
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Harnessing enzymes for greener polymerisations: advances in chain and step growth processes
Emily G. Dixon
 *a,
Luisa Ciano
a,
Anca Pordea
b,
Karen Alvey
c and
Vincenzo Taresco
*a
*a,
Luisa Ciano
a,
Anca Pordea
b,
Karen Alvey
c and
Vincenzo Taresco
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: emily.dixon1@nottingham.ac.uk; vincenzo.taresco@nottingham.ac.uk
bFaculty of Engineering, University of Nottingham, University Park, Nottingham NG7 2RD, UK
cSchool of Pharmacy, University of Nottingham, University Park, Nottingham NG7 2RD, UK
First published on 10th June 2025
Abstract
Synthetic organic polymers face increasing scrutiny due to their environmental impact, as most industrial monomers are petrol-based. Key concerns include fossil fuel consumption, greenhouse gas emissions, reliance on non-renewable feedstocks, insufficient recycling and the use of toxic and harmful catalysts. To align with the United Nations Sustainable Development Goals (SDGs) and Anastas and Warner's 12 Principles of Green Chemistry, the polymer industry must embrace a total revolution. Biocatalysis offers a promising route to achieve these goals. This review encourages the polymer community to adopt enzymatic catalysis for functional polymer synthesis, showcasing enzymes as powerful tools with green credentials that should be integrated into the field. To meet field-specific nomenclature we categorise enzymatic polymerisation based on the more general mechanisms, namely step polymerisation (SP), chain polymerisation (CP), and ring-opening polymerisation (ROP), reflecting the recent refinement of ‘step-growth’ and ‘chain-growth’ nomenclature by the IUPAC's Subcommittee on Polymer Terminology. This classification aims to engage readers with interests directed towards polymerisation processes and direct utilisation of the enzymes to specific reagents or classes of monomers. By presenting examples and using familiar mechanistic categorisations in a tutorial-style review, we aim to empower polymer chemistry researchers to embrace enzymatic catalysis as a sustainable and innovative tool. We conclude the review by highlighting recent innovative works, discussing challenges and proposing future opportunities.
 Emily Dixon | Emily Dixon completed her MSci in Chemistry with a Year in Industry at the University of Nottingham in 2023, where she also received a Salters’ Institute Graduate Award. She is now pursuing a PhD as part of the University of Nottingham's CDT in Sustainable Chemistry, focusing on tandem enzymatic polyester synthesis under the supervision of the co-authors of this review. |
 Luisa Ciano | Luisa Ciano is an Assistant Professor at the University of Nottingham. She obtained her Master's Degree in Pharmaceutical Chemistry and Technology from the University of Naples “Federico II” in 2009 and her PhD in Chemistry from the University of York in 2013. Following some PDRA positions at the University of York and the University of Manchester, she was awarded an Anne McLaren Research Fellowship in 2019 by the University of Nottingham, and became an Assistant Professor in 2022. Her research focuses on the study and characterisation of (metallo)enzymes for polymer (e.g. polysaccharides and plastic) degradation. |
 Anca Pordea | Anca Pordea received her PhD in 2008 working with Prof. Thomas R. Ward on the development of artificial metalloenzymes (University of Neuchâtel, Switzerland). She did postdoctoral research with Prof. Benjamin G. Davis (University of Oxford, UK), as a Swiss National Science Foundation fellow, and with Prof. Reinhard Neier (University of Neuchâtel). She joined the University of Nottingham in 2012 as an assistant, then associate professor. Her research interests are in enzyme engineering, to deliver sustainable alternatives for chemicals production and for the degradation of polymer waste. |
 Karen Alvey | Karen Alvey is an Anne McLaren Fellow in the School of Pharmacy at the University of Nottingham. A qualified pharmacist, she completed her PhD in 2014 on the formulation of protein therapeutics. Her current research focuses on enhancing medicine stability to improve patient access, employing interdisciplinary approaches that integrate formulation development, policy, and systems thinking. Prior to her Fellowship, she managed the Nanoscale and Microscale Research Centre at Nottingham, leading a team of researchers and collaborating with industry to provide access to advanced analytical instrumentation. |
 Vincenzo Taresco | Vincenzo Taresco is a Nottingham Research Fellow and Assistant Professor in the School of Chemistry at the University of Nottingham (UoN), earned his PhD in polymer chemistry from Sapienza University of Rome in 2014. He began his postdoctoral research in the School of Pharmacy at UoN and became a senior research officer (group leader) in the School of Chemistry, also in UoN, in 2019. By the end of 2022, he received the prestigious Nottingham Research Fellowship. His research group focuses on creating sustainable, functionalised, and multi-responsive polymeric devices by integrating bio-renewable moieties into polymer backbones. |
1. Introduction
1.1 Introduction to biocatalysis
Nature's catalysts (biocatalysts) facilitate biochemical reactions without undergoing change themselves and include functional proteins (enzymes) or nucleic acids (ribozymes). Biocatalysts are defined by IUPAC as “an enzyme or enzyme complex consisting of, or derived from, an organism or cell culture (in cell-free or whole-cell forms) that catalyses metabolic reactions in living organisms and/or substrate conversions in various chemical reactions”.1 Since antiquity, society has unknowingly utilised enzymes to catalyse reactions, with zymase, the enzyme in yeast responsible for converting glucose into ethanol, playing a key role in wine production since 6000 BCE.2 Millenia later, the use of enzymes in both the manufacture and in products themselves has grown exponentially: food and drink, pharmaceutical, detergent, polymer and waste management represent just a few examples of industries exploiting the use of enzymes.3Enzymes offer a compelling alternative to traditional chemical catalysts, providing several notable advantages such as functioning under mild reaction conditions (e.g., moderate temperatures, near-neutral pH, ambient pressure, and operation in aqueous and organic media), exhibiting high chemo-, regio-, and enantioselectivity, and being derived from renewable sources. These characteristics could contribute to reduced energy requirements and process intensification, which may support more efficient and potentially less resource-intensive chemical transformations.4,5 In addition, enzymes are non-toxic, making them inherently ideal for physiological and environmental applications, i.e., active pharmaceutical ingredients (APIs), food ingredients and agrochemical synthesis.6 Finally, the selectivity attainable through biocatalysis integrates both directing and catalytic behaviour within a single reagent, allowing access to products not easily attainable through chemical synthesis.7 Early implementations of in vitro biocatalysis were focused on fine chemicals, with the synthesis or resolution of enantiopure small molecules.8 Following this, widespread adoption of biocatalytic transformations accelerated significantly in the 1990s, with the rise of recombinant DNA technology and the growth of directed evolution. The quantity and specificity of enzymes available for chemical synthesis has since become vast, allowing for new frontiers in biotechnology, medicine and materials science through novel enzymatic mechanisms and synthetic cascades.7,9 One particularly pertinent industrial example of small molecule production lies within the biocatalytic synthesis of the antidiabetic sitagliptin, avoiding a high pressure, asymmetric hydrogenation using a rhodium-based chiral catalyst and avoiding additional purification steps to improve enantiomeric excess and purity, as well as safety and sustainability.7,10
Whilst the field of biocatalysis within the polymer industry is growing, the number of industrially utilised processes is small when compared to small organics – most implementation is within biological recycling and depolymerisation rather than synthesis. The climate crisis has intensified the need to develop renewable fuels and feedstocks for chemical synthesis, alongside efforts to valorise waste materials. Enzymatic degradation of natural polymers, such as waste lignocellulose for bioethanol production, is a well-established field,11 and in recent years scientific efforts have been directed towards addressing plastic pollution and the environmental impact of synthetic polymers, shifting the focus towards enzymatic polymer degradation and recycling rather than synthesis. As examples, the French biochemistry company CARBIOS and Australian Samsara Eco are set to open the world's first polyethylene terephthalate (PET) and nylon-6,6 biorecycling plants, respectively.12,13
The obvious examples of enzymatic polymer synthesis are encountered in nature. Natural biopolymers such as polynucleotides (DNA, RNA), polypeptides (gelatin, collagen), polysaccharides (cellulose, chitosan, starch) and others (lignin, rubber, polyhydroxyalkanoates) are all produced in living cells through biocatalysis. Their biosynthetic pathways generally require complex cofactors and/or derivatised substrates (such as coenzyme A, for the synthesis of polyhydroxyalkanoates; or nucleotide sugars, for the synthesis of polysaccharides), which makes the in vitro application of such syntheses quite challenging.14 Nonetheless, there are numerous applications in industries such as textiles, biomedicine and pulp/paper that involve enzymatic synthesis and modification of some of the natural polymers in vitro, using promiscuous or engineered enzymes capable to accept simpler substrates. This topic is outside the scope of this review, though it is highlighted in other reviews.15,16,17
Kobayashi, a pioneer and leading contributor to the field of enzymatic polymer synthesis, defines enzymatic polymerisation as the “in vitro chemical polymer synthesis via nonbiosynthetic (nonmetabolic) pathways catalysed by an isolated enzyme”.4 This review will follow this definition and focus on in vitro biocatalysis of synthetic polymers. Therefore, this review will focus on the use of enzymes for the synthesis and modification of synthetic polymers, specifically using isolated enzymes rather than whole cell biocatalysis.18
Despite increasing interest over the past 30 years, enzymatic polymer synthesis remains an underdeveloped field with a lot of untapped potential. By showcasing some recent advancements in the enzymatic synthesis of synthetic polymers in this tutorial-style review, the authors hope that groundbreaking advancements similar to those obtained in polymer degradation can be made within polymer synthesis, to enable a circular economy for the future.
1.2 Enzyme classification
The Enzyme Commission (EC) classifies enzymes into six types according to their function. Of these classes, oxidoreductases, and hydrolases are most commonly used in enzymatic polymer synthesis as they successfully enable the chemistry required for different types of polymerisation reactions, as well as being compatible with generic conditions required for polymerisation.19 Oxidoreductases typically catalyse the formation of (hydr)oxyl radical species, which further catalyse radical polymerisations, whilst transferases and hydrolases are involved in the formation of carboxylic acid derivatives (esters, amides), leading to polyesters and polyamides (Table 1).| Enzyme class | Reaction type | Representative enzymes | Polymers produced | Sections |
|---|---|---|---|---|
| Oxidoreductases | Oxidation leading to radicals (hydroxyl, alkoxyl, phenoxyl, enoxyl) | Peroxidases | Vinyl-based polymers | 2, 6, 7 |
| Laccases | Polyphenols | |||
| Glucose oxidase | ||||
| Pyranose oxidase | ||||
| Transferases | Acyl transfer | Carboxytransferases | Polyesters | 3, 4 |
| Acyltransferases | ||||
| Hydrolases | Reversible hydrolysis/alcoholysis: | Lipases | Polyesters | 3, 4, 7 |
| - (Trans)esterification | Esterases | Polyamides | ||
| - Amide formation | Cutinases | |||
| Peptidases |
Enzymes involved in synthetic polymer synthesis may be classified as “direct” or “indirect” depending on their interaction with monomers.
Direct enzymes accept monomers into the catalytic active site, to initiate the polymerisation. Hydrolases such as lipase are mostly encountered in this category (Fig. 1A). Radical-forming oxidoreductases such as horseradish peroxidase (HRP) can also act as direct enzymes, by forming the radical species of the monomers, which subsequently polymerise.
 | ||
| Fig. 1 Schematic representation of direct and indirect enzymatic polymerisation reactions. (A) Direct enzymes performing an example polycondensation (step) reaction using generic diol and diacid as monomers/substrate. Monomer(s) interact directly with the active site of the enzyme and undergo transformation into a polyester (the same enzyme can also catalyse the ROP process, not reported in the figure). (B) Indirect enzymes performing an example free-radical polymerisation (FRP) whereby a radical generating species (e.g. acetylacetone) and an oxidant (e.g. H2O2) interact with the enzyme to produce a radical initiator that allows for transformation of vinyl monomers into polymers (chain). | ||
Radical generating enzymes may also act as indirect enzymes, when the polymerisation is initiated by the generation of species exogenous to the monomer, called catalytic radical mediators. This is also described as initiase behaviour. An example of this is the polymerisation of vinyl monomers catalysed by HRP, where radical mediators are employed to generate the initiating species, avoiding direct interaction of the enzyme and monomer (Fig. 1B).20,21 This review seeks to explore how both of these mechanistic approaches have been utilised to catalyse the formation of synthetic polymers.
In some cases, direct enzymes can also function as indirect enzymes via the implementation of catalytic radical mediators. This is true in the case of some peroxidases, such as horseradish peroxidase (HRP), which acts directly in the oxidative polymerisation of aromatics, and indirectly in the polymerisation of vinyl monomers. In the latter case, radical mediators are employed to generate the initiating species, avoiding direct interaction of the enzyme and monomer – this may also be described as initiase behaviour.20,21
Lipases, such as CaLB, are triacyl glyceride hydrolases traditionally used in aqueous media for the hydrolysis of fatty acid esters into free fatty acids and glycerol, which is also their biological function. Their active site is typically composed of a catalytic triad containing a nucleophilic serine residue that operates via hydrogen bond activation with histidine and aspartate.33 Their mechanism employs an acylation step followed by subsequent deacylation (Fig. 2A). Lipases have a very large hydrophobic pocket surrounding the active site to accommodate their natural substrates, which are typically very hydrophobic and form biphasic droplets in water. This allows them to adsorb on the surface of the glyceride drops to perform the catalysis, a mechanism called interfacial activation. To ensure their stability in aqueous media, the hydrophobic pocket is covered with a polypeptide “lid”, which opens to accommodate the hydrophobic substrate.23
 | ||
| Fig. 2 (A) Catalytic triad mechanism of a (poly)condensation reaction in the active site of CaLB. (B) Catalytic residues Ser105, Asp187 & Ser105 within CaLB's active site (PDB: 1LBT). (C) Surface view of CaLB cocrystallised with Tween 80, showing the partially solvent exposed binding region. | ||
Lipases are reversible, which means they can also catalyse esterification/transesterification reactions. Their ability to tolerate hydrophobic environments makes them stable in organic solvent systems, which is particularly valuable when using them in the synthetic direction, to form hydrophobic molecules.34,35 Furthermore, lipases can retain their functionality in almost-anhydrous solvents, which can drastically shift the thermodynamic equilibrium towards condensation rather than hydrolysis.36
In contrast to many other lipases, the hydrophobic substrate binding site of the active site of CaLB only has a small lid, resulting in increased exposure of the active site to the solvent (Fig. 2C).37 This, combined with two mobile α-helices at the active site (α5 and α10), enables CaLB to be active towards an extensive range of substrates.37,38 Studies have demonstrated that the activation energy of the active site of CaLB is lower in less polar solvents, which causes differences in the activity of the enzyme depending on the solvent system used due to solvent effects in the active site region.33
The ability of lipases to adsorb on hydrophobic surfaces also enables their immobilisation on supports, which is a highly attractive strategy to allow their recovery and reuse, but also to increase their stability. Immobilisation occurs by interfacial activation, which keeps the enzyme in the open conformation, allowing substrate access. This results in immobilised lipases retaining activities close to their non-immobilised, free forms. N435 is the most successful example of a commercialised immobilised enzyme.23
 | ||
| Scheme 1 Catalytic cycle of HRP in phenolic and vinyl polymerisations utilising H2O2 and acetylacetone as mediator species. Horizontal black lines represent porphyrin ring. | ||
1.3 The case for enzymatic polymerisation
Enzymatic catalysis has gained significant attention in the polymer science community as a green alternative to traditional polymer synthesis and functionalisation. In addition to the general benefits mentioned earlier in this review, biocatalysis enables precise control of polymer architecture, molar masses (Mn, Mw), dispersity (Đ), and end group functionality.41 Furthermore, metals commonly used in chemical polymer catalysis can pose toxicity risks in polymeric products, which is particularly undesirable given their widespread use in biomedical applications. Finally, the use of traditional chemical catalysts often does not offer any selectivity of multifunctional monomers, leading to less control over polymer topography.42,43Consequently, the use of biocatalysis for the synthesis of polymers has been explored for the past three decades, after the birth of modern polymer chemistry in the early 20th century.44 Enzymatic polymer synthesis remains an emergent but fast growing field within polymer chemistry (Fig. 3).
 | ||
| Fig. 3 Number of publications against search terms, accessed from SCOPUS on 21/01/2025. Search fields set to title, abstract or key words. | ||
This significant literature presence may be attributed to the growing scrutiny towards synthetic organic polymers for their environmental impact throughout their life cycle, particularly as the majority of industrially utilised monomers are petrol-based. Consumption of fossil fuels, macro- and micro-debris (both in terrestrial and marine environments), greenhouse gas generation, non-renewable feedstocks and insufficient recycling encompass the main reasons for such scrutiny. In 2019 alone, the Organization for Economic Cooperation and Development (OECD) estimated that total greenhouse gas emissions related to fossil-based plastics were 1.8 gigatonnes of CO2 equivalent (Gt CO2e), or 3.7% of global emissions, which are projected to more than double by 2060.45
Evidently, there is a need to revolutionise the polymer industry in order to be aligned with the United Nations Sustainable Development Goals (SDGs) and Anastas and Warner's 12 Principles of Green Chemistry46 to foster a greater level of sustainability. Biocatalysis provides a valuable pathway towards achieving these objectives.
Whilst many reviews have been published in the field of enzymatic polymerisation,4,19,21,32,47 this review provides insight into enzymatic polymerisation by the mechanism employed, namely chain polymerisation (CP), ring-opening (ROP) polymerisation and step polymerisation (SP). These terms reflect the recent refinement of previous ‘step-growth’ and ‘chain growth’ nomenclature by the Subcommittee on Polymer Terminology (SPT) in the Polymer Division of the IUPAC, who recently published a perspective discussing the matter.48 ROP is generally defined as chain-growth but due to the ubiquity and broad literature based on enzymatic ROP (eROP) polymer synthesis, it is included in a separate section (section 3).
2. Chain polymerisation
Chain polymerisation (CP) is defined by the IUPAC as “chain reactions where the propagation steps occur by reaction between monomer(s) and active site(s) on the polymer chains with regeneration of the active site(s) at each step”.49 Monomers are generally unsaturated and the reaction can proceed radically or ionically after the active site is generated by an external initiator.50 This review covers radical based polymerisation only as this process has been demonstrated to be possible via biocatalytic methods.2.1 Free radical polymerisation (FRP)
Free radical polymerisation (FRP) is the most common mechanism employed for the synthesis of industrial vinyl-based polymers through a chain-growth mechanism, however, it offers little control over polymer microstructure. Classified by three stages – initiation, propagation and termination -free radical polymerisation (FRP) is simple and versatile, with approximately 50% of global plastic materials manufactured using FRP.51 It offers tolerance of a wide variety of vinyl monomers, solvents and reaction conditions. However, FRP fundamentally suffers from a lack of control in both polymer properties and also safety (the onset of Trommsdorff–Norrish effect).52 The tendency of radicals to self-terminate limits the number of active chains and results in broad dispersity. Since the 1990s, new methods of controlling radical polymerisation were developed, and are covered in sections 2.2 & 2.3. The general mechanism of FRP can be seen in Scheme 2 below.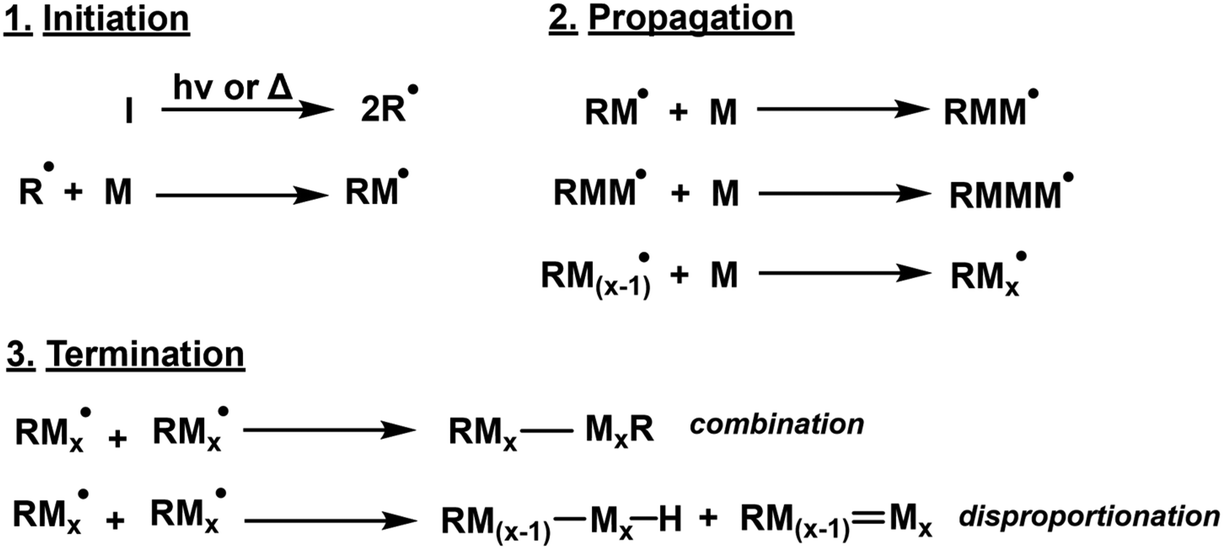 | ||
| Scheme 2 Generic FRP mechanism whereby I, R and M represent initiator, radical species and monomer respectively. | ||
Issues of control within radical polymerisations have been tackled by living or controlled radical polymerisation (CRP), now known as reversible-deactivation radical polymerisation (RDRP). RDRP provides a means of controlling radical polymerisation whilst offering control over polymer properties, such as well-defined and predictable molar mass, low dispersity, selective end group functionality, diverse polymer architecture and characteristics of living polymerisation.55 Subclasses of RDRP include reversible addition–fragmentation chain transfer polymerisation (RAFT), atom transfer radical polymerisation (ATRP) and nitroxide-mediated polymerisation (NMP).56 RDRP also permits the polymerisation of unprotected, functional vinyl monomers whilst employing gentle reaction conditions and a wide range of monomers and/or solvents which include water and supercritical CO2 (ScCO2).56 ATRP involves a reversible deactivation of propagating radicals to form dormant species that can be reactivated catalytically, whilst RAFT involves a degenerative radical transfer between propagating radicals and a dormant species.57 NMP is considered the simplest of the three techniques and involves the use of a stable radical (nitroxide) to reversibly deactivate propagating polymer radicals.58 Furthermore, RDRP methods have been used extensively in the preparation of copolymers in a wide range of architectures.58 Consideration towards the nature of both the initiator and monomers is required for a sustainable future of RDRP, as majority currently employed are fossil derived.55
Naturally, oxidoreductases dominate the field of enzymatic radical polymerisations for redox and radical generation ability, with laccases and peroxidases featuring as the most widely investigated classes.62
Horseradish peroxidase (HRP) is the most commonly used enzyme in this field and can catalyse the polymerisation and crosslinking of phenol derivatives, as well as vinyl monomers in the presence of H2O2 (see Scheme 1).4,63 The polymerisation of vinyl monomers using HRP was first reported in 1992 by Derango et al.64 and elucidated in 2000 by Gross et al., who proved that the keto-enoxy radicals of the β-diketone reducing agent generated by the enzyme are the radical species responsible for initiation of vinyl monomers.65 HRP is a haem-containing indirect enzyme that catalyses oxidation of a radical-generating mediator species through concurrent reduction of hydrogen peroxide to water, to initiate polymerisation of vinyl monomers.
Commonly, an acetylacetone (ACAC), H2O2/HRP ternary initiating system is employed, and has been shown to successfully initiate polymerisation of acrylamide, styrene, methacrylate(s), 4-acryloylmorpholine and 2-vinyl naphthalene.66,67 Such reactions can be conducted in ambient conditions in aqueous solution, or in water/organic mixture/emulsion.
There has been much discussion within the literature regarding the composition and necessity of the three components of the ternary system. Earliest experiments by Derango et al. suggested successful polymerisation of acrylamide without the use of a mediator.64 Later, Emery et al. demonstrated that all 3 components were essential,68 with Durand et al.,69 finding that not only is the ternary system essential, but H2O2 in high concentration causes enzymatic degradation, and that the ratio between components is critical when considering competition between initiation involving ACAC and inhibition caused by H2O2. More recently, in 2020, the work of Wang et al.,70 found that a HRP/ACAC binary system alone can produce polyacrylamide, polyacrylic acid and poly(methyl methacrylate) without the addition of H2O2, suggesting that the mechanism proposed in Scheme 1 requires further elucidation.
Aside from solution polymerisation, similar approaches are also used in the formation of hydrogels: Liao et al. demonstrated this application in the preparation of polyacrylamide gel for biological applications, involving both polymerisation and cross-linking of acrylamide and N,N′-methylenebisacrylamide (MBA), traditionally catalysed by toxic N,N,N′,N′-tetramethylethylenediamine/ammonium persulphate (TEMED/APS). This work showed that the ternary enzymatic system could completely replace the chemical method, with the enzymatically prepared gel demonstrating greater discrimination of larger proteins due to increased pore size.71
Zhang et al. produced MBA-based hydrogels for controlled release of aspirin, instead using cellulose acetoacetate (CAA) as mediator, demonstrating the ability of HRP to utilise more sterically hindered mediators during initiation.72 The work of the Gaitzsch group has demonstrated HRP-catalysed synthesis of poly(N-isopropyl acrylamide) (PNiPAAm) with HRP/ACAC/H2O2 to garner PNiPAAm with Mn of approx. 500 kDa and Đ = 1.3 after only 1 minute in water at room temperature. The authors further illustrated that modified HRP could be entrapped within the hydrogel in a microfluidic chip to perform eFRP within microfluidic reactors.73 HRP eFRP has also been used in the synthesis of polymers on surfaces, such as the grafting of methyl methacrylate and butyl acrylate onto starch,74,75 silica particles,76 and poly(ethylene glycol)dimethacrylate (PEGDMA) onto silk sericin.77
As mentioned, H2O2, the native substrate of HRP, can deactivate the enzyme. To combat this, H2O2 can be introduced slowly over time, or a H2O2 generating enzyme can be implemented to enable continuous generation of H2O2 in situ, without rapid increase in concentration.
Glucose oxidase (GOx) is an oxidoreductase that generates H2O2 through the oxidation of glucose to gluconic acid using O2 as oxidising agent, via reduction of the flavin adenine dinucleotide (FAD) cofactor.78 Whilst early examples employed the use of Fenton chemistry with GOx to provide the hydroxy radicals required for initiation,79 combining it with HRP provides an entirely enzymatic approach for the generation and utilisation of H2O2 in situ, for example in the synthesis of polyphenols (Scheme 3).40 Additionally, the HRP/GOx bienzymatic initiation system provides an effective means of sequestering molecular oxygen—an issue that typically hinders radical polymerisation—thereby enabling the potential for synthesis to be conducted under ambient atmospheric conditions (see section 2.2.2). However, despite this advantage, the dependence on the costly flavin adenine dinucleotide (FAD) cofactor may limit the economic viability of this approach for large-scale oxygen sequestration.
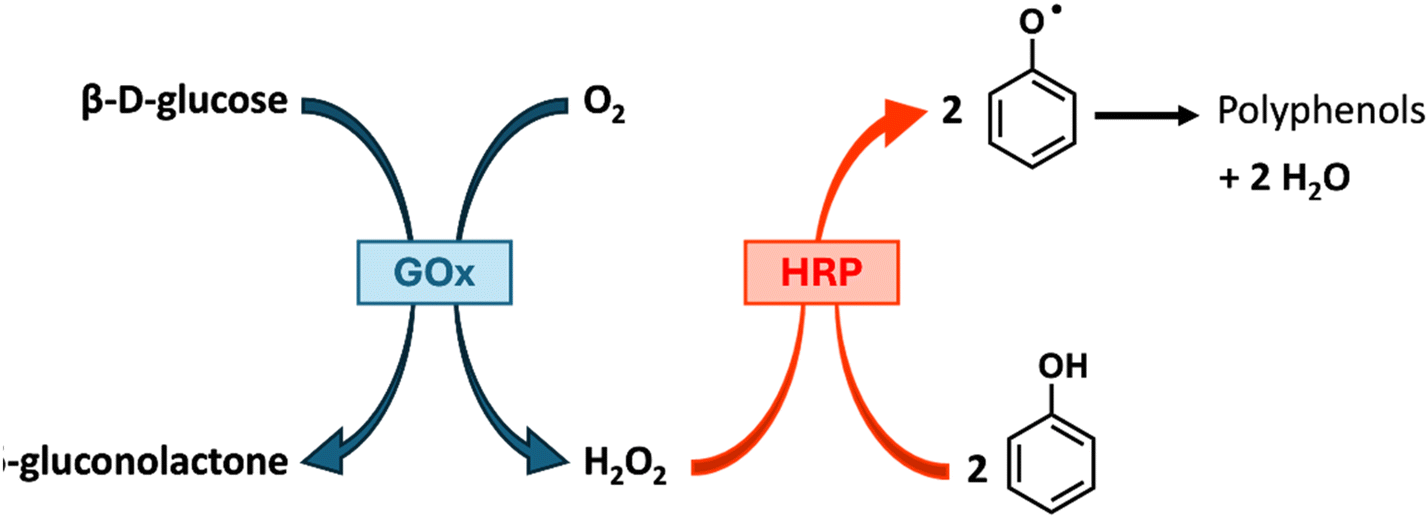 | ||
| Scheme 3 GOx/HRP mediated radical initiation/polymerisation of phenol. | ||
Currently, the use of HRP in industry is currently limited by several factors, including the need to be isolated from plant roots, its low specific activity and yield, as well as its relatively low stability.80 Although some progress has been made in the recombinant production of HRP,81,82 further development in large-scale microbial fermentation is necessary to make biocatalysis a more attractive option for the bulk polymer industry. Moreover, HRP suffers from deactivation after a certain number of turnovers and shows low tolerance to organic solvents and the high concentrations of organic monomers typically encountered in radical polymerisation processes. Enzyme immobilisation offers a potential solution by improving the stability of HRP and enabling its recovery and reuse after reactions.83
In this regard, the success Novozymes (now Novonesis following a recent merger) achieved with immobilised CaLB (Novozym435) may be attributed to the heterologous expression of the enzyme in Aspergillus niger rather than the native organism.
Laccases, another type of oxidoreductase, catalyse the oxidation of phenolics via the reduction of molecular oxygen to water. As multicopper oxidases, laccases contain at least 4 copper sites, with the type 1 copper site (categorised by a single copper ion coordinated with imidazole nitrogens of two histidine residues and a thiolate of a cysteine residue) responsible for monoelectronic substrate oxidation, whilst the remaining sites reduce molecular oxygen to water.84 Scheme 4 shows a simplified laccase oxidation of coniferyl alcohol, a phenol derivative that can be further polymerised to synthetic lignin oligomers.85
 | ||
| Scheme 4 Simplified laccase oxidation of coniferyl alcohol, generating 4 radical species available for polymerisation. Adapted from ref. 85, with permission from Elsevier, copyright 2011. | ||
Laccases may be considered a direct enzyme as the monomer interacts with the enzyme active site itself. In some cases, laccase has catalysed the synthesis of the same polymers as HRP with similar results.21,86 Being copper-based renders laccases ideal for ATRP, and is covered further in this review (section 2.3). As with peroxidases, the substrate scope of laccases can be increased by mediator species as electron shuttles. Within the literature, laccases are widely employed in the valorisation of renewable feedstock lignin, particularly for depolymerisation.87 The use of laccases is much less widespread than peroxidase-initiated polymerisations, as a limitation may be the ease of using liquid peroxide rather than bubbling air through the reaction, particularly on an industrial scale. As highlighted in a recent review by Kumar et al., investigating alternative enzymes to horseradish peroxidase (HRP) for radical initiation continues to be a crucial and underexplored area of research.88
We have provided an initial overview of enzymatic FRP, including some explanatory examples to illustrate how it may offer a greener alternative to traditional FRP. This method enables the production of the same materials as chemical synthesis with lower energy requirements and without the use of often toxic initiators. Additionally, the elimination of high temperatures and photo-irradiation makes it an ideal process for synthesising light and temperature-sensitive materials. For readers interested in more detailed examples and analyses, we refer to other literature that covers eFRP in greater depth.21,62,63,89
2.2 RAFT polymerisation
 | ||
| Scheme 5 General RAFT polymerisation mechanism using a thiocarbonylthio CTA. Note kinetic terms are omitted for simplicity. | ||
Essentially, RAFT is a free-radical polymerisation with the inclusion of a highly reactive dithioester, trithiocarbonate, dithiocarbonate (xanthate) or dithiocarbamate transfer agent (RAFT or chain transfer agent, CTA) that separates during chain transfer to generate both a new radical and thio-species. An external radical generating initiator is also required (commonly thermal azo-initiator AIBN), with radicals generated via heat or light.56 RAFT is frequently employed for the synthesis of block copolymers with extremely high monomer conversion (up to 100%), by incorporation of at least two monomers via a sequential polymerisation process.90 However, exogenous traditional RAFT initiators such as AIBN, benzoyl peroxide and potassium persulfate require high thermal input and are inherently hazardous. Recently, alternative strategies have emerged for greener, safer RAFT polymerisations, such as direct photoactivation of CTAs, electrochemical methods, ultrasound, cationic activation and enzymatic initiation.91
An interested reader is guided to the following reviews for further insights on the latest developments in RAFT polymerisation.91–93
2.2. Enzymatic RAFT polymerisation
In RAFT polymerisations, HRP can utilise hydrogen peroxide as an electron acceptor to catalyse the oxidation of ACAC to produce ACAC radicals employed as radical initiators.94 When ACAC radicals are combined with an appropriate CTA and monomer(s), RAFT polymerisation can occur.90,95 Enzymes must retain activity during polymerisation and be compatible with the RAFT agent of choice for successful polymerisation.96 In addition, pH regulation of such systems is essential for enzyme stability and activity, as well as initiation efficiency (as the enol form of ACAC is responsible for radical generation) and to avoid hydrolysis of RAFT agents.95,97Despite tolerating a wide variety of functionalities, RDRP reactions are typically conducted under inert atmosphere due to being highly oxygen-sensitive.98 Oxygen species can quench carbon-centred radicals by generating peroxy radical species, which are ineffective at polymer propagation.60,64,98 Uyama et al. also demonstrated this through use of a bienzymatic GOx/HRP system for the FRP of phenols.40
Both Zhang et al.94 and Danielson et al.20,99 have demonstrated successful RAFT synthesis of both homo and block copolymers in water using HRP/ACAC systems with N,N-dimethylacrylamide (NN-DMA) as a representative acrylamide monomer, replicating conventional RAFT synthesis with typical azo initiators. Danielson et al. rapidly (<30 minutes) produced well-defined polyacrylamides with high conversion (>93%), as well as complex architectures including a protein(lysozyme)–polymer (PNIPAAam)conjugate using the H2O2/HRP/ACAC ternary system. Living characteristics (linear increase of molar mass with conversion) typical of RAFT synthesis were demonstrated with the HRP-initiated system, and the enzyme retained approximately 75% of its activity post-polymerisation.99
An et al. expanded on the early work of Uyama et al. using HRP in FRP, and utilised GOx in a GOx-HRP cascade reaction to generate H2O2 in situ for use in RAFT. This also produced poly(NN-DMA) with high conversion (92%) in ca. 35 minutes (Fig. 4).94
 | ||
| Fig. 4 Proposed mechanism of HRP initiated RAFT polymerisation. Pn & Pm represent propagating polymer chains whilst R and Z are substituents on the CTA. Reproduced from ref. 94, with permission from American Chemical Society, copyright 2015. | ||
More recently, An et al. synthesised ultrahigh molecular weight (UHMW) and multiblock polymers via oxygen-tolerant RAFT polymerisation using enzymatic cascade catalysis of HRP and pyranose-2-oxidase (P2Ox).100 P2Ox is more efficient at using O2 than GOx to generate H2O2 for ACAC radical production, due to having significantly lower Michaelis–Menten constant (KM) for D-glucose and O2, where the Michaelis–Menten constant is defined as the concentration of substrate at which the reaction rate is half its maximum value.100
Similarly, Tan et al. have also utilised a ternary approach using a GOx/H2O2/ACAC system for the preparation of nano-objects with different morphologies based on RAFT dispersion polymerisation of 2-hydroxypropyl methacrylate (2-HPMA) in water, with subsequent polymerisation-induced self-assembly (PISA) (Scheme 6).101
 | ||
| Scheme 6 HRP-initiated aqueous RAFT dispersion polymerisation of HPMA. Reproduced from ref. 101 with permission from Wiley, copyright 2018. | ||
Methoxy poly(ethylene glycol) (mPEG)-based macromolecular chain transfer agent (macro-CTA) was used to mediate the process and yielded high (almost 100%) monomer conversion within 30 minutes and produced spheres, worms and vesicles of mPEG-poly(HPMA) in an open vessel. A control synthesis without GOx was performed by deoxygenating the HRP-only system with nitrogen and, in this case, only 70% monomer conversion was achieved after 60 minutes, proving that GOx mediation provides a successful means of oxygen-tolerance during RAFT synthesis.101 Such mild conditions to create nano-objects render enzyme-mediated RAFT-PISA ideal for the incorporation/encapsulation of biologics such as DNA, RNA and proteins, whilst elimination of hazardous initiator species promotes safer synthesis. While it may seem that such mild synthesis is only achievable with hydrophilic monomers in aqueous conditions, GOx has been shown to be stable in a large range of organic/aqueous solvent mixtures whilst retaining high enzyme activity.102 In the case of HRP, it has been found that increasing organic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aqueous solvent ratio for 1,4-dioxane, ethanol, THF and chloroform has a significant effect on enzyme activity, with activity in THF being the worst.103 Future research in this area would benefit from the exploration of more organic solvent-tolerant alternatives to HRP, or engineering of HRP itself, in order to expand the scope of monomers and polymerisation techniques available for more widespread use.
:aqueous solvent ratio for 1,4-dioxane, ethanol, THF and chloroform has a significant effect on enzyme activity, with activity in THF being the worst.103 Future research in this area would benefit from the exploration of more organic solvent-tolerant alternatives to HRP, or engineering of HRP itself, in order to expand the scope of monomers and polymerisation techniques available for more widespread use.
For readers interested in more detailed examples and explanations, exploring different reaction conditions and possible applications, we refer to the recent work of Kumar et al., which covers e-RAFT in more depth.88
2.3 Atom transfer radical polymerisation (ATRP)
 | ||
| Scheme 7 ATRP Mechanism (note charges and counterions are omitted for simplicity). Pn, X, Mt, m, L and M represent the alkyl chain, halide, transition metal, oxidation state and monomer respectively., adapted from ref. 57, with permission from the American Chemical Society, copyright 2012. | ||
One limitation of early ATRP is the requirement for the high concentration of transition metal catalyst, often stoichiometric with respect to the initiator, in order to overcome radical termination. Whilst techniques to improve catalytic activity and subsequent purification have been employed, even more stringent conditions would be required to remove transition metals to sub-ppm levels for polymer use in biomedical or electronic applications.57,104 Furthermore, transition metal catalysts are difficult to remove post-synthesis, and prolonged and unsustainable purifications are required (e.g. silica columns).57
Classic examples of ATRP include the polymerisation of styrene, acrylates, and methacrylates, which are often catalysed by copper(I) complexes. This process can yield polymers with a degree of polymerisation (DP) reaching up to 100 and a dispersity (Đ) of almost 1 (monodispersity).105
Primarily, the use of enzymes in ATRP eliminates the need for toxic and non-renewable transition metal catalysts, whilst also providing a route of synthesis that is oxygen tolerant.89
Bruns et al. were the first to utilise haem-proteins in ARTP, when N-isopropyl acrylamide formation was catalysed by HRP with an alkyl bromide initiator under the conditions of ARGET (activators regenerated by electron transfer).106 ARGET ATRP involves the regeneration of the activator species during the polymerisation process, reducing the need for excess catalyst by employing non-radical forming reducing agents such as sodium ascorbate (Scheme 8).106,108
 | ||
| Scheme 8 HRP initiated ARGET ATRP of N-isopropyl acrylamide. | ||
In the study of Bruns et al., PNIPAAm was synthesised in pH 6.0 buffer solution, yielding Đ of 1.44 and Mn of 99.9 kDa after 2.5 h; such low dispersity indicates a controlled radical polymerisation and the polymerisation was shown to be pH dependent.106
The Bruns group explored the HRP-catalysed polymerisation of N-vinylimidazole (PNVIm), a reaction previously unachievable by conventional methods due to the lack of resonance stabilization in propagating radicals. This instability led to premature chain termination and undesirable chain transfer reactions.109 Whilst RAFT polymerisation had shown only limited success, attempts at chemical ATRP failed entirely. The successful enzymatic polymerisation (Mn = 2.5 kDa, Đ = 2.20) highlights the potential of enzymes as initiators where traditional chemistry falls short. Moreover, imidazole-based monomers and polymers exhibit strong metal-ion binding, stripping metal ions from traditional ATRP catalysts. Residual metal contamination can result in discolouration and undesirable material properties—particularly problematic in biomedical contexts (due to toxicity) and electronic applications (due to interference from ionic species). Notably, the Bruns group achieved the removal of HRP by filtration over neutral aluminium oxide, yielding entirely metal-free PNVIm.
Di Lena et al. demonstrated analogous ATRP synthesis using laccase to polymerise polyethylene glycol methacrylate (PEGMA) with high control by utilising ascorbic acid and an alkyl bromide. This process rendered poly-PEGMA with an Mn of 272 kDa and Đ of 2.43 after one hour, however conversion was only 20%. The proposed mechanism suggests that ascorbic acid reduces at least one copper centre within the laccase to Cu(I), which subsequently initiates polymerisation via halogen abstraction typical of ATRP activation.107
One interesting application of ATRPase activity lies in the work of Wang et al., who performed ATRP on the surface of an SiO2-Br nanoparticle (NP) with N-acryloyl-L-lysine as monomer. Utilisation of an enzyme here allowed for polymerisation of monomers containing carboxy groups, which is not suitable for traditional transition metal ATRP catalysis, further solidifying the validity of biocatalysis within this remit.110
Furthermore, contrary to transition metal catalysis, the metal centres in active sites of ‘ATRPases’ are tightly bound, limiting contamination in the resultant polymer, producing a cleaner, more environmentally friendly and less toxic product, satisfying multiple principles of green chemistry and allowing for a greater range of polymer applications.46,89 A clear illustration of this is the use of ATRPases in the synthesis of hydrogels for tissue engineering and drug delivery, where otherwise multiple post-polymerisation purification steps would be required for the removal of residual transition metal catalysts.
3. Ring opening polymerisation (ROP)
3.1 Chemical ROP
Ring opening polymerisation (ROP) is most commonly applied to cyclic ester (lactones), cyclic amide (lactams) and cyclic ether monomers. The majority of examples are chain growth mechanisms, which may be ionic, radical or metathesis based, however the mechanism of some ROP reactions is complicated and transcends the boundary of this simple definition.50,111 Similar to RDRP, ROP requires a catalyst (traditionally metal based) and initiator (commonly an alcohol). With chemical ROP of small/medium size rings, (e.g. ε-caprolactone, ε-CL) the driving force of polymerisation is the large negative enthalpy associated with the alleviation of ring strain.112 However, for large ring systems (e.g. macrolides), the polymerisation is entropically driven due to decreased ring constrain and, with classic organometallic catalysts, only slowly produces low molar mass polymers.112,113 More generally, organometallic complexes have been found to offer the best control over chemical ROP. Tin(II) octanoate [Sn(Oct)2], which is a toxic and harmful compound, is commonly used as a catalyst for the synthesis of polycaprolactone (PCL), however the reaction requires high temperatures which promotes both intra- and intermolecular esterification, increasing Đ. Furthermore, the metal catalyst is difficult to remove post-polymerisation.114 On the contrary, enzymatic ROP (eROP) offers a versatile, metal-free approach to the synthesis of ring-based monomers with varying size and functionality.3.2 Enzymatic ROP (eROP)
 | ||
| Scheme 9 Mechanism of lipase to produce aliphatic polyesters. | ||
The reader is directed to the following reviews for an in-depth discussion and history of eROP.21,116 The review of Engel et al. comprehensively compares both solution and bulk eROP of both substituted and unsubstituted lactones.116
The first studies of ε-CL and δ-valerolactone (δ-VL) eROP were carried out by Uyama and Kobayashi, in bulk with a range of lipases over a period of days.117 Lipase from Pseudomonas fluorescens (Pf) produced polyesters with high conversion from both monomers (PCL: 92% conv., Mn = 7.7 kDa, Đ = 2.4, PVL = 95% conv., 1.6 kDa, Đ = 2.4). This work was later expanded by them and demonstrated that: (1) Pf-lipase showed higher catalytic activity towards macrolides than it did with ε-CL, which opposes the trend in ring strain;118 (2) the rate of eROP of macrolides (namely pentadecalactone) could be dramatically increased, thereby increasing conversion, Mn and improving Đ, by switching to a solvent based system and immobilised lipase.119
Both substituted and unsubstituted lactones have been successfully polymerised via eROP, with ε-caprolactone and ω-pentadecalactone being the most widely investigated.116
Investigations have been performed into the nature of the initiator of eROP, as carefully designed initiators can provide routes to complex polymer architectures through end-group functionalisation. The Martinelle group utilised this approach by using 2-hydroxyethyl methacrylate (HEMA) as an initiator in eROP of both ω-pentadecalactone (PDL) and ε-CL using N435 (CaLB) in the hope of producing a polymer with HEMA end groups for later functionalisation (Scheme 10).120 However, their success was limited due to CaLB also catalysing the hydrolysis of the HEMA initiator, causing transesterification and a mixture of polymeric products for both monomers.120
 | ||
| Scheme 10 CaLB catalysed ROP of PDL (m = 11) and CL (m = 2), initiated by HEMA (product a). Transesterification products (b, c and d) formed from methacrylate transfer from HEMA end group of polymer to hydroxy end group, and (e) polyester transfer to the hydroxyl group of the residual 1–2-ethanediol end group. | ||
The Howdle group proposed a solution to this problem by replacing HEMA with its amide analogue, N-hydroxymethyl acrylamide (HEAA), to tune the polymerisation with N435 and minimise side reactions with the initiator (as transamidation is less likely to occur).121 Subsequently, free radical copolymerisation with polyethylene glycol methacrylate (PEGMA) was performed to create polymeric nanoparticles for drug delivery applications. Following this success, it is evident that further investigation into the use of amides with lipases could render promising results for the polymer community for the use of multifunctional molecules and final functionalised/active polymers.
A powerful use of eROP lies in its combination with other polymerisation techniques, both enzymatic and chemical, in the creation of novel functional polymers. Known as tandem polymerisation, subsequent polymerisation/functionalisation may be considered a cascade reaction if it occurs in a single pot without isolation or purification of intermediates. Cascade reactions, by combining multiple sequential steps in a single process without the need for intermediate purification, significantly enhance the efficiency of polymer synthesis, reducing energy consumption, waste generation, and the use of hazardous reagents, thereby advancing greener and more sustainable methodologies.
A comprehensive review of chemoenzymatic polymerisation using lipases is given by Yang et al.122 Heise et al., reported the first tandem chemoenzymatic polymerisation, by utilising a bifunctional initiator containing both hydroxy and activated bromide groups, to permit eROP of ε-CL followed by chemical ATRP of styrene in block copolymer synthesis in high yield (90–95%).123 A challenge lies in the design of a one-pot dual catalytic process without the isolation of intermediates, requiring compatibility of both systems. This has been achieved by kinetic compartmentalisation (different activation temperatures of the enzymatic and chemical catalysts, respectively)124 and with careful reaction design (Scheme 11). This is particularly the case in simultaneous chemoenzymatic eROP/ATRP processes, as the Cu catalysts required can result in enzyme inhibition.125 Simultaneous eROP/RAFT polymerisation has been successfully demonstrated in the synthesis of block copolyesters, retaining good control and enzyme activity without intermediate isolation and purification.126 Dove et al. also exploited this methodology in the synthesis of poly(ω-pentadecalactone)-b-poly(acrylate) diblock copolymers.127
 | ||
| Scheme 11 Combined enzymatic ATRP/eROP to produce general diblock copolyester through three routes. Route (A) Simultaneous eROP/ATRP. Route (B) eROP with bifunctional initiator, followed by ATRP. Route (C) ATRP with bifunctional initiator, followed by eROP. Reproduced from ref. 21, with permission from the American Chemical Society, copyright 2016. | ||
This chemoenzymatic combination strategy has been expanded to nonradical polymerisation in tandem eROP/ring-opening metathesis polymerisation (eROP/ROMP).128 This reaction, though, may not be considered a true cascade reaction as the polymeric precursors made via ROMP are isolated prior to the enzymatic step.
More recently, eROP of ε-CL using norbornene alcohol (NB) initiator and CaLB (N435) has been performed in continuous flow to form NB-PCL macromonomer, proceeding with metal-catalytic ROMP to produce polyNB-g-PCL bottlebrush polymers.129 A wholly enzymatic tandem, one-pot approach was demonstrated by the work of the Martinelle group, who performed eROP of ω-pentadecalactone (PDL) and 6-mercapto-1-hexanol as initiator, followed by functionalisation with γ-thiobutyrolactone or vinyl acrylate, all with CaLB (N435).130 Wu et al. have also presented a fully enzymatic tandem approach using two consecutive enzymes to prepare an optically pure polymeric prodrug. In this example, ε-CL was ring opened by CaLB, initiated by a bulky ibuprofen containing initiator, followed by chain extension by alkaline protease from Bacillus subtilis to catalyse further polycondensation and obtain a higher molar mass product.131 Whilst the hydrolase enzyme family also includes esterases and cutinases, the literature is dominated by the use of lipases. Only few reports of polyester synthesis using an esterase have been published, and these are limited to eROP of ε-caprolactone and δ-valerolactone.132–135
eROP should be seen as a powerful tool for producing hybrid oligomers/polymers, which have further utility in subsequent controlled radical polymerisation or other functionalisation processes (see section 5).136 This hybrid approach provides the possibility to achieve unprecedented architectures and chemical domains with controlled degradability and applicability in various fields. The use of eROP should be integrated into multiple hybrid reactions, which have so far been primarily combined by chemoenzymatic means.
Furthermore, combinations of multiple enzymes with multiple polymerisation mechanisms could be further explored, including the use of an initase, e.g. HRP, to catalyse RDRP following eROP to enhance the green credential of polymerisation processes in series as well as post-functionalisation polymerisations.
 | ||
| Fig. 5 Enantiomers of lactide monomers and polymers produced through ROP. | ||
PLA, a renewable and biodegradable aliphatic polyester, is extensively researched and emerges as a promising alternative to traditional petrochemical polymers like PET in consumer and biomedical applications.139
Whilst it can be made directly from the polycondensation of lactic acid, ROP of lactide in bulk is preferred industrially due to increased control over the polymerisation, which results in lower dispersity, higher molar mass and high levels of end-group fidelity. Tin(II) octoate is most often used an initiator for chemically synthesised PLA.137,138 ROP of lactide in bulk requires temperatures higher than those needed for other lactones (often above 100 °C) due to the high melting point of lactide. eROP of lactide has been explored in the literature using a variety of lipases, and there is much discussion around the choice of solvent (or bulk), the specific lipase employed, enzyme enantioselectivity, and is the subject of multiple reviews.116,140,141 For example, Omay et al. synthesised PDLLA using both free lipase and N435 in dry toluene at 80 °C, achieving molar mass (Mn) of 21 kDa and 26 kDa respectively, but at relatively low conversion (<45%).142 Previously, Matsumura et al. had reported no activity of N435 towards eROP of D,L-lactide when in bulk rather than in organic solvent.89 Evidently, reaction conditions and the selectivity of the lipase play an important role in eROP of lactides but, regardless, eROP of lactide is noted to be generally a slow process and may limit widespread adoption.137,143,144
 | ||
| Fig. 6 Structures of lactams and some representative nylons. | ||
Nylon 6, structurally similar to nylon 6,6, is easier to synthesise chemically from ROP of ε-caprolactam, anionic ROP is usually employed to carry out this reaction, whilst eROP of lactams offers a route of limiting organic reagents and hence toxic side-products.145 Many aliphatic polyamides are manufactured on an industrial scale, with nylon 6 being the dominant aliphatic polyamide commercially produced.146
First reports on eROP with both substituted and unsubstituted β-lactams using N435 in toluene were given by Kazlauskas and Loos groups and, whilst successful, only yielded low molar mass oligomeric polyamides (when reported).147,148 This is due to the low solubility of the nylon-3 polymer formed in toluene, as well as competition between chain-elongation and termination.147,148 The same effect was also noticed when the Loos group attempted enzymatic ring opening copolymerisation of ε-caprolactone and β-lactam using N435 to produce copolymers of PCL and poly β-lactam – whilst polymerisation was successful, only oligomeric products were also obtained.149 However, despite limited success in achieving high molar mass polyamides from eROP of lactides, N435 has also shown promise in synthesis of lactam precursors in the installation of epoxide on (+)-3-carene.149,150
Recent work by Benea et al. has involved the enzymatic synthesis of novel poly(ester amide)s using CaLB (N435) from ε-caprolactam (ε-CLM) and a range of biomass-derived hydroxyacids (HA) whilst investigating the effect of (co)monomer ratio and performing reactions both in bulk and toluene (Scheme 12).151
 | ||
| Scheme 12 Enzymatic linear co(polyester amide) synthesis of ε-caprolactam(ε-CLM) and a range of hydroxyacids. X and Y represent the number of hydroxy acid and ε-CLM units randomly incorporated in the copolymer structure, respectively. | ||
Broadly, polymer molar mass and dispersity were similar in both toluene and bulk; the most notable differences lie in the choice of HA, and copolymerisation of ε-CLM with a hydroxyacid improved conversion of ε-CLM relative to homopolymerisations alone (10%, DP = 3.0). In the cases of L-malic acid, 3-hydroxybutyric acid, copolymerisation in both bulk and toluene at ratios 2:1, 1:1 and 1:2, respectively, only achieved better monomer conversion, but low DP (<4). 16-hydroxyhexadecanoic acid performed the best of all HAs in terms of molar mass.
Considering the ability of lipases to ring open both lactones and lactams, an interesting direction for further exploration lies within copolymerisation149 and the development of a library of possible monomers to tailor final polymer mechanical properties and (bio)degradation capability.
4. Step growth polymerisation
Common step-growth polymers include polyesters, polyamides, polyureas, polycarbonates, polyurethanes and polysulphides.50 Polymerisation occurs step-wise and usually liberates a small molecule (e.g. water, polycondensation). However, some step-growth mechanisms do not produce a condensate, such as polyurethane production from a diisocyanate and a diol.50 For conformity to the broader literature, the following sections of the review are discussed by polymer functionality.4.1 Polyesters
Synthetic polyesters are abundant in the polymer industry, and most are derived from petrochemical monomers. One of the most familiar examples is the aromatic polyester PET, synthesised from ethylene glycol and terephthalic acid, commonly manufactured into plastic drinks bottles, and notably non-biodegradable.152 Chemically, polyesters are typically made from dicarboxylic acid/diols or diacyl chloride/diol (AA-BB polycondensation), as well as self-condensation of hydroxy acids (AB polycondensation), transesterification of diesters and ring-opening polymerisation of lactones (sections 3.2.1 and 3.2.2).52 Such synthesis often requires metallic catalysts and high temperatures from 150–280 °C which can lead to unwanted side reactions such as cross-linking or isomerisation, as well as exclusion as thermally sensitive moieties (vinyl, epoxy groups and biologics).153For all these limitations, the introduction of enzymatic step-growth catalysed processes may be considered revolutionary in reducing the use of pollutant and toxic catalysts, improving final degradability and minimising side reactions due to their high specificity and the mild conditions required. Polyesters are most commonly synthesised by lipases and, as previously mentioned in section 3.2, the most widely used is CaLB, a fungal lipase.19 Other lipases have been implemented in polyester synthesis and include porcine pancreatic lipase (PPL) and lipases from Candida rugosa (Cr), Candida cylindracea (Cc), Aspergillus niger (An), Penicillium roqueforti (Pr), Burkholderia cepacia (Bc) and Pseudomonas fluorescens (Pf).113
Additionally, the use of lipase aligns with the increasing use of biomass-derived monomers and the use of greener solvents.31,154,155 This strong interest in advancing green chemistry is increasingly apparent in the literature for the synthesis of bio-based polymers towards a circular economy (led by environmental changes, reduction of oil-based resources and the political recognition of environmental impact) (Fig. 7).156–159
 | ||
| Fig. 7 Biomass derived diacid/diol/polyol monomers for use in step-growth enzymatic esterification reactions, note the undefined stereochemistry of 1,4:3,6-dianhydrohexitols represents 3 different possible stereoisomers. | ||
 | ||
| Scheme 13 Lipase synthesised polyesters formed by (a) polycondensation of dicarboxylic acids or their esters with a diol (AA-BB polycondensation) and (b) self-polycondensation with oxyacids or their esters (AB polycondensation). | ||
In the case of aliphatic polyesters, a popular route to enzymatic step-growth polyester synthesis is to use activated esters such as dimethyl or divinyl esters rather than diacids, with the reaction proceeding via transesterification.162–167 In the case of divinyl, the unstable enol leaving group is irreversibly tautomerized to acetaldehyde and evaporates, driving the equilibrium towards the polymer product. Furthermore, evaporation (either at ambient pressure or under vacuum) of volatile byproducts avoids the energy intensive removal of water/condensate or the use of molecular sieves, normally adopted in case of alcohol or water removal.4 Whilst it is of concern that increasing acetaldehyde concentration decreases enzyme efficiency, it has been demonstrated that this effect on CaLB is limited, and the lipase remains stable.168
This approach can be particularly useful in some cases, particularly when the corresponding diacid does not react at all or reacts sparingly when catalysed by an enzyme. For example, the reaction of adipic acid with 1,4-butanediol catalysed by Lipase P from P. fluorescens in isopropyl ether did not yield any polymer (polybutylene adipate, PBA), yet PBA in moderate yield was synthesised when the analogous activated ester, divinyl adipate, was employed.169 The diacid reaction was also performed with CaLB in solvent (toluene) and in bulk and produced only short chain oligomers of PBA in both cases.170
Multiple research groups have proved that high molar mass aliphatic polyesters can be achieved using activated ester monomers in solution, without the need for vacuum, azeotropic distillation or molecular sieves.171,172
Several research groups have employed an enzymatic approach when producing biodegradable, amphiphilic polyesters bearing free secondary hydroxyl groups by using different activated diesters, polyol glycerol and catalysed by CaLB (N435) in bulk and vary organic solvents.171,173 The chemo- and regioselectivity of the lipase towards primary alcohols supresses branching through polymerisation occurring at the secondary glyceryl hydroxyl.23 This avoids low yield of linear polymer, tedious and unfavourable protection–deprotection via multi-step chemical processes, and prevents unwanted crosslinking when working with multifunctional reagents.173 It has been reported that less than 10% pendant branching was measured when polyglycerol adipate was synthesised at 50 °C in THF, starting from DVA and glycerol (Scheme 14), whilst greater molar mass (Mw = 32.5 kDa) and moderate dispersity (Đ) were also noted compared to synthesis at higher temperatures.171 The remaining secondary hydroxy group can subsequently be modified to improve drug encapsulation and polymer self-assembly for the production of NPs for drug delivery applications.174
 | ||
| Scheme 14 Poly(glycerol adipate) synthesis from divinyl adipate (DVA)/Glycerol, catalysed by CaLB (Novozym 435). | ||
Similarly, Russel et al. utilised N435 in the bulk polycondensation of divinyl adipate and 1,4 butanediol to produce polybutylene succinate (PBS), at a similar weight percentage of enzyme and the same temperature, yielding polymer with molar mass Mw = 23.2 kDa.175 Perin et al. studied the polycondensation of glycerol and a small library of aliphatic dicarboxylic acids catalysed by CaLB Polyesters structurally similar to glycerides, with different degree of branching, were produced.164 These polyesters vary in amphiphilicity and physical properties, ranging from hydrophilic and amorphous to hydrophobic and semicrystalline, depending on the diacid used. Higher molar mass polyesters are obtained in bulk reactions with longer diacids, such as sebacic and dodecanedioic acids, compared to solution reactions with shorter diacids, like adipic and suberic acids. The resulting polymer amphiphilicity allows for self-assembly of nanoparticles. The same research group has studied in detail the CaLB-catalysed polycondensation of glycerol and sebacic acid in polar solvents. It has been demonstrated that the enzymatic process involves the formation and consumption of glyceridic species and acyl migration, which esterifies the secondary hydroxyl of glycerol. Enzymatic esterification of primary hydroxyls occurs up to carboxylic acid conversions of 0.60–0.75, with acyl migration becoming the rate-limiting step beyond this point. The hydrogen bonding accepting ability of solvents dictates the enzymatic catalysis rate, while polymer–solvent interaction governs polymer chain growth, with acetonitrile and acetone showing different effects on polymer properties.163 The chemo- and regio-selectivity of the lipase towards primary alcohols supresses branching through polymerisation occurring at the secondary glyceryl hydroxyl.23
The examples above were chosen to demonstrate the regio- and chemo-selective benefits of using lipase in aliphatic polyester synthesis particularly with a trifunctional monomer. Linear polymers can be produced whilst avoiding protection/deprotection steps required for analogous organic methods. The reader is directed to the following reviews for a holistic overview of enzymatic polyester synthesis from bio-based building blocks.19,176
4.1.1.1 The special case of polyhydroxyalkanoates. Although this review primarily focuses on synthetic polymers, it is important to acknowledge the class of natural polyesters known as polyhydroxyalkanoates (PHAs) in the context of enzymatically catalysed polymer synthesis.
PHAs represent the only known example in polymer chemistry where an enzyme has naturally evolved to catalyse direct condensation polymerisation. These hydroxy fatty acid-based polyesters serve as intracellular energy and carbon storage materials in microorganisms and can be produced at scale via bacterial fermentation.177
When isolated, PHAs exhibit material properties similar to conventional petrochemical-derived plastics, but offer significant advantages including tuneable degradation rates, intrinsic non-toxicity, biocompatibility, and derivation from renewable resources. These features have driven their application in biomedical fields such as drug delivery and tissue engineering, as well as in emerging areas including agriculture and biofuel production.177 However, their broader industrial adoption is constrained by high production costs, largely due to energy- and material-intensive downstream processing and isolation of PHAs from the intracellular matric, and may be done by such as solvent extraction, enzymatic or chemical digestion, and mechanical cell disruption, all of which may pose challenges for efficient scale-up.178
Polyhydroxybutyrate (PHB) is the most commonly synthesised PHA due to its simple monomer structure, ease of microbial production, and the extensive availability of well-characterised production strains.
In vivo PHB synthesis proceeds through different pathways depending on the carbon source.14 When starting from glucose, the pathway involves its conversion into pyruvate, followed by transformation into acetyl coenzyme A (acetyl-CoA), from which PHB is synthesised in three enzymatic steps (Fig. 8). Acetyl-CoA is a central metabolite present in all forms of life and serves as a crucial precursor for the biosynthesis of PHB, as well as other valuable compounds such as n-butanol, isoprenoids, and fatty acids.179
 | ||
| Fig. 8 (A) Structures of general PHAs and PHB. (B) Acetyl-CoA generation from both acetyl-CoA synthetase and pyruvate dehydrogenase pathways. ATP, adenosine triphosphate; AMP, adenosine monophosphate. (C) Biosynthetic pathway of PBH production from acetyl-CoA: (i) condensation of two acetyl-CoA units into acetoacetyl-CoA via PhA (an acetyl-CoA acyltransferase), (ii) Acetoacetyl-CoA is reduced to (R)-3-hydroxybutybutyryl-CoA via PhaB (an acetoacetyl-CoA reductase), through the oxidation of one equivalent of NADPH cofactor. (iii) PhC (a PHA synthase) irreversibly polymerises (R)-3-hydroxybutyryl-CoA molecules to form PHB, whilst releasing acetyl-CoA molecules.180 | ||
Scaling up microbial synthesis of polyhydroxyalkanoates (PHAs) offers a promising bioplastic alternative, but challenges such as low yield, high production costs, and complex intracellular regulation hinder industrial viability. Whilst the production cost of conventional plastics is approximately $1250 USD per tonne, the cost of producing PHAs remains significantly higher, ranging from $4000 to $15000 per tonne.181
From a sustainability standpoint, the process is further constrained by the intensive use of energy and costly cofactors (e.g., ATP, NAD(P)+, CoA), as well as the requirement for additional coenzymes and regeneration systems to maintain metabolic balance.14,182 While advances in strain engineering, alternative substrates, and extraction methods have made progress, cell-free in vitro systems are emerging as a more controllable and potentially more sustainable approach by decoupling PHA biosynthesis from microbial growth, reducing energy demands, and enabling targeted cofactor recycling.182–184
Alkotaini et al. have developed an innovative NADH regeneration method that bypasses competition with common food-based carbon sources such as glucose and lactate. Their bioelectrocatalytic strategy enables the reduction of NAD+ to NADH directly on a glassy carbon electrode, facilitating the in vitro synthesis of PHB while eliminating the high costs associated with glucose dehydrogenase and glucose.180
Another recent advancement involves the creation of an in vitro synthetic enzymatic biosystem (ivSEBs) capable of producing PHB from acetyl-CoA without requiring ATP, using starch-derived maltodextrin as the sole substrate.185 However, although this method reduces ATP dependency, it necessitates 17 individual enzymes, which may present significant energetic and practical challenges, potentially limiting its industrial adoption in sectors currently dominated by petrochemical polymers.
Whilst these advances are promising, further research is needed to simplify and improve in vitro biosynthesis systems to make them more efficient and cost-effective for industrial use. Expanding these approaches beyond PHB to other PHAs with varied monomers could enhance material diversity and application potential. Continued work on enzyme engineering, cofactor recycling, and substrate optimization will be key to driving industry adoption of sustainable bioplastics.
A common way to incorporate an aromatic component is to use an aromatic (rigid) diol with an aliphatic (flexible) diacid/diester (or vice versa), a so called ‘hard/soft’ approach. Furthermore, enzymatic aromatic polyester synthesis is typically performed in organic solvent rather than bulk polymerisation that is often seen for aliphatic polyesters, as the higher temperatures required to melt aromatic monomers is often above the optimum temperature of the enzyme. However, (semi)aromatic polyesters have recently been synthesised in bulk using CaLB (N435) at 70 °C.188
Uyama et al. combined the activated aromatic diester divinyl isophalate and 1,6-hexanediol using a range of different lipases of different origins.189 In toluene, CaLB rendered a polymer with the highest yield and molar mass (74%, Mw = 8.8 kDa), followed by lipases from M. meihei and P. fluorescens, with yields of 40% and 30%, and molar mass of 3.1 kDa and 4.3 kDa, respectively.189 As CaLB was most successful, it was used to catalyse the reaction of divinyl terephthalate in the same study with a range of 1,n-alkanediols, of which 1,6-hexanediol was the most successful, achieving a yield of 72% and molar mass of 2.9 kDa. Generally, polymers employing a high level of aromaticity often have lower molar mass due to increasing hydrophobicity of the growing chain during synthesis, depending on the solvent system employed. In this case, polymers containing divinyl terephthalate generally had lower molecular mass than the isophthalate analogue (Fig. 9), as the solubility of the para-substituted diester is lower.
 | ||
| Fig. 9 Structures of divinyl isophthalate and divinyl terephthalate. | ||
Although the diol component can contain an aromatic moiety, this study found it was a less successful route, with only 1,4-benzenedimethanol reacting with divinyl sebacate in the presence of CaLB, whilst dihyroxybenzenes, catechol, resorcinol, and hydroquinone did not react.189
Pellis et al. performed a similar reaction, using the same lipase with dimethyl sebacate in place of the divinyl analogue. Reaction parameters and resultant polymer properties can be seen in Table 2 below.
| References | mmol monomer (monomer ratio) | Solvent | CaLB | Vacuum | Temp. (°C) | Duration (h) | Mw (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|
| Pellis (2020) | 8.0 (1:1) |
Diphenyl ether | Immob. | Yes | 85 | 48 | 31.1 | 4.6 |
| Uyama (1999) | 2.0 (1:1) |
Toluene | Free | No | 75 | 90 | 10.5 | — |
Notably, Pellis et al. achieved a higher molar mass despite the liberation of methanol rather than acetaldehyde by using a dimethyl rather than divinyl ester, which may be explained by the employment of higher temperature and a vacuum to promote evaporation of methanol and hence drive the equilibrium towards the product. However, aromatic vinyl esters have been found to suffer from long-term instability and have still not yet been successfully commercialised, limiting their implementation.189
More recently, multiple groups (notably Loos & Pellis) have had more success by using furan-based monomers as replacements for the terephthalic acid (TA) component in aromatic polyesters and, by doing so, through the less common route of aromaticity in the diol, which is also catalysed by CaLB (N435).190–192 Furthermore, polymerisation of 2,5-furandicarboxylic acid (FDCA) monomers/derivatives via enzymatic catalysis allows for lower reaction temperatures than chemical methods (typically multi-stage polymerisation processes using metal oxide catalysts, above 190 °C),193 thus enhancing sustainability through reduced energy consumption and preventing thermal decomposition of the monomer.194
In the same study as the dimethyl sebacate/1,4-benzene dimethanol polymerisation, Pellis et al. studied the polycondensation of a range of aliphatic methyl esters with furan containing diols 2,5-bis(hydroxymethyl)furan and 3,4-bis(hydroxymethyl)furan (2,5-BHMF & 3,4-BHMF, Scheme 15).190 This study built on earlier work by the Loos group, which had previously only utilised the 2,5-isomer.192
 | ||
| Scheme 15 Enzymatic polycondensation using either furanic diols or methyl diesters, from the work of Pellis et al.190 and Silvianti et al.195 | ||
Lower polymer molar mass and recovered yield was achieved with all diesters when 3,4-BHMF is used as the diol, therefore CaLB may be more selective for the 2,5 substituted furan. However, polyesters derived from 3,4-BHMF exhibited higher thermal stability. The group of Loos expanded on this further by introducing the furanic component within the diester also, via bio-based dimethyl 2,5- (and 2,4)-furandicarboxylate (2,5- and 2,4-DMFDCA) and aliphatic diols ranging from 6 to 12 methylene units in length (Scheme 15).
The effect of isomeric substitution was shown to influence both the DP, crystallinity and architecture of the final polymers, with CaLB preferentially polymerising the asymmetrical 2,4-DMFDCA isomer due to preferential steric interactions in the enzymes active site. Contrastingly to the 2,5-isomer, the 2,4 diester showed less cyclic polymer species in the final product, again due to monomer asymmetry. The work of the Loos group demonstrated that switching from only an aliphatic diol to a combination of aromatic/aliphatic diols can increase the resulting polymer molar mass. We speculate that this is likely due to more favourable interactions with the hydrophobic pocket of the enzymes active site with the aromatic furanic species. Additionally, it has been shown that 2,5-DMFDCA can be polymerised with both aliphatic diols (methylene chain length ranging from 8 to 12) and 2,5-BHMF using CaLB (N435) in a range of biobased solvents, namely p-cymene, pinacolone and D-limonene.196 Among these, p-cymene yielded the most favourable results in terms of polymer yield, molar mass, and visual appearance of the resulting materials. This enhanced performance is attributed to its relatively high logP value (3.47), which likely reduces enzyme deactivation by limiting solvent–enzyme interactions. Moreover, it is hypothesised that the aromatic nature of certain solvents may play a protective role by shielding the active site of CaLB from interactions with aromatic repeat units in the polymer, thereby mitigating product–enzyme interactions and potential polymer degradation. Such advancements in the use of biobased solvents are significant not only from a sustainability standpoint, but also in contributing to a deeper understanding of how solvent selection influences the physicochemical properties of the resulting polymeric materials.
Alternatively, these reactions may be prepared in bulk, however this often limits thermal and mechanical properties of resulting linear aromatic polyesters due to their low molar mass and melting points.188,197 Therefore, the work of the Loos group has recently advanced previous work on Diels–Alder chemistry of BHMF-based polyesters197,198 in order to create covalent adaptable networks (CANs) with a biobased bismaleimide through [4 + 2] cycloaddition between the furan and maleimide (Fig. 10). This approach facilitates the dynamic breaking of covalent crosslinking bonds, thereby enabling the recycling of polymeric materials. The use of enzymatic polymerisation with bio-based monomers in bulk, coupled with reversible cross-linking, offers a promising pathway to produce sustainable, biobased commodity plastics. However, this approach could benefit from further optimisation, particularly in terms of reducing reaction times and temperatures as well as use of solvent for isolation and purification. An interested reader is directed to the review of Annatelli et al. for further insights into alternative furanic monomers for biobased polyesters.199
 | ||
| Fig. 10 (A) Enzymatic polymerisation of 1,5-BHMF with a range of aliphatic dimethyl esters in bulk. (B) Reaction (via solvent casting) of the BHMF-based polyesters with a maleimide group. (C) Schematic overview of the thermoreversible Diels–Alder cross-linking of furanic polyester units with the bismaleimide units. Reproduced from ref. 197, with permission from the American Chemical Society, copyright 2025. | ||
In addition to solvent selection, an emerging green strategy in enzymatic polyester synthesis involves the utilisation of biomass-derived monomers, particularly those sourced from lignin, to introduce aromatic units into the polyester backbone. Lignin, a complex and abundant aromatic biopolymer, serves as a key structural component of plant cell walls and represents a valuable renewable feedstock for the development of functionalised polyesters.
Often through bioconversion by metabolically engineered organisms, lignin can yield monomers 2,4-pyridinedicarboxylic acid/2,5-pyridinedicarboxylic acid, which are analogues of terephthalate.200,201 Pellis et al. successfully synthesised the two pyridine diesters with a range of diols (1,4-butanediol, 1,6-hexanediol and 1,8-octanediol) in both solventless and diphenyl ether systems, catalysed by CaLB (Scheme 16).202
 | ||
| Scheme 16 Polycondensation of 2,4 (a) and 2,5-dipyridinecarboxylic acid (b) with differing chain length alkyldiols.202 | ||
In this case, solution phase synthesis produced higher molar mass polymers as the reaction media would solidify during the bulk polymerisation process, as expected molar mass increased with increasing length of the diol. The two pyridine-based regioisomers generated polyesters with similar molar mass and dispersity than their benzyl analogues, suggesting their suitability as a replacement for terephthalate-based polymers. Interestingly, thermal characterisation showed that 2,4-pyridinedicarboxylic acid and analogous diethyl isophthalate produced amorphous polymers whilst the 2,5-derivatives were more crystalline, independent of the diol used. Similarly to the examples in section 4.1.1, CaLB (N435) has been employed in the synthesis of poly(cresyl adipate-cohexyl adipate) (PCHA) to render a semi-aromatic polyester with pendant hydroxy group remaining unreacted, which is unattainable through traditional metal-based catalysis. The molar mass of the resulting polymer was limited, achieving a maximum of Mw = 5.6 kDa, Mn = 2.7 kDa and Đ = 2.1.203
4.2 Polyamides
There is a vast abundance of polyamides in industry, with the polyamide market size valued at $32 bn in 2021,204 indicating that there is a large market to which enzymatic synthesis of polyamides could contribute. Lower prevalence in literature (compared to polyesters) may be attributed to the high melting temperature (Tm) and the poor solubility of polyamides in many organic solvents.146 Similarly to polyesters, CaLB appears to be the most prevalent enzyme employed in the less reported enzymatic polyamide synthesis. Despite the vast array of literature on the use of lipases such as CaLB for polyester synthesis (section 4.1.2), fewer examples are present when it comes to polyamides.205Despite these obstacles, it has been shown that CaLB (N435) catalysed the polyamidation of diethyl sebacate and 1,8-diaminooctane in diphenyl ether at a range of temperatures, with highest conversion of 65% at 90–110 °C.206 Even when preheating N435 up to 150 °C prior to polymerisation, amide bond conversion remained at 50%, proving the robustness of the immobilised lipase at higher temperatures and therefore its potential in high temperature polyamide synthesis.
In the case of nylon-6,6, in particular, there is much focus on the monomers employed for polyamide synthesis, such as adipic acid, 1,6 hexamethylenediamine (for nylon-6,6) and 6 aminocaproic acid (ε-caprolactam precursor), rather than the synthesis itself (established in the early 1930s by Carothers of DuPont).44 This is due to the fossil-based nature of the monomers which has piqued interest in their biosynthetic (including enzymatic) production from renewable carbon sources, and several biobased polyamides are commercially available.146,207–209
Despite the lower prevalence in literature, polyamides have been produced through enzymatic step-growth polymerisation; Cheng et al. were the first to synthesise high molar mass aliphatic polyamides from lipase, using a range of lipases, divinyl esters and amine monomers.210 Analogously to polyesters, self-polycondensation to produce polyamides can also be produced by A–B type monomers (ω-aminocarboxylic acids), and CaLB has also been successfully used in such polymerisation.210–212
Since then, most investigation into the enzymatic synthesis of polyamides has been performed by the Loos group, whilst CaLB remains the most popular enzyme used for such synthesis.146,149,159,192,205,211 However, the group were the first to synthesise polyamides through polycondensation using cutinase from Fusarium solani pisi immobilised in two ways (adsorption onto Lewatit, the same acrylic resin CaLB is immobilised onto in N435, and as a cross-linked enzyme aggregate, CLEA), with diethyl sebacate and a range of aliphatic diamines (Scheme 17).213 Cutinases are hydrolases, the same enzyme family as lipase, and contain the typical Ser-His-Asp catalytic triad in their active site, therefore activity in polymerisation reactions can be expected to be very similar – a comprehensive review of their activity in polymerisation compared to CaLB was published by the Gardossi group.214
 | ||
| Scheme 17 Nylon synthesis from diethyl sebacate and 1,4-butane diamine (m = 2), 1,6-hexanediamine (m = 4) and 1,8-diaminooctane (m = 6) from Stavila et al.213 | ||
Highest conversion in the one-step polycondensation (no second vacuum step) was shown by CLEA cutinase and demonstrated the highest monomer conversion for both m = 4 and m = 6 diamines of the three enzyme systems (Scheme 17). The conversion was lowest for m = 2, demonstrating that both cutinase and N435 possess higher activity towards longer chain diamines, in agreement with previous literature.213 Conversion for m = 6 (diethyl sebacate) can be directly compared to previous work by the Landfester group,206 who noted higher monomer conversions (>65%) with their one-step polymerisation using N435 at 100 °C compared to any of the cutinase systems performed at 70 °C, under the same reduced pressure (100 mmHg), suggesting some temperature dependence on the reaction kinetics independent of enzyme activity.
To further improve conversion, inspiration could be taken from analogous polyester synthesis by using a vinyl ester (e.g. divinyl sebacate) to produce volatile byproducts such as acetaldehyde and drive the equilibrium towards polymerisation, eliminating the need for successive vacuum steps, prolonged reaction times and use of molecular sieves.
Whilst enzymatic polycondensation of aliphatic polyamides holds promise, it faces significant challenges in competing with traditional chemical routes industrially. The use of acidic monomers that inherently function as catalysts, combined with the ability to conduct polymerisation in bulk, makes the enzymatic approach less favourable from both a cost and sustainability standpoint. This may also justify the limited body of literature available in this space, as the economic and practical advantages of conventional methods remain dominant here.
Following this, Loos et al. expanded on their earlier work by using immobilised cutinase systems to the synthesis of aliphatic oligoamides using p-xylylenediamine and dimethyl terephthalate as aromatic monomers, and diethyl sebacate (DES) and 1,8-diaminooctane as aliphatic monomers (Scheme 18).218
 | ||
| Scheme 18 Mixed aliphatic/aromatic polyamides synthesised by both cutinase and lipase systems.218 | ||
No amide bonds were formed between aromatic and aliphatic monomers with any enzyme system when using the same conditions as the previous study.213 Therefore, reaction duration and temperature were increased; even with this change, the best conversion for both 1 and 2 was with CaLB, and previously well-performing immobilised cutinase only achieved 18% and 3% conversion for the one-step synthesis of 1 and 2, respectively. However, CLEA cutinase did result in the same or higher degree of polymerisation (DP) despite showing lower monomer conversion. Synthesis 3 in Scheme 18 was oligomeric, with the steric hindrance of the diamide product that had already formed in the enzyme's active site preventing higher molar mass products.218
More recently, successful synthesis of furan-containing semi-aromatic polyamides has been demonstrated, using 2,5-DMFCA and several heteroatom-containing aliphatic diamines (Scheme 19).219
 | ||
| Scheme 19 FDCA-heteroatom containing polyamides synthesised using N435.219 | ||
Whilst the polyphthalamides were successfully synthesised by N435, the highest molar mass (Mw) achieved was 16.6 kDa, and this was only in moderate yield (37%). Higher yields were noted for DETA and EDDA, however such polymers were more oligomeric in length. Highest catalytic activity was found towards diamine monomers containing ether rather than amine heteroatom linkages, and synthesis in bulk rather than toluene was generally preferred.
More recent comparable work by Yang et al. involves using 2,5-DMFDCA and 1,10-diaminodecane in bulk using TBD (triazabicyclodecene) rather than lipase, and achieved higher molar mass polymers (Mw up to 66.1 kDa).220
These cases demonstrate limited success in the use of lipase for aromatic polyamide synthesis, as polymers synthesised to date suffer from relatively low monomer conversion and low molar mass. Similarly to analogous aromatic polyester synthesis (section 4.1.2), the current literature presence for enzymatically synthesised polyamides is limited to semi-aromatic (at least one aliphatic monomer), rather than fully aromatic polyamides (e.g. Kevlar), due to the necessity of employing a primary diol/diamine to enable catalysis by the lipase.
5. Chemoenzymatic methods
Modification and functionalisation of polymers using enzymes is widely reported in the literature, however less so for synthetic polymers than natural polymers, e.g. polysaccharides, where lipases are commonly employed to graft onto cellulosic hydroxy groups.21,221 Modifying pendant groups on both natural and synthetic polymers provides a means of accessing different physicochemical properties, expanding the field of polymer application.32 Post-polymerisation enzymatic modification of synthetic polymers can be achieved by a variety of enzyme classes and through a range of different routes, such as end group modification, grafting onto the polymer backbone and crosslinking/Michael addition of unsaturated polymers.32,222,223 The use of enzymes to aid in post polymerisation modification (PPM) avoids unwanted side reactions that can occur from the harsh conditions required for more classical chemo-modification, such as isomerisation and crosslinking (when undesired).222 Most literature exploring these methods employ a chemo-enzymatic approach, using enzymes for polymer synthesis and conventional chemistry for the modification step.222 For example, the work of Wu et al. demonstrated that N435-catalysed synthesis of azido-functional polyesters, followed by subsequent azide–alkyne click chemistry for the installation of alkene-end functionalised PEG, to create acid-degradable amphiphilic graft copolymers (Scheme 20).224 | ||
| Scheme 20 Azido-functionalised polyesters synthesised by N435 and PPM performed via azide–alkyne click chemistry, adapted from ref. 224, with permission from the Royal Society of Chemistry. | ||
Additionally, alkene functionality on polymers provides vast opportunity for modification and includes crosslinking (thermo- and photo-), Michael additions (aza- and thio), epoxidation and more. Pendant C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds can also provide a handle for functionalisation orthogonal to the polymer backbone – the use of itaconic acid as a diacid in lipase-catalysed polyester synthesis is a popular bio-mass derived monomer for this approach due to its dual functionality, the CC of which has been used for cross linking or Michael addition of pendant groups orthogonally.222,225
C bonds can also provide a handle for functionalisation orthogonal to the polymer backbone – the use of itaconic acid as a diacid in lipase-catalysed polyester synthesis is a popular bio-mass derived monomer for this approach due to its dual functionality, the CC of which has been used for cross linking or Michael addition of pendant groups orthogonally.222,225
This has also been achieved by the quantitative end-functionalisation of PEG catalysed by N435 to install end-vinyl groups that can later be utilised in further functionalisation and additive manufacturing.223
6. Considerations for enzymatic polymerisation
From a sustainability perspective, future enzymatic polymerisation processes must meet several criteria to be considered genuinely “green”. These include (but are not limited to) the use of renewable monomers, valorisation of waste materials, employment of green or solvent-free media, low energy input, reduced or eliminated isolation and purification steps, efficient catalyst reuse, and the production of biodegradable materials. In this review, we highlight representative examples where enzymatic catalysis has been integrated with one or more of these principles, with the aim of supporting the development of more sustainable and efficient strategies in green polymer synthesis.Whilst enzymatic catalysis emerges as a particularly promising approach, its integration into polymer chemistry demands careful evaluation of factors such as enzyme reusability, stability over multiple cycles, loading, cost, and operational requirements. Assessing the sustainability of enzymatic processes requires a holistic methodology that accounts for energy efficiency, waste reduction, carbon footprint, and economic viability. Whilst standardised metrics for enzyme-based green chemistry are still in development, frameworks such as Life Cycle Assessments (LCAs), ISO standards, and established green chemistry metrics offer a robust basis for evaluation.
Therefore, future developments should focus on advancing these aspects to more effectively harness enzymatic catalysis in synthetic polymer production and to deepen our understanding of its advantages over traditional catalytic methods.
Some key aspects regarding the down-stream processing of enzymatically catalysed polymerisations, in particular enzyme separation and reuse in multiple cycles, are discussed in sections 6.1 and 6.2.
6.1 Enzyme separation
Commercially available, isolated enzymes can be used in either their free (homogenous) or immobilised (heterogeneous) forms. A key consideration is the separation of the enzyme from the polymer product post-polymerisation, as well as the retention of its catalytic activity for potential reuse. The most common strategy is heterogeneous application—immobilising the enzyme on a solid support (e.g., CaLB as N435)—which facilitates removal via simple filtration. However, the high viscosity of polymer products, particularly in bulk polymerisations, often necessitates the addition of solvent to aid filtration, which is undesirable from a green chemistry perspective.In contrast, recovering free enzymes is more challenging due to their similar molecular weight to the polymer chains, making size-based separation methods—such as precipitation, ultrafiltration, chromatography, or dialysis—less effective. Some success has been reported using filtration over neutral aluminium oxide.109 Whilst it may be tempting to argue that residual enzymes, being chemically benign and present at low concentrations, could be tolerated in the final material, this is not universally applicable. In particular, metalloenzymes may impart undesirable properties to the polymer, such as conductivity or toxicity, which are especially problematic in biomedical or electronic applications.
6.2 Enzyme reuse
Enzyme reuse is a critical factor in evaluating the sustainability and economic viability of enzymatic polymerisation processes. Again due to ease of separation, immobilised enzymes are most easily reused – the work of Lentz et al. demonstrated no loss in CaLB's reactivity over 10 reaction cycles in the eROP of ε-CL using HEMA (Fig. 11) (see section 3.2.1).121 Nasr et al. also noted similar robustness of N435 upon successive polymerisation cycles of the PC between 1,6-hexanediol and diethyl adipate.226 | ||
| Fig. 11 Mean percentage conversion of CL and HEAA after 5 h of reaction (65 °C, 2-MeTHF, 10 wt% N435, ε-CL concentration of 0.143 g mL−1) whilst recycling the N435 beads (experiments run in triplicate). Adapted from ref. 121, with permission from the Royal Society of Chemistry, copyright 2022. | ||
To the best of the authors knowledge, analogous studies have not been completed for the reuse of free, homogenous enzymes in polymerisation reactions, likely because of their difficulty in separation and recovery. Although enzymes such as HRP which are commercially available as lyophilised powders have been immobilised and applied successfully in the polymerisation of ethylene glycol dimethacrylate (EGDMA) (HRP on Si wafers),227 and phenol (HRP on calcium alginate beads) although the latter showed more effective polymerisation when free rather than immobilised.228
7. Outlook
The rapid evolution of enzymatic polymerisation is opening new avenues for the design of sustainable materials. As outlined throughout this review, significant progress has been made in developing greener processes by leveraging enzyme selectivity, mild reaction conditions, and the potential for renewable feedstock integration. However, to realise the full potential of enzymatic polymerisation in industrial and high-performance applications, further innovations in process design, enzyme engineering, and system integration are required. In this context, the comprehensive review by Velasquez et al. provides an excellent overview of the broader challenges associated with the implementation of biobased and biodegradable polymers (BBPs) for a more sustainable future.229 Building on this foundation, an emerging and particularly exciting area within the literature is the chemoenzymatic synthesis and modification of polymers, as highlighted in sections 3.2 and 5 of this review. A promising extension of this strategy lies in the fully enzymatic, multi-step synthesis of complex macromolecules through biocatalytic pathways alone.Over the past decade, systems biocatalysis—the in vitro orchestration of enzyme cascades—has gained momentum in polymer chemistry. These systems enable one-pot syntheses without the need for intermediate purification, offering advantages in efficiency and sustainability. For further insights, readers are referred to recent reviews in this area.230–232 In polymer production, this approach has been exemplified by the work of the Bornscheuer group, which reported the enzymatic synthesis of (oligo)caprolactone from cyclohexanol using a cascade of alcohol dehydrogenase (ADH), cyclohexanone monooxygenase (CHMO), and lipase.233 Although this yielded only low-molecular-weight products due to compatibility issues among the enzymes under green conditions, subsequent optimisation via a fed-batch process improved molar mass—albeit still requiring extraction of an intermediate (6-hydroxyhexanoic acid).234
In a related development, a chemoenzymatic pathway employing a three-step, lipase-mediated reaction from cyclohexanone has been reported. This system utilized immobilized Trichosporon laibacchii lipase to catalyse the in situ formation of peracids, enabling Baeyer–Villiger oxidation to ε-caprolactone (ε-CL), followed by enzymatic ring-opening polymerisation (eROP) to polycaprolactone (PCL). This approach capitalized on the compatibility of reaction conditions and the catalytic promiscuity of the enzyme, thereby reducing both the financial and environmental costs of multistep chemical synthesis while improving overall process efficiency.
A similar strategy—employing a single enzyme for successive or simultaneous steps—was demonstrated by Thompson et al., who combined polycondensation (PC) and ROP mechanisms using Candida antarctica lipase B (CaLB) to tailor the thermal and mechanical properties of amorphous poly(glycerol adipate)-based polymers for drug-loaded microspheres.235 A more intricate example was reported by Benea et al., who synthesised novel polyesters and poly(ester amide)s from fully bio-resourced monomers via sequential eROP/PC steps, using a single enzyme, CaLB (N435). Architectural variations afforded by this approach enabled tuneable biodegradability and material performance.
The recent expansion of enzymatic polymerisation into poly(ester amide) synthesis is of particular note. These hybrid materials combine the desirable properties of polyamides (mechanical strength, thermal stability, toughness, flexibility) and polyesters (biodegradability, low glass transition temperature, non-toxicity), rendering them promising candidates for biomedical, agricultural, and high-performance material applications. Their improved solubility, compared to polyamides alone, further enhances their processability.217
Looking ahead, the development of both random and block (ter)polymers via fully enzymatic, multi-step strategies represents a compelling avenue for future research. These complex architectures could be tailored for specific end-uses in medicine, cosmetics, or sustainable packaging. Using a single, multifunctional enzyme and appropriately designed monomers for multiple sequential steps—particularly when the enzyme is immobilised and reused—may drastically reduce reliance on diverse catalysts and simplify process logistics. Further advancements in enzyme design, particularly de novo design, could provide a route towards this, much like it has for other serine hydrolase lipases similar to CaLB that have been designed computationally.236
These advances collectively highlight the transformative potential of enzyme-driven polymerisation strategies. Despite challenges in enzyme compatibility, process integration, and scalability, the convergence of chemoenzymatic and fully enzymatic techniques marks a significant step toward sustainable polymer synthesis. As the field continues to evolve, enzyme-enabled methodologies are poised to play a crucial role in meeting the material demands of a greener future.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
EGD acknowledges EPSRC/SFI CDT in Sustainable Chemistry – Atoms 2 Products (EP/S022236/1) for their funding support. VT and KEA would like to thank the University of Nottingham for their Nottingham Research Fellowships and Anne McLaren Research Fellowship.References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford, 1997, Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8 Search PubMed.
- H. Feldmann, in Yeast: Molecular and Cell Biology, ed. H. Feldmann, Wiley, Weinheim, 2nd edn, 2012, ch. 1, p. 17 Search PubMed.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS PubMed.
- J. Chapman, A. Ismail and C. Dinu, Catalysts, 2018, 8, 238 CrossRef.
- K.-E. Jaeger, T. Eggert, A. Eipper and M. T. Reetz, Appl. Microbiol. Biotechnol., 2001, 55, 519–530 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France and A. P. Green, et al., Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore and K. Baldenius, et al., Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- R. Buller, S. Lutz, R. J. Kazlauskas and R. Snajdrova, et al., Science, 2023, 382, eadh8615 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff and J. C. Moore, et al., Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- H. Guo, Y. Zhao, J.-S. Chang and D.-J. Lee, Bioresour. Technol., 2023, 367, 128252 CrossRef CAS PubMed.
- CARBIOS, CARBIOS and De Smet Engineers & Contractors enter engineering partnership for construction of world's first PET biorecycling plant, https://www.carbios.com/newsroom/en/carbios-and-de-smet-engineers–contractors-enter-engineering-partnership-for-construction-of-worlds-first-pet-biorecycling-plant/, 15 February 2024, (accessed 24/02/2024).
- S. Eco, Samsara Eco and NILIT team up to build world-first nylon 6,6 recycling facility, https://www.samsaraeco.com/news/samsara-eco-and-nilit-team-up-to-build-world-first-nylon-66-recycling-facility, 27 August 2024, (accessed 24/04/2025).
- H. Dong, X. Yang, J. Shi and C. Xiao, et al., Polymers, 2023, 15, 2333 CrossRef CAS PubMed.
- M. Faijes and A. Planas, Carbohydr. Res., 2007, 342, 1581–1594 CrossRef CAS PubMed.
- C. Jönsson, R. Wei, A. Biundo and J. Landberg, et al., ChemSusChem, 2021, 14, 4028–4040 CrossRef PubMed.
- A. K. Singh, H. M. N. Iqbal, N. Cardullo and V. Muccilli, et al., Int. J. Biol. Macromol., 2023, 242, 124968 CrossRef CAS PubMed.
- M. Kumar, R. Rathour, R. Singh and Y. Sun, et al., J. Cleaner Prod., 2020, 263, 121500 CrossRef CAS.
- V. Hevilla, A. Sonseca, C. Echeverría and A. Muñoz–Bonilla, et al., Macromol. Biosci., 2021, 21, 2100156 CrossRef CAS PubMed.
- A. P. Danielson, D. B. Van-Kuren, J. P. Bornstein and C. T. Kozuszek, et al., Polymers, 2018, 10, 741 CrossRef PubMed.
- S.-i. Shoda, H. Uyama, J.-i. Kadokawa and S. Kimura, et al., Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- L. Gustini, B. A. J. Noordover, C. Gehrels and C. Dietz, et al., Eur. Polym. J., 2015, 67, 459–475 CrossRef CAS.
- C. Ortiz, M. L. Ferreira, O. Barbosa and J. C. S. dos Santos, et al., Catal. Sci. Technol., 2019, 9, 238–242 RSC.
- B. Chen, J. Hu, E. M. Miller and W. Xie, et al., Biomacromolecules, 2008, 9, 463–471 CrossRef CAS PubMed.
- X. Tian, S. Zhang and L. Zheng, Enzyme Microb. Technol., 2016, 84, 32–40 CrossRef CAS PubMed.
- M. Hans, H. Keul and M. Moeller, Macromol. Biosci., 2009, 9, 239–247 CrossRef CAS PubMed.
- J. W. Peeters, O. van Leeuwen, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2005, 38, 5587–5592 CrossRef CAS.
- S. Dai, L. Xue, M. Zinn and Z. Li, Biomacromolecules, 2009, 10, 3176–3181 CrossRef CAS PubMed.
- U. Piotrowska and M. Sobczak, Molecules, 2015, 20, 1–23 CrossRef PubMed.
- M. D'Almeida Gameiro, A. Goddard, V. Taresco and S. M. Howdle, Green Chem., 2020, 22, 1308–1318 RSC.
- F. Silvianti, D. Maniar, L. Boetje and A. J. J. Woortman, et al., ACS Polym. Au, 2023, 3, 82–95 CrossRef CAS.
- M. Nikulin and V. Švedas, Molecules, 2021, 26, 2750 CrossRef CAS PubMed.
- C. Li, T. Tan, H. Zhang and W. Feng, J. Biol. Chem., 2010, 285, 28434–28441 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3192–3196 CrossRef CAS PubMed.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- A. Kumar, K. Dhar, S. S. Kanwar and P. K. Arora, Biol. Proced. Online, 2016, 18, 2 CrossRef PubMed.
- T. Zisis, P. L. Freddolino, P. Turunen and M. C. F. van Teeseling, et al., Biochemistry, 2015, 54, 5969–5979 CrossRef CAS PubMed.
- J. Pleiss, M. Fischer, M. Peiker and C. Thiele, et al., J. Mol. Catal. B:Enzym., 2000, 10, 491–508 CrossRef CAS.
- J. N. Rodríguez-López, D. J. Lowe, J. Hernández-Ruiz and A. N. P. Hiner, et al., J. Am. Chem. Soc., 2001, 123, 11838–11847 CrossRef PubMed.
- H. Uyama, H. Kurioka and S. Kobayashi, Polym. J. (Tokyo, Jpn.), 1997, 29, 190–192 CrossRef CAS.
- Á. S. Olalla, V. H. Talavera, D. L. García and E. G. Torres, et al., Eur. Polym. J., 2022, 170, 111173 CrossRef.
- K. L. Wooley, C. J. Hawker, R. Lee and J. M. J. Fréchet, Polym. J. (Tokyo, Jpn.), 1994, 26, 187–197 CrossRef CAS.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- American Chemical Society, The Establishment of Modern Polymer Science by Wallace H. Carothers, 2000.
- OECD, Global Plastics Outlook, 2022.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- S. Kobayashi, Polym. Adv. Technol., 2015, 26, 677–686 CrossRef CAS.
- C. H. Chan, J.-T. Chen, W. S. Farrell and C. M. Fellows, et al., Polym. Chem., 2022, 13, 2262–2227 RSC.
- C. M. Fellows, R. G. Jones, D. J. Keddie and C. K. Luscombe, et al., Pure Appl. Chem., 2022, 94, 1093–1147 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to Polymers, CRC Press, Third edn., 2011 Search PubMed.
- D. Braun, Int. J. Polym. Sci., 2009, 2009, 893234 Search PubMed.
- M. P. Stevens, Polymer Chemistry: An Introduction, Oxford University Press, Third edn., 1999 Search PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1937, 59, 241–253 CrossRef CAS.
- Y. Gao, D. Zhou, J. Lyu and S. A, et al., Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS PubMed.
- P. B. V. Scholten, D. Moatsou, C. Detrembleur and M. A. R. Meier, Macromol. Rapid Commun., 2020, 41, 2000266 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- G. Parravano, J. Am. Chem. Soc., 1951, 73, 183–184 CrossRef CAS.
- G. Ng, S. W. Prescott, A. Postma and G. Moad, et al., Macromol. Chem. Phys., 2023, 224, 2300132 CrossRef CAS.
- T. Lalot, M. Brigodiot and E. Maréchal, Polym. Int., 1999, 48, 288–292 CrossRef CAS.
- F. Hollmann and I. W. C. E. Arends, Polymers, 2012, 4, 759–793 CrossRef.
- S. R. Zavada, T. Battsengel and T. F. Scott, Int. J. Mol. Sci, 2016, 17, 195 CrossRef PubMed.
- R. A. Derango, L.-c. Chiang, R. Dowbenko and J. G. Lasch, Biotechnol. Tech., 1992, 6, 523–526 CrossRef CAS.
- B. Kalra and R. A. Gross, Biomacromolecules, 2000, 1, 501–505 CrossRef CAS PubMed.
- A. Singh, D. Ma and D. L. Kaplan, Biomacromolecules, 2000, 1, 592–596 CrossRef CAS PubMed.
- K. J. Rodriguez, M. M. Pellizzoni, R. J. Chadwick and C. Guo, et al., in Methods in Enzymology, ed. N. Bruns and K. Loos, Academic Press, 2019, vol. 627, pp. 249–262 Search PubMed.
- O. Emery, T. Lalot, M. Brigodiot and E. Maréchal, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3331–3333 CrossRef CAS.
- A. Durand, T. Lalot, M. Brigodiot and E. Maréchal, Polymer, 2000, 41, 8183–8192 CrossRef CAS.
- W. Wang, X. Fan, Q. Wang and J. Xu, et al., New J. Chem., 2020, 44, 194–199 RSC.
- C. Liao, T. Li, F. Chen and S. Yan, et al., RSC Adv., 2024, 14, 2182–2191 RSC.
- Y. Zhang, Y. Song, B. Wang and Z. Mao, et al., Eur. Polym. J., 2024, 212, 113047 CrossRef CAS.
- C. Jiao, D. Appelhans, B. Voit and N. Bruns, et al., Polym. Chem., 2024, 16, 742–750 RSC.
- S. Wang, Q. Wang, X. Fan and J. Xu, et al., Carbohydr. Polym., 2016, 136, 1010–1016 CrossRef CAS PubMed.
- S. Wang, J. Xu, Q. Wang and X. Fan, et al., Process Biochem., 2017, 59, 104–110 CrossRef CAS.
- H. Fukushima, M. Kohri, T. Kojima and T. Taniguchi, et al., Polym. Chem., 2012, 3, 1123 RSC.
- H. Hu, L. Wang, B. Xu and P. Wang, et al., J. Biomed. Mater. Res., Part B, 2020, 108, 2643–2655 CrossRef CAS PubMed.
- V. Leskovac, S. Trivić, G. Wohlfahrt and J. Kandrač, et al., Int. J. Biochem. Cell Biol., 2005, 37, 731–750 CrossRef CAS PubMed.
- H. Iwata, Y. Hata, T. Matsuda and Y. Ikada, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1217–1218 CrossRef CAS.
- C. B. Lavery, M. C. MacInnis, M. J. MacDonald and J. B. Williams, et al., J. Agric. Food Chem., 2010, 58, 8471–8476 CrossRef CAS PubMed.
- F. W. Krainer and A. Glieder, Appl. Microbiol. Biotechnol., 2015, 99, 1611–1625 CrossRef CAS PubMed.
- T. Gundinger and O. Spadiut, J. Biotechnol., 2017, 248, 15–24 CrossRef CAS PubMed.
- S. Nanayakkara, Z. Zhao, A. F. Patti and L. He, et al., ACS Sustainable Chem. Eng., 2014, 2, 1947–1950 CrossRef CAS.
- S. M. Jones and E. I. Solomon, Cell. Mol. Life Sci., 2015, 72, 869–883 CrossRef CAS PubMed.
- T. Kudanga, G. S. Nyanhongo, G. M. Guebitz and S. Burton, Enzyme Microb. Technol., 2011, 48, 195–208 CrossRef CAS PubMed.
- R. Ikeda, J. Sugihara, H. Uyama and S. Kobayashi, Macromolecules, 1996, 29, 8702–8705 CrossRef CAS.
- M. B. Agustin, D. M. de Carvalho, M. H. Lahtinen and K. Hilden, et al., ChemSusChem, 2021, 14, 4615–4635 CrossRef CAS PubMed.
- S. Kumar, R. Gaddala, S. Thomas and J. Schumacher, et al., Polym. Chem., 2024, 15, 2011–2027 RSC.
- R. Li, W. Kong and Z. An, Angew. Chem. Int. Ed., 2022, 61(26), e202202033 CrossRef CAS PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- M. D. Nothling, Q. Fu, A. Reyhani and S. Allison-Logan, et al., Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- J. Wan, B. Fan and S. H. Thang, Chem. Sci., 2022, 13, 4192–4224 RSC.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC.
- B. Zhang, X. Wang, A. Zhu and K. Ma, et al., Macromolecules, 2015, 48, 7792–7802 CrossRef CAS.
- Q. Xu, Y. Zhang, X. Li and J. He, et al., Polym. Chem., 2018, 9, 4908–4916 RSC.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- D. Teixeira, T. Lalot, M. Brigodiot and E. Maréchal, Macromolecules, 1999, 32, 70–72 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- A. P. Danielson, D. B. Van Kuren, M. E. Lucius and K. Makaroff, et al., Macromol. Rapid Commun., 2016, 37, 362–367 CrossRef CAS PubMed.
- Z. Liu, Y. Lv and Z. An, Angew. Chem., Int. Ed., 2017, 56, 13852–13856 CrossRef CAS PubMed.
- J. Tan, Q. Xu, X. Li and J. He, et al., Macromol. Rapid Commun., 2018, 39, 1700871 CrossRef PubMed.
- D. K. Schneiderman, J. M. Ting, A. A. Purchel and R. Miranda, Jr., et al., ACS Macro Lett., 2018, 7, 406–411 CrossRef CAS PubMed.
- P. Singh, R. Prakash and K. Shah, Talanta, 2012, 97, 204–210 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- T. E. Patten, J. Xia, T. Abernathy and K. Matyjaszewski, Science, 1996, 272, 866–868 CrossRef CAS PubMed.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS PubMed.
- Y.-H. Ng, F. Di Lena and C. L. L. Chai, Polym. Chem., 2011, 2, 589–594 RSC.
- K. Matyjaszewski, H. Dong, W. Jakubowski and J. Pietrasik, et al., Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- C. Fodor, B. Gajewska, O. Rifaie-Graham and E. A. Apebende, et al., Polym. Chem., 2016, 7, 6617–6625 RSC.
- M. Qi, H. Pan, H. Shen, X. Xia, C. Wu, X. Han, X. He, W. Tong, X. Wang and Q. Wang, Angew. Chem. Int. Ed, 2020, 59, 11748 CrossRef CAS PubMed.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- L. van der Mee, F. Helmich, R. de Bruijn and J. A. J. M. Vekemans, et al., Macromolecules, 2006, 39, 5021–5027 CrossRef CAS.
- I. K. Varma, A.-C. Albertsson, R. Rajkhowa and R. K. Srivastava, Prog. Polym. Sci., 2005, 30, 949–981 CrossRef CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- R. Dwivedi, S. Kumar, R. Pandey and A. Mahajan, et al., J. Oral Biol. Craniofac. Res., 2020, 10, 381–388 CrossRef PubMed.
- J. Engel, A. Cordellier, L. Huang and S. Kara, ChemCatChem, 2019, 11, 4983–4997 CrossRef CAS.
- H. Uyama and S. Kobayashi, Chem. Lett., 1993, 22, 1149–1150 CrossRef.
- H. Uyama, K. Takeya and S. Kobayashi, Bull. Chem. Soc. Jpn., 1995, 68, 56–61 CrossRef CAS.
- H. Uyama, M. Kuwabara, T. Tsujimoto and S. Kobayashi, Polym. J. (Tokyo, Jpn.), 2002, 34, 970–972 CrossRef CAS.
- M. Takwa, Y. Xiao, N. Simpson, E. Malmström, K. Hult, C. E. Koning, A. Heise and M. Martinelle, Biomacromolecules, 2008, 9, 704–710 CrossRef CAS PubMed.
- J. C. Lentz, R. Cavanagh, C. Moloney, K. Kortsen, H. R. Fowler, B. F. Pin, P. L. Jacob, E. Krumins, C. Clark, F. Machado, N. Breitkreuz, B. Cale, A. R. Goddard, J. D. Hirst, V. Taresco and S. M. Howdle, Polym. Chem., 2022, 13, 6032–6045 RSC.
- Y. Yang, J. Zhang, D. Wu and Z. Xing, et al., Biotechnol. Adv., 2014, 32, 642–651 CrossRef CAS PubMed.
- U. Meyer, A. R. A. Palmans, T. Loontjens and A. Heise, Macromolecules, 2002, 35, 2873–2875 CrossRef CAS.
- B. A. C. van As, P. Thomassen, B. Kalra and R. A. Gross, et al., Macromolecules, 2004, 37, 8973–8977 CrossRef CAS.
- M. de Geus, L. Schormans, A. R. A. Palmans and C. E. Koning, et al., J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4290–4297 CrossRef CAS.
- K. J. Thurecht, A. M. Gregory, S. Villarroya and J. Zhou, et al., Chem. Commun., 2006, 4383–4385, 10.1039/B611626D.
- R. L. Pflughaupt, S. A. Hopkins, P. M. Wright and A. P. Dove, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3326–3335 CrossRef CAS.
- S. Xiang, Q. Zhang, G. Zhang and W. Jiang, et al., Biomacromolecules, 2014, 15, 3112–3118 CrossRef CAS PubMed.
- K. Chen, W. Han, X. Hu and Y. Liu, et al., Chem. Eng. J., 2022, 437, 135284 CrossRef CAS.
- M. Takwa, N. Simpson, E. Malmström, K. Hult and M. Martinelle, Macromol. Rapid Commun., 2006, 27, 1932–1936 CrossRef CAS.
- X. Qian, J. Wang, Y. Li, X. Lin and Q. Wu, Macromol. Rapid Commun., 2014, 35, 1788–1794 CrossRef CAS PubMed.
- J. Ma, Q. Li, B. Song and D. Liu, et al., J. Mol. Catal. B: Enzym., 2009, 56, 151–157 CrossRef CAS.
- M. Wang, H. Shi, D. Wu and H. Han, et al., Molecules, 2014, 19, 9838–9849 CrossRef PubMed.
- H. Ren, Z. Xing, J. Yang and W. Jiang, et al., Molecules, 2016, 21, 796 CrossRef PubMed.
- A. Cao, T. Okamura, C. Ishiguro and K. Nakayama, et al., Polymer, 2002, 43, 671–679 CrossRef CAS.
- X. Guo, B. Choi, A. Feng and S. H. Thang, Macromol. Rapid Commun., 2018, 39, 1800479 CrossRef PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- J. Pretula, S. Slomkowski and S. Penczek, Adv. Drug Delivery Rev., 2016, 107, 3–16 CrossRef CAS PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- H. Zhao, J. Chem. Technol. Biotechnol., 2018, 93, 9–19 CrossRef CAS PubMed.
- P. McMichael, X. Schultze, H. Cramail and F. Peruch, Polym. Chem., 2023, 14, 3783–3812 RSC.
- D. Omay and Y. Guvenilir, Biocatal. Biotransform., 2013, 31, 132–140 CrossRef CAS.
- S. Matsumura, K. Mabuchi and K. Toshima, Macromol. Rapid Commun., 1997, 18, 477–482 CrossRef CAS.
- H. Zhao, G. A. Nathaniel and P. C. Merenini, RSC Adv., 2017, 7, 48639–48648 RSC.
- M. Varghese and M. W. Grinstaff, Chem. Soc. Rev., 2022, 51, 8258–8275 RSC.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef PubMed.
- S. Park, E. Forró, H. Grewal, F. Fülöp and R. J. Kazlauskas, Adv. Synth. Catal., 2003, 345, 986–995 CrossRef CAS.
- L. W. Schwab, R. Kroon, A. J. Schouten and K. Loos, Macromol. Rapid Commun., 2008, 29, 794–797 CrossRef CAS.
- E. Stavila, G. O. R. Alberda van Ekenstein, A. J. J. Woortman and K. Loos, Biomacromolecules, 2014, 15, 234–241 CrossRef CAS PubMed.
- P. N. Stockmann, D. Van Opdenbosch, A. Poethig and D. L. Pastoetter, et al., Nat. Commun., 2020, 11, 509 CrossRef CAS PubMed.
- I. C. Benea, I. Kántor, A. Todea and A. Pellis, et al., React. Funct. Polym., 2023, 191, 105702 CrossRef CAS.
- T. Muringayil Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, CSCEE, 2024, 9, 100673 CAS.
- A. R. Goddard, E. A. Apebende, J. C. Lentz and K. Carmichael, et al., Polym. Chem., 2021, 12, 2992–3003 RSC.
- C. Post, D. Maniar, V. S. D. Voet and R. Folkersma, et al., ACS Omega, 2023, 8, 8991–9003 CrossRef CAS PubMed.
- I. C. Benea, D. Dăescu, A. Todea and L. Nagy, et al., Int. J. Biol. Macromol., 2024, 282, 137046 CrossRef CAS PubMed.
- A. Pellis, E. Herrero Acero, V. Ferrario and D. Ribitsch, et al., Trends Biotechnol., 2016, 34, 316–328 CrossRef CAS PubMed.
- C. Vilela, A. F. Sousa, A. C. Fonseca and A. C. Serra, et al., Polym. Chem., 2014, 5, 3119–3141 RSC.
- Q. Zhang, M. Song, Y. Xu and W. Wang, et al., Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- Y. Jiang, A. Woortman, G. Van Ekenstein and K. Loos, Biomolecules, 2013, 3, 461–480 CrossRef PubMed.
- S. Okumura, M. Iwai and Y. Tominaga, Agric. Biol. Chem., 1984, 48, 2805–2808 CAS.
- S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS PubMed.
- G. B. Perin and M. I. Felisberti, Macromolecules, 2020, 53, 7925–7935 CrossRef CAS PubMed.
- G. B. Perin and M. I. Felisberti, Biomacromolecules, 2022, 23, 2968–2975 CrossRef CAS PubMed.
- G. B. Perin and M. I. Felisberti, Macromolecules, 2023, 56, 6968–6977 CrossRef CAS.
- P. L. Jacob, L. A. Ruiz Cantu, A. K. Pearce and Y. He, et al., Polymer, 2021, 228, 123912 CrossRef CAS.
- P. L. Jacob, B. Brugnoli, A. Del Giudice and H. Phan, et al., J. Colloid Interface Sci., 2023, 641, 1043–1057 CrossRef CAS PubMed.
- E. Axioti, E. G. Dixon, M. Reynolds-Green and E. C. H. Alexander, et al., Colloids Surf., B, 2024, 236, 113828 CrossRef CAS PubMed.
- H. K. Weber and K. Faber, Methods Enzymol., 1997, 286, 509–518 CAS.
- H. Uyama and S. Kobayashi, Chem. Lett., 1994, 23, 1687–1690 CrossRef.
- F. Binns, P. Harffey, S. M. Roberts and A. Taylor, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2069–2080 CrossRef CAS.
- V. Taresco, R. G. Creasey, J. Kennon and G. Mantovani, et al., Polymer, 2016, 89, 41–49 CrossRef CAS.
- S. Kobayashi, Proc. Jpn. Acad., Ser. B, 2010, 86, 338–365 CrossRef CAS PubMed.
- S. M. E. Swainson, I. D. Styliari, V. Taresco and M. C. Garnett, Polymers, 2019, 11, 1561 CrossRef CAS PubMed.
- A. Wongrakpanich, H. Bui Thi Thu, K. Sakchaisri and V. Taresco, et al., J. Drug Delivery Sci. Technol., 2024, 95, 105610 CrossRef CAS.
- A. K. Chaudhary, J. Lopez, E. J. Beckman and A. J. Russell, Biotechnol. Prog., 1997, 13, 318–325 CrossRef CAS.
- C. Dourado Fernandes, B. Francisco Oechsler, C. Sayer and D. de Oliveira, et al., Eur. Polym. J., 2022, 169, 111132 CrossRef CAS.
- A. V. Samrot, S. K. Samanvitha, N. Shobana and E. R. Renitta, et al., Polymers, 2021, 13, 3302 CrossRef CAS PubMed.
- D. Heinrich, M. H. Madkour, M. A. Al-Ghamdi and I. I. Shabbaj, et al., AMB Express, 2012, 2, 59 CrossRef PubMed.
- J. Nielsen, mBio, 2014, 5(6), e02153 CrossRef CAS PubMed.
- B. Alkotaini, S. Abdellaoui, K. Hasan and M. Grattieri, et al., ACS Sustainable Chem. Eng., 2018, 6, 4909–4915 CrossRef CAS.
- M. R. Kosseva and E. Rusbandi, Int. J. Biol. Macromol., 2018, 107, 762–778 CrossRef CAS PubMed.
- P. H. Opgenorth, T. P. Korman and J. U. Bowie, Nat. Chem. Biol., 2016, 12, 393–395 CrossRef CAS PubMed.
- P. H. Opgenorth, T. P. Korman and J. U. Bowie, Nat. Commun., 2014, 5, 4113 CrossRef CAS PubMed.
- F. Li, X. Wei, L. Zhang and C. Liu, et al., Angew. Chem., 2022, 134, e202111054 CrossRef.
- X. Wei, X. Yang, C. Hu and Q. Li, et al., Nat. Commun., 2024, 15, 3267 CrossRef CAS PubMed.
- GlobalData, Production capacity of polyethylene terephthalate worldwide from 2014 to 2024 (in million metric tons), Statista, 2020.
- B. Shojaei, M. Abtahi and M. Najafi, Polym. Adv. Technol., 2020, 31, 2912–2938 CrossRef CAS.
- C. Post, D. Maniar, J. A. Jongstra and D. Parisi, et al., Green Chem., 2024, 26, 8744–8757 RSC.
- H. Uyama, S. Yaguchi and S. Kobayashi, Polym. J., 1999, 31, 380–383 CrossRef CAS.
- A. Pellis, S. Weinberger, M. Gigli and G. M. Guebitz, et al., Eur. Polym. J., 2020, 130, 109680 CrossRef CAS.
- A. Bazin, L. Avérous and E. Pollet, Eur. Polym. J., 2021, 159, 110717 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrović and K. Loos, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- M. G. Gert-Jan, S. Laszlo and D. Matheus Adrianus, Comb. Chem. High Throughput Screening, 2012, 15, 180–188 CrossRef PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca and M. Matos, et al., Polym. Chem., 2015, 6, 5961–5983 RSC.
- F. Silvianti, D. Maniar, B. Agostinho and T. C. de Leeuw, et al., Biomacromolecules, 2024, 25, 2792–2802 CrossRef CAS PubMed.
- F. Silvianti, D. Maniar, T. C. de Leeuw and J. van Dijken, et al., RSC Sustainability, 2024, 2, 3436–3450 RSC.
- C. Post, P. van den Tempel, P. Herrera Sánchez, D. Maniar, R. K. Bose, V. S. D. Voet, R. Folkersma, F. Picchioni and K. Loos, ACS Sustainable Chem. Eng., 2025, 13, 3543–3553 CrossRef CAS PubMed.
- V. Froidevaux, M. Borne, E. Laborbe and R. Auvergne, et al., RSC Adv., 2015, 5, 37742–37754 RSC.
- M. Annatelli, J. E. Sánchez-Velandia, G. Mazzi and S. V. Pandeirada, et al., Green Chem., 2024, 26, 8894–8941 RSC.
- E. M. Spence, L. Calvo-Bado, P. Mines and T. D. H. Bugg, Microb. Cell Fact., 2021, 20, 15 CrossRef CAS PubMed.
- H. Gómez-Álvarez, P. Iturbe, V. Rivero-Buceta and P. Mines, et al., Bioresour. Technol., 2022, 346, 126638–126638 CrossRef PubMed.
- A. Pellis, J. W. Comerford, S. Weinberger and G. M. Guebitz, et al., Nat. Commun., 2019, 10, 1762 CrossRef PubMed.
- J. Seithümmer, M. Öztürk, D. S. Wunschik and J. Prießen, et al., Biotechnol. J., 2022, 17, 2100452 CrossRef PubMed.
- Straits Research, Polyamide Market: Information by Product (Polyamide 6, Polyamide, 66, Bio-Based Polyamide, Specialty Polyamide), Application (Engineering Plastics, Fibres), and Region - Forecast till 2030, 2023.
- I. Baum, B. Elsässer, L. W. Schwab and K. Loos, et al., ACS Catal., 2011, 1, 323–336 CrossRef CAS.
- L. Ragupathy, U. Ziener, R. Dyllick-Brenzinger, B. von Vacano and K. Landfester, J. Mol. Catal. B:Enzym., 2012, 76, 94–105 CrossRef CAS.
- T. Polen, M. Spelberg and M. Bott, J. Biotechnol., 2013, 167, 75–84 CrossRef CAS PubMed.
- N. S. Kruyer and P. Peralta-Yahya, Curr. Opin. Biotechnol., 2017, 45, 136–143 CrossRef CAS PubMed.
- T. P. Fedorchuk, A. N. Khusnutdinova, E. Evdokimova and R. Flick, et al., J. Am. Chem. Soc., 2020, 142, 1038–1048 CrossRef CAS PubMed.
- H. N. Cheng, Q. Gu and W. W. Maslanka, US Pat., 6677427B1, 2004 Search PubMed.
- E. Stavila and K. Loos, J. Renewable Mater., 2015, 3, 268–280 CrossRef CAS.
- F. Poulhès, D. Mouysset, G. Gil and M. P. Bertrand, et al., Tetrahedron: Asymmetry, 2012, 23, 867–875 CrossRef.
- E. Stavila, R. Z. Arsyi, D. M. Petrovic and K. Loos, Eur. Polym. J., 2013, 49, 834–842 CrossRef CAS.
- V. Ferrario, A. Pellis, M. Cespugli, G. M. Guebitz and L. Gardossi, Catalysts, 2016, 6, 205 CrossRef.
- R. N. Lima, C. S. dos Anjos, E. V. M. Orozco and A. L. M. Porto, Mol. Catal., 2019, 466, 75–105 CrossRef CAS.
- Y. Poojari and S. J. Clarson, Macromolecules, 2010, 43, 4616–4622 CrossRef CAS.
- A. Douka, S. Vouyiouka, L.-M. Papaspyridi and C. D. Papaspyrides, Prog. Polym. Sci., 2018, 79, 1–25 CrossRef CAS.
- E. Stavila, G. O. R. Alberda van Ekenstein and K. Loos, Biomacromolecules, 2013, 14, 1600–1606 CrossRef CAS PubMed.
- D. Maniar, K. F. Hohmann, Y. Jiang and A. J. J. Woortman, et al., ACS Omega, 2018, 3, 7077–7085 CrossRef CAS PubMed.
- S. Xie, J. Yang, X. Wang and J. Yang, Eur. Polym. J., 2022, 162, 110932 CrossRef CAS.
- N. Karaki, A. Aljawish, C. Humeau and L. Muniglia, et al., Enzyme Microb. Technol., 2016, 90, 1–18 CrossRef CAS PubMed.
- T. J. Farmer, J. W. Comerford, A. Pellis and T. Robert, Polym. Int., 2018, 67, 775–789 CrossRef CAS.
- S. Sen and J. Puskas, Molecules, 2015, 20, 9358–9379 CrossRef CAS PubMed.
- W.-X. Wu, N. Wang, B.-Y. Liu and Q.-F. Deng, et al., Soft Matter, 2014, 10, 1199 RSC.
- A. Pellis, P. A. Hanson, J. W. Comerford and J. H. Clark, et al., Polym. Chem., 2019, 10, 843–851 RSC.
- K. Nasr, J. Meimoun, A. Favrelle-Huret and J. D. Winter, et al., Polymers, 2020, 12, 1907 CrossRef CAS PubMed.
- A. F. Naves, A. M. Carmona-Ribeiro and D. F. S. Petri, Langmuir, 2007, 23, 1981–1987 CrossRef CAS PubMed.
- N. Pradeep, A. Anupama and U. Hampannavar, J. Environ. Earth Sci., 2012, 2, 31–36 Search PubMed.
- S. T. R. Velasquez, Q. Hu, J. Kramm and V. C. Santin, et al., Angew. Chem., Int. Ed., 2025, 64, e202423406 CrossRef CAS PubMed.
- J. Nazor, J. Liu and G. Huisman, Curr. Opin. Biotechnol., 2021, 69, 182–190 CrossRef CAS PubMed.
- J. M. Sperl and V. Sieber, ACS Catal., 2018, 8, 2385–2396 CrossRef CAS.
- Z. Wang, B. Sundara Sekar and Z. Li, Bioresour. Technol., 2021, 323, 124551–124551 CrossRef CAS PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol and U. Menyes, et al., Angew. Chem., Int. Ed., 2015, 54, 2784–2787 CrossRef CAS PubMed.
- C. Scherkus, S. Schmidt, U. T. Bornscheuer and H. Gröger, et al., ChemCatChem, 2016, 8, 3446–3452 CrossRef CAS.
- C. J. Thompson, D. Hansford, S. Higgins and G. A. Hutcheon, et al., J. Microencapsul., 2006, 23, 213–226 CrossRef CAS PubMed.
- A. Lauko, S. J. Pellock, K. H. Sumida and I. Anishchenko, et al., Science, 2025, 388, eadu2454 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |