
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetrazine-based linkers as intrinsically tagged alternatives for click functionalization of metal–organic frameworks
Guillermo
Gómez-Tenés†
 ,
Pol
Gimeno-Fonquernie†
,
Alechania
Misturini†
,
Clara
Chinchilla-Garzón
,
Víctor
Carratalá
,
Larisha
Cisneros
and
Carlos
Martí-Gastaldo
*
,
Pol
Gimeno-Fonquernie†
,
Alechania
Misturini†
,
Clara
Chinchilla-Garzón
,
Víctor
Carratalá
,
Larisha
Cisneros
and
Carlos
Martí-Gastaldo
*
Functional Inorganic Materials Team, Instituto de Ciencia Molecular (ICMol), Universitat de València, Paterna 46980, Spain. E-mail: carlos.marti@uv.es
First published on 14th October 2024
Abstract
Reticular chemistry has in the post-synthetic modification (PSM) of frameworks one of the most versatile tools to adapt the systems’ physicochemical properties to the specific requirements which are imposed by their application in different contexts. We can safely say that PSM methodologies in all their variants are currently one of the main resources that reticular chemists turn to when they need to diversify a framework compositionally. Practically all these modifications require the integration of functional groups appended to the organic linkers in the framework, either by direct synthesis or by post-synthetic exchange. The reactivity of these tags allows, at a subsequent stage, covalent modification of the framework under conditions that ideally respect its structural integrity. In this perspective article we introduce the use of tetrazine-based linkers as intrinsically tagged alternatives to integrate PSM with click chemistry reactivity. This strategy is ideally suited to molecular frameworks, as it combines very mild modification conditions with direct control over the organisation of built-in appendices and the acknowledged potential of click chemistry to build framework libraries.
 Guillermo Gómez-Tenés | Guillermo Gómez-Tenés received his BSc in Chemistry and MSc in Molecular Nanoscience and Nanotechnology from the Universitat of València in 2022 and 2023 respectively. Then, he obtained an FPU fellowship from the Spanish government in 2023 and joined the Funimat research group as a PhD student to focus on the active encapsulation of guests in reticular materials under the supervision of Dr Carlos Martí-Gastaldo and Dr Natalia M. Padial. |
 Pol Gimeno-Fonquernie | Born and raised in Barcelona (Spain), Pol Gimeno-Fonquernie obtained a bachelor's degree in chemistry from the Autonomous University of Barcelona in 2017, followed by a Master's in Materials Science from the University of Barcelona in 2019. In 2023, he completed a PhD in Chemistry at the University of Adelaide (Australia). Currently, he holds a postdoctoral position at the Institute of Molecular Science in Valencia. His research focuses on the study and modification of crystalline porous materials using X-ray crystallography. |
 Alechania Misturini | Alechania Misturini is a postdoctoral researcher at the Institut de Ciència Molecular in the Universitat de València, Spain. She received a PhD in sustainable chemistry at the Universitat Politècnica de València (2022). Her master's degree in physical chemistry (2019), and bachelor's degrees in chemistry (2016) and chemistry education (2017) were obtained at the Universidade Federal de Santa Catarina, Brazil. She currently investigates the flexible response of Metal-Organic Frameworks, and adsorptive and separation processes with nanoporous materials using computational methods. |
 Carlos Martí-Gastaldo | Carlos Martí-Gastaldo obtained his PhD from the University of València in 2009. Following this, he joined the University of Liverpool, initially as a Marie Curie Postdoctoral Fellow and subsequently as a University Research Fellow of the Royal Society. In 2014, he transitioned to the Institute of Molecular Science (ICMol) as a Ramón y Cajal Fellow, where he established the Functional Inorganic Materials research group. He has since been awarded both an ERC Starting Grant in 2016 and a Consolidator Grant in 2022. His research primarily focuses on the reticular design of molecular frameworks, with applications in molecular recognition, catalysis, and environmental remediation. |
Introduction
Metal–Organic Frameworks (MOFs) have garnered significant attention in chemistry and materials science due to their high degree of tunability.1,2 By leveraging the richness and versatility of organic chemistry, a MOF with specific properties can be synthesized by creating specific organic linkers and assembling it with the appropriate metal nodes. For instance, to synthesize a hydrophobic MOF, one can design an organic linker containing a hydrophobic functional group like a carborane, and then assemble it into a MOF structure.3 This approach has led to the synthesis of many materials with diverse physical and chemical properties. However, this approach is limited by two main factors. Firstly, most functional groups are unstable under the harsh assembly conditions required for MOFs, which often involve high temperatures and very acidic media. Secondly, modifications to the organic linker can alter its solubility, bulkiness and geometry, making it difficult to achieve the desired topology, and in some cases, making impossible to crystallize the corresponding MOF architecture.Post-synthetic modifications, introduced by Cohen in 2007,4 allow to perform modifications to MOFs after they have been assembled.5–7 This technique has expanded MOF tunability by overcoming the limitations of the direct approach, thereby broadening the scope of MOF functionalization (Fig. 1). PSM requires milder conditions compared to MOF assembly and preserves the original topology. Instead of synthesizing an organic linker with the desired functionality, the linker is synthesized with a functional group, referred to as a tag, which allows the incorporation of the desired residue via a covalent reaction after the MOF is assembled (Fig. 1d). The number of tags that can withstand MOF assembly conditions is still limited, but several functional groups can be covalently incorporated into the same tag. Another option is to use tags which have been protected beforehand8 to facilitate the assembly of the material. However, a deprotection step is required later, making the process longer and more prone to affect the crystallinity of the system (Fig. 1c). The synthesis of MOFs using tagged ligands allows for very homogeneous functionalization of the MOF structure with a broad range of functional groups. However, controlling the topology can be challenging, and in some cases, it may be impossible to obtain an isoreticular structure of the MOF with the untagged linker.
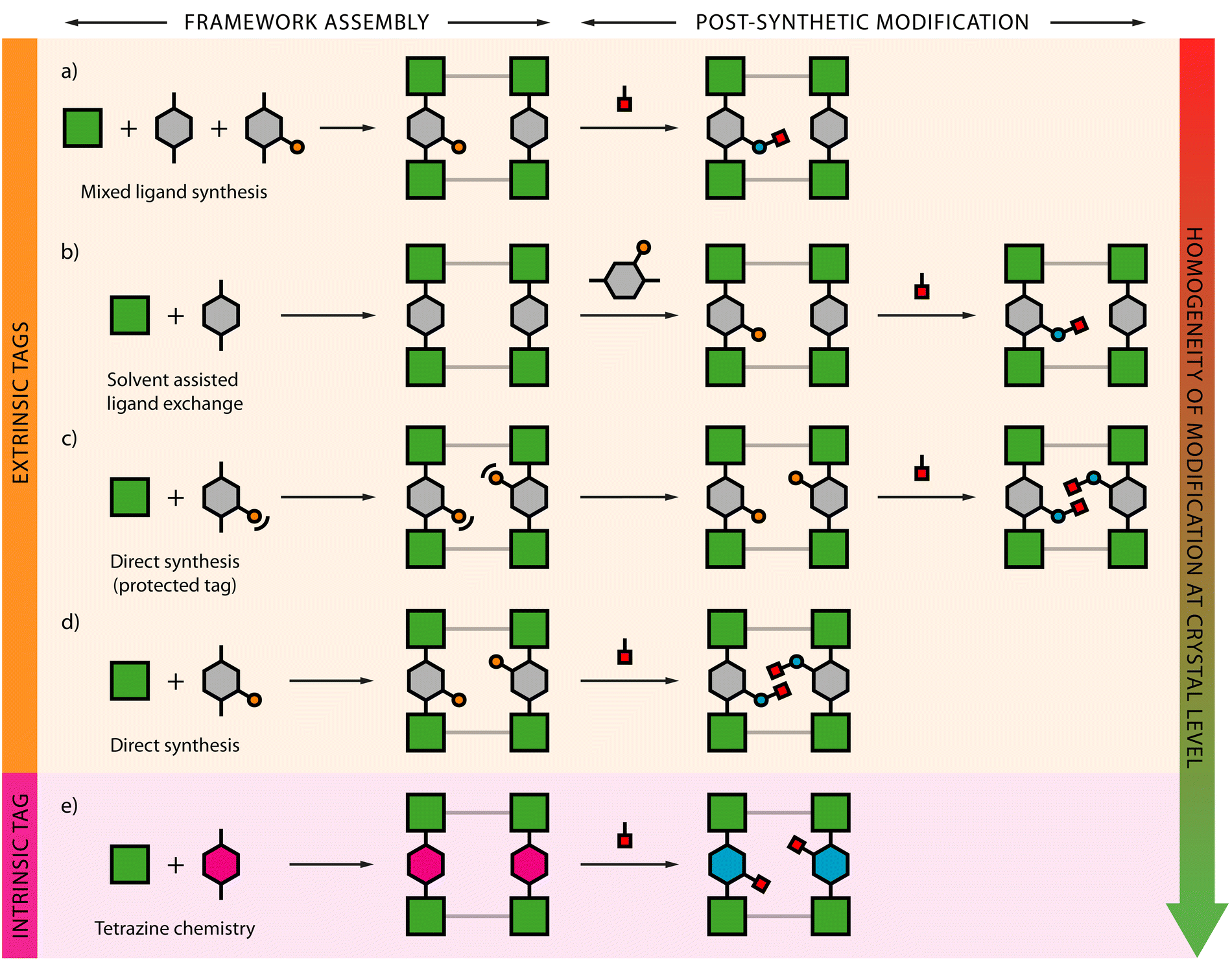 | ||
| Fig. 1 Schematic representation of covalent PSM strategies using (a) mixed-ligand synthesis approach, (b) solvent assisted ligand exchange, direct synthesis with (c) protected and (d) unprotected tags, and (e) tetrazine click chemistry. | ||
Solvent-assisted ligand exchange (SALE) is a type of PSM that can sometimes overcome this limitation9,10 (Fig. 1b). In this process, the desired MOF is firstly synthesized with the untagged linker to form the desired topology. After assembly, the untagged linker is partially exchanged with the tagged linker, allowing the functionalization of the framework. This approach provides better control over the MOF topology while still enabling the desired functionalization. However, it results in a non-homogeneous distribution of the functional groups. In some cases, SALE can be simplified by adding a mixture of tagged and untagged linkers during the MOF assembly step11 (Fig. 1a). In this scenario, the untagged linker directs the MOF topology, while the tagged linker allows for MOF functionalization. However, this method also results in a non-homogeneous distribution of the functional groups.
Despite these four PSM approaches expand the scope of MOF tunability and functionality, none of them provides both good topological control and homogeneous functionalization. Additionally, the tags required for PSM are commonly appended to the backbone of the linker, and when the desired functionality is added to the tag, the resulting group can become very bulky. This bulkiness is a limitation in MOFs with low porosity, causing pore blockage or making quantitative functionalization impossible due to steric effects.
In recent years, the use of 1,2,4,5-tetrazine as a tag for the PSM of MOFs has emerged as a powerful approach which addresses several of those limitations. The geometrical similarity of the tetrazine moiety to a phenyl ring allows for the synthesis of intrinsically tagged linkers without appended groups, which facilitates framework assembly compared to other alternatives reliant on linker substitution (Fig. 1e). Functional groups are incorporated via click chemistry, which occurs under very mild conditions and without the need of a catalyst. In this feature article we introduce basic concepts on the reactivity of tetrazines in inverse electron-demand Diels Alder reaction, review the frameworks compatible with this post-functionalization chemistry, analyze several examples that highlight the potential of this approach and the particularities intrinsic to the heterogeneous reactivity imposed by the use of solid supports, and conclude with our personal views on the current drawbacks and future directions that might support further development in this context.
Compatibility of tetrazine linkers with MOF assembly
The great success achieved by using click chemistry in bioconjugation12 or natural products syntheses,13 among others, has caused a lot of interest in exporting it to the field of reticular chemistry. One of the first examples with this aim is the incorporation of azide–alkyne tags, as reported by Sada and co-workers.14 This route enables the quantitative conjugation of a broad range of molecules by azide–alkyne 1,3-cycloaddition reactions under milder conditions when compared to other covalent PSM, such as amine condensation. Moreover, it does not involve the use of protecting groups and the reactive tag is stable during MOF synthesis. However, it is worth noting that this reaction typically requires the use of Cu(I) as catalyst15 to facilitate the click reaction, generating potential issues such as contamination, and concerns regarding biocompatibility. Once more, this kind of PSM also requires pending groups, carrying the problems already discussed.Tetrazines, concretely 1,2,4,5-tetrazine, have emerged as a solution to most problems generated by classical PSM. 1,2,4,5-Tetrazine reacts under an inverse electron-demand Diels–Alder (iEDDA) reaction,16 which was pioneered by Bertozzi and honoured with the 2022 Nobel Prize in Chemistry.17 The reaction proceeds via a [4+2] cycloaddition between an electron-poor diene (tetrazine) and an electron-rich alkene or alkyne (dienophile). Depending on the dienophile's nature, different iEDDA reactions can take place. For instance, reaction with an alkene gives a 4,5-dihydropyridazine, which can transform into its two 1,4-dihydropyridazine tautomers (Scheme 1a). These can be subsequently oxidized to give the pyridazine form. A different reaction undergoes when using alkynes, giving pyridazine directly (Scheme 1b). There are other special cases such as what happens with 2,5-norbornadiene (NBD). Here, the pyridazine form is obtained after a second retro Diels–Alder reaction (Scheme 1c).
 | ||
| Scheme 1 Reactions between a tetrazine ligand and (a) an alkene, (b) an alkyne or (c) 2,5-norbornadiene as dienophiles. | ||
Notably, these reactions are catalyst-free, work under mild conditions, exhibit rapid kinetics, and occur with high selectivity, yielding N2 as the only by-product. Once the tetrazine rings are included in the backbone of a MOF, this can be easily tuned via a click reaction. The functionalized MOF provides then a homogeneous distribution of the incorporated functional group with a high molecular control over where it is anchored. The ability to synthesize dienophiles with different functional groups according to established methods, makes this a powerful option for the synthesis of libraries of functionalized frameworks.
From a geometrical standpoint, organic linkers containing a tetrazine moiety can be easily designed by replacing a phenyl ring in common organic linkers used in MOF synthesis with a tetrazine moiety. The geometrical similarity between these two groups allows this exchange without altering the linker's geometry, flexibility, or bulkiness. Introducing the tetrazine moiety by replacing the phenyl ring, rather than appending it to the linker backbone, makes these linkers an intrinsically tagged alternative.
Linkers bearing binding groups such as pyridine, carboxylates18 or azolates19 have been employed to stitch secondary building units (SBUs)20 together into extended framework structures. These linkers are of relatively high symmetry and commonly built from rigid unsaturated hydrocarbon fragments. As previously mentioned, a phenyl ring can be replaced by the tetrazine moiety without altering the linker's symmetry or connectivity. However, the nature of this central ring is drastically changed from an electron-rich (Fig. 2a) to an electron deficient (Fig. 2e) aromatic system. On the one hand, this ring becomes more reactive, facilitating post-synthetic modifications. On the other hand, tetrazine's π-acidity can make the connector groups less activated hence weakening the linker-metal bonds. This can potentially affect the stability of the framework or even complicate its assembly during synthesis.
 | ||
| Fig. 2 (a)–(h) Molecular electrostatic potential surfaces for (a) benzene and (e) 1,2,4,5-tetrazine rings and different MOF ligands, using BP86-D3/Def2-TZVP21–23 level of theory in Orca package24 (version 5.0.4). Anchoring sites containing pyridine (b) and (f), benzoic acid (c) and (g) or pyrazole (d) and (h) groups and benzene (middle row) or 1,2,4,5-tetrazine (bottom row) rings as the central aromatic portions. Scale unit in a.u. (i) Representative MOF architectures containing pyridine- and carboxylate-based linkers with intrinsic tetrazine tags, and respective CCDC identifiers. Several crystallographic structures composed only by low valence metal sites and 3,6-di(4-pyridyl)-1,2,4,5-tetrazine (pytz) are one-dimensional porous coordination polymers.25 SUPBEN26 and DOCVEZ27 are three-dimensional structures formed by pytz and thiocyanate or hexafluorosilicate groups, respectively, coordinated to the metal nodes. Further topological diversity is achieved by the combination of pytz and different carboxylate linkers, as exemplified by the two-dimensional coordination sheets of IWAJEW,28 RIHJOJ29 and IBOKOD,30 and the three-dimensional structures BUWGEG30 and WOGHEG.31 Fewer MOFs are reported in the literature by the combination of 4,4′-(1,2,4,5-tetrazine-3,6-diyl)dibenzoic acid ligands and higher valence metal sites, as exemplified by LIXPOA32 and UiO-68-TZDC structure, which was reported almost simultaneously by Martí-Gastaldo (AKEKIN)33,34 and Loeb (FAHJEH)33,34 groups. The further tunning of these anchoring points can provide improvements on the stability and synthesis conditions. Although azolate-based structures were not found in the literature, we point out the 3,6-bis(1H-pyrazol-4-yl)-1,2,4,5-tetrazine linker as a promising candidate for the synthesis of new MOFs with intrinsic tetrazine tags. | ||
The type of connector can also modulate the electronic structure of the linker's central ring (Fig. 2b–d and f–h) due to its conjugated π-system, which can be an additional strategy to control the framework's response to PSM, while avoiding the formation of weak interactions to the metal nodes. Furthermore, when connectors are added to the tetrazine ring, there is no immediate impact on its planarity, but once assembled into a framework, distortions may occur. Studies have shown that a slight distortion in the tetrazine ring enhances the iEDDA reaction, while excessive distortion can hinder it.35,36
The first example of an inorganic supramolecular architecture containing a tetrazine motif was reported by Schröder and co-workers in 1997. By using 3,6-di(4-pyridyl)-1,2,4,5-tetrazine (pytz) as tetrazine organic linker, they synthesized an Ag(I) coordination polymer.37 A few years later, the same group reported a Cd(II) MOF {[Cd2(4,4′-pytz)3(μ-NO3)(NO3)3(MeOH)]} using the same organic linker.38 Despite these two early examples, the synthesis of MOFs containing tetrazine motifs was not reported extensively until 2008. Since then, the synthesis of MOFs containing tetrazine moieties can be classified into two different groups, depending on the nature of the metal binding groups (Fig. 2i).
MOFs based on nitrogenated tetrazine linkers have been built from the assembly of pyridines and M(II) metals (Fig. 2f). Most of the examples reported use 3,6-di(4-pyridyl)-1,2,4,5- tetrazine (pytz) as a pillaring agent for layered architectures.
Chen and co-workers reported the first example of a tetrazine MOF that exhibited porosity (WOGHEG structure in Fig. 2i).31 Using a mixed linker approach they synthesized a Zn(II) layer-pillared MOF containing pytz as pillars connected to the axial position of the Zn(II) paddle-wheel. The 3-fold interpenetrated structure showed great hydrogen uptake. Interestingly, these properties were mainly attributed to the small pore size of the structure and there was no mention on the effect of the tetrazine moiety in the hydrogen uptake.
Following a similar strategy, Kitagawa and co-workers synthesized three isostructural Zn(II) chain-pillared 2D-MOFs.28 Each of the structures had a different pillar linker with a similar length but different functionality. CID-21 was pillared by 1,4-bis(4-pyridyl)benzene (bdp), CID-22 by the tetrazine linker pytz (IWAJEW structure in Fig. 2i) and CID-23 by 1,4-bis(4-pyridyl)acetylene (bpa). CID-22 showed the highest methanol and water uptake of all three due to the high affinity of the tetrazine group with polar electron-donating molecules.
Kepert and co-workers used the same pytz linker as a pillar in a Fe(II)/Au(I) Hofmann system.39 The framework presented a spin-crossover behaviour that could be modified via PSM of the tetrazine moiety. By reacting the 1,2,4,5-tetrazine moiety with 2,5-norbonadiene to form the 1,2-diazine moiety, the spin crossover of the MOF shifted from 277 K to 178 K.
Apart from direct synthesis MOF formation, tetrazine MOFs have also been synthesized via PSM. An example was reported by Kaskel and co-workers following a two-step approach. They firstly synthesized a Cu(II) MOF (DUT-71) containing 4,4′,4′′,4′′′-(1,3-phenylenebis(azanetriyl)tetrabenzoate) (mpbatb) as the organic linker. In a second step they introduced via PSM the organic linker pytz and to form the mixed ligand framework containing tetrazine motifs (DUT-74).40
This first group showed the potential of tetrazine MOFs in several applications. However, they also presented some limitations. First, its chemical stability was limited due to the relatively weak interaction between the pyridine groups and M(II) metals. Secondly, most of the synthesized MOFs have low dimensionality, forming either layers or chains with limited pore accessibility. Following with the general trend in the area, efforts were made to extend the synthesis of MOFs to polycarboxylic tetrazine linkers. In 2015, Rodríguez-Diéguez described early attempts to synthesise the potassium framework [K2(44btz)]n,41 isoreticular to IRMOF-16 and built from the 4,4′-(1,2,4,5-tetrazine-3,6-diyl)dibenzoic acid (H244dbtz).42
The first example of a tetrazine MOF based on high-valent metals for higher chemical and thermal stability was not reported until 2019 by Hong-Cai Zhou and coworkers.32 Reaction of H244dbtz with Al(III) led to the assembly of AlTz-53 (LIXPOA in Fig. 2i), based on interconnected square-lattice (sql) nets. AlTz-53 contains infinite linear zigzagging chains of [Al(X)(COO)2]n (where X = OH or solvent) containing AlO4(OH)2 octahedra, in which four oxygen atoms from four carboxyl groups and two bridging oxygen atoms are coordinated to the aluminium(III) centre. Interestingly, the desolvation of AlTz-53 in DEF resulted in a lattice transformation to a hierarchically porous isomer (AlTz-68) with interconnected kagome (kgm) nets. The second example was presented almost simultaneously by the Martí-Gastaldo and Loeb groups.33,34 Reacting H244dbtz with a Zr(IV) salt they synthesized a zirconium MOF (UiO-68-TZDC) with a face-centered cubic (fcu) topology isostructural to UiO-68 (AKEKIN/FAHJEH in Fig. 2i). Martí-Gastaldo and co-workers presented a comprehensive study on the effect of the tetrazine moiety in the framework properties and chemical stability. As discussed previously, the substitution of a phenyl ring for a tetrazine moiety significantly impacts the MOF properties. UiO-68-TZDC shows a significantly lower chemical stability compared to UiO-68. For instance, the work reveals how soaking UiO-68 in a water:methanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v%) solution for 24 hours shows a minimal loss of crystallinity, while UiO-68-TZDC shows a drastic loss. By synthesizing a mixed linker framework via SALE they were able to improve the chemical stability of UiO-68-TZDC while preserving its optical properties.
:1, v/v%) solution for 24 hours shows a minimal loss of crystallinity, while UiO-68-TZDC shows a drastic loss. By synthesizing a mixed linker framework via SALE they were able to improve the chemical stability of UiO-68-TZDC while preserving its optical properties.
To our knowledge, AlTz-53, AlTz-68 and UiO-68-TZDC are the only three examples of carboxylate donor tetrazine MOFs. We argue future development should target linkers that can keep the electron-deficient character of the tetrazine while stablishing stronger coordination bonds with the metal nodes to overcome the stability and synthetic problems previously mentioned. According to our computational analysis of MEP surfaces, linkers based on pyrazolate donors (Fig. 2i) stand out as promising candidates for the development of the next generation of tetrazine MOFs.
Tetrazine-enabled click functionalization
The functionalization of materials via tetrazine-enabled click chemistry has already been reported for several materials as polymers,43 metal–organic cages44 and organic macrocycles.45 This paper intends to highlight the state of the art and opportunities for the functionalization of porous molecular frameworks with focus on the versatility of this approach and its potential application in the MOF field.In 2011, Cohen and co-workers showed for the first time an iEDDA reaction for the PSM of MOFs.46 However, their approach did not involve starting with a framework built from tetrazine linkers. Instead, they presented a 2-step approach starting from a parent framework containing pending amine groups, UMCM-1-NH2, and a mixed linker (1:1 BDC:NH2-BDC) IRMOF-3 (Fig. 3a). In the first step, they conducted a condensation reaction between the amine group and pentenoic anhydride to form an amide bond, introducing free olefin groups into the framework. Subsequently, they reacted the free olefin group with dimethyl-1,2,4,5-tetrazine-3,6-dicarboxylate (TDC) to create the 1,4-dihydropyridazine cycloaddition product appended to the framework. Although this 2-step process allows the functionalization of MOFs, it normally would lead to very long pending groups that could block MOF porosity or the full functionalization of the framework. This was already evidenced in the original work, that reports an incomplete transformation of olefinic moieties upon iEDDA reaction of IRMOF-3-NH2 with 100% NH2-BDC.
 | ||
| Fig. 3 (a) Schematic representation of the tandem approach for PSM of UMCM-1-NH2 (top) and mixed IRMOFs (bottom) with a tetrazine in an inverse-electron-demand Diels–Alder (iEDDA) reaction. Adapted from ref. 46 with permission from ACS, copyright 2011. (b) Synthetic routes to introduce superhydrophilicity into AlTz-68. From the defective structure of AlTz-53 (purple framework), AlTz-68-C18 (purple framework with green external surface) can be obtained by a two-step process for a selective external modification. Also in a two-step approach, AlTz-68-C18* (green framework) is produced by a click modification after the lattice rearrangement of defective AlTz-53. Adapted from ref. 47 with permission from Cell Press, copyright 2020. (c) Control of JUK-20 flexibility via click chemistry, by modulating the bulkiness of the anchoring dienophile. Adapted from ref. 30 with permission from ACS, copyright 2021. (d) Crystals of UiO-68-TZDC before (pink) and after (dark pink) reaction with fullerene (top). View of the H244dbtz-C60 units that result from the covalent grafting of fullerene (in green) to tetrazine connectors (bottom). Adapted from ref. 48 with permission from Wiley, copyright 2022. (e) MOF hybrid crystals prepared by conjugation of nanotubes to the crystal surface to form a permeable coating (left). Proposed linkage of CNT sidewall to the surface of tetrazine rings with the nanotube sidewall acting as electron-rich dienophiles (grey) for the formation of pyridazine rings (yellow) in the binding sites (top right). SEM Images of the crystals of UiO-68-TZDC after the reaction with carbon nanotubes (bottom right). Adapted from ref. 49 with permission from Wiley, copyright 2023. | ||
Direct iEDDA PSM examples in MOFs containing tetrazine linkers have only emerged in recent years, with a scarce number of examples. Zhou and co-workers managed to tailor the hydrophobicity of a MOF by functionalizing a tetrazine MOF, appending alkane chains via click chemistry (Fig. 3b).47 By iEDDA cycloaddition of dienophiles to AlTz-68 they attached two different alkane chains, C6 and C18. While C6 penetrated the entire framework, reducing porosity, C18 was exclusively attached to the surface resulting in a hydrophobic surface respectful with the porosity of the framework. Following a similar approach, Lin and co-workers functionalized UiO-68-TZDC with C18 (UiO-68-TZDC-C18) to alter its surface hydrophobicity.50 Additionally, they demonstrated that a composite of UiO-68-TZDC-C18 with 5% loading of polyaniline (PANI) exhibited excellent anticorrosive properties.
Functional group addition via iEDDA also grants control over the framework flexibility. Matoga and co-workers synthesized a mixed linker Cd MOF containing tetrazine groups (JUK-20, IBOKOD structure in Fig. 2i),30 which exhibited a 2-fold interpenetration with high flexibility (Fig. 3c). By anchoring different dienophiles with varying degrees of bulkiness, they could modulate the rigidity of the framework. In a subsequent study, they demonstrated that the addition of these dienophiles also modified its luminescent properties and the adsorption of CO2 and vapours of methanol, ethanol, isopropanol, D2O, and H2O.51 Chang and colleagues similarly illustrated how the incorporation of different functional groups via iEDDA reaction can modulate CO2 adsorption.52
By using UiO-68-TZDC and its Hf analogue (HfTz-68), Demel and colleagues showcased the versatility of functionalization via click reaction in tetrazine MOF PSM.53 They incorporated three different organic molecules (quinine, thioquinine, and the organometallic complex CuNONO) to the tetrazine frameworks via iEDDA.
Framework chemical diversification was further explored by Martí-Gastaldo and co-workers, who not only demonstrated the versatility of the technique but also provided fundamental insights into the reaction (Fig. 3d).48 They conducted an extensive study to optimize iEDDA in UiO-68-TZDC, using 2,5-norbornadiene (NBD) as a model substrate. Under optimized conditions of 40 °C for 16 hours with a 3:1 NBD:MOF ratio, they preserved MOF crystallinity and morphology. Moreover, through DFT calculations, they observed a decrease in energy of the tetrazine linker within the MOF compared to the free linker, resulting in the narrowing of the gap between frontier orbitals and an increase in reaction rate, enabling milder reaction conditions and the easier insertion of a wide variety of alkene molecules. They demonstrated this by anchoring a broad range of dienophiles, which all result in quantitative transformations into pyridazine and 1,4-dihydropyridazine derivatives. Additionally, they proved the versatility of this strategy by anchoring commercial C60 to the framework. In a subsequent study, they used this click functionalization to wrap UiO-68-TZDC crystals with thin layers of nanotubes. These crystal composites display high hydrophobicity that helps retaining the porosity intact after water treatment, combined with the electrical conductivity provided by the nanotube coating (Fig. 3e).
The abovementioned studies mainly employ iEDDA reactions to achieve functionalized materials for the desired application. However, the chemistry of the iEDDA reactions involving tetrazine MOFs has not been fully unveiled yet compared to the thorough studies available for organic reactivity in solution.16 For instance, very little has been discussed about second order rate constants, which determine the kinetics of the reactions. This is more challenging for heterogeneous liquid–solid phase reactions with MOFs that will require additional efforts to establish specific iEDDA reactivity trends for varying frameworks and dienophiles.
The impact of the dienophile nature on the iEDDA reaction rates was preliminary investigated for JUK-20 modified MOFs.30 The authors evaluated dienophiles containing different electron-donor groups (ethyl-vinyl ether, eve, and 5-norbornene-2-methanol, noh), and different molecular bulkiness to evaluate the angular stress effects (norbornene, nor, and noh), as well as cyclohexene (chx). The iEDDA reactions took from several hours (chx) to a few minutes (noh) to be carried out, with rates following the order chx < eve < nor < noh, which highlights the strong impact of both angular stress and electron-donor groups.
Changes in the dienophile size/bulkiness, pore openings and channel shape of the framework can also affect the diffusion rate and iEDDA reactivity.52 The energy difference between the frontier orbitals of the dienophile and the reticulated tetrazine units is also a factor to consider.54 Different kinetics can be expected for iEDDA reactions taking place with tetrazine rings embedded or not in a framework. In the case of UiO-68-TZDC, the iEDDA proceeds much faster for the MOF that the free linker in equivalent conditions, and pseudo-first order reactions are observed in both cases.48 DFT calculations showed that the MOF has a LUMO energy 3.8 kcalmol−1 lower that the free ligand, which reduces its gap to the HOMO of the dienophile, thus enhancing the reaction rate.
Another key factor which hasn’t been fully explored in MOFs is the importance of the class of iEDDA reaction involved in the functionalization of the framework (Scheme 1). Reaction with alkenes for the formation of non-planar 1,4-dihydropyridazines with sp3 carbons can induce structural transformations in the crystal. This has been beautifully exemplified recently by Pilgrim and co-workers55 in metal–organic cages, where they induce transformations in their systems via click reactions with different dienophiles. To our knowledge, although several MOFs have been modified to give the 1,4-dihydropyridazine ligand form, the possible structural modifications have been widely overlooked.
Conclusions and outlook
Despite the synthesis of tetrazine architectures was reported more than 20 years ago, the PSM of tetrazine MOFs via iEDDA has just started to be explored in the last five years. The general aspects summarized here highlight the potential of tetrazine linkers as universal tags to diversify the chemistry of molecular frameworks according to the organic methods already developed for click chemistry. Based on the mildness of the conditions used, and the possibility of preparing multiple dienophiles compatible with this type of covalent chemistry, tetrazine-based click functionalization ventures as an excellent option to prepare framework libraries with unlimited pore chemistries. However, little information is given in the study on the reaction rates and how this is affected in the solid-state. There should be more studies to evaluate how the pore size, framework topology, the angular stress of the tetrazine moiety or the diffusion of dienophiles through the MOF pores affect the reaction rate of iEDDA in the solid state. Additionally, the structural transformations induced by iEDDA reactions within MOFs, particularly those forming 1,4-dihydropyridazine, should be studied more extensively, as they have been largely overlooked.We also argue that the main limitation to general application lies possibly in the small number of tetrazine MOFs still available, which are mainly limited to soft frameworks based on pyridine-type linkers and a much smaller number of more stable systems based on carboxylate coordination of tetravalent metals. While these last are thermally and chemically more stable, and thus perfectly compatible with further functionalization respectful with their structural integrity, the synthesis of additional examples seems to be limited by the required use of modulators for highly acidic media in which the tetrazine units are not stable, thus preventing further progress. To avoid this problem, we propose the development of an alternative generation of tetrazine linkers incorporating connectors based on all-nitrogen azoles as pyrazole (Hpz) or 1,2,4-triazole (Htz). Compared to carboxylate, the synthesis of azolate frameworks does not require such an acidic acid media and would be compatible with the tetrazine core. Even more, according to our preliminary analysis, the inductive effect of the pyrazole groups on the tetrazine ring could impoverish its electron deficient character to make it compatible also with complementary normal-electron-demand Diels–Alder reactions. This fact, combined with the resulting lower basicity of the pyrazole groups, could also facilitate the assembly of new families of tetrazine frameworks that, to the best of our knowledge, remain still unexplored and would help underpinning the potential of click functionalization routes.
Author contributions
All authors participated in the conceptualization of the work under C. M.-G. supervision. G. G.-T, P. G.-F. and A. M. led the writing of the original draft which was reviewed and edited by all authors.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the H2020 program (ERC-2021-COG-101043428), Generalitat Valenciana (PROMETEU/2021/054), Ministerio de Ciencia, Innovación y Universidades and Agencia Estatal de Investigación (CNS2022-135677). G.G.-T. and C.C.-G. thank the Spanish government for a FPU grant (FPU22/01246) and a FPI grant (PRE2021-098634) respectively. V.C. thanks the Generalitat Valenciana for a predoctoral grant (CIACIF/2022/381).References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- S. Tasleem, M. Tahir and W. A. Khalifa, Int. J. Hydrogen Energy, 2021, 46, 14148–14189 CrossRef.
- L. Gan, A. Chidambaram, P. G. Fonquernie, M. E. Light, D. Choquesillo-Lazarte, H. Huang, E. Solano, J. Fraile, C. Viñas, F. Teixidor, J. A. R. Navarro, K. C. Stylianou and J. G. Planas, J. Am. Chem. Soc., 2020, 142, 8299–8311 CrossRef PubMed.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368–12369 CrossRef PubMed.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef.
- R. J. Marshall and R. S. Forgan, Eur. J. Inorg. Chem., 2016, 4310–4331 CrossRef.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- A. M. Fracaroli, P. Siman, D. A. Nagib, M. Suzuki, H. Furukawa, F. D. Toste and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 8352–8355 CrossRef.
- M. Kim, J. F. Cahill, Y. Su, K. A. Prather and S. M. Cohen, Chem. Sci., 2011, 3, 126–130 RSC.
- D. Yu, Q. Shao, Q. Song, J. Cui, Y. Zhang, B. Wu, L. Ge, Y. Wang, Y. Zhang, Y. Qin, R. Vajtai, P. M. Ajayan, H. Wang, T. Xu and Y. Wu, Nat. Commun., 2020, 11, 927 CrossRef.
- M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- J. Zhang, V. Shukla and D. L. Boger, J. Org. Chem., 2019, 84, 9397–9445 CrossRef.
- Y. Goto, H. Sato, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2008, 130, 14354–14355 CrossRef.
- M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518–4519 CrossRef.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- C. Bertozzi, ACS Cent. Sci., 2023, 9, 558–559 CrossRef.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef PubMed.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- C. X. Bezuidenhout, C. Esterhuysen and L. J. Barbour, Chem. Commun., 2017, 53, 5618–5621 RSC.
- N. Kumar, S.-Q. Wang, S. Mukherjee, A. A. Bezrukov, E. Patyk-Kaźmierczak, D. O’Nolan, A. Kumar, M.-H. Yu, Z. Chang, X.-H. Bu and M. J. Zaworotko, Chem. Sci., 2020, 11, 6889–6895 RSC.
- W. Liang, P. M. Bhatt, A. Shkurenko, K. Adil, G. Mouchaham, H. Aggarwal, A. Mallick, A. Jamal, Y. Belmabkhout and M. Eddaoudi, Chem, 2019, 5, 950–963 Search PubMed.
- Y. Hijikata, S. Horike, M. Sugimoto, H. Sato, R. Matsuda and S. Kitagawa, Chem. – Eur. J., 2011, 17, 5138–5144 CrossRef.
- Y. Hijikata, S. Horike, M. Sugimoto, M. Inukai, T. Fukushima and S. Kitagawa, Inorg. Chem., 2013, 52, 3634–3642 CrossRef PubMed.
- D. Jędrzejowski, M. Pander, W. Nitek, W. Bury and D. Matoga, Chem. Mater., 2021, 33, 7509–7517 CrossRef.
- M. Xue, S. Ma, Z. Jin, R. M. Schaffino, G.-S. Zhu, E. B. Lobkovsky, S.-L. Qiu and B. Chen, Inorg. Chem., 2008, 47, 6825–6828 CrossRef CAS.
- S.-H. Lo, L. Feng, K. Tan, Z. Huang, S. Yuan, K.-Y. Wang, B.-H. Li, W.-L. Liu, G. S. Day, S. Tao, C.-C. Yang, T.-T. Luo, C.-H. Lin, S.-L. Wang, S. J. L. Billinge, K.-L. Lu, Y. J. Chabal, X. Zou and H.-C. Zhou, Nat. Chem., 2020, 12, 90–97 CrossRef PubMed.
- B. Lerma-Berlanga, C. R. Ganivet, N. Almora-Barrios, S. Tatay, Y. Peng, J. Albero, O. Fabelo, J. González-Platas, H. García, N. M. Padial and C. Martí-Gastaldo, J. Am. Chem. Soc., 2021, 143, 1798–1806 CrossRef CAS PubMed.
- B. H. Wilson, C. S. Vojvodin, G. Gholami, L. M. Abdulla, C. A. O’Keefe, R. W. Schurko and S. J. Loeb, Chem, 2021, 7, 202–211 CAS.
- I. Novianti, T. Kowada and S. Mizukami, Org. Lett., 2022, 24, 3223–3226 CrossRef.
- D. Svatunek, M. Wilkovitsch, L. Hartmann, K. N. Houk and H. Mikula, J. Am. Chem. Soc., 2022, 144, 8171–8177 CrossRef PubMed.
- M. A. Withersby, A. J. Blake, N. R. Champness, P. Hubberstey, W. Li and M. Schröder, Angew. Chem., Int. Ed. Engl., 1997, 36, 2327–2329 CrossRef.
- M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey and M. Schröder, J. Am. Chem. Soc., 2000, 122, 4044–4046 CrossRef.
- J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 10164–10168 CrossRef.
- P. Müller, F. M. Wisser, V. Bon, R. Grünker, I. Senkovska and S. Kaskel, Chem. Mater., 2015, 27, 2460–2467 CrossRef.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef PubMed.
- A. J. Calahorro, B. Fernández, C. García-Gallarín, M. Melguizo, D. Fairen-Jimenez, G. Zaragoza, A. Salinas-Castillo, S. Gómez-Ruiz and A. Rodríguez-Diéguez, New J. Chem., 2015, 39, 6453–6458 RSC.
- C. F. Hansell, P. Espeel, M. M. Stamenović, I. A. Barker, A. P. Dove, F. E. D. Prez and R. K. O’Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- B. S. Pilgrim, D. A. Roberts, T. G. Lohr, T. K. Ronson and J. R. Nitschke, Nat. Chem., 2017, 9, 1276–1281 CrossRef CAS.
- Q.-H. Guo, J. Zhou, H. Mao, Y. Qiu, M. T. Nguyen, Y. Feng, J. Liang, D. Shen, P. Li, Z. Liu, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 5419–5428 CrossRef CAS PubMed.
- C. Chen, C. A. Allen and S. M. Cohen, Inorg. Chem., 2011, 50, 10534–10536 CrossRef CAS.
- L. Feng, S.-H. Lo, K. Tan, B.-H. Li, S. Yuan, Y.-F. Lin, C.-H. Lin, S.-L. Wang, K.-L. Lu and H.-C. Zhou, Matter, 2020, 2, 988–999 CrossRef.
- B. Lerma-Berlanga, C. R. Ganivet, N. Almora-Barrios, R. Vismara, J. A. R. Navarro, S. Tatay, N. M. Padial and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2022, 61, e202208139 CrossRef CAS.
- B. Lerma-Berlanga, N. M. Padial, M. Galbiati, I. Brotons-Alcázar, J. Albero, H. García, A. Forment-Aliaga, C. R. Ganivet and C. Martí-Gastaldo, Adv. Funct. Mater., 2023, 33, 2302246 CrossRef CAS.
- B.-H. Li, S.-L. Wang, S. Pal, P. B. So, G.-Y. Chen, W.-J. Huang, Y.-L. Hsu, S.-Y. Kuo, J.-M. Yeh and C.-H. Lin, Microporous Mesoporous Mater., 2021, 325, 111319 CrossRef CAS.
- D. Jędrzejowski, M. Ryndak, J. J. Zakrzewski, M. Hodorowicz, S. Chorazy and D. Matoga, ACS Appl. Mater. Interfaces, 2023, 15, 25661–25670 CrossRef.
- Y.-J. Zhang, H.-X. Nie, M.-H. Yu and Z. Chang, J. Solid State Chem., 2021, 300, 122257 CrossRef CAS.
- M. Vinu, K. Sivasankar, S. Prabu, J. Han, C. Lin, C. Yang and J. Demel, Eur. J. Inorg. Chem., 2020, 461–466 CrossRef CAS.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef PubMed.
- M. R. Black, S. Bhattacharyya, S. P. Argent and B. S. Pilgrim, J. Am. Chem. Soc., 2024, 146, 28233–28241 Search PubMed.
Footnote |
| † These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2024 |