the previous section. The ASOC1μν term seems to correlate directly with the Δg shift in a rather proportional manner (Fig. S7†). It could be assumed that this term dominates the changes in the spin–orbit coupling component, making the anisotropy the principal contributor. Overall, calculations on idealised [V(NH3)x]4+ are consistent with the main factors determining the three terms for the hyperfine tensor identified for 1–12 real compounds. In addition, results on model systems provide new hints into how the coordination geometry can influence the Fermi contact term, as a lower coordination environment tends to enhance this contribution.
Conclusions
In the first part of this study employing DFT methods a benchmarking of hyperfine coupling has been performed using mononuclear transition metal complexes of the first series proposed as spin qubits. The best results are obtained by DFT methods employing the hybrid functional B3LYP together with methods including relativistic effects such as x2c or ZORA and quadruple-ζ or decontracted basis sets to provide a good description of the internal s orbitals. This methodology also provides good values for g, which can be considered predictive of the two essential quantities obtained from the continuous-wave EPR spectra. Larger discrepancies with the experimental values are obtained if other hybrid functionals such as PBE0 or ωB97X and basis sets of a lower quality are used. Due to the varying magnitude of spin–orbit coupling and the multi-configurational nature of lanthanide ground states, a multi-configurational method is necessary rather than DFT methods for studying complexes with these metals.The second part is devoted to a comprehensive analysis of HFC modulation in CuII and VIV molecular spin qubits, examining the distinct contributions of Fermi contact, spin–dipolar, and spin–orbit-coupling terms across various coordination environments. Our results reveal that geometric configuration plays a critical role in determining HFC behaviour. This is evidenced by compounds 2, 3 (pseudo-octahedral coordination) and 4 (vanadyl) which, despite sharing VIV centres, exhibited markedly different HFC characteristics, non-axial and axial, respectively, due to their different coordination spheres and resulting SOMO orbitals.
We identified that pseudo-octahedral VIV systems, as 2 and 3, undergoing Jahn–Teller distortions serve as particularly promising candidates for spin qubit applications by breaking spin-Hamiltonian axiality. Systems with trigonal distortion exhibit an enhanced potential for orbital mixing with the dz2 orbital, resulting in low axial components of the hyperfine coupling. Our extended model analysis further revealed that trigonal planar, trigonal prismatic, and square planar configurations, where the z-axis remains relatively unperturbed by ligand interactions, offer favourable conditions for low-energy dz2 orbitals to be the SOMO for d1 configuration.
The HFC components exhibited distinctive patterns: Fermi contact contributions were predominantly negative, reflecting spin polarisation of inner-shell s electrons respect to the positive spin density of the SOMO. Their magnitude decreases in VIV systems as coordination number increases due to nullifying effects from valence shell contributions. Spin–dipolar interactions displayed axial relation dependence on the SOMO, which could be disrupted through Jahn–Teller distortions. Spin–orbit coupling contributions showed direct correlation with Δg, with highly anisotropic molecules that could produce greater HFC shifts with more rhombic character. Based on these findings, we propose strategic approaches for HFC engineering in molecular spin qubits or quantum sensors. Previous work by Luis and colleagues demonstrated that achieving universal quantum operations requires hyperfine interactions that are neither purely axial nor purely isotropic, establishing specific relationships between the anisotropic parameters.87 One promising strategy would involve enhancing the perpendicular HFC component while leveraging spin–dipolar contributions to differentiate the perpendicular axes. The metal centre can then be selected to fine-tune spin–orbit coupling effects through g-value modulation, with careful consideration of the anisotropy of the system. Alternatively, symmetry breaking via Jahn–Teller distortion provides a pathway to unequal spin–dipolar contributions, resulting in rhombic systems where spin–orbit coupling further distinguishes the three HFC components. These insights provide a foundation for the rational design of molecular spin qubits and quantum sensors with tailored HFC properties by understanding the interplay between geometric structure, electronic configuration, and the various contributions to HFC.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Joan Cardona: conceptualisation, methodology, formal analysis, investigation writing – original draft, review & editing. Àlex Solé: conceptualization, investigation, writing – review & editing. Pablo Mella: conceptualisation, data curation. Daniel Aravena: conceptualisation, supervision, resources, validation, writing – review & editing. Javier Ruiz-Hidalgo: conceptualisation, supervision, resources, writing, funding acquisition – review & editing, funding acquisition. Silvia Gómez-Coca: conceptualisation, supervision, resources, writing – review & editing, funding acquisition. Eliseo Ruiz: conceptualisation, supervision, resources, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Ministerio de Ciencia e Innovación (project PID2020-117142GB-I00, PID2021-122464NB-I00, TED2021-129593 B-I00, CNS2023-144561 and Maria de Maeztu CEX2021-001202-M) is acknowledged. We also acknowledge the Generalitat de Catalunya for the 2021-SGR-00286 grant. E. R. also acknowledges the Generalitat de Catalunya for an ICREA Academia grant. We thank CSUC for the computational resources. P. M. thanks Universidad de Santiago de Chile, USACH, Project DICYT code 022242AP_Postdoc.Notes and references
- M. Atzori and R. Sessoli, The Second Quantum Revolution: Role and Challenges of Molecular Chemistry, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- E. Coronado, Molecular magnetism: from chemical design to spin control in molecules, materials and devices, Nat. Rev. Mater., 2019, 5, 87–104 CrossRef.
- M. J. Amdur, K. R. Mullin, M. J. Waters, D. Puggioni, M. K. Wojnar, M. Gu, L. Sun, P. H. Oyala, J. M. Rondinelli and D. E. Freedman, Chemical control of spin–lattice relaxation to discover a room temperature molecular qubit, Chem. Sci., 2022, 13, 7034–7045 RSC.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, Millisecond Coherence Time in a Tunable Molecular Electronic Spin Qubit, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed.
- E. Knill, R. Laflamme and G. J. Milburn, A scheme for efficient quantum computation with linear optics, Nature, 2001, 46–52 CrossRef CAS PubMed.
- C. J. Yu, M. J. Graham, J. M. Zadrozny, J. Niklas, M. D. Krzyaniak, M. R. Wasielewski, O. G. Poluektov and D. E. Freedman, Long Coherence Times in Nuclear Spin-Free Vanadyl Qubits, J. Am. Chem. Soc., 2016, 138, 14678–14685 CrossRef CAS PubMed.
- R. Blatt and D. Wineland, Entangled states of trapped atomic ions, Nature, 2008, 453, 1008–1015 CrossRef CAS PubMed.
- P. D. Gregory, J. A. Blackmore, S. L. Bromley, J. M. Hutson and S. L. Cornish, Robust storage qubits in ultracold polar molecules, Nat. Phys., 2021, 17, 1149–1153 Search PubMed.
- J. Clarke and F. K. Wilhelm, Superconducting quantum bits, Nature, 2008, 453, 1031–1042 CrossRef CAS PubMed.
- R. Hanson and D. D. Awschalom, Coherent manipulation of single spins in semiconductors, Nature, 2008, 453, 1043–1049 CrossRef CAS PubMed.
- G. Aromí, D. Aguilà, P. Gamez, F. Luis and O. Roubeau, Design of magnetic coordination complexes for quantum computing, Chem. Soc. Rev., 2012, 41, 537–546 RSC.
- L. Childress and R. Hanson, Diamond NV centers for quantum computing and quantum networks, MRS Bull., 2013, 38, 134–138 CrossRef CAS.
- Y. C. Liu, Y. C. Dzeng and C. C. Ting, Nitrogen Vacancy-Centered Diamond Qubit: The fabrication, design, and application in quantum computing, IEEE Nanotechnol. Mag., 2022, 16, 37–43 Search PubMed.
- D. P. DiVincenzo, The Physical Implementation of Quantum Computation, Fortschr. Phys., 2000, 48, 771–783 CrossRef.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, 2001 Search PubMed.
- M. S. Fataftah, M. D. Krzyaniak, B. Vlaisavljevich, M. R. Wasielewski, J. M. Zadrozny and D. E. Freedman, Metal-ligand covalency enables room temperature molecular qubit candidates, Chem. Sci., 2019, 10, 6707–6714 RSC.
- A. M. Ariciu, D. H. Woen, D. N. Huh, L. E. Nodaraki, A. K. Kostopoulos, C. A. P. Goodwin, N. F. Chilton, E. J. L. McInnes, R. E. P. Winpenny, W. J. Evans and F. Tuna, Engineering electronic structure to prolong relaxation times in molecular qubits by minimising orbital angular momentum, Nat. Commun., 2019, 10, 3330 CrossRef PubMed.
- D. Aravena and E. Ruiz, Spin dynamics in single-molecule magnets and molecular qubits, Dalton Trans., 2020, 49, 9916–9928 RSC.
- M. Atzori, E. Morra, L. Tesi, A. Albino, M. Chiesa, L. Sorace and R. Sessoli, Quantum Coherence Times Enhancement in Vanadium(IV)-based Potential Molecular Qubits: the Key Role of the Vanadyl Moiety, J. Am. Chem. Soc., 2016, 138, 11234–11244 CrossRef CAS PubMed.
- S. Chicco, G. Allodi, A. Chiesa, E. Garlatti, C. D. Buch, P. Santini, R. De Renzi, S. Piligkos and S. Carretta, Proof-of-Concept Quantum Simulator Based on Molecular Spin Qudits, J. Am. Chem. Soc., 2023, 146, 1053–1061 CrossRef PubMed.
- S. Chicco, A. Chiesa, G. Allodi, E. Garlatti, M. Atzori, L. Sorace, R. De Renzi, R. Sessoli and S. Carretta, Controlled coherent dynamics of [VO(TPP)], a prototype molecular nuclear qudit with an electronic ancilla, Chem. Sci., 2021, 12, 12046–12055 RSC.
- C. Godfrin, A. Ferhat, R. Ballou, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Operating Quantum States in Single Magnetic Molecules: Implementation of Grover's Quantum Algorithm, Phys. Rev. Lett., 2017, 119, 187702 CrossRef CAS PubMed.
- R. Hussain, G. Allodi, A. Chiesa, E. Garlatti, D. Mitcov, A. Konstantatos, K. S. Pedersen, R. De Renzi, S. Piligkos and S. Carretta, Coherent Manipulation of a Molecular Ln-Based Nuclear Qudit Coupled to an Electron Qubit, J. Am. Chem. Soc., 2018, 140, 9814–9818 CrossRef CAS PubMed.
- E. Moreno-Pineda, C. Godfrin, F. Balestro, W. Wernsdorfer and M. Ruben, Molecular spin qudits for quantum algorithms, Chem. Soc. Rev., 2018, 47, 501–513 RSC.
- V. Rollano, M. C. de Ory, C. D. Buch, M. Rubín-Osanz, D. Zueco, C. Sánchez-Azqueta, A. Chiesa, D. Granados, S. Carretta, A. Gomez, S. Piligkos and F. Luis, High cooperativity coupling to nuclear spins on a circuit quantum electrodynamics architecture, Commun. Phys., 2022, 5, 246 CrossRef.
- Ö. Üngör, T. M. Ozvat, Z. Ni and J. M. Zadrozny, Record Chemical-Shift Temperature Sensitivity in a Series of Trinuclear Cobalt Complexes, J. Am. Chem. Soc., 2022, 144, 9132–9137 CrossRef PubMed.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Clarendon Press, Oxford, 1970 Search PubMed.
- F. Neese and E. I. Solomon, Interpretation and Calculation of Spin-Hamiltonian Parameters in Transition Metal Complexes, in Magnetism: Molecules to Materials IV, WIley-VCH, 2002, ch. 9, pp. 345–466 Search PubMed.
- K. Bader, D. Dengler, S. Lenz, B. Endeward, S. D. Jiang, P. Neugebauer and J. van Slageren, Room temperature quantum coherence in a potential molecular qubit, Nat. Commun., 2014, 5, 5304 CrossRef CAS PubMed.
- S. Lenz, K. Bader, H. Bamberger and J. van Slageren, Quantitative prediction of nuclear-spin-diffusion-limited coherence times of molecular quantum bits based on copper(ii), Chem. Comm., 2017, 53, 4477–4480 RSC.
- C. E. Jackson, C.-Y. Lin, S. H. Johnson, J. van Tol and J. M. Zadrozny, Nuclear-spin-pattern control of electron-spin dynamics in a series of V(iv) complexes, Chem. Sci., 2019, 10, 8447–8454 RSC.
- C.-J. Yu, S. von Kugelgen, D. W. Laorenza and D. E. Freedman, A Molecular Approach to Quantum Sensing, ACS Cent. Sci., 2021, 7, 712–723 CrossRef CAS PubMed.
- M. J. Graham, M. D. Krzyaniak, M. R. Wasielewski and D. E. Freedman, Probing Nuclear Spin Effects on Electronic Spin Coherence via EPR Measurements of Vanadium(IV) Complexes, Inorg. Chem., 2017, 56, 8106–8113 CrossRef CAS PubMed.
- N. P. Kazmierczak, R. Mirzoyan and R. G. Hadt, The Impact of Ligand Field Symmetry on Molecular Qubit Coherence, J. Am. Chem. Soc., 2021, 143, 17305–17315 CrossRef CAS PubMed.
- M. Atzori, S. Benci, E. Morra, L. Tesi, M. Chiesa, R. Torre, L. Sorace and R. Sessoli, Structural Effects on the Spin Dynamics of Potential Molecular Qubits, Inorg. Chem., 2018, 57, 731–740 CrossRef CAS PubMed.
- M. L. Munzarová, P. Kubáček and M. Kaupp, Mechanisms of EPR Hyperfine Coupling in Transition Metal Complexes, J. Am. Chem. Soc., 2000, 122, 11900–11913 CrossRef.
- K. G. Dyall, Interfacing relativistic and nonrelativistic methods. IV. One- and two-electron scalar approximations, J. Chem. Phys., 2001, 115, 9136–9143 CrossRef CAS.
- J. Autschbach, Relativistic Effects on Electron–Nucleus Hyperfine Coupling Studied with an Exact 2-Component (X2C) Hamiltonian, J. Chem. Theory Comput., 2017, 13, 710–718 CrossRef CAS PubMed.
- P. Verma and J. Autschbach, Relativistic Density Functional Calculations of Hyperfine Coupling with Variational versus Perturbational Treatment of Spin–Orbit Coupling, J. Chem. Theory Comput., 2013, 9, 1932–1948 CrossRef CAS PubMed.
- S. Gohr, P. Hrobárik, M. Repiský, S. Komorovský, K. Ruud and M. Kaupp, Four-Component Relativistic Density Functional Theory Calculations of EPR g- and Hyperfine-Coupling Tensors Using Hybrid Functionals: Validation on Transition-Metal Complexes with Large Tensor Anisotropies and Higher-Order Spin–Orbit Effects, J. Phys. Chem. A, 2015, 119, 12892–12905 CrossRef CAS PubMed.
- D. Peng and M. Reiher, Exact decoupling of the relativistic Fock operator, Theor. Chem. Acc., 2012, 131, 1081 Search PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, Relativistic total energy using regular approximations, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, Relativistic regular two-component Hamiltonians, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- J. Autschbach and B. Pritchard, Calculation of molecular g-tensors using the zeroth-order regular approximation and density functional theory: expectation value versus linear response approaches, Theor. Chem. Acc., 2011, 129, 453–466 Search PubMed.
- M. Barysz and A. J. Sadlej, Infinite-order two-component theory for relativistic quantum chemistry, J. Chem. Phys., 2002, 116, 2696–2704 CrossRef CAS.
- B. Sandhoefer and F. Neese, One-electron contributions to the g-tensor for second-order Douglas–Kroll–Hess theory, J. Chem. Phys., 2012, 137, 094102 CrossRef CAS PubMed.
- M. Reiher, Douglas–Kroll–Hess Theory: a relativistic electrons-only theory for chemistry, Theor. Chem. Acc., 2006, 116, 241–252 Search PubMed.
- A. Wolf, M. Reiher and B. A. Hess, The generalized Douglas–Kroll transformation, J. Chem. Phys., 2002, 117, 9215–9226 CrossRef CAS.
- E. Malkin, I. Malkin, O. L. Malkina, V. G. Malkin and M. Kaupp, Scalar relativistic calculations of hyperfine coupling tensors using the Douglas–Kroll–Hess method with a finite-size nucleus model, Phys. Chem. Chem. Phys., 2006, 8, 4079–4085 RSC.
- D. Peng and M. Reiher, Local relativistic exact decoupling, J. Chem. Phys., 2012, 136, 244108 CrossRef.
- W. Kutzelnigg and W. Liu, Quasirelativistic theory equivalent to fully relativistic theory, J. Chem. Phys., 2005, 123, 241102 CrossRef PubMed.
- Y. J. Franzke and J. M. Yu, Hyperfine Coupling Constants in Local Exact Two-Component Theory, J. Chem. Theory Comput., 2022, 18, 323–343 CrossRef CAS PubMed.
- S. Ghassemi Tabrizi, A. V. Arbuznikov, C. A. Jimenez-Hoyos and M. Kaupp, Hyperfine-Coupling Tensors from Projected Hartree-Fock Theory, J. Chem. Theory Comput., 2020, 16, 6222–6235 CrossRef CAS PubMed.
- Y. J. Franzke, F. Bruder, S. Gillhuber, C. Holzer and F. Weigend, Paramagnetic Nuclear Magnetic Resonance Shifts for Triplet Systems and Beyond with Modern Relativistic Density Functional Methods, J. Phys. Chem. A, 2024, 128, 670–686 CrossRef CAS PubMed.
- P. Pollak and F. Weigend, Segmented Contracted Error-Consistent Basis Sets of Double- and Triple-zeta Valence Quality for One- and Two-Component Relativistic All-Electron Calculations, J. Chem. Theory Comput., 2017, 13, 3696–3705 CrossRef CAS PubMed.
- I. Malkin, O. L. Malkina, V. G. Malkin and M. Kaupp, Relativistic two-component calculations of electronic g-tensors that include spin polarization, J. Chem. Phys., 2005, 123, 244103 CrossRef PubMed.
- L. Visscher and K. G. Dyall, Dirac–Fock Atomic Electronic Structure Calculations using Different Nuclear Charge Distributions, At. Data Nucl. Data Tables, 1997, 67, 207–224 CrossRef CAS.
- A. Wodynski and M. Kaupp, Density Functional Calculations of Electron Paramagnetic Resonance g- and Hyperfine-Coupling Tensors Using the Exact Two-Component (X2C) Transformation and Efficient Approximations to the Two-Electron Spin–Orbit Terms, J. Phys. Chem. A, 2019, 123, 5660–5672 CrossRef CAS PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- Y. J. Franzke, L. Spiske, P. Pollak and F. Weigend, Segmented Contracted Error-Consistent Basis Sets of Quadruple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations, J. Chem. Theory Comput., 2020, 16, 5658–5674 CrossRef CAS PubMed.
- F. Weigend, F. Furche and R. Ahlrichs, Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr, J. Chem. Phys., 2003, 119, 12753–12762 CrossRef CAS.
- E. V. R. de Castro and F. E. Jorge, Accurate universal Gaussian basis set for all atoms of the Periodic Table, J. Chem. Phys., 1998, 108, 5225–5229 CrossRef CAS.
- T. Tsuchiya, M. Abe, T. Nakajima and K. Hirao, Accurate relativistic Gaussian basis sets for H through Lr determined by atomic self-consistent field calculations with the third-order Douglas–Kroll approximation, J. Chem. Phys., 2001, 115, 4463–4472 CrossRef CAS.
- M. Schindler and W. Kutzelnigg, Theory of magnetic susceptibilities and NMR chemical shifts in terms of localized quantities. II. Application to some simple molecules, J. Chem. Phys., 1982, 76, 1919–1933 CrossRef CAS.
- L. C. de Camargo, M. Briganti, F. S. Santana, D. Stinghen, R. R. Ribeiro, G. G. Nunes, J. F. Soares, E. Salvadori, M. Chiesa, S. Benci, R. Torre, L. Sorace, F. Totti and R. Sessoli, Exploring the Organometallic Route to Molecular Spin Qubits: The [CpTi(cot)] Case, Angew. Chem., Int. Ed., 2021, 60, 2588–2593 CrossRef CAS PubMed.
- K. Bader, M. Winkler and J. van Slageren, Tuning of molecular qubits: very long coherence and spin–lattice relaxation times, Chem. Comm., 2016, 52, 3623–3626 RSC.
- C.-Y. Lin, T. Ngendahimana, G. R. Eaton, S. S. Eaton and J. M. Zadrozny, Counterion influence on dynamic spin properties in a V(iv) complex, Chem. Sci., 2019, 10, 548–555 RSC.
- D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, Minimal Distortion Pathways in Polyhedral Rearrangements, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed.
- J. Cirera, E. Ruiz and S. Alvarez, Shape and Spin State in Four-Coordinate Transition-Metal Complexes: The Case of the d6 Configuration, Chem.–Eur. J., 2006, 12, 3162–3167 CrossRef CAS PubMed.
- D. Koutsandreas, S. Evangelos, P. Fotios and V. and Assimakopoulos, On the selection of forecasting accuracy measures, J. Oper. Res. Soc., 2022, 73, 937–954 CrossRef.
- S. Kossmann, B. Kirchner and F. Neese, Performance of modern density functional theory for the prediction of hyperfine structure: meta-GGA and double hybrid functionals, Mol. Phys., 2007, 105, 2049–2071 CrossRef CAS.
- O. L. Malkina, B. Schimmelpfennig, M. Kaupp, B. A. Hess, P. Chandra, U. Wahlgren and V. G. Malkin, Spin–orbit corrections to NMR shielding constants from density functional theory. How important are the two-electron terms?, Chem. Phys. Lett., 1998, 296, 93–104 CrossRef CAS.
- O. L. Malkina, J. Vaara, B. Schimmelpfennig, M. Munzarová, V. G. Malkin and M. Kaupp, Density Functional Calculations of Electronic g-Tensors Using Spin−Orbit Pseudopotentials and Mean-Field All-Electron Spin−Orbit Operators, J. Am. Chem. Soc., 2000, 122, 9206–9218 CrossRef CAS.
- J. Vaara, K. Ruud, O. Vahtras, H. Ågren and J. Jokisaari, Quadratic response calculations of the electronic spin–orbit contribution to nuclear shielding tensors, J. Chem. Phys., 1998, 109, 1212–1222 CrossRef.
- D. Goldfarb and S. Stoll, EPR Spectroscopy: Fundamentals and Methods, 2018 Search PubMed.
- J. R. Harmer, in eMagRes, 2016, vol. 5, pp. 1493–1514 Search PubMed.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- E. Ruiz, J. Cirera and S. Alvarez, Spin density distribution in transition metal complexes, Coord. Chem. Rev., 2005, 249, 2649–2660 CrossRef CAS.
- A. Szabó and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Dover Publications, Mineola (N. Y.), 1996 Search PubMed.
- R. J. Deeth, Discrete variational Xα calculations of the spectra of chlorocuprate(II) complexes: a detailed comparison with experiment and the cellular ligand field model, J. Chem. Soc., Dalton Trans., 1990, 355–363 RSC.
- J. Weber, A. Goursot, E. Penigault, J. H. Ammeter and J. Bachmann, Electronic structure of metallocene compounds. 3. Comparison of the results of multiple-scattering X.alpha. calculations with various electronic observables of cobaltocene, J. Am. Chem. Soc., 1982, 104, 1491–1506 CrossRef CAS.
- I. Gimeno, A. Urtizberea, J. Román-Roche, D. Zueco, A. Camón, P. J. Alonso, O. Roubeau and F. Luis, Broad-band spectroscopy of a vanadyl porphyrin: a model electronuclear spin qudit, Chem. Sci., 2021, 12, 5621–5630 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02500a |
| This journal is © The Royal Society of Chemistry 2025 |