DOI:
10.1039/D5SC02645H
(Edge Article)
Chem. Sci., 2025,
16, 11841-11848
Hexamethylbenzene elimination enables the generation of transient, sterically unhindered multiply bonded boron species†
Received
9th April 2025
, Accepted 16th May 2025
First published on 19th May 2025
Abstract
We present a method for the generation of boron-containing unsaturated small molecules via hexamethylbenzene elimination. The fragmentation precursors are obtained through bond insertion into phenyl boranorbornadiene (PhB(C6Me6), 1). Compound 1 undergoes 1,1-insertion with 2,6-xylyl isocyanide, affording a boron-doped bicyclo[2.2.2]octa-2,5-diene 2. Heating 2 in toluene results in the formation of a base-stabilized boraketenimine PhB(CNxyl)2 (i.e., borylene diisocyanide) as an intermediate via retro-Diels–Alder reaction. Surprisingly, PhB(CNxyl)2 dimerizes to give a boron-doped 6-membered ring (PhB)2C4(CNxyl)64. The reaction of 1 with trimethylamine N-oxide and phenyl azide yields triphenyl boroxine and a BN4 ring, respectively, implying the involvement of transient oxoborane (PhB![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) and iminoborane intermediates (PhBNPh), respectively. Furthermore, boranorbornadiene also undergoes 2,3-insertion with mesityl isocyanate (MesNCO), affording a fused 6/5-membered heterocycle 11. This insertion profile is analogous to the insertion of phenyl azide into 1.
O) and iminoborane intermediates (PhBNPh), respectively. Furthermore, boranorbornadiene also undergoes 2,3-insertion with mesityl isocyanate (MesNCO), affording a fused 6/5-membered heterocycle 11. This insertion profile is analogous to the insertion of phenyl azide into 1.
Introduction
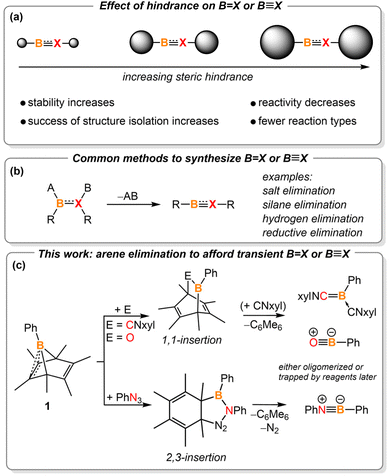
Molecules with boron-containing unsaturated bonds are known as key synthetic intermediates for accessing boron-doped heterocycles, which are important in the fields of catalysis,1 medicinal chemistry,2 luminescent materials,3 and chemical sensors.4 In addition, they are fascinating mediators for small molecule activation due to the intrinsic instability of the bonds compared to their carbon-based counterparts.5 The synthesis and reactivity profiles of compounds containing boron–second-row element unsaturated bonds are well established. They are commonly isolated by attaching kinetically stabilizing bulky substituents to the boron moiety (Fig. 1a).5i,6 The enhanced steric hindrance prevents the target species from oligomerizing or reacting with reagents used in the synthesis.5f,g However, the steric bulk also decreases the reactivity of these molecules, limiting the discovery of new reaction types.7 Thus, developing methods to cleanly generate transient, non-sterically hindered small molecules with unsaturated boron multiple bonds is the key to unlocking reactivity patterns that are currently hidden.
 |
| | Fig. 1 (a) Effects of steric hindrance on multiply bonded boron species; (b) common methods to synthesize boron-containing double bonds or triple bonds; (c) hexamethylbenzene elimination enables the generation of boron unsaturated bonds (this work). | |
Removing two substituents from boron and its adjacent atom is the most common method to construct boron unsaturated bonds (i.e., elimination, Fig. 1b).5i,6b,8 While the eliminated substituent on boron is often a halide or hydride, the adjacent atom's substituent varies depending on its electronegativity. For example, salt or silane elimination often occurs in the construction of compounds containing BN and BO unsaturated bonds.6b,8b,9 Recently, we used arenes, specifically benzene and anthracene, as the leaving group to synthesize diazoborane10 and transient borylene intermediates.11 Such arene extrusion reactions represent a mild approach to elimination processes, where aromatization would provide the extra driving force necessary for boron-centered elimination chemistry. We postulated that such arene extrusion reactions could also be applied to synthesizing transient, sterically unhindered multiply bonded boron species.
Boranorbornadienes are boron analogues of norbornadienes.12 In 1988, Fagan reported the synthesis of phenyl-substituted hexamethyl-boranorbornadiene PhB(C6Me6). Recently, we reported the synthesis of the halo-substituted versions (RB(C6Me6), R = Cl, Br), which were used to generate carbene-ligated haloborylenes as reactive intermediates.11 Herein, we report the generation of transient base-stabilized boraketenimine (PhB(CNxyl)2), oxoborane (PhBO), and iminoborane (PhBNPh) via hexamethylbenzene elimination (Fig. 1c). Dimerization of a boraketenimine is reported here for the first time, illustrating the privilege of non-sterically hindered boron unsaturated species. In addition, boron-involved retro-Diels–Alder reactions and Cope rearrangements are also disclosed in this work.
Results and discussion
Generating a transient boraketenimine
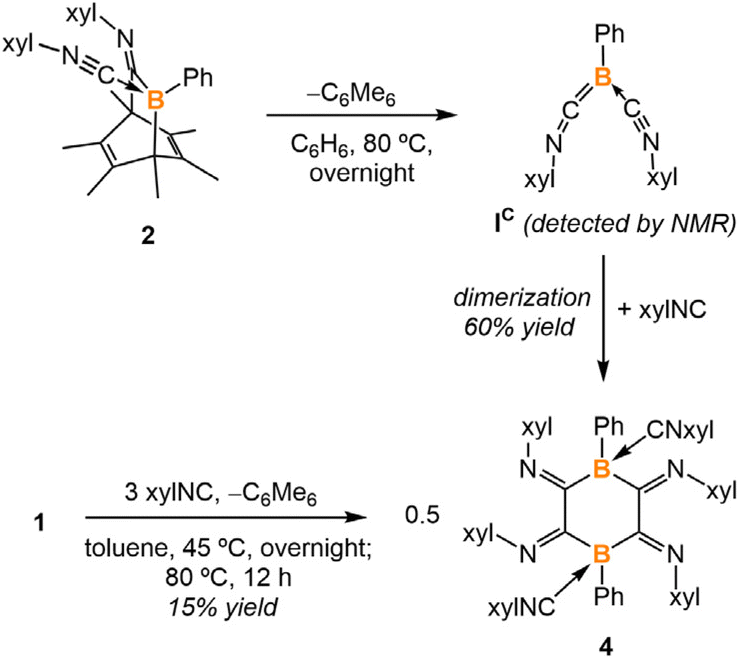
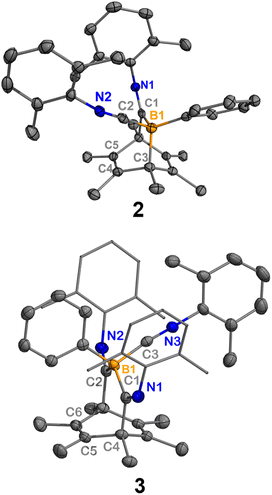
In the first experiment, we targeted the preparation of a precursor for a boraketenimine containing a B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond.13 It should be mentioned that the stabilization of boraketenimines typically demands bulky aryl substituents.14 The reaction of PhB(C6Me6) (1) with 2,6-xylyl isocyanide (xylNC) (2 equiv.) in toluene afforded two new boron species according to the 11B NMR spectrum, one with a singlet at −17.8 ppm and one with a singlet at −13.5 ppm (Scheme 1). The reaction was monitored by NMR spectroscopy, and it required three days to go to completion. Storing the concentrated reaction mixture at −35 °C yielded block-shaped crystals and a few plate-shaped crystals. Single-crystal X-ray diffraction (SC-XRD) analysis of the block-shaped crystal revealed a boron-doped bicyclo[2.2.2]octa-2,5-diene 2 (Fig. 2). It is rationalized that compound 2 is generated from 1 through the insertion of an isocyanide followed by the coordination of another isocyanide to the boron. In the solid-structure of 2, the B1–C2 bond (1.588(2) Å) is remarkably shorter than the B1–C1 and B1–C3 single bonds (1.638(2) Å, 1.679(2) Å, respectively). The C1–N1 and C2–N2 distances are 1.281(2) Å and 1.159(2) Å, which fall within the range of CN double bonds and CN triple bonds, respectively.15 SC-XRD analysis of the plate-shaped crystal revealed a boron-doped bicyclo[3.2.2]nona-6,8-diene 3, which represents the product of a double isocyanide insertion into 1. The B1–C1 and B1–C2 distances are 1.673(3) Å and 1.671(3) Å, respectively, comparable to the B1–C1 distance in 2.
C double bond.13 It should be mentioned that the stabilization of boraketenimines typically demands bulky aryl substituents.14 The reaction of PhB(C6Me6) (1) with 2,6-xylyl isocyanide (xylNC) (2 equiv.) in toluene afforded two new boron species according to the 11B NMR spectrum, one with a singlet at −17.8 ppm and one with a singlet at −13.5 ppm (Scheme 1). The reaction was monitored by NMR spectroscopy, and it required three days to go to completion. Storing the concentrated reaction mixture at −35 °C yielded block-shaped crystals and a few plate-shaped crystals. Single-crystal X-ray diffraction (SC-XRD) analysis of the block-shaped crystal revealed a boron-doped bicyclo[2.2.2]octa-2,5-diene 2 (Fig. 2). It is rationalized that compound 2 is generated from 1 through the insertion of an isocyanide followed by the coordination of another isocyanide to the boron. In the solid-structure of 2, the B1–C2 bond (1.588(2) Å) is remarkably shorter than the B1–C1 and B1–C3 single bonds (1.638(2) Å, 1.679(2) Å, respectively). The C1–N1 and C2–N2 distances are 1.281(2) Å and 1.159(2) Å, which fall within the range of CN double bonds and CN triple bonds, respectively.15 SC-XRD analysis of the plate-shaped crystal revealed a boron-doped bicyclo[3.2.2]nona-6,8-diene 3, which represents the product of a double isocyanide insertion into 1. The B1–C1 and B1–C2 distances are 1.673(3) Å and 1.671(3) Å, respectively, comparable to the B1–C1 distance in 2.
 |
| | Scheme 1 Reaction of PhB(C6Me6) (1) with 2 equivalents of 2,6-xylyl isocyanide (xylNC) affords 2 in 81% yield and 3 as a side product. | |
 |
| | Fig. 2 Molecular structures of 2 and 3. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: 2: B1–C1 1.638(2), B1–C2 1.588(2), C1–N1 1.281(2), C2–N2 1.159(2), B1–C3 1.679(2), C3–C4 1.528(2), C4–C5 1.334(2), B1–C1–N1 131.1(1), B1–C2–N2 177.3(1); 3: B1–C1 1.673(3), B1–C2 1.671(3), B1–C3 1.593(3), C1–N1 1.290(3), C2–N2 1.280(2), C3–N3 1.154 (3), C4–C5 1.529(3), C5–C6 1.329(3), B1–C1–N1 127.1(2), B1–C2–N2 126.7(2), B1–C3–N3 172.8(2). | |
Despite various attempts, the formation of the side-product 3 could not be prevented, nor could 3 be generated quantitatively. Increasing the equivalents of xylNC (e.g., 4 eq.) led to a notable enhancement in the yield of 3 (15% NMR yield). However, extension of the reaction time did not result in the conversion of 2 to 3. The treatment of 1 with equimolar xylNC at room temperature resulted in the complete consumption of xylNC and half consumption of boranorbornadiene 1, suggesting that the isocyanide insertion step is the rate-determining step, and the coordinated isocyanide becomes unreactive.
Heating a benzene solution of 2 at 80 °C overnight led to a color change from yellow to dark red and the precipitation of yellow crystals. The 1H NMR spectrum of the crude reaction mixture indicated the formation of free hexamethylbenzene (2.13 ppm in C6D6). SC-XRD analysis of the yellow crystals yielded the structure of a tetraiminyl-1,4-diboracyclohexane 4. In the molecular structure of 4, the B1–C1 and B1′–C2 distances are measured to be 1.603(2) Å and 1.640(2) Å, respectively, which fall within the range of B–C single bonds (Fig. 3). The C1–C2 distance of 1.502(2) Å lies within the range of C–C single bonds.15,16 The geometry of the B1–C1–C2–B1′–C1′–C2′ 6-membered ring is comparable to the chair conformation of cyclohexane. Compound 4 is insoluble in common solvents such as benzene, toluene, dichloromethane, and THF, preventing NMR spectroscopic analysis at room temperature. However, heating 4 in toluene-D8 at 105 °C allowed for the observation of a singlet at −10.9 ppm in the 11B NMR spectrum.
 |
| | Fig. 3 Molecular structure of 4. Hydrogen atoms and all dimethylphenyl groups in the bottom representation have been omitted for clarity to show the central diboracyclohexane unit. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–C1 1.642(2), C1–C2 1.520(2), C1–N1 1.282(2), B1–C3 1.599(2), C3–N3 1.151(2), B1–C1–C2 115.5(1), B1–C3–N3 168.5(2), N1–C1–C2 126.0(1), B1–C1–C2–B1′−53.6(2). | |
It is hypothesized that a retro-Diels–Alder reaction occurred on 2 at the elevated temperature, producing hexamethylbenzene and boraketenimine (xylNC)2BPh as an intermediate (Scheme 2). Indeed, monitoring the reaction by 11B NMR spectroscopy revealed an intermediate at 11Bδ = −17.0 ppm (Fig. S30†), comparable to the bulker boraketenimine TpB(CNMe)2 (Tp = 2,4,6-triisopropylphenyl) reported by Braunschweig (11Bδ = −18.8 ppm).6a Moreover, compound 4 was prepared via the reaction of compound 1 with xylNC (3 equiv.). However, the one-pot reaction results in significantly lower yields of 4 (15%) compared to the two-step synthesis (60%).
 |
| | Scheme 2 Synthesis of the tetraiminyl-1,4-diboracyclohexane 4. | |
Density Functional Theory (DFT) calculations indicate that the energy barrier of this retro-Diels–Alder reaction is 28.8 kcal mol−1, which is surmountable at the given temperature. As indicated by the different N–C–N angles in the optimized structure (Fig. 4, bottom), the intermediate IC is best described as a boraketenimine stabilized by coordination with a neutral isocyanide. Though the following steps, which finally led to the formation of 4, are experimentally elusive, DFT computations suggest a possible pathway comprising C–C coupling, ring contraction, and ring expansion (Fig. 4). The barrier of the C–C coupling process is computed to be 28.4 kcal mol−1, comparable to that of the retro-Diels–Alder step. The subsequent ring contraction and ring expansion represent the formal coupling of the other pair of carbons. We will show that a strong Lewis acid could abstract the coordinated isocyanide in 2. Upon formation of the dimerized product IVC, two Lewis acidic boron centers could abstract two isocyanides from 2, resulting in the isolated product 4. Overall, the Gibbs free energy of the dimerized product IVC is 24.8 kcal mol−1 lower than its monomer IC.
 |
| | Fig. 4 Energy profiles were calculated for the reaction of 2 to 4. The relative Gibbs free energies (in red) and electronic energies (in blue) are calculated at 298 K and given in kcal mol−1. The calculation is at M06-2X-D3/6-311g** level of theory with applied solvation models in toluene. | |
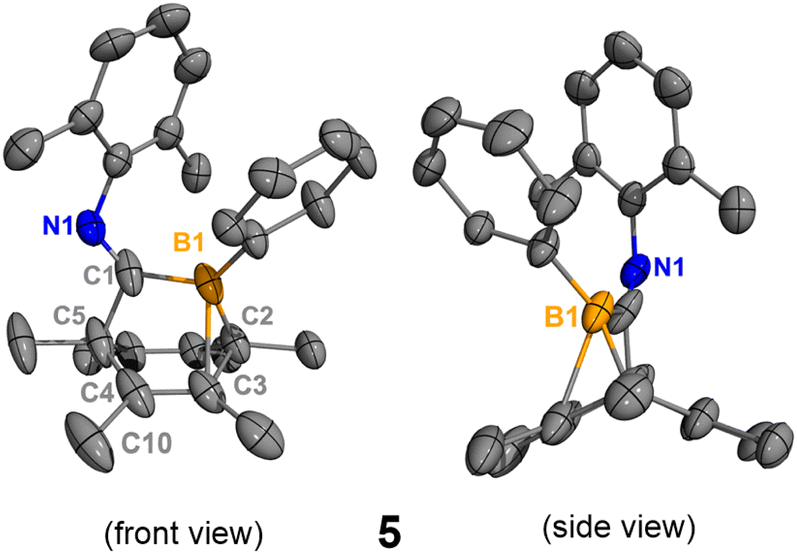
In order to obtain the uncoordinated bora-bicyclo[2.2.2]octa-2,5-diene, aiming to generate a base-free boraketenimine, a strong Lewis acid was used to abstract the isocyanide from the boron center (Scheme 3). As indicated by 11B NMR spectroscopy, the treatment of 2 with equimolar B(C6F5)3 resulted in the clean formation of B(C6F5)3·xylNC (11Bδ: −20.9 ppm) and the uncoordinated bora-bicyclo[2.2.2]octa-2,5-diene 5 (11Bδ: 41.7 ppm). While pure 5 does not crystallize on its own, storing a concentrated hexanes solution of the reaction mixture at −35 °C for 15 days gave a few yellow crystals, determined to be 5 and 6 by SC-XRD analyses. Each unit cell contains one molecule of 5 and one molecule of 6 (Fig. S32†). The B1–C1 (1.584(3) Å) and B1–C2 (1.573(3) Å) distances in 5 are remarkably shorter than those in 2 and 3 (Fig. 5). Interestingly, the B1–C3 distance in 5 is 2.057(3) Å, indicating a weak interaction between the electrophilic B1 and C3C4 double bond. This CC double bond is slightly longer than those in 2 and 3 (5, C3C4 1.358(3) Å; 2, C4C5 1.334(2) Å; 3, C5C6 1.329(3) Å), and the boron atom in 5 is more distorted away from the C1–C2–C5 plane (C5–C1–B1–C2 31.4°). Compound 6 is not detectable by NMR spectroscopy due to its insufficient amount in the reaction mixture, and its formation may result from the C–H activation of 5. In addition, the isocyanide adduct 2 was regenerated from the treatment of 5 with equimolar xylNC. Therefore, compounds 2 and 5 are interconvertible by treating them with the respective Lewis acid or base.
 |
| | Scheme 3 Abstraction of isocyanide ligand by a Lewis acid to generate 5. | |
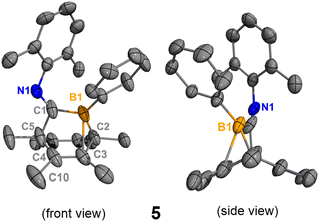 |
| | Fig. 5 Molecular structure of 5. Hydrogen atoms have been omitted for clarity. The molecular structure of 6 is in the ESI (Fig. S32†). Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–C1 1.584(3), B1–C2 1.578(3), B1–C3 2.057(3), C2–C3 1.544(3), C3–C4 1.358(3), C4–C5 1.529(4), C4–C10 1.503(3), C1–B1–C2 109.6(1), C1–B1–C3 95.2(1), B1–C2–C3 82.4(1), C5–C1–B1–C2 31.4(2). | |
Compound 5 does not undergo retro-Diels–Alder reaction to release a base-free boraketenimine (PhB = CNxyl) at elevated temperatures; instead, it decomposes into multiple unidentifiable species. To test the ability of compound 5 to act as a synthon for the base-free boraketenimine, 5 was treated with cyclooctyne and mesityl nitrile N-oxide (Scheme 4). Based on 1H NMR spectroscopy, the reaction of both reagents did not lead to the elimination of hexamethylbenzene. In both reactions, compound 5 reacts as 1,3-dipole, with boron representing the electrophilic site and nitrogen the nucleophilic site. The 1,3-dipole undergoes (3 + 2) cycloaddition with cyclooctyne and (3 + 3) cycloaddition with mesityl nitrile N-oxide. The following 1,2-migration of the 5-membered ring and 3,2-migration of the 6-membered ring afford a norbornadiene derivative 7 and a norcaradiene derivative 8, respectively. The solid-state structures of 7 and 8 were obtained via SC-XRD analysis (Fig. S34 and S35†).
 |
| | Scheme 4 Treatment of compound 5 with cyclooctyne and mesityl nitrile N-oxide. | |
Generating a transient oxoborane
Following the study of the reactivity with isocyanide, we anticipated that the reaction of 1 with trimethylamine N-oxide (TMAO) would afford a precursor to elusive oxoborane species. Currently, there are only a handful of examples for generating transient oxoborane, and a free oxoborane (RBO) has not been isolated.17
In contrast to the reaction of 1 with isocyanide, the reaction between 1 and TMAO in THF was very fast, and both starting materials were consumed within an hour at room temperature (Scheme 5). The 1H NMR spectrum of the reaction mixture displayed a singlet at 2.13 ppm in C6D6, indicating the formation of hexamethylbenzene. After removing hexamethylbenzene from the reaction mixture by sublimation, the remaining solids were analyzed by 11B NMR spectroscopy in CDCl3, showing a singlet at 29.0 ppm. The 1H NMR spectrum is in accordance with triphenylboroxine (9) in CDCl3. It is known that the formation of 9 results from the trimerization of oxoboranes.18 However, our attempts to trap the oxoborane intermediate with other reagents were unsuccessful (Fig. S31†).
 |
| | Scheme 5 Generation of phenyl oxoborane intermediate and its trimerization. | |
DFT calculations suggest that compound 1 and TMAO could first form a Lewis adduct IO. The geometry of IO is pre-organized for oxygen insertion and trimethylamine leaving (Fig. S37†). This oxygen insertion step is very exergonic (−73.5 kcal mol−1), and its energy barrier for accessing the intermediate IIO is only 19.9 kcal mol−1. The release of oxoborane from hexamethylbenzene constitutes a retro-Diels–Alder reaction viaTS3O. Surprisingly, the energy barrier of this retro-Diels–Alder reaction is remarkably low (13.5 kcal mol−1) compared to that of the boraketenimine leaving step (28.8 kcal mol−1), indicating that the formation of oxoborane is very favorable (Fig. 6).
 |
| | Fig. 6 Proposed intermediates and transition states for the reaction of 1 to 9. The relative Gibbs free energies (in red) and electronic energies (in blue) are calculated at 298 K and given in kcal mol−1. The calculation is at M06-2X-D3/6-311g** level of theory with applied solvation models in toluene. | |
Generating a transient iminoborane
Compared to boraketenimine and oxoborane, iminoborane is more well-studied.19 Transient iminoborane is commonly generated via N2 loss from the corresponding azidoborane (R2BN3) precursors.20 To examine whether the hexamethylbenzene platform could generate transient iminoborane, compound 1 was treated with phenyl azide at 50 °C (Scheme 6). The 1H NMR spectrum of the reaction mixture indicated the quantitative generation of hexamethylbenzene. The boron-containing species was separated from the mixture by washing with hexanes and recrystallization. SC-XRD analysis displays a BN4 5-membered ring 10, known from (3 + 2) cycloaddition between iminoborane PhBNPh and phenyl azide.8b,21 The center boron in 10 gives a resonance of 25.4 ppm in the 11B NMR spectrum (Fig. 8). The nucleus-independent chemical shift (NICS) values of compound 10 (NICS(0) = −7.1; NICS(1) = −7.2) indicate that it possesses a certain degree of aromaticity.22 It should be noted that no intermediates were observed by NMR spectroscopy during the formation of 10.
 |
| | Scheme 6 Generation of diphenyl iminoborane intermediate and its trapping with phenyl azide to afford compound 10. | |
DFT calculations were performed to better understand the mechanism of iminoborane generation (Fig. 7). Compound 1 and phenyl azide could first form a Lewis acid-base adduct IN. Phenyl azide would act as a nitrene source to afford IIIN as an intermediate, and the iminoborane evolves a retro-Diels–Alder reaction from IIIN, analogous to the formation of (xylNC)2BPh from 2. However, the overall barrier of this reaction pathway is too high to consider (45.4 kcal mol−1). Alternatively, the adduct IN could undergo a [3,3]-sigmatropic rearrangement, leading to the formation of a bicyclic 6-fused-5 membered ring IIN. This [3,3]-sigmatropic rearrangement is an extension of the Cope rearrangement (Fig. S36†). Intermediate IIN is extremely unstable and could subsequently undergo fragmentation to hexamethylbenzene, dinitrogen, and iminoborane by overcoming a very low barrier (TS2N, 3.6 kcal mol−1). The overall energy barrier for this alternative pathway is only 28.7 kcal mol−1, which is surmountable at the given temperature.
 |
| | Fig. 7 Proposed intermediates and transition states for the reaction from 1 to 10. The relative Gibbs free energies (in red) and electronic energies (in blue) are calculated at 298 K and given in kcal mol−1. The calculation is at M06-2X-D3/6-311g** level of theory and applied solvation models in toluene. | |
 |
| | Fig. 8 Molecular structures of 10 and 11 in the solid-state. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: for 10, B1–N1 1.432(2), B1–N4 1.425(2), N1–N2 1.382(2), N2–N3 1.276(2), N3–N4 1.387(2), B1–N1–N2 109.9(1), N1–N2–N3 109.9(1), N1–B1–N4 100.7(1); for 11, B1–N1 1.423(2), N1–C1 1.401(1), C1–O1 1.211(2), C1–C2 1.533(2), B1–N1–C1 112.1(1), N1–C1–O1 124.1(1). | |
In order to assess the feasibility of the second reaction pathway, compound 1 was treated with mesityl isocyanate, which is isoelectronic to phenyl azide. We proposed that treating 1 with isocyanate resulted in an analogous reaction to that of phenyl azide, while the bicyclic analog could not undergo hexamethylbenzene fragmentation at room temperature. Indeed, the reaction of 1 with mesityl isocyanate led to the expected 2,3-insertion product, affording bicyclic 11 (Scheme 7 and Fig. 8). Heating 11 at 50 °C showed no obvious fragmentation to hexamethylbenzene, iminoborane, and carbon monoxide.
 |
| | Scheme 7 Synthesis of compounds 11. | |
Conclusion
In conclusion, we have demonstrated that hexamethylbenzene elimination is a new synthetic method for constructing transient sterically unhindered boron-containing unsaturated bonds. The 1,1-insertion of isocyanide and an oxygen atom into boranorbornadiene results in boron-doped bicyclo[2.2.2]octa-2,5-dienes, which readily undergo retro-Diels–Alder reactions to afford the corresponding boraketenimine and oxoborane as intermediates. The 2,3-insertion of mesityl isocyanate and phenyl azide is reminiscent of a boron-involved Cope rearrangement. The fragmentation of the phenyl azide insertion product yields iminoborane, a species that readily undergoes (3 + 2) cycloaddition with phenyl azide. These reactions are interesting from a fundamental synthetic chemistry perspective, and the benzene extrusion strategy is helpful for the generation of novel boron-containing unsaturated species.
Data availability
Experimental details, characterization, and computational details, including Fig. S1–S37 and Tables S1–S13,† X-ray crystallographic data for 2, 3, 4, 5, 6, 7, 8, 10, and 11 (CIF). CCDC identification codes 2299088–2299093, 2416228 and 2416229 are associated with the supplementary crystallographic data for this paper. These data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/data_request/cif.
Author contributions
C. Z. conceptualized the project, synthesized and characterized the compounds, wrote the draft together with. P. D. and B. Y. E. T. R. J. G. and C. C. C. supervised the project, acquired financial support for the project, and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 2247252 (CCC) and No. 2046544 (RJG). The authors acknowledge Research Computing at Massachusetts Institute of Technology (Engaging Cluster, Supercloud) for providing computational resources and technical support that have contributed to the results reported within this publication. The authors thank Junyi Wang for discussing computational results.
Notes and references
-
(a) J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC;
(b) G. Kumar, S. Roy and I. Chatterjee, Org. Biomol. Chem., 2021, 19, 1230–1267 RSC;
(c) V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5 CrossRef CAS;
(d) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC;
(e) E. von Grotthuss, S. E. Prey, M. Bolte, H. W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082–6091 CrossRef CAS PubMed.
-
(a) A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC;
(b) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed;
(c) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed;
(d) K. Nedunchezhian, N. Aswath, M. Thiruppathy and S. Thirugnanamurthy, J. Clin. Diagn. Res., 2016, 10, ZE01–ZE04 Search PubMed;
(e) M. Z. H. Kazmi, O. M. Schneider and D. G. Hall, J. Med. Chem., 2023, 66, 13768–13787 CrossRef CAS PubMed.
-
(a) S. Agnoli and M. Favaro, J. Mater. Chem. A, 2016, 4, 5002–5025 RSC;
(b) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS;
(c) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS;
(d) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC;
(e) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed;
(f) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC;
(g) X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed;
(h) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC;
(i) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CrossRef CAS.
-
(a) E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC;
(b) S. Muhammad, M. R. S. A. Janjua and Z. Su, J. Phys. Chem. C, 2009, 113, 12551–12557 CrossRef CAS;
(c) K. S. Thanthiriwatte and S. R. Gwaltney, J. Phys. Chem. A, 2006, 110, 2434–2439 CrossRef CAS PubMed;
(d) Y. Tokoro, A. Nagai, K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 550–555 CrossRef CAS PubMed;
(e) C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed;
(f) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed;
(g) W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, A. Kaur, D. J. D. Wilson and R. J. Gilliard, Chem.–Eur. J., 2019, 25, 12512–12516 CrossRef CAS PubMed.
-
(a) M. Arrowsmith, J. Bohnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287–14292 CrossRef CAS PubMed;
(b) W. Lu, K. Xu, Y. Li, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 15691–15695 CrossRef CAS PubMed;
(c) L. Zhu and R. Kinjo, Angew. Chem., Int. Ed., 2022, 61, e202207631 CrossRef CAS PubMed;
(d) L. Zhu and R. Kinjo, Angew. Chem., Int. Ed., 2023, 62, e202306519 CrossRef CAS PubMed;
(e) L. Zhu and R. Kinjo, Chem. Commun., 2023, 59, 10436–10439 RSC;
(f) E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2023, 145, 7107–7112 CrossRef CAS PubMed;
(g) J. Li, Z. Lu and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 23691–23697 CrossRef CAS PubMed;
(h) H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS PubMed;
(i) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 CrossRef CAS PubMed.
-
(a) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed;
(b) H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345–347 CrossRef CAS PubMed.
-
(a) A. M. Borys, E. F. Rice, G. S. Nichol and M. J. Cowley, J. Am. Chem. Soc., 2021, 143, 14065–14070 CrossRef CAS PubMed;
(b) A. N. Price, G. S. Nichol and M. J. Cowley, Angew. Chem., Int. Ed., 2017, 56, 9953–9957 CrossRef CAS PubMed.
-
(a) W. Lu, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2017, 56, 9829–9832 CrossRef CAS PubMed;
(b) R. Guo, T. Li, R. Wei, X. Zhang, Q. Li, L. L. Liu, C. H. Tung and L. Kong, J. Am. Chem. Soc., 2021, 143, 13483–13488 CrossRef CAS PubMed.
- R. Guo, X. Zhang, T. Li, Q. Li, D. A. Ruiz, L. L. Liu, C. H. Tung and L. Kong, Chem. Sci., 2022, 13, 2303–2309 RSC.
- C. Zhang, C. C. Cummins and R. J. Gilliard Jr, Science, 2024, 385, 327–331 CrossRef CAS PubMed.
- C. Zhang, R. J. Gilliard Jr and C. C. Cummins, Chem. Sci., 2024, 15, 17873–17880 RSC.
-
(a) F. Lindl, X. Guo, I. Krummenacher, F. Rauch, A. Rempel, V. Paprocki, T. Dellermann, T. E. Stennett, A. Lamprecht, T. Brückner, K. Radacki, G. Bélanger-Chabot, T. B. Marder, Z. Lin and H. Braunschweig, Chem.–Eur. J., 2021, 27, 11226–11233 CrossRef CAS PubMed;
(b) C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132–5139 CrossRef CAS;
(c) F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Organometallics, 2014, 34, 229–235 CrossRef;
(d) P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS.
-
(a) C. Chen, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 19905–19911 CrossRef CAS PubMed;
(b) A. B. Cuenca and E. Fernandez, Chem. Soc. Rev., 2021, 50, 72–86 RSC;
(c) M. Eaton, Y. Zhang and S. Y. Liu, Chem. Soc. Rev., 2024, 53, 1915–1935 RSC;
(d) M. M. Olmstead, P. P. Power, K. J. Weese and R. J. Doedens, J. Am. Chem. Soc., 1987, 109, 2541–2542 CrossRef CAS;
(e) M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854–6856 CrossRef CAS.
-
(a) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469–4473 CrossRef PubMed;
(b) T. J. Hadlington, T. Szilvasi and M. Driess, Chem. Sci., 2018, 9, 2595–2600 RSC;
(c) M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schafer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem.–Eur. J., 2018, 24, 6843–6847 CrossRef CAS PubMed.
-
(a)
P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement—A Crystallographer's Guide to SHELXL, Oxford University Press, 2006 CrossRef;
(b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- A. Maercker, W. Brieden, T. Schmidt and H. D. Lutz, Angew. Chem., Int. Ed., 2003, 28, 477–478 CrossRef.
-
(a) B. Pachaly and R. West, J. Am. Chem. Soc., 1985, 107, 2987–2988 CrossRef CAS;
(b) M. Ito, N. Tokitoh and R. Okazaki, Tetrahedron Lett., 1997, 38, 4451–4454 CrossRef CAS;
(c) C. R. P. Millet, D. R. Willcox, G. S. Nichol, C. S. Anstoter and M. J. Ingleson, Angew. Chem., Int. Ed., 2024, e202419094 Search PubMed;
(d) H. F. Bettinger, Organometallics, 2007, 26, 6263–6267 CrossRef CAS;
(e) Y. K. Loh, K. Porteous, M. A. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141(20), 8073–8077 CrossRef CAS PubMed;
(f) H. Dolati, L. Denker, B. Trzaskowski and R. Frank, Angew. Chem., Int. Ed., 2021, 60, 4633–4639 CrossRef CAS PubMed.
- A. Stoy, M. Harterich, R. D. Dewhurst, J. O. C. Jimenez-Halla, P. Endres, M. Eysselein, T. Kupfer, A. Deissenberger, T. Thiess and H. Braunschweig, J. Am. Chem. Soc., 2022, 144, 3376–3380 CrossRef CAS PubMed.
-
(a) E. R. Lory and R. F. Porter, J. Am. Chem. Soc., 1973, 95, 1766–1770 CrossRef CAS;
(b) G. Maier, S. Pfriem, U. Schäfer and R. Matusch, Angew. Chem., Int. Ed., 1978, 17, 520–521 CrossRef;
(c) P. Paetzold, C. V. Plotho, G. Schmid, R. Boese, B. Schrader, D. Bougeard, U. Pfeiffer, R. Gleiter and W. Schüfer, Chem. Ber., 2006, 117, 1089–1102 CrossRef.
-
(a) C. Zhang, X. Liu, J. Wang and Q. Ye, Angew. Chem., Int. Ed., 2022, 61 Search PubMed;
(b) P. Paetzold and R. Truppat, Chem. Ber., 1983, 116, 1531–1539 CrossRef CAS;
(c) T. Thiess, G. Belanger-Chabot, F. Fantuzzi, M. Michel, M. Ernst, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 15480–15486 CrossRef CAS PubMed.
-
(a) D. Prieschl, G. Bélanger-Chabot, X. Guo, M. Dietz, M. Muller, I. Krummenacher, Z. Lin and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 1065–1076 CrossRef CAS PubMed;
(b) T. T. Liu, J. Chen, B. T. Guan, Z. Lin and Z. J. Shi, Chem.–Eur. J., 2023, 29, e202203676 CrossRef CAS PubMed;
(c) R. Guo, X. Zhang, T. Li, Q. Li, D. A. Ruiz, L. L. Liu, C. H. Tung and L. Kong, Chem. Sci., 2022, 13, 2303–2309 RSC;
(d) M. Nutz, B. Borthakur, R. D. Dewhurst, A. Deißenberger, T. Dellermann, M. Schäfer, I. Krummenacher, A. K. Phukan and H. Braunschweig, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef CAS PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Philipp
Dabringhaus
,
Bi Youan E.
Tra
,
Robert J.
Gilliard
Jr
,
Philipp
Dabringhaus
,
Bi Youan E.
Tra
,
Robert J.
Gilliard
Jr