
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics of the coordination effect and ionic transport in TiO2-filled poly(ethylene carbonate)-based electrolytes†
Wei Tan a,
Kento Kimurab and
Yoichi Tominaga*a
a,
Kento Kimurab and
Yoichi Tominaga*a
aGraduate School of Bio-Applications and Systems Engineering (BASE), Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo 184-8588, Japan. E-mail: ytominag@cc.tuat.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Nakacho, Koganei, Tokyo 184-8588, Japan
First published on 13th May 2025
Abstract
Solid composite electrolytes based on poly(ethylene carbonate) and rutile TiO2 are investigated via molecular dynamics simulations. The incorporation of TiO2 induces the formation of a space charge layer at the polymer–filler interface and facilitates low-resistance Li+ hopping, significantly altering Li+ coordination environments and leading to excellent local Li+ mobility.
Broader contextThis is the first report on the characterization of ionic mobility and solvation structure in Li salt-concentrated poly(ethylene carbonate) (PEC) electrolytes with TiO2 using molecular modeling, calculations and simulations based on molecular dynamics, the radial distribution function and the free volume theory. Polymer electrolytes have attracted much attention as ion-conductive soft materials for novel battery systems because of their safety compared with liquid electrolytes, flexibility and light weight. We have already reported that highly salt-concentrated PEC electrolytes exhibit unique ion-conductive behavior (high conductivity, a high Li transference number t+, excellent oxidation stability above 5 V, etc.) in the solid state. Moreover, we found that the composite electrolytes with inorganic fillers such as TiO2 exhibit excellent properties such as increased Li+ self-diffusion coefficients and t+ values greater than 0.8. We believe that the presence of aggregated ions with moderately complex structures leads to favorable performance in such PEC-based electrolytes and that the interfacial ionic structure between the electrolyte phase and the TiO2 surface may play an important role in enhancing ionic transport. We therefore focus on the computational characterization of PEC electrolyte models over a wide range of salt concentrations to understand the unusual solvation structure of ions. |
Introduction
Since their commercial debut by Sony in the 1990s, lithium-ion batteries (LIBs) have become essential to modern society, serving as the primary power source for mobile phones, laptops, drones, and other portable electronic devices.1–3 With continuous advancements in large-scale production and significant cost reductions, the energy density and specific energy of LIBs have surpassed 300 W h kg−1 and 750 W h L−1, respectively, with further improvements driven by progress in materials science and manufacturing.4,5 However, the growing electric vehicle (EV) market demands even higher energy densities, targeting over 500 W h kg−1, to achieve driving ranges exceeding 1000 kilometres.6Despite this increasing demand for high-capacity LIBs, several challenges remain, particularly concerning the lithium metal anode.7,8 The weak bonding between lithium atoms not only facilitates the growth of lithium dendrites but also significantly increases the risks of internal short circuits and safety hazards.9–11 Over the past decade, researchers have proposed various strategies to mitigate these issues, including advanced separators, electrolyte optimization, engineering of the interphase layer, and structural modifications of the anode.12–15 Nevertheless, increasing the energy density inherently raises safety concerns. Therefore, replacing flammable liquid electrolytes with solid-state electrolytes has become a crucial focus for enhancing safety while meeting the demands for higher performance.
Solid electrolytes offer significant potential not only in mitigating dendrite formation but also in addressing critical instability in LIBs.16,17 Among them, solid polymer electrolytes (SPEs), utilizing organic macromolecules such as poly(ethylene oxide) (PEO) or poly(ethylene carbonate) (PEC) as the polymer matrix, exhibit distinct advantages.18,19 These include low density, excellent interfacial compatibility with electrodes, and favourable mechanical flexibility, making SPEs particularly attractive for applications where weight reduction is essential, such as in EVs.
However, despite these advantages, a major challenge remains: the room-temperature ionic conductivity of SPEs typically falls within the range of 10−6 to 10−5 S cm−1.20,21 To overcome this limitation, extensive research has focused on developing solid composite electrolytes (SCEs) by introducing inorganic fillers into the polymer matrix. These fillers such as TiO2 help lower the glass transition temperature of the polymer, disrupt its crystalline regions and promote the dissociation of lithium salts, thereby enhancing the segmental motion of polymer chains and increasing the concentration of free Li cations, significantly improving their mobility.22–24 By synergistically combining the flexibility of polymers with the mechanical robustness of inorganic fillers, SCEs achieve better interfacial contact with electrodes while effectively suppressing lithium dendrite growth, paving the way for safer and more efficient high-energy-density batteries.
Nevertheless, a considerable gap still exists between the current performance of these materials and their theoretical potential, continuously driving researchers to seek breakthroughs through innovative technical strategies. One such approach is molecular dynamics (MD) simulations. MD simulations enable large-scale modelling that provides detailed and intuitive representations of lithium salt dissociation and ion transport dynamics.25,26 This allows researchers to gain a clear understanding of how the introduction of fillers and their surface modifications affect Li+ transport behaviour at the molecular level. Previous studies have demonstrated that incorporating TiO2 into various polymer-based SCEs can significantly enhance metal ion conductivity and increase the Li+ transport number.27,28 Building on these findings, our study focuses on the role of TiO2 in a system consisting of a PEC-based matrix and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI). Composite models comprising PEC, LiTFSI and rutile TiO2 were constructed through a multi-step process, as described in ESI.1,† with the separate models shown in Fig. 1. Moreover, MD simulations were employed to investigate the ionic behaviours in the SCE systems. This study systematically analysed the changes in the Li+ coordination environment and ionic conductivity resulting from TiO2 incorporation, providing an in-depth evaluation of how space charge layer (SCL) formation influences ion transport within the PEC-based composite electrolyte.
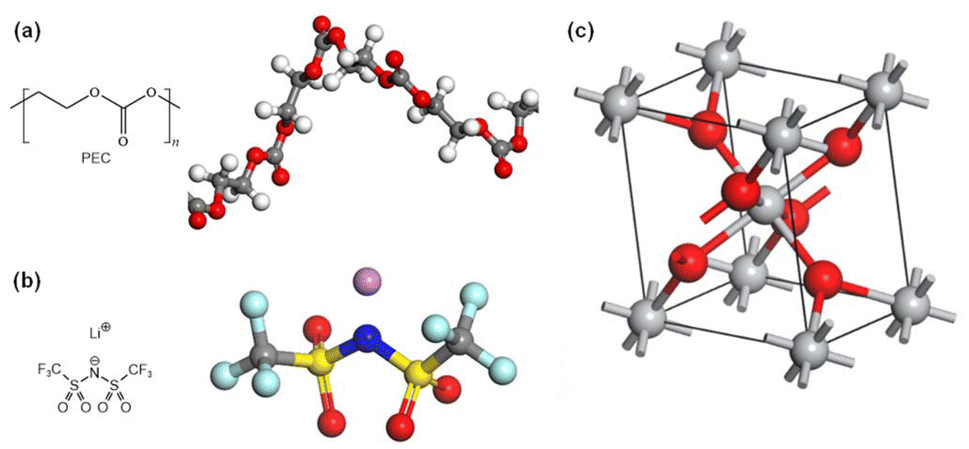 | ||
| Fig. 1 Chemical structures and molecular models of (a) a PEC chain, (b) LiTFSI and (c) a unit cell of rutile TiO2. | ||
All simulations conducted in this work, including model construction and structure optimization, were performed using the Materials Studio29 (MS) software package and its associated modules. MD simulations were executed using the Forcite module. Utilizing the COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies) II force field, the Forcite module functions as a powerful molecular mechanics engine, enabling efficient energy calculations, structure optimization, and MD simulations, making it well suited for large-scale polymeric systems. COMPASS II is known for its extensive parameterization, combining empirical data with results from ab initio HF/6-31G* calculations to achieve high accuracy in simulating condensed-phase materials.30,31 Numerous studies have demonstrated the applicability of COMPASS and Forcite in polymer and electrolyte research. For example, Amrhar et al. investigated the adsorption mechanisms of anionic dyes on the TiO2 (110) surface,32 and Ibrahim et al. simulated a graphene membrane/TiO2 bilayer nanocomposite to enhance the electronic and mechanical properties of the graphene membrane.33 These examples underscore the reliability and versatility of COMPASS II and support its use in this study for accurately capturing the structural and dynamic properties of the PEC-based electrolyte.
To balance accuracy and computational efficiency, appropriate functionals and methods were meticulously chosen for each calculation step, as detailed in ESI.2.† In addition, coordination structures and ionic mobility within the system were systematically analysed based on radial distribution function (RDF) and mean-square displacement (MSD) results (theoretical basis provided in ESI.3†). These data were extracted from MD simulations conducted under the NVT ensemble (where the number of atoms, volume, and temperature are conserved) and the NVE ensemble (where the number of atoms, volume, and energy are conserved), ensuring reliable characterization of dynamic behaviours.
Results and discussion
Following MD simulations, significant changes in the spatial distribution of ions within the electrolyte model were observed, as illustrated in Fig. 2. The initial model, constructed using a Monte Carlo-based method, exhibited a homogeneous spatial arrangement of the PEC backbone and both ion species (Li+ and TFSI−). However, after extensive 2000 ps NVT MD simulations, a substantial fraction of TFSI anions migrated toward the TiO2 surface, forming a distinct interfacial region. This behaviour indicates a strong adsorption affinity of the rutile TiO2 (110) surface for TFSI anions. Concurrently, partial displacement of the PEC backbone toward the TiO2 surface was observed, albeit to a lesser extent compared to the TFSI anions. The adsorbed TFSI anions and PEC segments collectively formed an interface, leaving a narrow gap between this interface and the bulk region, corresponding to a SCL. In contrast, Li cations were predominantly localized within the bulk and SCL regions, avoiding proximity to the TiO2 surface.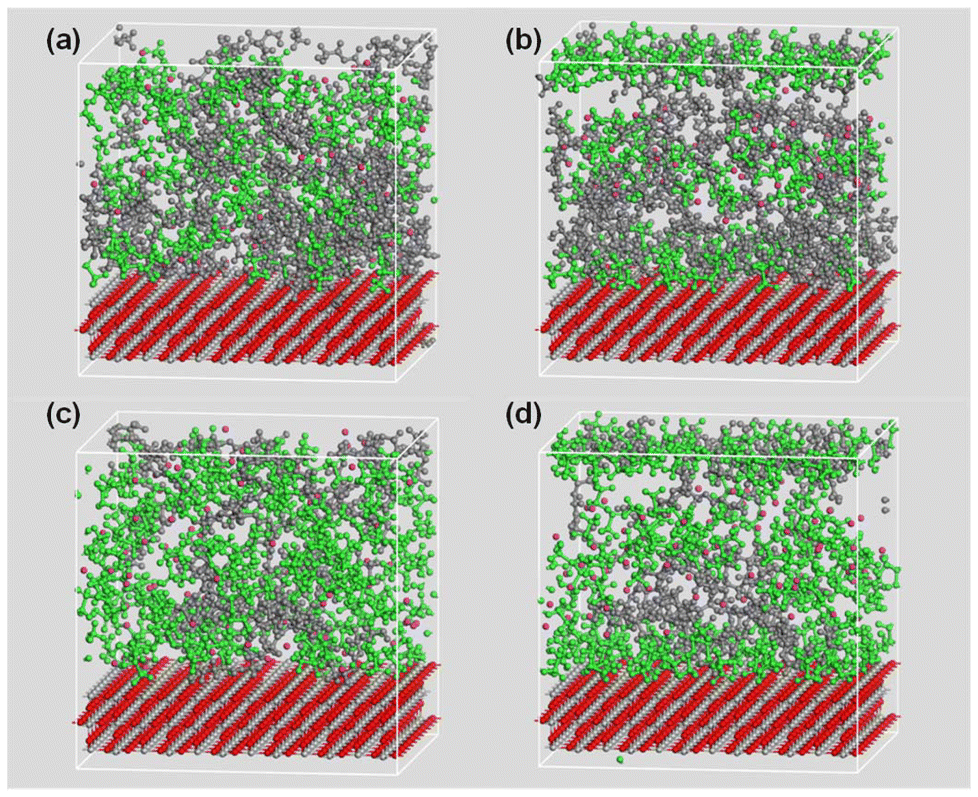 | ||
| Fig. 2 TiO2-filled PEC/LiTFSI SCE models under different conditions: (a) initial configuration of the 40 mol% model; (b) 40 mol% model after 2000 ps NVT MD simulation; (c) initial configuration of the 120 mol% model; and (d) 120 mol% model after 2000 ps NVT MD simulation. TFSI anions are shown in green, PEC chains in grey and Li cations in purple. | ||
The RDFs of Li cations in PEC/LiTFSI electrolytes were calculated before and after TiO2 addition, as shown in Fig. 3. Oxygen atoms from TFSI anions were denoted as O(TFSI), while carbonyl oxygen atoms from PEC were labelled as O(PEC). The results show a significant reduction in the coordination number (CN) between Li cations and O(TFSI) in both 40 mol% and 120 mol% salt concentration systems following TiO2 incorporation. This reduction indicates that TiO2 effectively suppresses Li–O(TFSI) coordination, thereby promoting Li+ dissociation within the electrolyte. Additionally, a rightward shift of the second RDF peak is observed in TiO2-containing systems, reflecting increased distances between TFSI anions, which is consistent with the formation of interfacial gaps in the model. For O(PEC), at a 40 mol% salt concentration, TiO2 addition enhances Li–PEC interactions, evidenced by a leftward shift of the first RDF peak, suggesting stronger coordination. However, at 120 mol% salt concentration, the observation is counterintuitive and warrants further investigation: Li cations appear to completely lose interaction with PEC carbonyl oxygen atoms. This observation suggests that, within this interfacial system, the coordination environment of Li cations is dominated solely by O(TFSI).
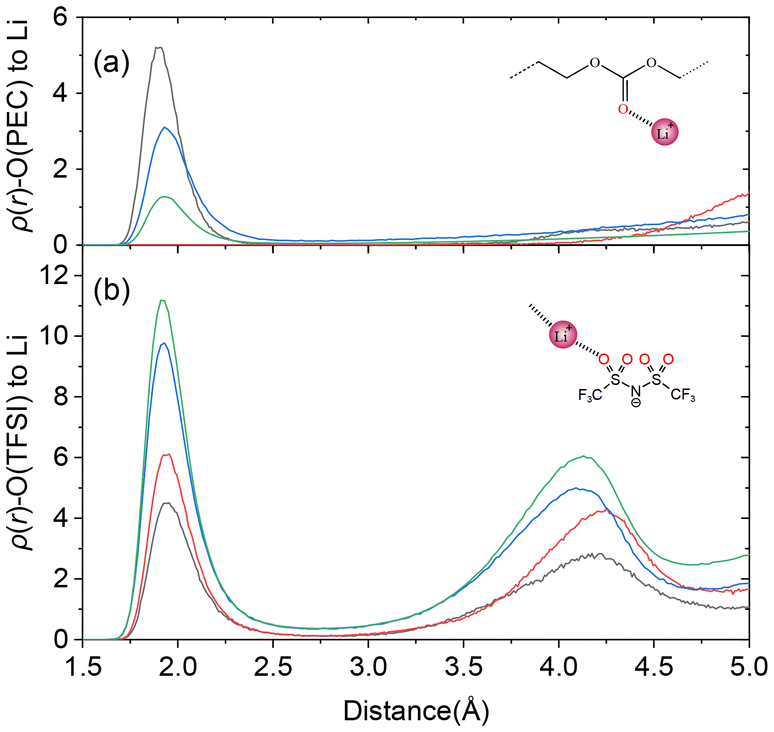 | ||
| Fig. 3 RDFs simulated for PEC/LiTFSI SPE and TiO2-filled SCE models between Li cations and oxygen atoms from (a) carbonyl groups in polymer chains and (b) TFSI anions. RDF curves are coloured based on salt concentration: 40 mol% SCE (black), 120 mol% SCE (red), 40 mol% SPE (blue) and 120 mol% SPE (green). | ||
In addition, we calculated the RDFs between Ti atoms on the TiO2 (110) surface and the two types of oxygen atoms, O(TFSI) from TFSI anions and O(PEC) from the polymer, as shown in Fig. 4. The RDF profiles reveal that Ti exhibits adsorption interactions with both O(TFSI) and O(PEC), with a notably stronger affinity for O(TFSI). As the salt concentration increases from 40 mol% to 120 mol%, the adsorption of O(TFSI) is further enhanced, while the interaction with O(PEC) weakens. This trend is attributed to the increased relative abundance of TFSI anions at higher salt concentrations. Furthermore, the adsorption distances between Ti and both O(TFSI) and O(PEC) are approximately 2.7 Å, which is notably longer than the average Ti–O bond length within the rutile TiO2 lattice (1.96 Å),34 suggesting physisorption rather than chemisorption.
 | ||
| Fig. 4 RDFs simulated for TiO2-filled PEC/LiTFSI SCE models between Ti atoms and oxygen atoms from (a) carbonyl groups in polymer chains and (b) TFSI anions. RDF curves are coloured based on salt concentration: 40 mol% (black) and 120 mol% (red). | ||
Based on the RDF data, we further calculated the CNs of Li cations corresponding to each RDF curve, as summarized in Table S1.† Following TiO2 addition, the total CNs of Li cations decrease to 2.61 (40 mol%) and 1.79 (120 mol%), compared to 4.03 and 3.85 in the TiO2-free systems. At a salt concentration of 40 mol%, TiO2 addition sharply reduces the Li–O(TFSI) CN from 2.97 to 1.39, while slightly increasing the Li–O(PEC) CN from 1.06 to 1.22. In contrast, at 120 mol% salt concentration, TiO2 incorporation significantly decreases both Li–O(TFSI) and Li–O(PEC) CNs, with the latter dropping to 0. These reductions indicate weakened coordination interactions, which are expected to enhance Li+ mobility. The formation of the SCL likely facilitates low-resistance hopping migration of Li cations, thereby improving ionic conductivity.35,36 Notably, in the 120 mol% SCE system, Li+ migration through the SCL occurs with minimal interaction with PEC, deviating from conventional hopping mechanisms. Additionally, the Ti–O(TFSI) CNs are 1.01 and 1.26 for the 40 mol% and 120 mol% systems, respectively, while the Ti–O(PEC) CNs decrease from 0.46 to 0.23. Notably, the total number of adsorbed oxygen atoms (from both TFSI anions and PEC) per surface Ti atom remains nearly constant at approximately 1.5 across both concentrations, suggesting that the overall adsorption capacity is robust and largely unaffected by salt concentration.
Due to Ti-induced adsorption interactions, the mobility of TFSI anions is inevitably impacted. To quantify this effect, TFSI anions were classified into two groups based on whether their oxygen atoms were adsorbed by Ti: TFSI(interface) and TFSI(bulk). The MSDs of Li+ and both TFSI− groups in the TiO2-containing PEC/LiTFSI system were subsequently calculated, as shown in Fig. 5. The results demonstrate that Li cations exhibit the highest MSD, followed by TFSI(bulk), while TFSI(interface) displays a significantly lower MSD than both, indicating restricted mobility due to surface adsorption.
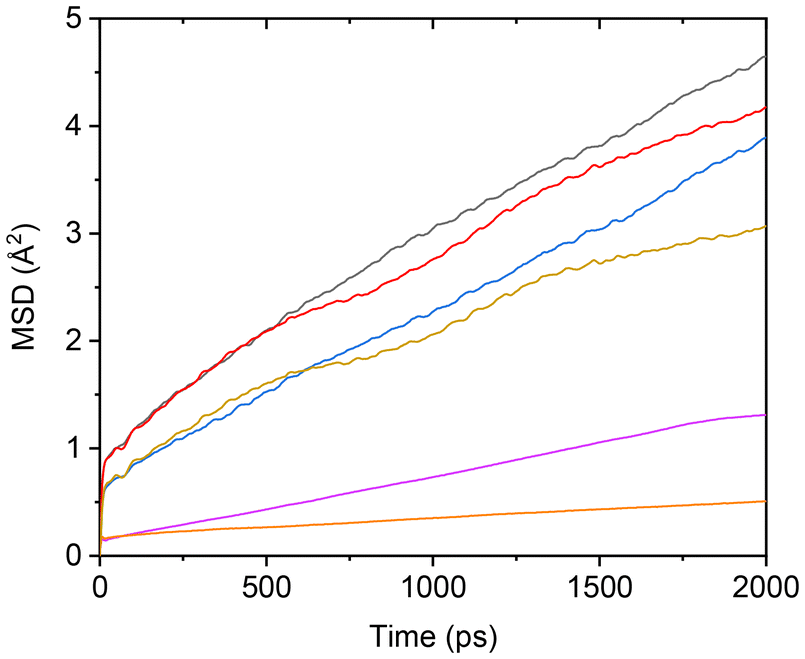 | ||
| Fig. 5 MSDs simulated for TiO2-filled PEC/LiTFSI SCE models under different conditions. Data are shown for Li+ at salt concentrations of 40 mol% (black) and 120 mol% (red), TFSI(bulk) at salt concentrations of 40 mol% (blue) and 120 mol% (dark yellow), and TFSI(interface) at salt concentrations of 40 mol% (purple) and 120 mol% (orange). | ||
Using the MSD data, we further calculated the ionic conductivity and transport numbers for each ion species. The logarithmic conductivity denoted as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ and transport number results are summarized in Tables S2 and S3.† The results reveal that TiO2 incorporation significantly enhances Li+ conductivity. Notably, at a 40 mol% salt concentration, the Li+ conductivity (2.12 × 10−4 S cm−1) exceeds that of the TiO2-free system at 120 mol%. Furthermore, the influence of salt concentration on Li+ conductivity is markedly reduced in TiO2-containing systems. Given the previously discussed stability of Ti adsorption interactions, the SCLs formed at 40 mol% and 120 mol% likely exhibit similar electrochemical properties, resulting in comparable resistance to Li+ migration. In contrast, TFSI− conductivity decreases substantially upon TiO2 addition. For TFSI(bulk), conductivity drops by approximately 20% at 40 mol% and 35% at 120 mol%, showing a clear declining trend. This decline may be attributed to the formation of the SCL, which compresses the free volume available for TFSI anions from the interface side. Reductions are even more pronounced for TFSI(interface), with conductivity decreasing by 53% at 40 mol% and 88% at 120 mol%, respectively. In TiO2-free systems, the Li+ transport numbers are 0.33 (40 mol%) and 0.55 (120 mol%). Upon TiO2 addition, these values increase sharply to 0.79 (40 mol%) and 0.85 (120 mol%), indicating that TiO2 suppresses anion migration, thereby altering the conduction mechanism and significantly enhancing the Li+ transport number.
σ and transport number results are summarized in Tables S2 and S3.† The results reveal that TiO2 incorporation significantly enhances Li+ conductivity. Notably, at a 40 mol% salt concentration, the Li+ conductivity (2.12 × 10−4 S cm−1) exceeds that of the TiO2-free system at 120 mol%. Furthermore, the influence of salt concentration on Li+ conductivity is markedly reduced in TiO2-containing systems. Given the previously discussed stability of Ti adsorption interactions, the SCLs formed at 40 mol% and 120 mol% likely exhibit similar electrochemical properties, resulting in comparable resistance to Li+ migration. In contrast, TFSI− conductivity decreases substantially upon TiO2 addition. For TFSI(bulk), conductivity drops by approximately 20% at 40 mol% and 35% at 120 mol%, showing a clear declining trend. This decline may be attributed to the formation of the SCL, which compresses the free volume available for TFSI anions from the interface side. Reductions are even more pronounced for TFSI(interface), with conductivity decreasing by 53% at 40 mol% and 88% at 120 mol%, respectively. In TiO2-free systems, the Li+ transport numbers are 0.33 (40 mol%) and 0.55 (120 mol%). Upon TiO2 addition, these values increase sharply to 0.79 (40 mol%) and 0.85 (120 mol%), indicating that TiO2 suppresses anion migration, thereby altering the conduction mechanism and significantly enhancing the Li+ transport number.
Conclusions
In this study, MD simulations were employed to investigate the effects of rutile-TiO2 incorporation on the interfacial structure and its subsequent influence on ionic coordination environments and mobility within PEC/LiTFSI electrolytes. Based on the simulation results, we propose that the addition of TiO2 nanoparticles induces the formation of interfacial regions with reduced resistance and enhanced Li+ transport properties within the PEC/LiTFSI SPE system. In the absence of TiO2, Li cations migrate via hopping mechanisms governed by the dynamic formation and dissociation of coordination with the PEC backbone and TFSI anions. However, upon encountering TiO2 nanoparticles, the presence of SCLs alters the ion transport pathway, leading Li cations to migrate preferentially via a modified hopping mechanism. In highly concentrated SCEs, Li+ migration within the interfacial region involves only 1 or 2 TFSI anions in each dynamic process. It is important to note that, although the simulations yielded high ionic conductivity and a Li+ transport number of up to 0.85, the constructed SCE models represent interfacial regions in close contact with TiO2 nanoparticles. These models thus characterize the local environment rather than the entire TiO2-filled composite electrolytes. The significantly improved ionic conductivity and elevated Li+ transport number observed in the interfacial regions of SCEs are attributed to the optimized interfacial pathways, which facilitate more efficient Li+ migration compared to the TiO2-free systems. These effects highlight the potential of rutile TiO2 nanoparticles in addressing critical challenges in solid-state batteries, including energy density limitations and safety concerns. The atomic-scale insights provided by this work underscore the importance of filler-engineered interfaces in optimizing polymer-based electrolytes. Future studies should focus on extending MD simulations to investigate surface defects and long-term interfacial stability, which could further advance the design of high-performance, durable solid-state battery systems.Author contributions
Conceptualization, W.T. and Y.T.; methodology, W.T.; software, W.T.; formal analysis, W.T.; writing – original draft, W.T.; writing – review and editing, K.K. and Y.T.; supervision, K.K. and Y.T.; and funding acquisition, Y.T. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by a Grant-in-Aid for Scientific Research (B) (23K23057) of JSPS KAKENHI and the Data Creation and Utilization-Type Material Research and Development Project (JPMXP1122712807) of MEXT, Japan.References
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 Search PubMed.
- P. V. Kamat, ACS Energy Lett., 2019, 4, 2757–2759 Search PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 Search PubMed.
- H. Li, Joule, 2019, 3, 911–914 CrossRef CAS.
- T. Waldmann, et al., J. Power Sources, 2020, 472, 228614 Search PubMed.
- J. Liu, et al., Nat. Energy, 2019, 4, 180–186 Search PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- Y. Gao, et al., Nano-Micro Lett., 2022, 14, 94 Search PubMed.
- I. Yoshimatsu, T. Hirai and J. Yamaki, J. Electrochem. Soc., 1988, 135, 2422 CrossRef CAS.
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS.
- M. Rosso, et al., Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS.
- D. Wang, et al., Adv. Sci., 2017, 4, 1600168 CrossRef PubMed.
- W. Xu, et al., Energy Environ. Sci., 2014, 7, 513–537 RSC.
- A. C. Kozen, et al., ACS Nano, 2015, 9, 5884–5892 CrossRef CAS PubMed.
- L. Fan, et al., Adv. Energy Mater., 2018, 8, 1802350 CrossRef.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- P. Bonnick and J. Muldoon, Energy Environ. Sci., 2022, 15, 1840–1860 RSC.
- J. Mindemark, et al., Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- F. Wu, et al., Energy Storage Mater., 2020, 33, 26–54 CrossRef.
- D. Zhou, et al., Chem, 2019, 5, 2326–2352 CAS.
- L. Yue, et al., Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- S. Duan, et al., Adv. Mater., 2024, 36, 2314120 Search PubMed.
- Y. Zheng, et al., Energy Storage Mater., 2023, 57, 540–548 Search PubMed.
- H. Zhang, et al., Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- M. Yao, et al., ACS Appl. Mater. Interfaces, 2021, 13, 47163–47173 CrossRef CAS PubMed.
- J. Zhou, et al., Energy Storage Mater., 2019, 22, 256–264 Search PubMed.
- N. Soontornnon, K. Kimura and Y. Tominaga, ACS Appl. Energy Mater., 2024, 7, 4190–4199 Search PubMed.
- M. Sasikumar, et al., J. Alloys Compd., 2021, 882, 160709 Search PubMed.
- BIOVIA Materials Studio 7.0, Dassault Systèmes, San Diego, 2014 Search PubMed.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 Search PubMed.
- H. Sun, et al., J. Mol. Model., 2016, 22, 47 Search PubMed.
- O. Amrhar, et al., J. Mol. Liq., 2023, 377, 121554 Search PubMed.
- Q. Ibrahim and R. Akbarzadeh, J. Mol. Model., 2020, 26, 165 CrossRef CAS PubMed.
- M. H. Samat, et al., Results Phys., 2016, 6, 891–896 Search PubMed.
- H. Qiu, et al., Angew. Chem., 2022, 134, e202113086 CrossRef.
- N. J. J. de Klerk and M. Wagemaker, ACS Appl. Energy Mater., 2018, 1, 5609–5618 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; Fig. S1; Tables S1–S3. See DOI: https://doi.org/10.1039/d5eb00078e |
| This journal is © The Royal Society of Chemistry 2025 |