
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChallenges and strategies for probing the composite interface of PEM electrolyzers and fuel cells using operando AP-XPS†
Rebecca Hamlyna,
Johannes Mahlb,
Xueqiang Zhanga,
Damon Englishb,
Terry McAfeeb and
Ethan J. Crumlin *ab
*ab
aChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: ejcrumlin@lbl.gov; Tel: +1 (510) 486 6235
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 29th May 2025
Abstract
Understanding the surface chemistry of electrocatalyst systems under operando conditions is central to revealing the electrocatalytic cell's working mechanisms. Determination of these catalytic processes on a molecular scale and the involved components is fundamental to streamlining material design for energy conversion and storage applications. X-ray photoelectron spectroscopy (XPS) is an established technique used to study the chemical and electronic states of materials. While the surface sensitivity of XPS is typically high, use of tender X-ray energies and technical advancements have allowed for the direct probing of solid–vapor and solid–liquid interfaces. However, protocols and documentation of experimental considerations for operando XPS probing of working electrolyzers and fuel cells remain scarce. Herein, we report an approach for the study of working polymer electrolyte membrane (PEM) electrolysis cells using ambient pressure X-ray photoelectron spectroscopy (AP-XPS). This approach directly probes the composite electrode surface on the membrane electrode assembly (MEA) in 100% relative humidity to establish a meaningful liquid layer for electrocatalysis. We carry out a systematic investigation from the cell constituent components to a fully assembled working operando electrolytic system and establish a method for AP-XPS study of the complex composite MEA, providing recommendations for data acquisition and component analysis.
1 Introduction
Sustainability efforts focused on the storage and conversion of energy in the form of fuels require the continued advancement of electrochemical cells.1–4 The advancement of these systems involves the development of cost-effective, robust materials that efficiently convert electrical and chemical energy.5 Many of the specific design details will continue to evolve based on our understanding of the dynamic chemical processes occurring at the electrolyte/electrode interface. These processes have historically been challenging to elucidate due to the limitations of the surface-sensitive spectroscopic techniques used to probe material surfaces, including interference due to particle inelastic mean free paths (IMPFs) and the need for (ultra-) high-vacuum conditions for operation of certain components.6 However, recent advancements in techniques such as X-ray photoelectron spectroscopy (XPS) have made significant strides in overcoming these challenges.XPS is a quantitative technique that probes the elemental and chemical speciation, and local potential of a sample at its surface.7–10 The incident X-rays cause photoionization of atoms in the material to emit photoelectrons with a measured kinetic energy, which is easily converted to binding energy via the photoelectric relationship
| hν = EB + EK + ΦS, | (1) |
Since its advent as a UHV technique, XPS has advanced into the ambient pressure (AP) regime, enabling the study of surface phenomena in the presence of gases at pressures into the mTorr range.11,12 This development to higher pressures is primarily achieved through the use of differential pumping chambers for rapid reduction of the pressure in the path of the photoelectron, and electrostatic lenses that guide it through a series of small apertures toward the detector.13
More recently, further technological advancement toward even higher pressures into tens and hundreds of Torr have facilitated the study of liquid and solid–liquid interactions. This leap has been facilitated by continued advancement of endstation design, specialized sample environments (such as photoelectron transmissible window cells and liquid jet systems) and in concert with synchrotron radiation sources that provide the photon flux and energies that facilitate to probe the high pressure environment.14–18 Such operando studies offer detailed insight into electrocatalytic systems at the working interface.9,16,19–22
Operando electrolysis and fuel cell studies require a continuous liquid layer on the sample of interest. For aqueous environments at room temperature, 100% relative humidity is achieved at a pressure of 20 Torr. Beamline 9.3.1 at the Advanced Light Source is able to reach these pressure and relative humidity conditions and is optimally designed to access the solid–liquid interface.9,21,23–25 This capability is, in part, enabled by the use of ‘tender’ X-rays (2–6 keV), which comprise photons of much higher energy than those of typical soft X-ray AP-XPS systems (0.2–1.5 keV). Tender X-rays yield photoelectrons with sufficiently high energy to escape the sample, liquid layer, and gaseous environment on their path to the detector.9,24,26 The ‘dip-and-pull’ method is applied for the analysis of bare bulk electrodes, which are submerged into a beaker of electrolyte and partially drawn back up to create a thin, continuous liquid layer in front of the analysis spot (Fig. 1A).9,24
 | ||
| Fig. 1 Schematic comparing two exposed-electrode experimental set-ups for operando electrochemical AP-XPS. (A) A ‘dip and pull’ 3-electrode system for model electrocatalyst studies in liquid electrolyte. (B) The operando cell with an opening for access to the working electrode on a composite catalyst-coated membrane (CCM). This may be used in a 2 or 3 electrode configuration. | ||
Some researchers have developed increasingly complex systems that involve cells with an X-ray permeable “sealing” mechanism to contain the liquid or minimize evaporation from the interface into vacuum. Examples include the use of a Si window thin enough (∼15 nm) for the escape of electrons27 or the deposition of catalyst particles onto layers of graphene supported on holey-SiNx, as pioneered by Kolmakov, et al.28–32 A more thorough review of these cells and systems can be found in the literature.9,17
These advancements play an integral role in our understanding of the reaction mechanisms on model electrocatalysts. In practice, the material components and assembly of electrolyzers and fuel cells tend to have much greater complexity than the electrode catalyst alone.1,33 These systems include use of ionomer and carbon in the catalyst ink, porous transport layers (PTLs) to aid in water flow and current collection, etc., each with their own effects on the overall functionality and efficiency of the cell.33–35 While it is not possible to include every component of a fuel cell stack in a system for XPS analysis, due to the limited escape depths of emitted photoelectrons, advancements in the analysis of applied systems are possible to further our understanding of these complex interfaces.
The preparation of the MEA is an important factor in the performance and local makeup of the electrode–electrolyte interface.35–39 Poor dispersal of components, especially the ionomer and catalyst, can lead to significant heterogeneities across a sample, resulting in ‘hot’ and ‘cold’ areas in terms of electrocatalytic performance.38,40,41 Further, with use, catalyst loss can occur through delamination and dissolution, as well as through the migration of components on and into the membrane.42–45 The degradation mechanisms in these composite systems remain a significant focus in the study of PEM electrolyzers, and this interest has driven the development of electrode systems for XPS analysis of membrane-based fuel cells and electrolyzers at synchrotron facilities.46–50
Here, we continue this effort and report the development of two-electrode cells for the operando XPS investigation of composite MEA electrolysis systems. Our goal is to be able to analyze composite electrodes made using conventional MEA processing techniques for use in commercial systems. We replicate the humid environment of that used in the "dip-and-pull" method (∼20 Torr) with an open window for access to the electrode–electrolyte interface, as shown in Fig. 1B. These composite devices allow us to achieve much higher current densities than the model system counterparts, better emulating those of commercial systems and likely affecting chemical speciation of the catalyst. Importantly, we are able to obtain information from electrode adsorbates, the polymer electrolyte or other binders in addition to the catalyst for a comprehensive understanding of the composite interface and component interactions that drive the chemistry. However, due to the complex nature of these samples and experimental conditions, the set up and data acquisition is challenging. In this work, we offer considerations for the cell design and an experimental approach for effective and representative study of composite MEA electrodes. This work first establishes that we have a full liquid layer at the working electrode surface, and then demonstrates operando functionality and electrochemically active catalyst components. This study presents and discusses findings related to material variability and electrochemical responses across a set of MEAs and their potential impacts on cell design. An integrally important aspect of the process, assessment and alleviation of beam damage, is also addressed, as this is known to affect the polymer electrolyte.51–55 Finally, we offer an experimental strategy for the collection of spectra to minimize this effect and ensure accurate and representative XPS analysis of the composite electrodes. This work serves as a guide for the materials chemistry community to a novel X-ray characterization approach for interrogating a wide range of complex material components, while navigating aspects of heterogeneity, electrical connectivity and effects of the beam.
2 Experimental methods
2.1 Materials and controls
A series of catalyst-coated membranes (CCMs) were used in this work. The CCMs are broadly categorized by their catalyst sources, including a custom commercial set from Fuel Cell Store, Inc. (FCS) and a laboratory-prepared set using Alfa Aesar (AA), Johnson Matthey (JM) and Tanaka Kikinzoku Kogyo (TKK) powder catalysts.The commercial Fuel Cell Store company's catalyst inks were prepared by mixing powder catalyst nanoparticles with DI-water, alcohol, and Nafion dispersion. The ink was deposited onto a Nafion 117 membrane with a loading of 3.0 mg cm−2 in the shape of a circular disk 5–6 mm in diameter.
The laboratory-prepared samples are larger CCMs used for electrochemical testing in fuel cell stacks with an electrode surface area of 2.5 cm2. Inks were prepared by mixing ionomer (Nafion D521), Vulcan carbon and powder catalyst in an aqueous solution with propanol (ionomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst ratio 0.116:1). Deposition was conducted either by spin coating or by spray coating with a sonicating tip for good component dispersion (SonoTek, ExactaCoat). Target loadings for the anode (Ir or IrOx) and cathode (PtC) were 0.40 mg cm−2 and 0.10 mg cm−2, respectively, and were confirmed via X-ray fluorescence spectroscopy.
:catalyst ratio 0.116:1). Deposition was conducted either by spin coating or by spray coating with a sonicating tip for good component dispersion (SonoTek, ExactaCoat). Target loadings for the anode (Ir or IrOx) and cathode (PtC) were 0.40 mg cm−2 and 0.10 mg cm−2, respectively, and were confirmed via X-ray fluorescence spectroscopy.
Control samples were prepared to assist in identification of peaks and relative percentages of components. The controls consisted of single materials (Ir polycrystalline foil, TKK IrOx powder, Nafion ionomer D521, Vulcan Carbon) or mixtures that were drop cast onto a stainless steel substrate.
2.2 Cell design and assembly
Two cells were constructed, both of which were composed of a metal front and back plate. A rendering of the front plate design is shown in Fig. 3D. The back plate serves as both an electrical conductor and the liquid flow delivery system, designed with a 1 mm relief channel for liquid flow across the back electrode. The direction of liquid flow is from the bottom to the top to encourage any bubbles formed during experimentation to flow up and out of the cell. A cut Viton sheet is used as a gasket to seal the cell and prevent leakages in vacuum.Cell #1 (open circle cell) is composed of steel and Pt-coated stainless steel to minimize corrosion under electrochemical conditions. The front plate opening is a 5 mm diameter circle (Fig. 3D). A small curved disk of stainless steel mesh was placed on top of the water channel and under the CCM to maintain electrical connectivity between the Pt cathode and the back plate in the event that the membrane stretches from any flow pressure.
Cell #2 (slit cell) is composed of titanium with a front plate opening consisting of 5 horizontal slits, each 30 mm long and 1 mm high. The flow channels at the back plate align with the front plate openings. The membrane facing side of the front plate is covered in insulating tape up to 0.5 mm from the slit openings to maximize the amount of electrochemical signal coming from the area accessible via XPS. Because of the insulating tape, absolute current values were decreased to 27% of the non-taped values. Calculation of current densities were multiplied by this factor to account for unused area. A rendering of this cell is included in Fig. S2.†
2.3 AP-XPS at ALS
Experiments were conducted at the tender X-ray AP-XPS beamline 9.3.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory.24 The beamline is equipped with a bending magnet for tunable X-rays within the tender range (∼2–6 keV) and a Si (111) double crystal monochromator. The analyzer is a Scienta Omicron R4000 HiPP-2.24 The chamber is also equipped with a Dyson residual gas analyzer (RGA) for mass spectrometry. For hydrated conditions, the chamber contained a beaker of degassed DI H2O to maintain 17–20 Torr vapor pressure using an analyzer cone aperture of 0.3 mm.2.4 Experimental procedure
In the present study, a two-electrode system was used to investigate the working mechanism of IrOx nanoparticle-catalyzed water splitting. The surface physicochemical properties of the anode side, before and during the oxygen evolution reaction (OER) were probed by AP-XPS under operando conditions. During measurement, fresh DI water flowed continuously through the cathode side of the CCMs at 3.0 mL min−1, and the electrochemistry was tested on-line. Survey spectra in addition to core-level photoemission of the F 1s, C 1s, O 1s, Ir 4f (and 5p), Ir 3d5/2, Pt 4f, Pt 3d5/2, S 1s and valence band (VB) were recorded at voltages between 0.0 and 2.0 V using a Biologic SP300 potentiostat. Under operational conditions, the anode was grounded to the spectrometer unless otherwise indicated.For data processing, snapshot spectra were subtracted by the detector background, and pixels converted to eV using a calibration curve. All spectra were then analyzed using the CasaXPS software package and Origin Pro.56 The regions were calibrated to the Fermi level or to the adventitious C 1s peak in the case of the control samples if there was no catalyst metal present.
3 Results and discussion
Section A of this results section establishes a functional operando system with a highlight of key attributes that help identify a working experimental setup. Sections 3.2–3.4 cover commonly encountered issues of variability, disconnectivity and beam damage, respectively. We show examples of these phenomena or a practical strategy to test for them and when applicable, offer an approach to resolve or minimize these effects.3.1 Establishment of a functional operando system
In this section, we establish a functional operando system for the study of membrane electrode assemblies. Our operando cell is a two-electrode system, in which water flow is delivered to the back of the membrane with the front side exposed for direct access to the MEA for incident X-rays and ejected electrons (Fig. 1B). A humidified environment in the chamber keeps the exposed area at the front hydrated. This is achieved by the use of a water bulb or reservoir, with a maintained ∼20 Torr water pressure, corresponding to 100% humidity at room temperature.Because hydration is critical to the proper functioning of any electrolyzer, the establishment of a full liquid layer on the surface of our catalyst-coated membrane is a necessary first step before operando study. To this end, we collected spectra spanning the full range of water pressures. Beginning with a dry MEA assembled in our operando cell, water was introduced up to full humidity and finally supplemented with water flow from the cathode side (Fig. S1†).
Calculations of the IMFPs of the emitted photoelectrons through various components and a comparison of relative signal intensities allow for estimating the water thickness on our catalyst layer.57,58 The process for these calculations is detailed, and results are summarized in Tables S1–S3.† Even before the start of water flow, the humid environment creates a full liquid layer of 5–6 nm.
With the establishment of a fully hydrated CCM, we can proceed with an electrochemical investigation. Fig. 2 displays the catalyst spectra with the application of potential via chronoamperometry across the electrolyzer cell. The Ir 4f spectrum at OCV has a line shape characteristic of metallic Ir, in which the 4f7/2 peak intensity is higher than that of the 4f5/2. As the potential increases from OCV to 2.0 V, the line shape changes significantly as the iridium is oxidized. Notably, the 4f5/2 peak becomes taller than the 4f7/2 peak already by 1.0 V, when we would expect a transition of most species to a 4+ state, and there is continued broadening of the higher binding energy side of the peaks at increasing potentials. Ultimately, at 2.0 V, a feature at ∼67 eV appears, indicative of a valence state higher than 4+, and the 4f7/2 peak position increases in binding energy by approximately 0.7 eV.
 | ||
| Fig. 2 (Left) Data from the working 2-electrode cell of a TKK MEA (0.4 mg cm−2 catalyst loading) with increasing anodic conditions (OCV – 2.0 V) from left to right. Top row is the operando AP-XPS Ir 4f spectra for 5 polarization conditions, taken in standard collection mode at different locations on the sample freshly exposed to the X-ray. The OCV data envelope (blue) is included in the other 4 anodic plots for comparison. The middle panel shows the corresponding data for the chronoamperometry applied voltage and measured current density. The bottom panel shows the partial pressure gas traces from the RGA. (Right) Images of the operando slit cell (cell #2) and a standard MEA. | ||
The third panel of the Fig. 2 displays the RGA gas trace spectra. There is a concomitant increase in the partial pressure of oxygen (32 amu) with potentials above 1.5 V, indicating successful oxygen evolution. The hydrogen and nitrogen spectra are included for reference. It should be noted that the hydrogen background trace is highly susceptible to fluctuations and is affected by any movement of the sample in and out of the analyzer cone. Ultimately, these data demonstrate the functional electrolysis of water and oxygen evolution with our two-electrode operando cell. Representative electrochemical data for the results shown in Fig. 2 are included in Fig. S3,† which resembles systems previously described.59–62
3.2 Variability and cell design considerations
Early studies began with a front plate geometry that had a single large, circular opening for access to the electrode interface (Fig. 3). Conveniently, this offered a large area to probe multiple locations on the sample, as is necessary to obtain representative results and assess beam damage. However, this posed some issues that remain an important consideration with regard to the nature of the sample and cell design choices. | ||
| Fig. 3 (A–C) Schematics depicting spot-to-spot movement across the exposed composite electrode in the circle cell #1 and corresponding Ir 3d5/2 spectra at various locations. Panels A and B depict spatial consistency and variability, respectively, of constituent signals from the catalyst ink. Panel C uses color shading from blue to green to depict variability in catalyst response to anodic voltage. Panel D shows renderings of the front plate geometry between the two cell designs, with a membrane bulging out of the circle cell #1. | ||
The need to move along the sample can introduce issues with variability, complicating data interpretation. The structure of the Nafion membrane alone exhibits microscale variability. The sulfonic acid head group can segregate and will often create channels for proton migration to form a semi-crystalline structure.63 Fresh and used or chemically degraded membranes will have component discrepancies at the surface compared to the bulk as well as across domains on the scale of tens of microns, as shown by microscopy and spectroscopy.64,65 This is the substrate upon which the catalyst ink is deposited for MEA fabrication. Depending on relative component concentrations and the catalyst ink deposition method, samples may exhibit varying degrees of spatial heterogeneity.41 Further heterogeneity can be introduced with heavy use through degradation phenomena such as migration, sintering and delamination.43
The probe area for our beam is approximately 250 (height) × 300 (width) μm, which is considerably larger than typical catalyst ink aggregates at hundreds of nanometers or even larger agglomerates that can reach micron scale.35,38,39 Thus, we generally expect that this probe size would average over such a large area that the signal would be fairly consistent across the surface. Such is the case for most samples encountered in our work thus far. However, we found that a subset of samples exhibit heterogeneity across the surface on a much larger scale. Fig. 3A and B depict exposed electrode areas with consistent and variable electrode components, respectively. An example of catalyst signal is shown spatially to the left with the extent of blue color, with the respective Ir 3d5/2 spectra to the right for three sampled locations. This effect can be extended to all components in the catalyst ink. Thus, at the very least we suggest an assessment of spatial heterogeneity and sampling in triplicate for conventional scanning modes.
Variability can also appear in the response to applied potential. While the data in Fig. 2 show continually oxidizing iridium with increasing potential, this is not necessarily the case for each spot sampled on all MEAs in all cell types. In keeping with the spatial variability seen in baseline components, a notable finding from our studies is that we can also see variability in catalyst response to applied potential as data is collected from spot to spot. This is exemplified in Fig. 3C, where spectra obtained from a TKK sample in two different spots exhibit different behavior under the same conditions, indicated in the schematic by the blue–green color gradation. The spectra were obtained while under an applied anodic potential of 1.5 V, where we would expect to see significant oxidation of the catalyst components. This is indeed the case for spot B (in green), but not so for spot A (in blue), which is notably metallic in contrast.
In the search for a reason for this discrepancy between sampling locations, we made attempts to probe several aspects concerning electrical conductivity and experienced potential of the catalyst at a given location. We began with assessing a given sampling location's distance from the current collector. As we have an open window to the electrode, it stands to reason that planar resistance across this window could contribute to a decrease in the actual potential felt by any given spot. We found that being closer to the edge can help with the effective delivery of potential and shuttling of electrons to the current collector, but this is not a consistent remedy to eliminate all discrepancies in catalyst response.
A final notable aspect of the front plate geometry is the tendency for membrane swelling over time. With elevated pressure at the cathode and flow fields inside of the cell, especially during hydrogen evolution, and lower relative pressure at the anode/in the chamber, the MEA would often bulge out of the plane of the front plate (Fig. 3D). This results in shadowing of some of the sample area from the shallow incidence angle of the beam and may result in mechanical instability. In an effort to address this issue and decrease the distance from probed location to current collector as described above, we redesigned the cell with a significant change to the front plate, as shown in Fig. 3D. Instead of one circular opening, with variable distance from the current collector, the new front plate features multiple slits. These slits allow for a probing location that is a constant distance from the plate, which can help to create a more uniform effective potential. A slit height of 1 mm also keeps the surface flat, ensuring better mechanical stability of the membrane.
3.3 Assessment of disconnected species
To effectively evaluate the variability in catalyst response to the applied potential, we took advantage of XPS sensitivity to potential to determine whether XPS would be able to help identify populations of particles that were electrically disconnected.21,23,66,67 In a typical experimental set up, the working electrode is grounded to the spectrometer so that the conductive electrode species will remain pinned to their binding energy location, and any changes in location and shape correspond to electrochemical changes at the sample surface. For this connection test, the working electrode was not grounded to the spectrometer and was only connected to the potentiostat.A control experiment was conducted using Au foils (Fig. 4). One foil (C, connected; shown in green) was connected to the potentiostat directly, and the second foil (D, disconnected; shown in orange) was floating freely. Seven Au 4f spectra are shown from dry to humid conditions with green spectra pertaining to the connected foil and orange spectra pertaining to the floating foil. In spectrum g1, the potentiostat is at 0.0 V v. ground (spectrometer), and the 4f7/2 peak is in its typical binding energy location at 84.2 eV. Upon application of −2.0 V in spectrum 2, the doublet shifts to the right by 2 eV. Spectrum g3 shows this same condition for the disconnected foil, which has undergone significant charging from the beam exposure, shifting the doublet far to the left.68 Once we move into humid conditions, this charging of the disconnected foil is largely but not entirely mitigated69 so that the disconnected spectra are near the 0.0 V binding energy location (spectra g5 and g7). The connected foil spectra shown in green consistently shift in binding energy at a rate of 1:1 with the application potential.
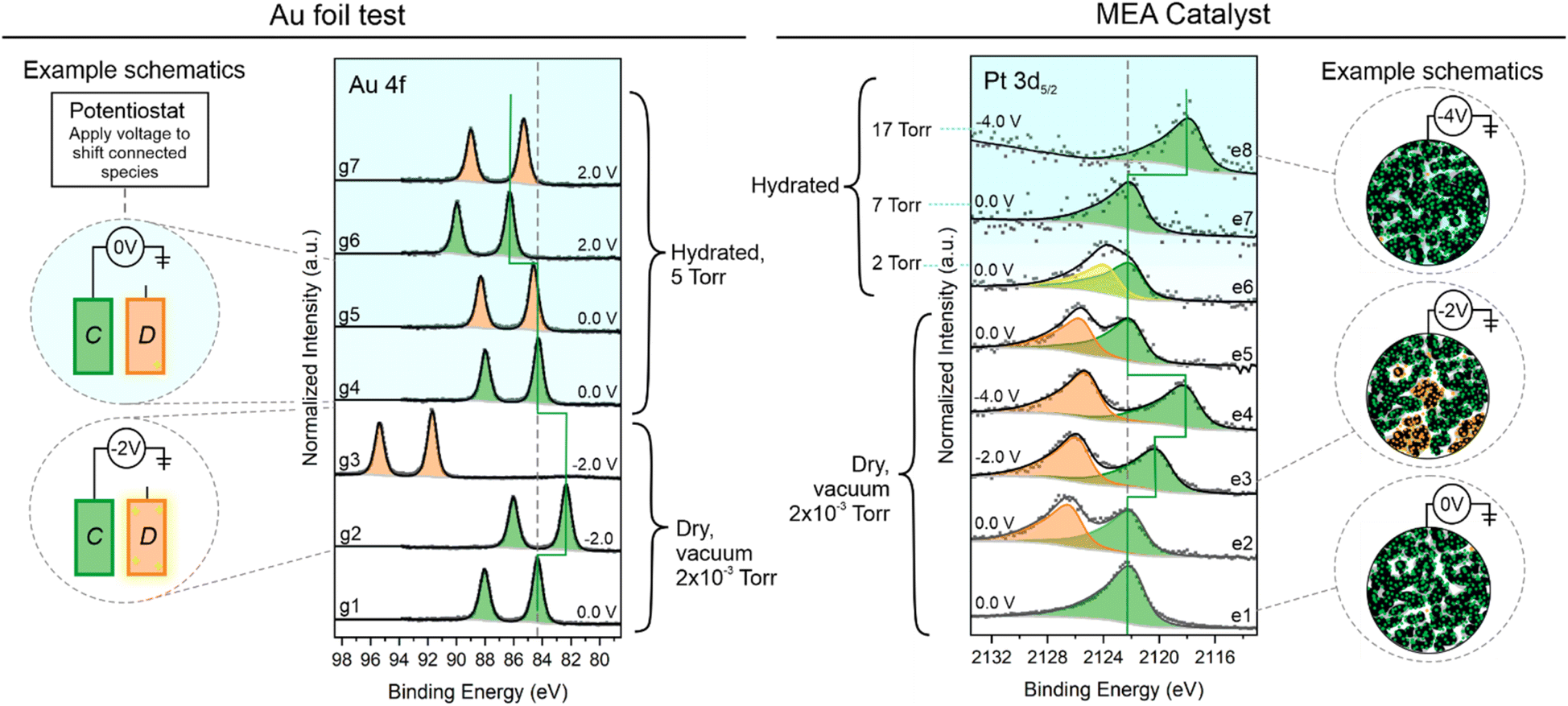 | ||
| Fig. 4 Particle connectivity trial as a function of potential and condition. Left: Au 4f spectra for an electrically connected (C) and disconnected (D) Au foil. Right: Pt 3d5/2 XPS results for trials on a Pt-based MEA. Example schematics of the electrical setup are to the side of the spectral regions for some conditions. | ||
We applied the same principle to the working electrode on the MEA, removing the bias across the cell by disconnecting the counter electrode from the potentiostat and applying a bias to the working electrode versus ground. In this way, we can follow the speciation in the Pt 3d5/2 region (or any core level attributed to an electrode expected to be conductive). In spectrum e1, there is a characteristic single asymmetric peak probing a connected area of the sample. When we move to another location on the sample, in spectrum e2, there is a second peak in orange at higher binding energy, pertaining to disconnected catalyst particles that are charging from the beam. In spectra e3 and e4, the green connected species are shifted away from the orange peak with applied potentials of −2.0 and −4.0 V, respectively. Once we begin to introduce water to the environment, from spectrum e5–e7, the charging of the disconnected species is healed, with the two peaks merging into one. Spectrum e5 begins in vacuum, spectrum e6 is ∼2 Torr H2O, and e7 is 7 Torr H2O, eliminating charging before reaching 100% relative humidity. Spectrum e8, fully hydrated at 17 Torr, exhibits the final test for connectivity with an application potential of −4.0 V, and we see a corresponding shift in the working electrode spectrum. The absence of peak splitting in spectrum e8 indicates that the introduction of water into the system has eliminated the disconnected species. This could be either due to swelling of the membrane improving the mechanical contact or spontaneous electrochemical polarization at the Pt surface from the surrounding electrolyte that can modulate metal on nonconductive supports.70
This is an important test to conduct for different experimental setups, as locally disconnected species will not contribute to the cell-scale electrochemical results, which can hinder analytical data interpretation. Yet more importantly, a splitting of the spectra, as shown in Pt 3d5/2 spectrum 2 or 6, could result in significant misinterpretation of the data, leading to erroneous conclusions. In some systems, this may serve as a helpful diagnostic tool for estimation of insulated components and for understanding chemical and mechanical breakdown of the system over time.
3.4 Beam effects
An important consideration in spectroscopy is the effect of exposure to the X-ray beam. The spectra on the left side of Fig. 5 show the effects of beam exposure in two components of the composite electrode over the course of several minutes. The beam stability of the catalyst was highly variable between different sample types. Some of the catalyst signals (FCS, TKK) were stable over the course of hours of beam exposure (as was the case for data shown in Fig. 2), while others (JM, AA) were much more sensitive. We encountered rapid loss of signal for some catalysts, an example of which can be seen in the left hand Ir 4f spectra.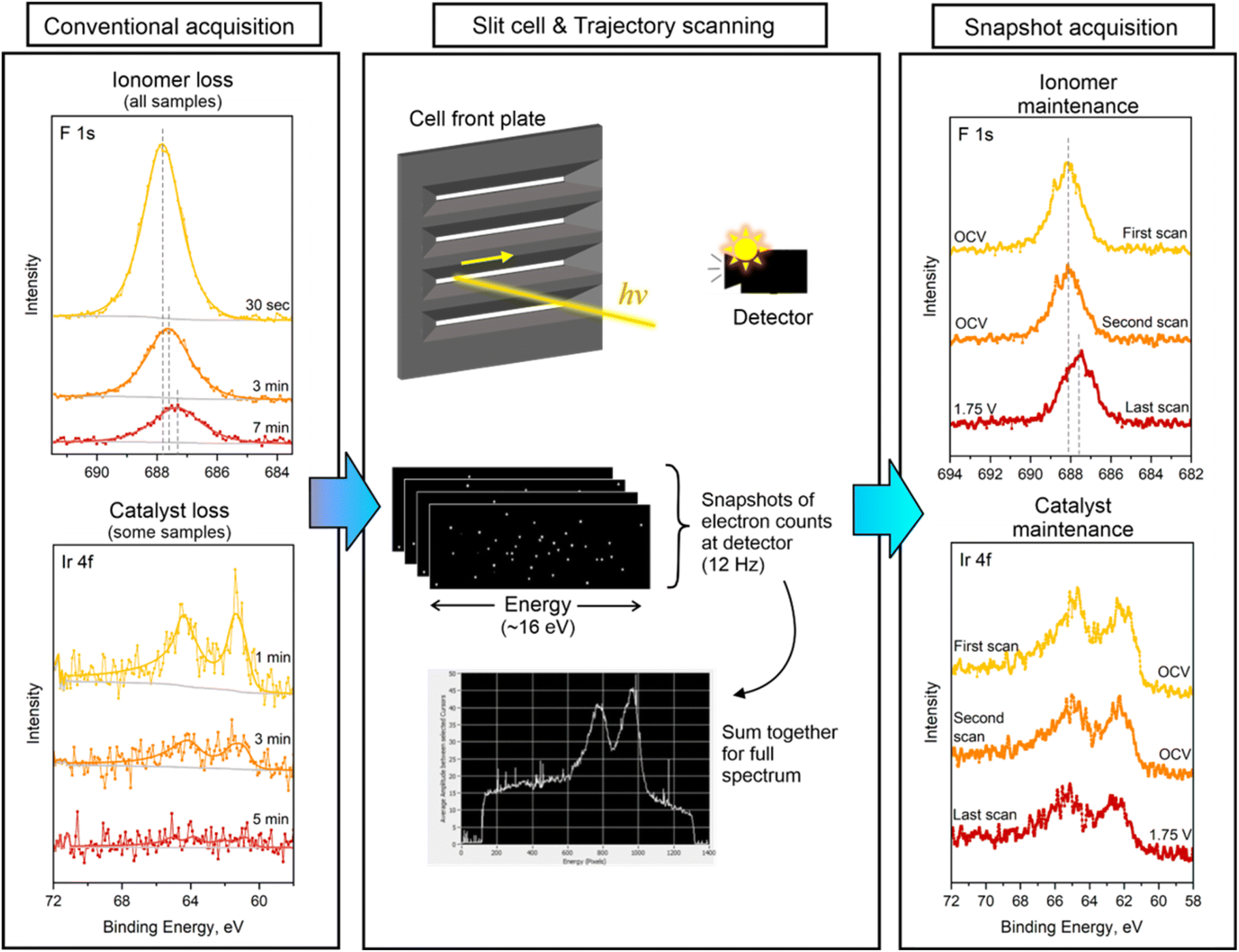 | ||
| Fig. 5 Beam effects and mitigation via trajectory scanning and snapshot collection mode. Left hand panel: conventional data acquisition leads to loss and a change in the electrode signal. Center: the cell #2 slit design keeps the sample flat and allows for use of trajectory scanning, while snapshot acquisition mode collects low-resolution spectra at a rate of 12 Hz, which are summed for higher quality spectra. Right panel: resulting stable signal from electrode components. | ||
Studies of the effect of X-ray beams on Nafion have indicated that it is often affected by beam damage, depending on the photon source.51–55 It is expected that with the brightness of a synchrotron, this is more likely. Indeed, significant damage is reliably seen in the polymer components of the catalyst ink for all samples. The C–Fx backbone of the ionomer is susceptible to beam-induced cleavage, and while we detected no trace of volatilized compounds in the mass spectra, ionomer features in the C 1s and F 1s regions exhibit large signal decay over time. Importantly, the binding energy of some peaks can shift as damage preferentially occurs to select species. This is seen in the F 1s region over the course of a few minutes for conventional data acquisition. As typical collection times for this mode take between 15 and 60 minutes for a given condition, this can easily result in incorrect assignment of species and hinder our ability to understand the ionomer–catalyst interaction in these electrodes. Finally, because the ionomer serves as the charge-carrying electrolyte essential to the operation of the electrolyzer, it is important to minimize exposure to the beam and (continuously) move the sample during data collection.
In general, we recommend starting analysis with a check for beam damage of each component individually and then in series on one spot to compare regions. For all spectral sets, we begin with the most sensitive regions first before proceeding to regions exhibiting greater beam stability. For samples with a stable catalyst signal, our collection order will begin with the polymer components (F 1s, C 1s) for spot-to-spot collection and will be repeated as necessary to evaluate changes evolved over the course of collection. We record both the exposure time for each scan and keep a map of probing locations on the sample for each study.
In addition to mindful collection of data with conventional acquisition modes, we have developed a second method of collection at beamline 9.3.1, colloquially referred to as "snapshot mode", that addresses many of the issues posed with beam damage and spatial variability. In the standard mode of data acquisition, the analyzer is used to sweep across the electron kinetic energy range of interest, so that each channel of the detector is used to collect electrons at a given kinetic energy for a period of time. These individual spectra are shifted accordingly before being averaged into the final spectrum. This process typically takes tens of seconds to several minutes (or longer) for a given region. In snapshot mode, the analyzer is fixed to an electron kinetic energy at the center of the energy region to be acquired, and the spread of electrons in their flight through the lens system and analyzer is collected across the multichannel detector, so that each channel becomes its own energy range.71 For our system, a pass energy of 200 eV gives an energy range across our detector of approximately 16 eV. With a camera sampling rate of 12 Hz, we are able to capture sub-100 ms spectra. By coupling the fast snapshot acquisition with sample movement in the established trajectory, resulting snapshot frames can be summed to give a spectrum that is a true average of the whole area probed, with very little beam exposure in any given location on the sample (central panel, Fig. 5). This gives us the ability to obtain more representative data and scan the same trajectory multiple times for good comparison between conditions. A typical trajectory comprised of one pass along the slit will have an estimated area of 4.5 mm2 (0.25 mm beam spot size × 18.0 mm trajectory length), but this can be doubled when using a serpentine path to approximately 9 mm2.
The right most panel of Fig. 5 shows the multiple spectra obtained for an experiment using snapshot mode on the same Alfa Aesar sample that displayed significant beam damage in the left-hand panels under conventional scanning mode. The first two spectra (yellow to orange) are in sequence at the same condition, showing no change in intensity or peak energy location. Only for the last scan in red, taken at the end of the experiment at 1.75 V, do we see a shift in the binding energy and peak shape for the fluorine and iridium spectra due to electrochemical changes rather than damage. The total time of exposure for a given location on the sample for this full experiment was less than 30 seconds, and the signal intensities from first to last scan did not suffer significant losses or beam-induced change.
While the snapshot-trajectory acquisition method assists in the collection of data averaged over a much larger area, there may be areas in the trajectory path that have variable signal or speciation of interest. In the spectrum processing, we are able to batch process snapshot frames, and thus isolate the spectra from a subset of the trajectory area if desired. This can be done with other core level regions from the same area.
4 Conclusions
In summary, this paper demonstrates an approach for the fastidious collection of operando XPS data on a fully composite PEM electrolyzer. We present factors that may introduce errors in the interpretation of electrolyzer and fuel cell complex composite electrodes with XPS analysis and provide suggestions for error mitigation. These factors include hydration of the MEA (establishment of operando conditions), beam exposure effects, artifacts from sample preparation, spatial variability, cell design, and electrical connectivity of electrode constituents. Researchers who conduct studies of these complex material composites must take care to obtain representative data and to avoid misinterpretation of the catalyst-coated membrane composition and response to applied potential. Fig. 6 summarizes these factors, accompanying considerations and recommendations, which help to inform and affect one another. | ||
| Fig. 6 Summary diagram of important considerations and recommendations for aspects affecting the successful acquisition of operando AP-XPS data on composite PEM electrolysis and fuel cells. | ||
First, the sample preparation and use history must be thoroughly detailed, as it pertains to the catalyst ink uniformity and the state of the membrane and electrodes. The cell design is an important consideration for several factors, including distance from current collectors/use of graphene, ease of movement across the surface during collection, and the effects of an open front plate where a PTL or GDL would be.
Effects of beam exposure are always relevant, especially if the polymer surrounding the catalyst is damaged, disabling ion transport and affecting the catalyst active state. A simple test for fully disconnected species is facile with XPS systems, leveraging the potential sensitivity of the analyzer, and can help mitigate serious misinterpretations of spectra in the event that these species exist in a given sample environment. We also recommend a combination of spot-to-spot collection (in triplicate at a minimum if any variance is found) and rastering trajectories with snapshot acquisition if available. Now that we have this new methodology, future work will be able to systematically evaluate the operando catalyst response to potential and also obtain direct spectroscopic feedback regarding the synthesis and manufacturing outcomes of the composite catalyst ink. This can include investigations into several tuneable aspects of the composite electrode: methods for catalyst synthesis, resulting particle sizes, phases and morphology, ink formulations and loading, methods for MEA fabrication, treatment, aging and so on. We hope this work will inspire and enable such pursuits by the broader research community. Continued establishment of rigorous protocols will improve characterization efforts, ensure the reliability of reported data, and support efforts toward improved composite electrochemical systems.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written with contributions from all authors. R. H. planned and executed the operando AP-XPS experiments, analysed the data, led the design for the operando slit cell, assisted in development of software for snapshot data analysis and drafted the manuscript. J. M. led development of software for the analysis of snapshot data. X. Z. developed the circle cell and contributed to early PEM studies that helped to form the basis for this work. T. M. assisted in development of operando cells and offered technical support. D. E. developed the software for the acquisition of snapshot data and trajectory capabilities. E. J. C. conceived, supervised, and planned the studies and the operando cell designs. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts.Acknowledgements
Funding provided by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office through the H2NEW Consortium Award No. DE-AC02-05CH11231. J. M. and E. J. C. were partially supported by an Early Career Award in the Condensed Phase and Interfacial Molecular Science Program, in the Chemical Sciences Geosciences and Biosciences Division of the Office of Basic Energy Sciences of the U.S. Department of Energy, under Contract No. DE-AC02-05CH11231. E. J. C was partially supported by the Condensed Phase and Interfacial Molecular Science Program (CPIMS) of the U.S. Department of Energy under contract No. DE-AC02-0511231. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.References
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- Q. Abbas, M. Mirzaeian, M. R. C. Hunt, P. Hall and R. Raza, Energies, 2020, 13, 5847 CrossRef CAS.
- B. S. Pivovar, M. F. Ruth, D. J. Myers and H. N. Dinh, Electrochem. Soc. Interface, 2021, 30, 61–65 CrossRef CAS.
- A. Badgett, M. Ruth and B. Pivovar, Economic Considerations for Hydrogen Production with a Focus on Polymer Electrolyte Membrane Electrolysis, Elsevier B. V., 2021 Search PubMed.
- A. Jablonski and C. J. Powell, J. Electron Spectrosc. Relat. Phenom., 1999, 100, 137–160 CrossRef CAS.
- K. Siegbahn and K. Vetenskaps-societeten i Uppsala, ESCA; Atomic, Molecular and Solid State Structure Studied by Means of Electron Spectroscopy, Almqvist & Wiksells, Uppsala, 1967 Search PubMed.
- S. Hofmann, Auger- and X-Ray Photoelectron Spectroscopy in Materials Science, 2013, vol. 148 Search PubMed.
- E. J. Crumlin, Z. Liu, H. Bluhm, W. Yang, J. Guo and Z. Hussain, J. Electron Spectrosc. Relat. Phenom., 2015, 200, 264–273 CrossRef CAS.
- D. E. Starr, M. Favaro, F. F. Abdi, H. Bluhm, E. J. Crumlin and R. van de Krol, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 106–115 CrossRef CAS.
- F. Reinert and S. Hüfner, New J. Phys., 2005, 7, 97 CrossRef.
- C. S. Fadley, J. Electron Spectrosc. Relat. Phenom., 2010, 178, 2–32 CrossRef.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872 CrossRef CAS.
- S. Kaya, H. Ogasawara, L. Å. Näslund, J. O. Forsell, H. S. Casalongue, D. J. Miller and A. Nilsson, Catal. Today, 2013, 205, 101–105 CrossRef CAS.
- H. Ogasawara, S. Kaya and A. Nilsson, Top. Catal., 2016, 59, 439–447 CrossRef CAS.
- J. J. Velasco Vélez, D. Bernsmeier, T. E. Jones, P. Zeller, E. Carbonio, C.-H. Chuang, L. J. Falling, V. Streibel, R. V. Mom, A. Hammud, M. Hävecker, R. Arrigo, E. Stotz, T. Lunkenbein, A. Knop-Gericke, R. Krähnert and R. Schlögl, Faraday Discuss., 2022, 236, 103–125 RSC.
- J.-J. Velasco-Vélez, L. J. Falling, D. Bernsmeier, M. J. Sear, P. C. J. Clark, T.-S. Chan, E. Stotz, M. Hävecker, R. Kraehnert, A. Knop-Gericke, C.-H. Chuang, D. E. Starr, M. Favaro and R. V Mom, J. Phys. D Appl. Phys., 2021, 54, 124003 CrossRef.
- M. Blum, L. Weinhardt, O. Fuchs, M. Bär, Y. Zhang, M. Weigand, S. Krause, S. Pookpanratana, T. Hofmann, W. Yang, J. D. Denlinger, E. Umbach and C. Heske, Rev. Sci. Instrum., 2009, 80, 123102 CrossRef CAS PubMed.
- K. A. Stoerzinger, W. T. Hong, E. J. Crumlin, H. Bluhm and Y. Shao-Horn, Acc. Chem. Res., 2015, 48, 2976–2983 CrossRef CAS PubMed.
- V. A. Saveleva and E. R. Savinova, Curr. Opin. Electrochem., 2019, 17, 79–89 CrossRef CAS.
- E. J. Crumlin, H. Bluhm and Z. Liu, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 84–92 CrossRef CAS.
- V. Shutthanandan, M. Nandasiri, J. Zheng, M. H. Engelhard, W. Xu, S. Thevuthasan and V. Murugesan, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 2–10 CrossRef CAS.
- M. Favaro, B. Jeong, P. N. Ross, J. Yano, Z. Hussain, Z. Liu and E. J. Crumlin, Nat. Commun., 2016, 7, 12695 CrossRef CAS PubMed.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- D. E. Starr, Z. Liu, M. Hävecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- D. E. Starr, H. Bluhm, Z. Liu, A. Knop-gericke and M. Hävecker, In Situ Charact Hetergeneous Catal, 2013, pp. 315–343 Search PubMed.
- T. Masuda, H. Yoshikawa, H. Noguchi, T. Kawasaki, M. Kobata, K. Kobayashi and K. Uosaki, Appl. Phys. Lett., 2013, 103, 111605 CrossRef.
- A. Kolmakov, D. A. Dikin, L. J. Cote, J. Huang, M. K. Abyaneh, M. Amati, L. Gregoratti, S. Günther and M. Kiskinova, Nat. Nanotechnol., 2011, 6, 651–657 CrossRef CAS PubMed.
- R. S. Weatherup, B. Eren, Y. Hao, H. Bluhm and M. B. Salmeron, J. Phys. Chem. Lett., 2016, 7, 1622–1627 CrossRef CAS PubMed.
- J. J. Velasco-Velez, V. Pfeifer, M. Hävecker, R. S. Weatherup, R. Arrigo, C. H. Chuang, E. Stotz, G. Weinberg, M. Salmeron, R. Schlögl and A. Knop-Gericke, Angew. Chem., Int. Ed., 2015, 54, 14554–14558 CrossRef CAS PubMed.
- L. J. Falling, R. V. Mom, L. E. Sandoval Diaz, S. Nakhaie, E. Stotz, D. Ivanov, M. Hävecker, T. Lunkenbein, A. Knop-Gericke, R. Schlögl and J. J. Velasco-Vélez, ACS Appl. Mater. Interfaces, 2020, 12, 37680–37692 CrossRef CAS PubMed.
- R. Mom, L. Frevel, J. J. Velasco-Vélez, M. Plodinec, A. Knop-gericke and R. Schlögl, J. Am. Chem. Soc., 2019, 141, 1–13 CrossRef PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. Lopez-Haro, L. Guétaz, T. Printemps, A. Morin, S. Escribano, P. H. Jouneau, P. Bayle-Guillemaud, F. Chandezon and G. Gebel, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- C. Qiu, Z. Xu, F. Y. Chen and H. Wang, ACS Catal., 2024, 14, 921–954 CrossRef CAS.
- A. Orfanidi, P. J. Rheinländer, N. Schulte and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F1254–F1263 CrossRef CAS.
- S. Takahashi, T. Mashio, N. Horibe, K. Akizuki and A. Ohma, ChemElectroChem, 2015, 2, 1560–1567 CrossRef CAS.
- S. Khandavalli, J. H. Park, N. N. Kariuki, D. J. Myers, J. J. Stickel, K. Hurst, K. C. Neyerlin, M. Ulsh and S. A. Mauger, ACS Appl. Mater. Interfaces, 2018, 10, 43610–43622 CrossRef CAS PubMed.
- N. N. Kariuki, A. T. Haug, J. H. Park, M. J. Lindell and D. J. Myers, J. Electrochem. Soc., 2022, 169, 104502 CrossRef CAS.
- Y. Komoda, K. Okabayashi, H. Nishimura, M. Hiromitsu, T. Oboshi and H. Usui, J. Power Sources, 2009, 193, 488–494 CrossRef CAS.
- S. M. Alia, K. S. Reeves, J. S. Baxter and D. A. Cullen, J. Electrochem. Soc., 2020, 167, 144512 CrossRef CAS.
- L. Hu, T. Van Cleve, H. Yu, J. H. Park, N. Kariuki, A. J. Kropf, R. Mukundan, D. A. Cullen, D. J. Myers and K. C. Neyerlin, J. Power Sources, 2023, 556, 232490 CrossRef CAS.
- S. M. Alia, K. S. Reeves, H. Yu, J. Park, N. Kariuki, A. J. Kropf, D. J. Myers and D. A. Cullen, J. Electrochem. Soc., 2022, 169, 054517 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- Y. Takagi, H. Wang, Y. Uemura, E. Ikenaga, O. Sekizawa, T. Uruga, H. Ohashi, Y. Senba, H. Yumoto, H. Yamazaki, S. Goto, M. Tada, Y. Iwasawa and T. Yokoyama, Appl. Phys. Lett., 2014, 105, 131602 CrossRef.
- L. Yu, Y. Takagi, T. Nakamura, O. Sekizawa, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa, G. Samjeské and T. Yokoyama, Phys. Chem. Chem. Phys., 2017, 19, 30798–30803 RSC.
- T. Nakamura, Y. Takagi, S. Chaveanghong, T. Uruga, M. Tada, Y. Iwasawa and T. Yokoyama, J. Phys. Chem. C, 2020, 124, 1–6 Search PubMed.
- L. Yu, Y. Takagi, T. Nakamura, T. Sakata, T. Uruga, M. Tada, Y. Iwasawa, S. Masaoka and T. Yokoyama, J. Phys. Chem. C, 2019, 123, 603–611 CrossRef CAS.
- Y. Takagi, H. Wang, Y. Uemura, T. Nakamura, L. Yu, O. Sekizawa, T. Uruga, M. Tada, G. Samjeské, Y. Iwasawa and T. Yokoyama, Phys. Chem. Chem. Phys., 2017, 19, 6013–6021 RSC.
- M. C. Militello and S. W. Gaarenstroom, Surf. Sci. Spectra, 2021, 10, 117 CrossRef.
- M. Schulze, M. Lorenz, N. Wagner and E. Gülzow, Fresenius. J. Anal. Chem., 1999, 365, 106–113 CrossRef CAS.
- C. Chen, G. Levitin, D. W. Hess and T. F. Fuller, J. Power Sources, 2007, 169, 288–295 CrossRef CAS.
- D. K. Paul, J. B. Giorgi and K. Karan, J. Electrochem. Soc., 2013, 160, F464–F469 CrossRef CAS.
- M. J. Dzara, K. Artyushkova, J. Foster, H. Eskandari, Y. Chen, S. A. Mauger, P. Atanassov, K. Karan and S. Pylypenko, J. Phys. Chem. C, 2024, 128, 8467–8482 CrossRef CAS PubMed.
- N. Fairley, V. Fernandez, M. Richard-plouet, C. Guillot-deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1993, 20, 77–89 CrossRef CAS.
- H. Shinotsuka, B. Da, S. Tanuma, H. Yoshikawa, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2017, 49, 238–252 CrossRef CAS PubMed.
- S. Siracusano, V. Baglio, S. A. Grigoriev, L. Merlo, V. N. Fateev and A. S. Aricò, J. Power Sources, 2017, 366, 105–114 CrossRef CAS.
- J. J. Velasco-Vélez, E. A. Carbonio, C. H. Chuang, C. J. Hsu, J. F. Lee, R. Arrigo, M. Hävecker, R. Wang, M. Plodinec, F. R. Wang, A. Centeno, A. Zurutuza, L. J. Falling, R. V. Mom, S. Hofmann, R. Schlögl, A. Knop-Gericke and T. E. Jones, J. Am. Chem. Soc., 2021, 143, 12524–12534 CrossRef PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef CAS PubMed.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75–83 CrossRef CAS PubMed.
- A. K. Friedman, W. Shi, Y. Losovyj, A. R. Siedle and L. A. Baker, J. Electrochem. Soc., 2018, 165, H733–H741 CrossRef CAS.
- W. Shi and L. A. Baker, RSC Adv., 2015, 5, 99284–99290 RSC.
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Nørskov, A. Nilsson and H. Ogasawara, Nat. Commun., 2013, 41(4), 1–6 Search PubMed.
- M. F. Lichterman, S. Hu, M. H. Richter, E. J. Crumlin, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain, T. Mayer, B. S. Brunschwig, N. S. Lewis, Z. Liu and H.-J. Lewerenz, Energy Environ. Sci., 2015, 8, 2409 RSC.
- M. A. Kelly, J. Electron Spectrosc. Relat. Phenom., 2010, 176, 5–7 CrossRef CAS.
- D. I. Patel, T. Roychowdhury, V. Jain, D. Shah, T. G. Avval, S. Chatterjee, S. Bahr, P. Dietrich, M. Meyer, A. Thißen and M. R. Linford, Surf. Sci. Spectra, 2019, 26, 016801 CrossRef.
- T. S. Wesley, M. J. Hülsey, K. S. Westendorff, N. B. Lewis, E. J. Crumlin, Y. Roman-Leshkov and Y. Surendranath, Chem. Sci., 2023, 14, 7154–7160 RSC.
- O. Höfert, C. Gleichweit, H. P. Steinrück and C. Papp, Rev. Sci. Instrum., 2013, 84, 093103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07085b |
| This journal is © The Royal Society of Chemistry 2025 |