DOI:
10.1039/D4CC03680H
(Highlight)
Chem. Commun., 2024,
60, 12270-12286
Asymmetric electrophilic functionalization of amino-substituted heteroaromatic compounds: a convenient tool for the enantioselective synthesis of nitrogen heterocycles
Received
22nd July 2024
, Accepted 24th September 2024
First published on 25th September 2024
Abstract
The catalytic asymmetric electrophilic functionalization of the less reactive N-heteroaromatic compounds has been reported using the approach of the introduction of an exocyclic amino substituent. This strategy has allowed enantioselective Friedel–Crafts alkylation in pyrazoles, isoxazoles and isothiazoles, as well as in aminoindoles, aminobenzofurans and aminobenzothiophenes. Several stereoselective methods have been used for the 1,4-addition or 1,2-addition of these heteroaromatic compounds to different electrophiles employing organocatalysts or chiral metal complexes. The activating exocyclic amino substituent has also been used as a nucleophile in tandem reactions, including formal cycloadditions ([3+2] and [3+3]), for the synthesis of highly functionalized chiral nitrogen heterocycles.
1. Introduction
Nitrogen heteroaromatic compounds are a significant class of organic molecules characterized by the presence of nitrogen atoms within their aromatic ring structures.1 The presence of nitrogen atoms in the aromatic ring modifies the electron distribution and reactivity of these compounds compared to their all-carbon counterparts, leading to a wide range of applications and functionalities. These kinds of compounds play a pivotal role in various fields, including medicinal and pharmaceutical chemistry,2 materials science,3 and agricultural chemistry,4 due to their unique chemical and physical properties. Moreover, N-heteroaromatic scaffolds are present in natural products (alkaloids), and play crucial roles in biological processes and exhibit a wide range of bioactivities.5
The versatility of N-heteroaromatic compounds extends to their synthetic utility, where they are employed as intermediates in organic synthesis, catalysts or ligands in coordination chemistry. Advances in synthetic methodologies have enabled the precise modification of these compounds, enhancing their applicability and leading to the development of novel materials, ligands or drugs. In this context, the enantioselective synthesis or the functionalization of N-heteroaromatic compounds are extremely important for synthetic organic chemistry.6 One of the most straightforward methods for the catalytic asymmetric functionalization of N-heteroaromatic compounds is enantioselective electrophilic functionalization. The most studied catalytic asymmetric electrophilic reactions of N-heterocycles are the reactions using indoles7 and pyrroles8 as nucleophiles. This particularity is due to the fact that pyrroles and indoles are electron-rich heteroaromatic compounds and the Friedel–Crafts alkylation and other electrophilic substitutions are favored. However, other important five membered nitrogen heterocycles such as pyrazoles9 or isoxazoles10 are less prone to Friedel–Crafts alkylation because of the electronic properties of these N-heterocycles (Fig. 1). Normally, pyrazoles and isoxazoles are less electron-rich aromatic heterocycles, therefore it is difficult to perform electrophilic substitutions with this kind of nitrogen heterocycle. One way to increase the reactivity of aromatic compounds is the introduction of exocyclic electron-rich groups such as hydroxyl, alkoxy or amino groups. These groups increase the electronic density on the aromatic rings making them more reactive for Friedel–Crafts alkylation or electrophilic substitution. The synthetic versatility conferred by the presence of an exocyclic amino group in N-heterocyclic compounds is remarkable, due to the existence of multiple nucleophilic sites. This feature presents a significant synthetic challenge, particularly in achieving regioselectivity. Moreover, the presence of these nucleophilic groups can also increase the molecular complexity of the synthesized compounds because they can permit domino or tandem reactions due to the higher number of nucleophilic sites in the molecule. In fact, the versatility of amino-exocyclic N-heteroaromatic compounds has made them very interesting scaffolds for the synthetic community and in medicinal chemistry in recent years. Despite the high versatility, catalytic enantioselective functionalization of these N-heterocyclic compounds is underdeveloped. Consequently, in this review, we want to collect the efforts of several research groups around the world working on the catalytic enantioselective functionalization of these amino-substituted N-heterocyclic compounds. Although there has been an increasing contribution to this topic since 2022, we are confident enough that the interest of the scientific community in these kinds of amino-exocyclic N-heterocycles will grow and more elegant approaches will be developed. Along these lines, the existing protocols for the catalytic enantioselective functionalization of 5-aminopyrazoles, 5-aminoisoxazoles and 5-aminoisothiazoles will be presented as well as related methodologies using 3-aminoindoles, 3-aminobenzofuranes and 3-aminobenzothiophenes. Thus, the main aim of this review is to provide a starting point for the development of potential synthetic methodologies for the functionalization of these nitrogen heterocycles. Henceforward, we have divided this exposition depending on the nature of the reaction in the asymmetric electrophilic functionalization of each N-heteroaromatic compound.
 |
| | Fig. 1 Nucleophilicity in N-heteroaromatic compounds. (a) Examples of electron-rich heteroaromatic compounds. (b) Examples of less electron-rich heteroaromatic compounds. | |
2. Electrophilic functionalization of 5-aminopyrazoles
5-Aminopyrazoles11 have gained the attention of medicinal chemistry researchers because this kind of nitrogen heterocycle is present in several compounds with biological activities such as antibacterial12 or antifugal13 agents. The synthetic versatility of 5-aminopyrazoles is very significant, due to the presence of several nucleophilic positions, making regioselectivity a synthetic challenge. The versatility of 5-aminopyrazoles has made them very interesting frameworks for the synthetic community and medicinal and pharmaceutical chemistry fields, and several reports have been described showing their electrophilic functionalization.14 Although the catalytic enantioselective functionalization of 5-aminopyrazoles has been scarcely described, we have divided the examples reported in the literature depending on the addition type.
2.1 Enantioselective 1,2-addition of 5-aminopyrazoles
In 2020, our research group reported the first organocatalytic enantioselective aminoalkylation of 1-substituted 5-aminopyrazoles 1 using cyclic imines 2 with a benzoxathiazine 2,2-dioxide structure as electrophiles, which are synthetic precursors of benzosulfamide heterocycles (Scheme 1).15 Utilizing a N-tert-butyl squaramide derived from quinine (Cat. A), we successfully obtained a range of chiral sulfamidates 3 bearing an aminopyrazole moiety, achieving good yields and moderate to excellent enantiomeric excesses. The reaction tolerated various substituents at different positions on the phenyl group of the cyclic imine, consistently yielding the desired products, except when a nitro group was present at position 6. Regarding the 5-aminopyrazole partner, both methyl and various phenyl derivatives attached to N1 were well accommodated, with the exception of a 4-methoxyphenyl ring, which resulted in a racemic compound. Alkyl groups at C3 of the aminopyrazole ring performed efficiently in the reaction, although the 3-phenyl derivative produced low yield and enantiomeric excess. Additionally, we explored the impact of substituents on the 5-amino group, finding optimal results with phenyl and 4-methyl groups. Conversely, the 4-methoxyphenylamino derivative showed low enantioselectivity, and the 4-bromophenylamino group led to a low yield. The absolute stereochemistry at the stereogenic center was determined to be (S) by X-ray analysis. The proposed mechanistic pathway is depicted in Scheme 1. The chiral squaramide A acts as a bifunctional organocatalyst activating both reaction partners in the reaction, making the pre-orientation required to achieve the stereoselectivity of the reaction.
 |
| | Scheme 1 Organocatalytic addition of 5-aminopyrazoles 1 to cyclic benzoxathiazine 2,2-dioxides 2 catalysed by a chiral squaramide. | |
The synthetic versatility of the exocyclic amino group was demonstrated by the synthesis of tetracyclic compounds 4a and 4b upon treatment of the chiral sulfamidates 3 with formaldehyde in acidic conditions (Scheme 2).
 |
| | Scheme 2 Synthetic transformations of 5-aminopyrazolyl sulfamidates. | |
Lin and colleagues at Zhejiang University utilized a chiral spirocyclic phosphoric acid (Cat. B) in the enantioselective synthesis of unnatural pyrazole-based α-amino acid derivatives through the reaction of N-aryl-5-aminopyrazoles 1 with β,γ-alkynyl-α-imino esters 5 (Scheme 3).16 The reaction yielded the corresponding products 6 in good to excellent yields (67–98%) and enantioselectivities (73–99% ee). The method was effective with aminopyrazoles having either an unsubstituted or monosubstituted amino group at C5. Moreover, a phenyl group and various alkyl substituents at position 3 of the 5-aminopyrazole, along with differently substituted aryl groups at N1, were well-tolerated, without significantly affecting the yield and enantioselectivity. For the alkynyl ketoester imine 5, both N-Boc and N-Cbz imines performed similarly. The reaction accommodated a variety of substituted aryl rings, heterocycles, and cycloalkyl groups. However, using methyl esters led to reduced enantioselectivity compared to ethyl or benzyl esters. Based on control experiments and the observed S stereochemistry, a plausible reaction pathway was proposed. Cat. B serves as bifunctional catalysts to activate both the 5-aminopyrazole and the imino ester through hydrogen bonding. Nucleophilic attack of the aminopyrazole to the imine would be directed to the Si-face of the imino group.
 |
| | Scheme 3 Phosphoric acid-catalyzed asymmetric addition of aminopyrazoles 1 to β,γ-alkynyl-α-imino esters 5. | |
The reaction performed at 1 mmol scale provided results consistent with those obtained at 0.1 mmol scale, indicating the robustness of the method. Additionally, the exocyclic NH2 group in the reaction products 6 facilitated further transformations, allowing the synthesis of chiral thiourea 7 and lactam 8 derivatives (Scheme 4).
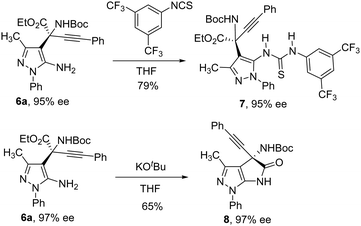 |
| | Scheme 4 Synthetic transformations of aminopyrazole-derived amino acids. | |
In 2023, Li and Zhao applied a binaphthol-derived phosphoric acid Cat. C in the reaction of 5-aminopyrazole derivatives 1 with cyclic ketimines 9 derived from 3H-indol-3-one, classifying it as an aza-Friedel–Crafts reaction.17 This reaction yielded 5-aminopyrazole-based indolin-3-ones 10 featuring a quaternary stereocenter. Indol-3-ones 9 with various aryl groups attached to C2 were tested, resulting in enantiomeric excesses above 90% in all cases except when substituted with an electron-donating methyl group, which led to lower enantiomeric excesses (Scheme 5). Indol-3-ones with substituents of different electronic nature at positions 4 or 5 in the homoaromatic ring also yielded good results. Furthermore, the reaction performed well with 5-aminopyrazoles 1 bearing an unsubstituted NH2 group at C5, a methyl group at C3, and a wide range of substituted aryl rings at N1. However, when a phenyl group was present at C3, the expected product was obtained with low enantiomeric excess. Additionally, the 5-benzylamino derivative of the imidazole exhibited good enantioselectivity but low yield. Based on the observed stereochemistry, a mechanistic pathway was proposed involving a transition state (TS) where dual hydrogen bonds between the chiral phosphoric acid and the substrates drive the 5-aminopyrazoles to attack the Si-face of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group of the ketimine 9.
N group of the ketimine 9.
 |
| | Scheme 5 Phosphoric acid-catalyzed reaction of 5-aminopyrazole 1 derivatives with cyclic ketimines 9. | |
2.2 Enantioselective 1,4-addition of 5-aminopyrazoles
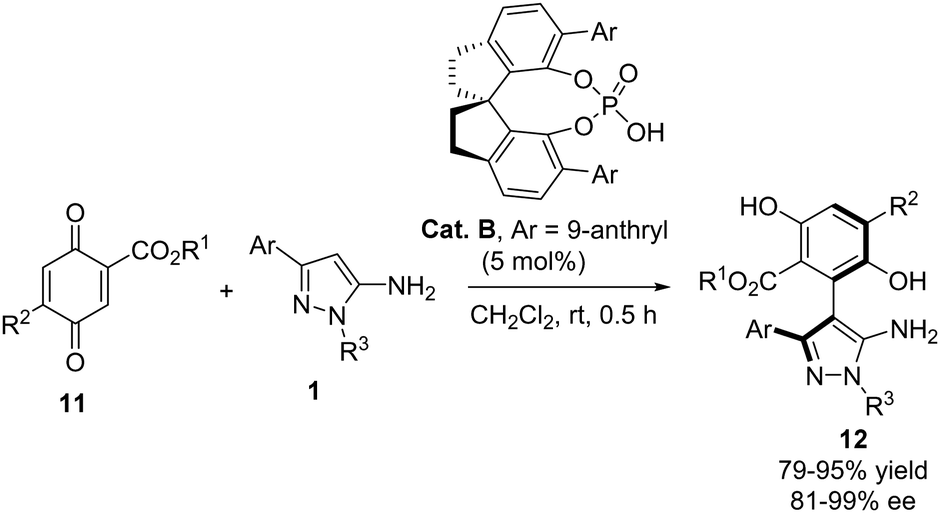
Recently, Xen and Li reported a highly enantioselective arylation of 3-aryl-5-aminopyrazoles 1 with quinone derivatives 11 (Scheme 6).18 The reaction, catalyzed by the chiral phosphoric acid Cat. B, involved the conjugate addition of the pyrazole to a double bond of the quinone, providing rapid access to an array of arylpyrazoles 12 with axial chirality. Generally, the reaction products were obtained with excellent yields and enantioselectivities. A broad variety of esters in the quinone partner were well tolerated, yielding excellent results, although substituent R2 was maintained as H except in one example with MeO. The 3-aryl substituent of the aminopyrazole could be a phenyl ring substituted with groups of varying electronic character or even a heterocyclic ring. Furthermore, different alkyl or aryl groups attached to N1 were tolerated.
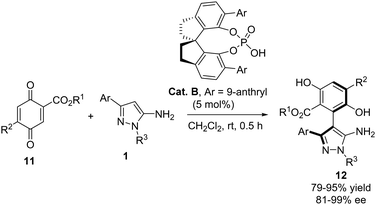 |
| | Scheme 6 Enantioselective addition of aminopyrazoles 1 to quinone derivatives 11 for the synthesis of arylpyrazoles with axial chirality. | |
The authors demonstrated the efficiency and applicability of the method by performing the reaction on a 1 mmol scale. Additionally, the catalyst load could be decreased to 1 mol% without affecting the yield or stereoselectivity of the reaction. The amino group of the reaction product 12a could be selectively acylated with p-bromobenzoyl chloride in the presence of two OH groups, without any loss of optical purity (Scheme 7).
 |
| | Scheme 7 Selective acylation of the amino group in arylpyrazoles 12. | |
Related to the above work, Zheng's group described in 2024 a highly atroposelective arylation of 3-alkyl-5-aminopyrazoles 1 with naphthoquinone ester derivatives 14 through a 1,4-addition reaction catalyzed by the chiral phosphoric acid (R)-Cat. C (Scheme 8).19 In general, the chiral naphthylpyrazoles 15 were obtained with excellent yields (up to 99%) and excellent enantioselectivities (up to 99%). An assortment of ester groups in the naphthoquinone partner were tolerated, although the best results were obtained with the methyl ester. Regarding the 5-aminopyrazole partner, the reaction tolerates substituents at N1 of the 5-aminopyrazole moiety, including cyclohexyl, isopropyl, benzyl, or substituted phenyl groups, all of which exhibited comparable yields and enantioselectivities in the reaction, although with the isopropyl group a decrease in the enantiomeric excess was observed. In order to obtain good enantioselectivities, a tBu group at C3 is mandatory, as evidenced by the lack of enantioselectivity observed when a methyl group is attached to this position, while 5-aminopyrazoles bearing an aryl group at C3 were not reported. The authors could scale-up the reaction to the 1 gram scale providing consistent results with those obtained at the 0.2 mmol scale, indicating the robustness of their synthetic methodology. The authors also proposed a mechanistic pathway where chiral phosphoric acid works as a bifunctional catalyst to activate both the naphthoquinone ester and the 5-aminopyrazole through hydrogen bonding.
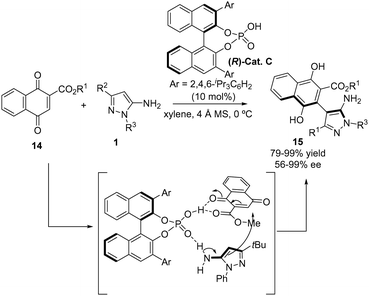 |
| | Scheme 8 Enantioselective addition of aminopyrazoles 1 to naphthoquinone derivatives 14 for the synthesis of naphthylpyrazoles with axial chirality. | |
2.3 Formal [3+3] cyclizations using 5-aminopyrazoles
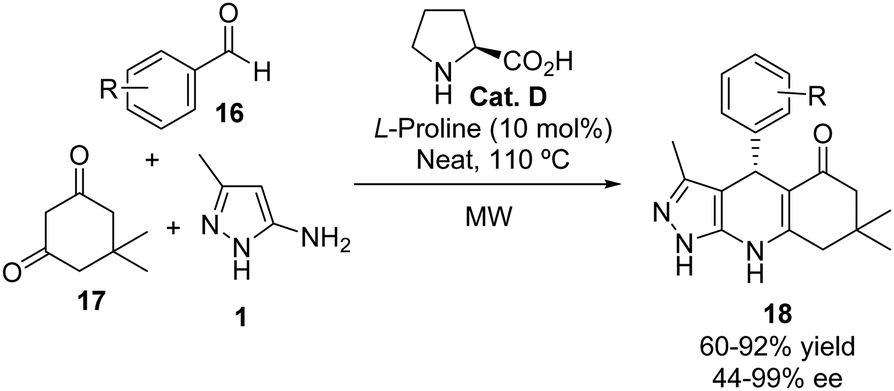
The reaction of 5-aminopyrazoles with conjugated alkenes has been extensively used in the enantioselective synthesis of pyrazolo[3,4-b]quinoline derivatives. The first example was reported by Myrboh in 2016.20 This group described a three-component reaction involving dimedone 17, 3-methyl-5-aminopyrazole 1, and a range of aromatic aldehydes 16. The reaction was catalyzed by L-proline (Cat. D, 10 mol%) in the absence of any solvent under microwave irradiation at 110 °C (Scheme 9). Various benzaldehyde derivatives bearing electron-withdrawing or electron-donating substituents at the ortho, meta, or para positions were tested, resulting in products with good yields (60–92%) and enantiomeric excesses ranging from 44–99%.
 |
| | Scheme 9 Proline-catalyzed three-component synthesis of pyrazolo[3,4-b]quinoline derivatives 18. | |
A plausible mechanism for the reaction is depicted in Scheme 10. Initially, a Knoevenagel reaction between the aldehyde and dimedone, mediated by Cat. D, occurs to form the corresponding enone 19. This is followed by the conjugate addition of the 5-aminopyrazole, forming a C–C bond. Subsequently, a nucleophilic attack of the amino group on the ketone takes place, followed by dehydration and tautomerization, which ultimately produces the final product 18. Remarkably, the authors observed a complete regioselectivity in the nucleophilic attack of the N-1-unprotected aminopyrazole. The authors did not indicate how the L-proline induces the stereoselectivity upon the nucleophilic attack of the aminopyrazole 1 to the enone 19.
 |
| | Scheme 10 Plausible mechanism for the proline-mediated three component synthesis of pyrazoloquinolinones. | |
In 2022, the group of X. Li developed a procedure for the construction of trifluoromethylated pyrazolo[3,4-b]pyridin-6-ones 21.21 The method involves the reaction of 3-(2-trifluoromethylene)oxindoles 20 and 1-aryl-5-aminopyrazoles 1via chiral phosphoric acid (Cat. E) and MgSO4 synergistic catalysis (Scheme 11). N-Aryl-5-aminopyrazoles bearing electron-donating and electron-withdrawing groups proved to be suitable reactants, yielding the corresponding products in moderate yields (50–90%) with high stereoselectivities (up to 95% ee and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). The best results were obtained when the substituents on the N-Ar ring were alkyl groups or groups attached to the meta position. However, when electron-donating or electron-withdrawing groups were attached to the ortho or para positions, the yield and stereoselectivity decreased.
:1 dr). The best results were obtained when the substituents on the N-Ar ring were alkyl groups or groups attached to the meta position. However, when electron-donating or electron-withdrawing groups were attached to the ortho or para positions, the yield and stereoselectivity decreased.
 |
| | Scheme 11 Enantioselective addition of aminopyrazoles 1 to 3-trifluoroethylidene oxindoles 20 for the synthesis of trifluoromethylated pyrazolo[3,4-b]pyridinones. | |
A phenyl ring at position 3 was tolerated, although the enantioselectivity was lower than with alkyl groups. Substituents on the 5-amino group decreased reactivity, resulting in low yield and stereoselectivity of the reaction products. Oxindole derivatives with substituents at positions 5 or 6 of the benzene ring were good substrates, providing better results when these substituents had an electron-donating character. However, substituents at positions 4 or 7 of the benzene ring caused a significant loss of enantioselectivity. The CF3 group could be substituted by other electron-withdrawing groups, although with lower enantioselectivity, except in the case of an ethoxycarbonyl group.
Based on experimental results and previous reports, the authors proposed a reaction mechanism that explains the synergistic integration of the chiral phosphoric acid and MgSO4. The N-aryl-5-aminopyrazole 1 forms a H bond with the catalyst and, simultaneously, the O–H and Lewis acid coordinates with the oxindole 20. The pyrazole substrate undergoes addition to the Re-face of the double bond generating intermediate Int. III, followed by intramolecular transamidation to afford the desired product (Scheme 12).
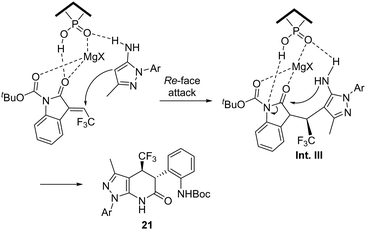 |
| | Scheme 12 Proposed reaction mechanism for the synthesis of trifluoromethylated pyrazolo[3,4-b]pyridinones 21. | |
The [3+3] annulation of 5-aminopyrazoles with enals under chiral carbene catalysis has been extensively studied by several research groups. The first examples were almost simultaneously published by Li's group,22 and Wang and Lang's group.23 These studies involved the reaction of 2-bromoenals 22 with 5-aminopyrazoles 1 to afford the corresponding cycloadducts 23 catalysed by the same kind of organocatalyst (Scheme 13).
 |
| | Scheme 13 Organocatalyzed addition reaction of 2-bromoenals 22 and 5-aminopyrazoles 1 under NHC catalysis. Examples of antimicrobial pyrazolo[3,4-b]pyridinones. | |
Under both catalytic systems, the reaction tolerated a wide range of 2-bromoenals 22 bearing alkyl, heteroaryl, and phenyl groups with diverse electronic properties and substitution patterns. In most reported examples, the reaction products were obtained in good yields with enantiomeric excesses well above 90%. Additionally, various alkyl and aryl substituents at position 3 of the imidazole ring provided good results with excellent enantioselectivities. N-Aryl and N-Me substituted imidazoles were suitable substrates for this transformation. Some of the products 23 prepared by Li were tested for antifungal properties. Chiral compounds bearing R2 = Ph, R3 = Me and R1 = 3-BrC6H4, 3-ClC6H4 or 2-MeOC6H4 showed excellent antifungal activity against Phytophora capsici and Colletotrichum fructicola.22
A mechanistic proposal made by Li is outlined in Scheme 14.22 Initially, 2-bromoenal 22 is activated by the NHC catalyst to form a Breslow intermediate Int. IV, which undergoes bromide elimination to generate an α,β-unsaturated acyl azolium intermediate (Int. V). The aminopyrazole 1 then undergoes nucleophilic conjugate addition from its Re face to the Si face of the β-C(sp2) group of the acyl azolium intermediate. Subsequent intramolecular lactam formation completes the process, yielding the pyrazolo[3,4-b]pyridone product 23 (Scheme 14).
 |
| | Scheme 14 Reaction mechanism proposed by Li for the enantioselective formal [3+3] cycloaddition.22 | |
The asymmetric synthesis of chiral pyrazolo[3,4-b]pyridin-6-ones from enals and 5-aminopyrazoles was recently reported by Zhenquian Fu's group in 2023.24 This transformation employs a chiral N-heterocyclic carbene (NHC) catalyst Cat. H, Cs2CO3 as the base, and 3,3′,5,5′-tetra-tert-butyldiphenoquinone (DQ) as an oxidant in THF (Scheme 15). A variety of enals 24 bearing alkyl groups, as well as diverse substituted aryl rings attached to the double bond, underwent annulation with 5-aminopyrazoles to afford the desired products in high yields and with excellent enantioselectivity (up to 98% ee in most cases). The 5-aminopyrazole component tolerated alkyl and aryl groups at position 4 of the ring, as well as various substitutions on the aromatic ring at N1, consistently delivering excellent outcomes.
 |
| | Scheme 15 NHC-catalyzed reaction of enals 24 and 5-aminopyrazole 1 reported by Fu. | |
A proposed reaction mechanism is depicted in Scheme 16. Initially, the nucleophilic addition of the N-heterocyclic carbene (NHC) to the enal forms the Breslow intermediate (Int. VIII). Upon oxidation, this intermediate transforms into an α,β-unsaturated acylazolium Int. IX. Subsequently, 1,2-addition of 5-aminopyrazole to the acylazolium species, followed by an aza-Claisen rearrangement, tautomerization, and subsequent intramolecular lactamization yields the desired chiral lactam 23. An alternative pathway involving a 1,4 addition of 5-aminopyrazole 1 followed by tautomerization, proton transfer, and intramolecular lactamization, could also potentially lead to the same product (Scheme 16).
 |
| | Scheme 16 Proposed mechanism for the oxidative NHC-catalyzed annulation. | |
Concurrently with this study, Duan and Qi reported a similar reaction employing slightly modified conditions, using tBuOMe as the solvent and a related NHC catalyst (Cat. ent G.) lacking the nitro group (Scheme 17).25 Enals 24 featuring a range of aromatic, heteroaromatic, alkenyl, or alkynyl substituents at the β-position of the double bond reacted with variously substituted 5-aminopyrazoles to afford the desired products 23 in high yields and with excellent enantiomeric excesses (85–97% ee). Enals bearing alkyl substituents at this position exhibited lower yields but maintained excellent enantioselectivity. Moreover, aminopyrazoles with different alkyl and aryl groups at C3 and various aromatic groups at N1 reacted with similarly outstanding outcomes (Scheme 17). The authors further expanded the scope of the reaction to include isatin-derived enals 25, yielding a variety of spirooxindoles 26 in good yields and with excellent enantioselectivity. This strategy accommodated a range of substituted isatin derivatives possessing diverse electronic properties, as well as either a benzyl or methyl protecting group at the N1 position of isatin. Additionally, 1,3-diphenyl-1H-pyrazol-5-amine performed well under the optimized reaction conditions.
 |
| | Scheme 17 NHC-catalyzed reaction of enals 24 with 5-aminopyrazoles 1 reported by Duan and Qi. | |
Very recently, a novel enantioselective synthesis of 1H-pyrazolo[3,4-b]pyridine 28 analogues starting from 5-aminopyrazoles 1 and α,β-unsaturated 2-acyl imidazoles 27 was disclosed (Scheme 18).26 In this transformation, a complex generated in situ from Ni(OTf)2 and a newly developed chiral C2-symmetric bipyridine-N,N′-dioxide ligand (Lig. A) catalyzed the Michael addition/cyclization sequence, delivering the corresponding adducts in 85–97% yield with up to 99% ee under mild conditions. While no 5-aminopyrazoles bearing an alkyl group at C3 were reported, derivatives featuring aryl groups of varying electronic properties, heteroaromatic rings, or a cyclopropyl group at this position reacted efficiently with high yields and excellent enantioselectivities (typically above 90% ee). Substituents at N1 of the indole moiety included tert-butyl, benzyl, or substituted phenyl groups, all of which exhibited comparable yields and enantioselectivities in the reaction. Regarding the acyl imidazole component, electron-rich and electron-poor aryl groups, heterocycles, and alkenes attached to the β-position of the double bond consistently provided excellent results. Furthermore, the reaction could be successfully scaled up to 1 mmol scale and the catalyst load decreased to 2 mol%, demonstrating robust performance without significant impact on reaction outcomes.
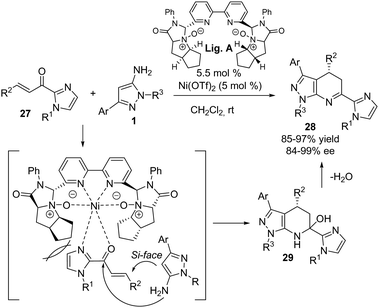 |
| | Scheme 18 Ni/N,N′dioxide-catalyzed asymmetric synthesis of 1H-pyrazolo[3,4-b]pyridine analogues 28. | |
To explain the stereochemical outcome of the reaction, the authors proposed a mechanism wherein the nickel catalyst activates the 2-acyl imidazole 27 through bidentate N,O-coordination facilitating the attack of the 5-aminopyrazole from the Si-face, leading to an intermediate 29 with high regioselectivity, which would generate the desired product after releasing water from compound 28.
3. Electrophilic functionalization of 5-aminoisoxazoles
5-Aminoisoxazole27 is another nitrogen heteroaromatic compound that has focused the attention of pharmaceutical, medicinal and agrochemical chemists in recent decades due to the biological activities presented by compounds that contain this scaffold.28 Therefore, the asymmetric synthesis of compounds bearing this N-heterocycle are remarkable for the synthetic community. However, only a few examples of enantioselective electrophilic functionalization of isoxazoles have been reported in the last few years.
3.1 Enantioselective 1,2-addition of 5-aminoisoxazoles
In 2021, the group of Zhang reported the addition of 5-aminoisoxazoles 30 to isatin-derived ketimines 31 (Scheme 19).29 The authors used a chiral phosphoric acid (Cat. I) as a catalyst for this aza-Friedel–Crafts reaction, accessing the desired chiral 3-disubstituted 3-amino-oxindoles 32 in high yields (84–99%) and moderate to excellent enantioselectivities (53–99% ee). The reaction tolerates the presence of several alkyl groups at the N-1 of the ketimines, as well as several substituents at the aromatic ring of 31. Regarding the 5-aminoisoxazoles 30, the enantiomeric excesses are excellent when the authors tried the reaction with 3-aryl-substituted isoxazoles. In order to obtain good enantioselectivities the exocyclic amino group should be secondary. The authors also provided a plausible stereochemical model for the reaction where the phosphoric acid acts as a bifunctional organocatalyst activating at the same time both reagents by hydrogen bonding. Moreover, they could scale-up the methodology to a 4 mmol reaction, using only 0.5 mol% of chiral catalyst and also perform several transformations.
 |
| | Scheme 19 Phosphoric acid catalyzed the enantioselective addition of 5-aminoisoxazoles 30 to isatin-derived ketimines 31. | |
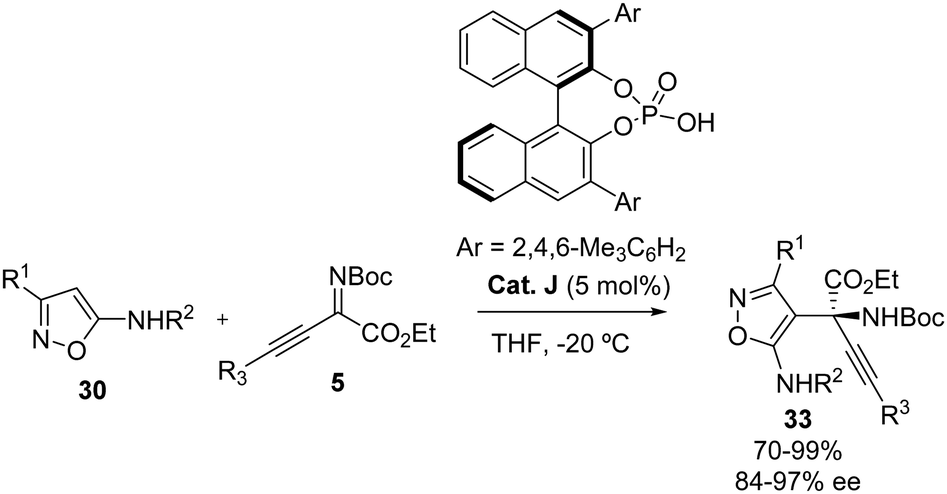
The Zhang group also described the addition of these nucleophiles 30 to β,γ-alkynyl-α-ketimino esters 5 using a Cat. J as the catalyst (Scheme 20).30 The corresponding α-isoxazole-α-alkynyl amino esters were obtained in high yields (94–99%) and good to excellent enantioselectivities (84–97% ee). Regarding the isoxazole 30, aryl or alkyl groups are tolerated at the C-3 position. Concerning the reactivity of the electrophile 5 under the optimized reaction conditions, the authors tested the reaction with different iminoesters with a variety of substituents at the alkyne moiety such as aryl rings, heterocycles or cycloalkyl groups. The –NHR group in the nucleophile 30 proved to be essential for selectivity, pointing towards a bifunctional mode of action of the catalyst. The proposed mechanistic model is very similar to the one presented in Scheme 19.
 |
| | Scheme 20 Enantioselective addition of 5-aminoisoxazoles 30 to β,γ-alkynyl-α-ketimino esters 5. | |
The enantioenriched products can be employed in different synthetic transformations, such as the iodocyclization in the presence of a silver(I) salt to obtain the corresponding heterocyclic compound 34 in moderate yield, taking advantage of the exocyclic amino group. The triple bond can be partially hydrogenated to give the Z-alkene 35 in good yield. Moreover, the protected aminoalcohol 36 can be prepared in good yield by reduction with NaBH4. In all these transformations, the optical purity was preserved (Scheme 21).
 |
| | Scheme 21 Synthetic transformations of an α-isoxazole-α-alkynyl amino ester. | |
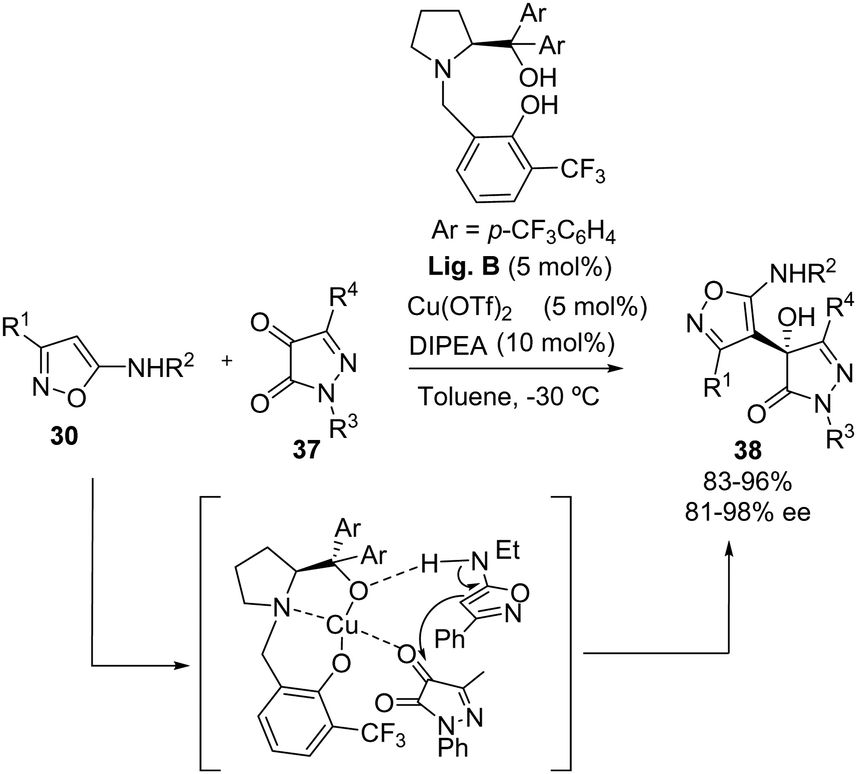
Metal catalysis can also be employed for the enantioselective addition of 5-aminoisoxazole, as reported by the group of Sun and Wang in 2024.31 In this case, the authors employed a Cu(II) complex with a prolinol-derived chiral ligand (Lig. B) to carry out the addition of 5-aminoisoxazoles 30 to pyrazole-4,5-diones 37 (Scheme 22). The corresponding compounds 38 with pyrazolone-isoxazole scaffolds were obtained in high yields (83–96%) and with excellent enantioselectivities in most of the cases (75–99% ee). The reaction exhibited comparable yields and enantioselectivies independently of the electronic character of the substituents (NO2, halogen, alkyl or MeO) or the position (meta or para) of the substituents in the aromatic ring attached at the N1 of 37. However, when the substituent was in ortho-position the enantiomeric excess decreased. The authors also tested a variety of 3-aryl-5-aminoisoxazoles 30, but did not show any example with an alkyl group at the C-3 position. In this report, the authors also observed a lack of reactivity when N,N-dimehtylamino isoxazole was employed, indicating an interaction between the nucleophile and the chiral catalyst. Moreover, when the prolinol hydroxyl in Lig. B was replaced with a methoxy group, only the racemic product was obtained, showing the importance of both hydroxyl groups at the ligand to generate the chiral complex. These hypotheses were supported by DFT calculations.
 |
| | Scheme 22 Enantioselective addition of 5-aminoisoxazoles 30 to pyrazole-4,5-diones 37. | |
3.2 Enantioselective 1,4-addition of 5-aminoisoxazoles
Isoxazole-containing heterotriarylmethanes can be prepared in an enantioselective manner, as reported by the group of Lin in 2023,32 by the addition of 30 to 3-methide-3H-pyrroles generated in situ (Scheme 23). The authors described the treatment of pyrrol-3-aryl carbinols 39 with a chiral spirocyclic phosphoric acid (Cat. K), which generates the highly reactive 3-methide-pyrrole (Int. XII) by dehydration, and subsequent Friedel–Crafts-type addition to generate the desired products 40 in good to high yields (71–97%) and with good enantioselectivities (78–93% ee). In this case, the methodology is efficient with –NH2 and –NHEt substituents on the isoxazole, but reactivity and stereocontrol are again compromised when –NEt2 is tested. The authors only tested 5-aminoisoxazoles with aryl substituents at C-3, and they observed that the corresponding products 40 were obtained with good results independently of the electronic character or the position of the substituents. The authors proposed a mechanism where the chiral phosphoric acid (Cat. K) acts as a bifunctional organocatalyst in the reaction.
 |
| | Scheme 23 Enantioselective addition of 5-aminoisoxazoles 30 to 3-methide-3H-pyrroles generated in situ. | |
A spirocyclic chiral phosphoric acid (Cat. L) was also employed by the group of Luo and Zhou for the atroposelective arylation33 of 5-aminoisoxazoles 30 with azonaphthalenes 41 (Scheme 24).34 This methodology enables the synthesis of a wide range of axially chiral heterobiaryl diamines 42 in generally good yields with excellent enantioselectivities (up to 99%). The reaction tolerates an assortment of aryl groups as well as an iPr substituent at the C-3 carbon of isoxazole. Control experiments demonstrated that the presence of the N–H group is essential for achieving high yield and atroposelectivity, and the substituent at this nitrogen can be an alkyl, benzyl or allyl group. Regarding the azonaphthalenes 41, the authors show a good compatibility of substituents (Br, Me or MeO) at 6 or 7 position of the naphthalene aromatic ring. They could scale-up the methodology to a 2 mmol reaction with similar results in terms of yield and enantioselectivity. Furthermore, a linear effect was observed, suggesting that a monomeric complex of the catalyst is likely involved in the process for inducing the asymmetry.
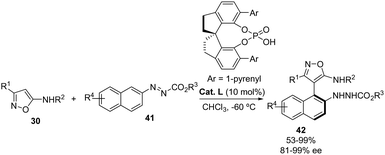 |
| | Scheme 24 Atroposelective arylation of 5-aminoisoxazoles 30 with azonaphthalenes 41 catalyzed by a chiral phosphoric acid. | |
3.3 [3+2] cyclizations using 5-aminoisoxazoles
5-Aminoisoxazoles can also be used as substrates in an enantioselective dearomative [3+2] annulation. In 2020, Wang, Zhang and their coworkers described a novel synthesis of chiral isoxazoline-fused dihydrobenzofurans 44 bearing two contiguous quaternary stereocenters (Scheme 25).35 This was achieved through the [3+2] annulation of 5-aminoisoxazoles 30 with quinone monoimines 43, catalyzed by a chiral phosphoric acid Cat. I. The products were obtained in moderate to good yields (35–87%) and with excellent enantioselectivities (81–94%). To determine the absolute configuration of the synthesized products, X-ray crystal structural analyses were employed. The stereochemistry of the products provided valuable insights into the reaction mechanism. Based on the absolute configuration of the [3+2] annulation products 44, the authors proposed a plausible reaction mechanism where the first step is the double activation of both electrophile and nucleophile by hydrogen bonding with Cat. I. 5-Aminoisoxazole 30 attacked quinone monoimine 43 to give intermediate Int. XIII, which immediately underwent aromatization to give phenol Int. XIV. Lastly, intramolecular acetalization generated the final product 44.
 |
| | Scheme 25 Enantioselective dearomative [3+2] annulation of 5-aminoisoxazoles 30 with quinone monoimines 43. | |
Notably, the approach also provides access to a variety of bridged isoxazoline-fused dihydrobenzofurans 46 through a tandem process (dearomative [3+2] annulation–cyclization) of ethyl 4-acetate-isoxazol-5-amines 45 with quinone imine 43, highlighting its versatility (Scheme 26).
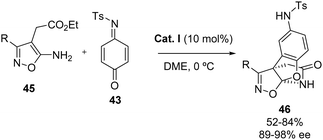 |
| | Scheme 26 Enantioselective synthesis of isoxazoline-fused dihydrobenzofurans. | |
3.4 Formal [3+3] cyclizations using 5-aminoisoxazoles
5-Aminoisoxazoles have been used to construct spiro-isoindolinone-dihydropyridines 48 through the cyclization with α-(3-isoindolinonyl) propargylic alcohols 47 (Scheme 27).36 This interesting process, catalyzed by a racemic phosphoric acid (Cat. M) and a silver salt, yielded the corresponding racemic compounds 48 with moderate to high yields and was reported by Zhang in 2024. The authors explored the asymmetric reaction of 30a with 47a using the chiral phosphoric acid Cat. C ((S)-TRIP) as a catalyst, but the product 48a was obtained with low enantiomeric excess (39% ee). The authors proposed a reaction mechanism based on the control experiments performed. The mechanism consists on protonation and dehydration of α-(3-isoindolinonyl) propargylic alcohol 47, forming an iminium Int. XV. This intermediated is then attacked by 5-aminoisoxazole, followed by cyclization promoted by silver salt leading to a spiro intermediate Int. XVI which undergoes ring cleavage resulting in the final aromatic product 48.
 |
| | Scheme 27 Formal [3+3] cycloaddition of 5-aminoisoxazoles 30 with α-(3-isoindolinonyl) propargylic alcohols 47. | |
Recently, a variety of enantioselective methodologies have emerged for formal [3+3] cycloadditions employing 5-aminoisoxazoles 30 as substrates. For example, Wang and coworkers described the synthesis of chiral six-membered ring N,O-hemiaminals 50 using an enantioselective functionalization of 30 with high yields, moderate to excellent diastereoselectivities and excellent enantiomeric excesses (Scheme 28).37 Specifically, the authors developed a Michael/N-hemiacetalization cascade reaction involving 5-aminoisoxazoles 30 and β,γ-unsaturated α-ketoesters 49, catalyzed by a chiral Cu(II) complex generated from Cu(OTf)2 and Lig. C. Under the optimized reaction conditions, the reaction tolerated a wide range of β,γ-unsaturated α-ketoesters 49 bearing heteroaryl and phenyl groups with diverse electronic properties and substitution patterns at the γ-position of the double bond. However, when the γ-substituent was an alkyl group the reaction did not take place. Additionally, various aryl substituents or a tBu group at position 3 of the isoxazole ring provided good results with good to excellent enantioselectivities (92–98% ee). Although, when a fluorine atom was placed at the para position, product 50 was isolated in 96% yield and 75% ee. Moreover, the authors could perform the reaction to 5 mmol scale obtaining excellent results. The authors proposed a reaction mechanism based on control experiments such as nonlinear experiments among others and DFT calculations.
 |
| | Scheme 28 Enantioselective Michael/N-hemiacetalization cascade reaction with 5-aminoisoxazoles and β,γ-unsaturated α-ketoesters. | |
The use of formal [3+3] cycloaddition reactions employing 5-aminoisoxazoles 30 has also been highlighted for synthesizing chiral dihydroisoxazolo[5,4-b]pyridin-6-ones 51, underscoring the versatility of 5-aminoisoxazoles as 1,3-bisnucleophiles (Scheme 29).38 This methodology utilizes α-bromoenals 22 catalyzed by a N-heterocyclic carbene (NHC) Cat. ent G, delivering the corresponding products 51 in high yields and excellent enantioselectivities. The authors described 42 examples where a wide range of 2-bromoenals 22 bearing alkyl, heteroaryl, and phenyl groups with diverse electronic properties and substitution patterns were used. In most described examples, the pyridin-6-ones 51 were obtained in good yields (up to 99%) with enantiomeric excesses well above 93%. Additionally, various alkyl, cycloalkyl, aryl and heteroaryl substituents at the C-3 of the isoxazole 30 delivered excellent enantioselectivities as well. However, when the substituent was a tBu group the corresponding product was obtained with low yield (19%). To demonstrate the practical application of this method, a scale-up reaction yielded the target product with comparable efficiency. Further synthetic transformations of the resulting compound were explored, including O-methylation, reduction of the lactam using LiAlH4, and N-benzylation, and all these transformations occurred in high yields and without detrimental effects on the optical purity. Moreover, the authors proposed a plausible mechanism similar to the one using aminopyrazoles.22,23
 |
| | Scheme 29 Enantioselective cycloaddition of 5-aminoisoxazoles 30 with α-bromoenals 22 using NHC catalysis. | |
4. Electrophilic functionalization of 5-aminoisothiazoles
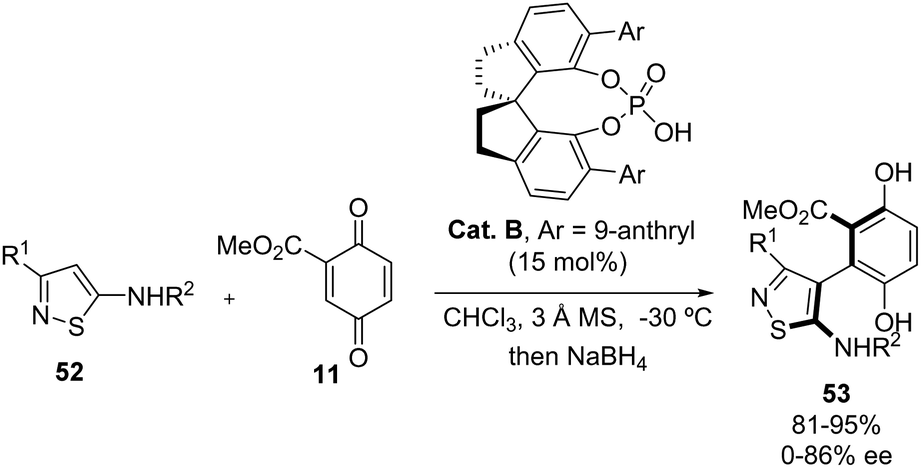
Isothiazole39 is another interesting nitrogen heteroaromatic scaffold important in medicinal or agrochemical industries. The enantioselective functionalization of 5-aminoisothiazoles 52 is underdeveloped and only one example has been reported so far. In 2023, Zhang's group developed a methodology to synthesize isothiazole-containing axially chiral biaryls 53 with moderate to good yields and enantioselectivities.40 This method utilizes chiral phosphoric acid catalysis (Cat. B) for the atroposelective33 arylation of 5-amino-isothiazoles 52 with methyl p-quinone carboxylate 11 (Scheme 30). In order to reduce the oxidized compound, sodium borohydride was added to the reaction mixture. The reaction only tolerates several 3-(hetero)aryl-5-amino-isothiazoles 52 obtaining good enantioselectivities (70–90% ee) and high yields (89–95%). However, if the aryl group at C-3 presented a substituent at the ortho position, the corresponding chiral biaryls 53 were obtained with very low enantioselectivities (0–15% ee). The structures of the products were confirmed via X-ray crystal structural analysis.
 |
| | Scheme 30 Atropoenantioselective arylation of 5-amino-isothiazoles with methyl p-quinone carboxylate. | |
5. Electrophilic functionalization of 3-aminobenzofuran
Benzofurans41 are significant electron-rich heteroaromatic compounds that are present in natural products and biologically active compounds. Habitually, electron-rich aromatic heteroaromatic compounds such as benzofuran can be used as nucleophiles to participate in asymmetric electrophilic functionalizations. However, most of these reactions are limited at the nucleophilic C3-position of the aromatic heterocyclic ring. A strategy for the asymmetric electrophilic aromatic substitution at the C2-position of these heteroaromatic molecules is the introduction of an exocyclic amino substituent at C-3. Several examples have been described for the enantioselctive functionalization of 3-aminobenzofuran.
5.1 Enantioselective 1,2-addition of 3-aminobenzofurans
A quinine-derived thiourea Cat. N was used by the group of Zhou and Xu in 2023 to obtain 3-aryl-3-aminooxindoles 55.42 In this case 3-aminobenzofurans 54 were reacted with isatin-derived ketimines 31 in an aza-Friedel–Crafts type reaction, obtaining the desired tetrasubstituted stereocenter-containing products in good to excellent yields (72–95%) and high to excellent enantioselectivities (48–97% ee) in most of the cases (Scheme 31). In order to have high enantioselectivities, the N-1 of compounds 31 should have a trityl (Tr) substituent. The reaction tolerates substituents at the 5, 6 or 7 position of the aromatic ring of 31 obtaining good enantioselectivities (86–92% ee), but the presence of strong electron-withdrawing groups such as the NO2 group decreased the enantioselectivity of the reaction (71% ee). Regarding the 3-aminobenxofuran derivatives 54, the authors studied the scope of the reaction with substituents at 5, 6 or 7, showing excellent enantioselectivities in all the cases (87–97% ee). The absolute configuration of the products was assigned by X-ray crystallography analysis. The author proposed that the chiral thiourea acts as a bifunctional organocatalyst activating at the same time the electrophile and the nucleophile by hydrogen bonding.
 |
| | Scheme 31 Organocatalytic 1,2-addition of 3-aminobenzofurans 54 to isatin-derived ketimines 31 catalyzed by a chiral thiourea. | |
The authors showed the robustness of their methodology by the scale-up of the reaction at the gram scale. Moreover, they used the exocyclic amino group to synthesize the spyrocyclic compound 56 through the treatment of product 55a with triphosgene (Scheme 32).
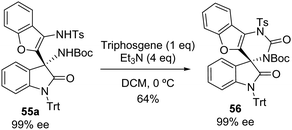 |
| | Scheme 32 Synthesis of a chiral spirocyclic compound 56 with triphosgene. | |
5.2 Enantioselective 1,4-addition of 3-aminobenzofurans
1,4-Conjugate addition products can be achieved for the reaction of 3-aminobenzofurans with α,β-unsaturated carbonyl compounds using metal catalysis. The group of Kang and Du took advantage of the ability of chiral-at-metal rhodium(III) complex Cat. O to activate 2-acylimidazoles 27 (Scheme 33), generating the desired 2-substituted 3-aminobenzofurans 57 in high yields (76–99%) and enantioselectivities (87–98% ee).43 3-Aminobenzofurans with substituents of different electronic nature at positions 5 or 6, as well as different protecting groups at the nitrogen such as Ts, Ns or Ms also yielded good results. Regarding the acyl imidazole component, electron-rich and electron-poor aryl substituents, heterocycles, alkenes, alkyl or CF3 attached to the β-position of the double bond delivered excellent results in terms of enantioselectivity (85–96% ee) and conversions. The authors also showed that 2-acylpyridine derivatives can be used as electrophiles obtaining good results. Gram scale reactions were also successful, delivering the desired product in 95% yield and 95% ee, with only 0.5 mol% catalyst loading. Interestingly, the acylimidazole moiety can be converted into an aldehyde or ester without compromising the optical purity of the sample. The authors proposed a plausible mechanistic pathway, where the rhodium catalyst activates the 2-acyl imidazole 27 through bidentate N,O-coordination facilitating the attack of the 3-aminobenzofuran 54 from the Re-face.
 |
| | Scheme 33 Chiral Rh-catalyst for the addition of 3-aminobenzofurans 54 to 2-acylimidazoles 27. | |
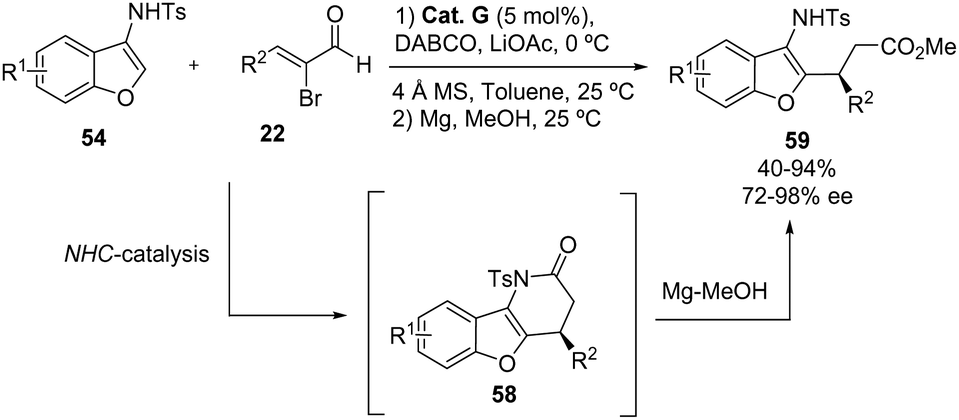
In a report presented by the group of Biju in 2020, an NHC catalyst was used for the enantioselective C2-functionalization of 3-aminobenzofurans 54 with 2-bromoenals 22 (Scheme 34).44 The intermediate chiral dihydropyridinones 58 were ring-opened with Mg/MeOH to obtain the corresponding δ-amino acid derivatives 59 in moderate to excellent yields (40–94%) and good to excellent enantioselectivities (72–98% ee). The authors tested the reaction with a wide range of bromoenals 22, with different aryl, heteroaryl and alkyl substituents at the β-position. Interestingly, if the treatment with Mg/MeOH is not performed, the chiral intermediates dihydropyridinones 58 can be isolated in high optical purity (74–98% ee), although with diminished yields (44–79%).
 |
| | Scheme 34 NHC-catalyzed reaction between 3-aminobenzofurans 54 and 2-bromoenals 22. | |
5.3 Formal [3+3] cyclizations using 5-aminobenzofurans
The first formal [3+3] cycloaddition through an enantioselective conjugate addition with 3-aminobenzofurans 54 to α,β-unsaturated aldehydes was reported by the group of Yang and Deng in 2018 (Scheme 35).45 A prolinol organocatalyst Cat. P served for the activation of enals 24 towards the nucleophilic addition of 3-aminobenzofurans, delivering the aza-[3+3] cycloaddition products 60 in high yields (49–95%) and with excellent enantioselectivities (91–99% ee). The method was effective with enals having either an aryl or an alkyl substituent at the β-position, although when the substituent was an alkyl group the corresponding products were obtained with moderate yields (49–55%). The authors also tested different substituted 3-aminobenzofuran derivatives 54 at C-5, C-6 and C-7 with excellent results in yields and perfect enantioselectivities (99% ee in almost all cases). The reaction also tolerates scale up to multi-gram amounts. The authors proposed a plausible transition state model where the chiral secondary amine Cat. P activated α,β-unsaturated aldehyde 24 generating the iminium ion Int. XVII which enabled a Si-face attack by 54 to give intermediate enamine/iminium Int. XVIII. This intermediate underwent a reversible enamine/iminium transformation followed by spontaneous hydrolysis and intramolecular cyclization to afford cyclic chiral hemiaminal 60.
 |
| | Scheme 35 Prolinol-catalyzed enantioselective addition of 3-aminobenzofurans 54 to enals 24. | |
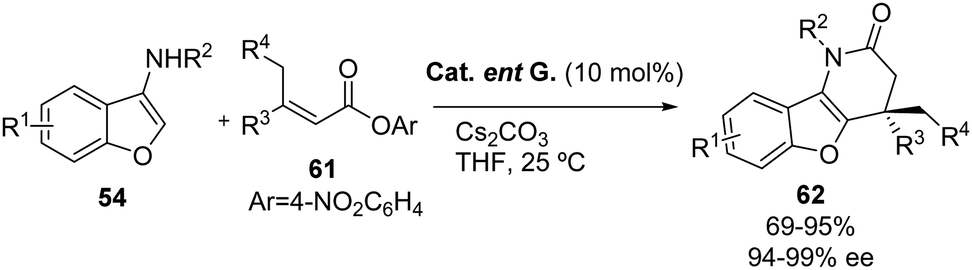
3-Aminobenzofuran nucleophiles 54 can also be reacted with β,β-disubstituted α,β-unsaturated esters 61 by means of NHC-catalysis, as reported by the group of Xu and Chen (Scheme 36).46 The final products 62 bearing a quaternary stereocenter were obtained in moderate to excellent yields (69–99%) and excellent enantioselectivities (94–99% ee). A number of esters 61 with different substitution patterns at the β-position were tested, obtaining good results independently of the electronic character of the substituents (94–99% ee). The reaction also tolerates substituents at the C-5 and C-6 of aminobenzofurans 54. The absolute configuration of the products was established by X-ray crystallographic analysis and a 3 mmol scale reaction was performed, obtaining the product in 91% yield and with 98% ee. The tosyl protecting group can be removed using Na/naphthalene and the obtained secondary lactam was treated with DIBAL-H generating a dihydropyridin-3-one by benzofuran ring opening.
 |
| | Scheme 36 NHC-catalyzed addition of 3-aminobenzofurans 54 to β,β-disubstituted α,β-unsaturated esters 61. | |
The authors propose a mechanistic pathway for the formal [3+3] cycloaddition reaction that is depicted in Scheme 37. The chiral NHC Cat. ent G activates the electrophile by the formation of an acylazolium Int. XIX. Then, the nucleophile adds selectively to the β-carbon of Int. XIX, generating Int. XX with a quaternary stereocenter. Subsequent lactamization of Int. XXI generates the observed δ-lactams 62 and regenerates the catalyst.
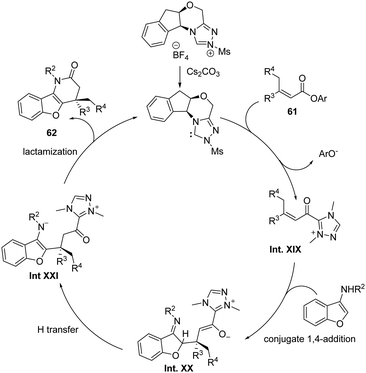 |
| | Scheme 37 Proposed mechanism for the synthesis of δ-lactams 62. | |
Zhou's group described in 2021 the reaction between 3-aminobenzofurans 54 with 2-alkynyl enones 63 to obtain tetracyclic compounds 64 using a quinine-derived thiourea Cat. N as the catalyst (Scheme 38).47 The corresponding dihydropyridines 64 were obtained in moderate to good yields (43–82%) and with high to excellent enantioselectivities (83–99% ee). The authors tested several 2-alkynyl enones 63. Analyzing the results, for the alkynyl moiety in substrate 63, both electron-rich and electron-deficient substitutions at the ortho, meta or para position in the aromatic rings were tolerated, giving products 64 in good yields with excellent ee values (up to 92%). When aliphatic substituents such as n-butyl and cyclopropyl were incorporated, due to the low reactivity, a higher temperature (60 °C) had to be applied to achieve better conversions, and the products were obtained with lower yields (43% and 50%, respectively), but with good enantiomeric excesses (92% ee and 83% ee, respectively). The ring size of substrate 63 was also evaluated under the standard reaction conditions, and cyclopentenone- and cycloheptenone-derived 2-(1-alkynyl)-2-alken-1-ones 63 gave the corresponding fused products 64 with slightly lower yields and enantioselectivities compared with the cyclohexanone. Substituents at C-5 and C-6 were tolerated in nucleophiles 54, and good results were obtained independently of the electronic character of the substituents. More than a gram of final product was obtained when the reaction was scaled-up to 3 mmol (71%, 95% ee).
 |
| | Scheme 38 Enantioselective formal [3+3] annulation reaction of 3-aminobenzofuran 54 with 2-alkynylenones 63 catalyzed by a chiral thiourea. | |
As reported by the group of Zhou and Xu, the formal [3+3] annulation occurs as depicted in Scheme 39 where the chiral thiourea Cat. N acts as a bifunctional organocatalyst. The conjugated addition of the heterocycle to the thiourea-activated enone forms a new C–C bond (Int. XXII) and after a proton transfer, an allenyl ketone intermediate Int. XXIII is formed. After a rearomatization, this intermediate (Int. XXIV) undergoes cyclization to form the new heterocycle Int. XXV, after the addition of the amino group to the sp carbon in the allene. Finally, γ-protonation of intermediate Int. XXV afforded the polycyclic 1,4-dihydropyridine 64 and releases the catalyst.
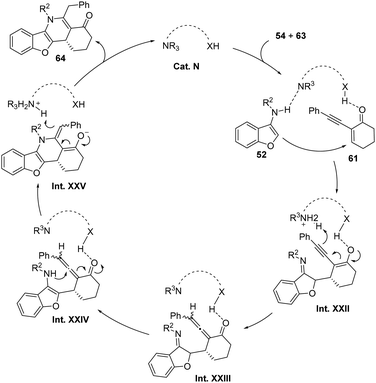 |
| | Scheme 39 Proposed mechanism for the formal [3+3] annulation with alkynyl enones. | |
In 2024, the group of Wang reported the reaction between 3-aminobenzofurans 54 and isatin-derived enals 25, using an NHC catalyst (Cat. ent G) under oxidative conditions (Scheme 40).48 3,4-Piperidinoyl spirooxindoles 65 with a quaternary stereocenter were obtained in good yields (80–89%) and high enantioselectivities (80–97% ee). This strategy permitted a wide range of substituted isatin derivatives possessing diverse electronic properties, as well as either a benzyl, phenyl or methyl protecting group at the N1 position of 25. Regarding the nucleophile 54, the authors only tested 3-aminobenzofurans substituted at C-5 and C-6. The authors suggested a mechanism in which the reactive α,β-unsaturated acylazolium is generated by oxidation of the Breslow intermediate formed by the addition of the catalyst to the enal similar to the mechanism depicted in Scheme 16.
 |
| | Scheme 40 NHC-catalyzed asymmetric formal [3+3] annulation between 3-aminobenzofurans 54 and isatin-derived enals 25. | |
6. Electrophilic functionalization of 3-aminobenzothiophene
Benzothiophene is another electron-rich heteroaromatic compound that exhibits interesting biological activities and is present in several drugs.49 Although benzothiophenes have an electron-rich heteroaromatic framework, they have been much less studied in electrophilic enantioselective functionalization than other heteroaromatic compounds. There are few examples reported in the literature using the strategy of the incorporation of an amino group at the C-3 position to increase the nucleophilicity of this heterocycle.
6.1 Enantioselective 1,4-addition of 3-aminobenzothiophenes
There is one example of the addition of 3-aminothiophene 66 to 2-acylimidazoles 27 (Scheme 41) catalysed by the chiral-at-metal rhodium(III) complex Cat. O obtaining the corresponding product 67 in 76% yield and 76% enantiomeric excess.43 Compared with 3-aminobenzofurans, 3-aminothiophene 66 gave poorer results.
 |
| | Scheme 41 Chiral Rh-catalyzed the asymmetric addition of 3-aminobenzothiophene 66 to 2-acylimidazoles 27. | |
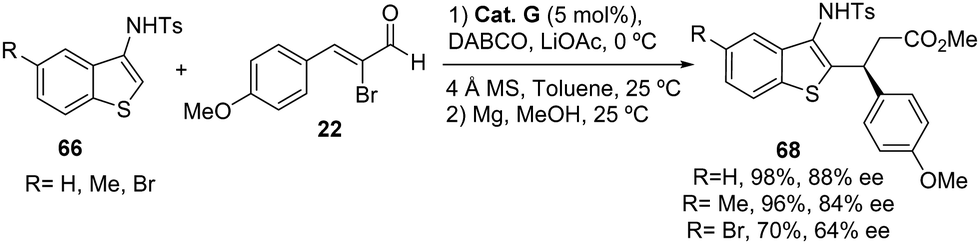
In the report presented by the group of Biju,44 for the enantioselective C2-functionalization of 3-aminobenzofurans 54 with 2-bromoenals 22, they also reported three examples using 3-aminothiphene derivatives 66 obtaining similar yields for compounds 68 but lower enantioselectivities (64–88% ee) than when 3-aminobenzofuran derivatives were used (Scheme 42).
 |
| | Scheme 42 NHC-catalyzed the reaction between 3-aminobenzothiophene 66 and 2-bromoenals 22. | |
6.2 Formal [3+3] cyclizations using 3-aminobenzothiophenes
In the reported methodology by Yang and Deng,45 the nucleophilic addition of 3-aminobenzothiophenes to enals catalyzed by the prolinol organocatalyst was also evaluated (Scheme 43). In this case, the authors re-optimised the reaction conditions using Cat. Q as an organocatalyst and EtOAc as the solvent and performing an acetylation to facilitate purification and characterization of the reaction products 69. Under these conditions, the corresponding [3+3]-cyclization products 69 were obtained with lower results compared with aminobenzofurans (81–87%, 81–85% ee).
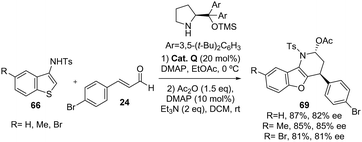 |
| | Scheme 43 Prolinol-catalyzed addition of 3-aminobenzothiophenes to enals 24. | |
In the described synthetic method by Wang in 2024,48 3-aminobenzothiophene was also evaluated as a nucleophile, in the [3+3] annulation with isatin-derived enals catalysed by chiral NHC catalyst Cat ent G. The corresponding 3,4-piperidinoyl spirooxindoles 70 were obtained in 80% yield with 91% ee (Scheme 44). This result is similar to that obtained with 3-aminobenzofuran.
 |
| | Scheme 44 NHC-catalyzed oxidative annulation between 3-aminobenzothiophene and isatin-derived enals. | |
7. Electrophilic functionalization of 3-aminoindole
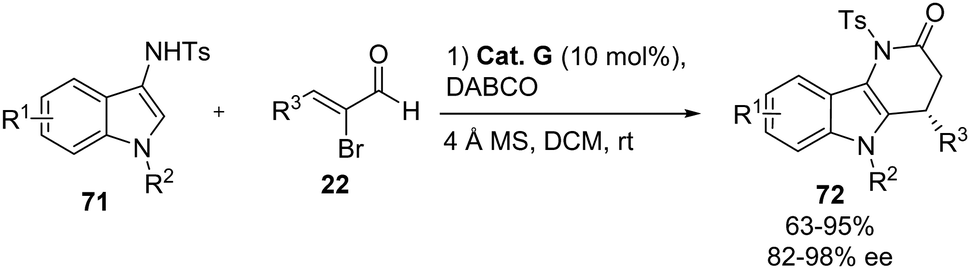
Indoles are the most studied heteroaromatic compound in the asymmetric functionalization with electrophiles,7 and the most reactive position in this scaffold is the C-3 carbon. However, there are several strategies to functionalize the C-2 position.50 The introduction of an amino group at the C-3 position can be used for the asymmetric functionalization at the C-2 position of indoles. In fact, there is one example using this strategy reported by Du and his group in 2022. They investigated the nucleophilic addition of 3-aminoindoles 71 to bromoenals 22 catalyzed by NHC catalyst Cat. G (Scheme 45).51 The corresponding δ-carbolinones 72 were obtained in moderate to excellent yields (63–95%) and excellent enantioselectivities in most of the cases (82–98% ee). In this methodology a wide range of α-bromoenals 22 were tested, obtaining the corresponding products 72 with good results when the β-substituents are aryl or heteroaryl groups, but the reaction did not take place when an alkyl group is present. Interestingly, the same products can be obtained using simple cinnamaldehyde under oxidative conditions, using 1 equivalent of a diphenoquinone (DQ) as the oxidant. The authors proved the scalability of the process up to 1 mmol scale and they showed that the tosyl protecting group can be removed by treatment with SmI2.
 |
| | Scheme 45 NHC-catalyzed oxidative [3+3] annulation between 3-aminoindoles 71 and α-bromoenals 22. | |
Conclusions
As summarized in this review, we have shown that highly remarkable and important nitrogen heteroaromatic compounds such as pyrazoles, isothiazoles and isoxazoles can be functionalized with electrophiles in an estereoselective way by the introduction of an exocyclic amino group. Several successful electrophilic functionalization methodologies for the enantioselective synthesis of these chiral nitrogen heterocycles have been described in the literature over the last four years. Most of the developed methods used organocatalysis to achieve this goal. Generally, the examples reported involve the nucleophilic 1,4-additions of these nitrogen heterocycles to α,β-unsaturated aldehydes using chiral NHC catalysis followed by an intramolecular lactam formation leading to a formal [3+3] cycloaddition. Interestingly, three atroposelective arylations have been described through organocatalytic 1,4-additions using quinone derivatives or azonaphthalenes as electrophiles. Moreover, several 1,2-additions to ketimines have been described. However, the number of electrophiles is still low (activated ketimines, quinone derivatives, α,β-unsaturated aldehydes, α,β-unsaturated 2-acyl imidazoles or β,γ-unsaturated α-ketoesters). Therefore, there is still room for the development of new electrophilic functionalization reactions using other electrophiles such as ketones and activated ketones, aldehydes, aldimines, nitroalkenes or simply α,β-unsaturated ketones. Regarding N-heteroaromatic compounds, aminopyrazoles and aminoisoxazoles have been the most explored nucleophiles, while the use of 5-aminoisothiazoles is still underdeveloped with only one example described in the literature. Therefore, there are future possibilities of research with this nitrogen heterocycle.
Author contributions
C. V. planned and supervised the project and G. B., A. M., M. M.-M., A. S.-M. and C. V. wrote the manuscript.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from Grant PID2020-116944GB funded by MCIN/AEI/10.13039/501100011033 and Grant CIAICO/2021/147 funded by Conselleria d′Educació, Universitats i Ocupació are acknowledged.
Notes and references
-
(a)
C. J. Moody, Advances in Nitrogen Heterocycles, AI Press Ltd., London, 1999 Search PubMed;
(b)
K. L. Ameta, R. Kant, A. Penoni, A. Maspero and L. Scapinello, N-Heterocycles Synthesis and Biological Evaluation, Springer, Singapore, 2022 CrossRef.
-
(a) E. Kabir and M. Uzzaman, Results Chem., 2022, 4, 100606 CrossRef CAS;
(b) M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC;
(c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
-
(a) D. Chen, S.-J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC;
(b) P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187–204 CrossRef CAS.
-
(a) C. Lamberth, Pest Manage. Sci., 2013, 69, 1106–1114 CrossRef CAS PubMed;
(b) D. Quiroga and E. Coy-Barrera, Organics, 2023, 4, 503–523 CrossRef CAS.
-
(a)
T. Aniszewski, Alkaloids: Chemistry, Biology, Ecology, and Applications, Elsevier, Oxford, 2015 Search PubMed;
(b) C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075–3081 CrossRef CAS.
-
(a) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Org. Biomol. Chem., 2019, 17, 3670–3708 RSC;
(b) M. G. Vinogradov, O. V. Turova and S. G. Zlotin, Adv. Synth. Catal., 2021, 363, 1466–1526 CrossRef CAS;
(c) N. Hoffmann, Photochem. Photobiol. Sci., 2021, 20, 1657–1674 CrossRef CAS PubMed;
(d) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156–1171 CrossRef CAS PubMed.
-
(a) G. Bartoli, G. Bencivennia and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC;
(b) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC;
(c) J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550–3567 RSC;
(d) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199–1222 CrossRef CAS;
(e) T. Ahmad, S. Khan and N. Ullah, ACS Omega, 2022, 7, 35446–35485 CrossRef CAS PubMed.
-
(a) D. Gaviña, M. Escolano, J. Torres, G. Alzuet-Piña, M. Sánchez-Roselló and C. del Pozo, Adv. Synth. Catal., 2021, 363, 3439–3470 CrossRef;
(b) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 2709–2762 CrossRef CAS.
-
(a) A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423–1463 CrossRef CAS;
(b) S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed;
(c) V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed;
(d) E. Kang, H. T. Kim and J. M. Joo, Org. Biomol. Chem., 2020, 18, 6192–6210 RSC.
-
(a) N. Agrawal and P. Mishra, Med. Chem. Res., 2018, 27, 1309–1344 CrossRef CAS PubMed;
(b) S. Fuse, T. Morita and H. Nakamura, Synthesis, 2017, 2351–2360 CAS;
(c) F. Hu and M. Szoztak, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS;
(d) S. Madhavan, S. K. Keshri and M. Kapur, Asian J. Org. Chem., 2021, 10, 3127–3165 CrossRef CAS.
-
(a) H. F. Anwar and M. H. Elnagdi, ARKIVOC, 2009, 198–250 Search PubMed;
(b) R. Aggarwal, V. Kumar, R. Kumar and S. P. Singh, Beilstein J. Org. Chem., 2011, 7, 179–197 Search PubMed;
(c) J. Fichez, P. Busca and G. Prestat, Targets Heterocycl. Syst., 2018, 22, 322–347 Search PubMed;
(d) A. Shaabani, M. T. Nazeri and R. Afshari, Mol. Diversity, 2019, 23, 751–807 CrossRef CAS PubMed.
-
(a) S. T. Moe, A. B. Thompson, G. M. Smith, R. A. Fredenburg, R. L. Stein and A. R. Jacobson, Bioorg. Med. Chem., 2009, 17, 3072–3079 CrossRef CAS PubMed;
(b) M. H. Helal, M. A. Salem and H. Aly, J. Heterocycl. Chem., 2017, 54, 2614–2626 CrossRef CAS;
(c) K. S. Gudmundsson, B. A. Johns and J. Weatherhead, Bioorg. Med. Chem. Lett., 2009, 19, 5689–5692 CrossRef CAS PubMed.
-
(a) P. Giori, M. Guarneri, D. Mazzota, G. Vertuani and C. Branca, Farmaco Sci., 1979, 34, 277–283 CAS;
(b) C. B. Vicentini, T. Poli, M. Manfrini, M. Guarneri, P. Giori and V. Brandolini, Farmaco Sci., 1987, 42, 133–143 CAS.
-
(a) R. Aggarwal and S. Kumar, Beilstein J. Org. Chem., 2018, 14, 203–242 CrossRef CAS PubMed;
(b) M. Lusardi, A. Spallarossa and C. Brullo, Int. J. Mol. Sci., 2023, 24, 7834 CrossRef CAS PubMed.
- L. Carceller-Ferrer, A. González del Campo, C. Vila, G. Blay, M. C. Muñoz and J. R. Pedro, Eur. J. Org. Chem., 2020, 7450–7454 CrossRef CAS.
- A. G. Woldegiorgis, Z. Han and X. Lin, Adv. Synth. Catal., 2022, 364, 274–280 CrossRef CAS.
- X.-X. Qiao, Y. He, T. Ma, C.-P. Zou, X.-X. Wu, G. Li and X.-J. Zhao, Chem. – Eur. J., 2023, 29, e202203914 CrossRef CAS PubMed.
- X. Gao, C. Li, L. Chen and X. Li, Org. Lett., 2023, 25, 7628–7632 CrossRef CAS PubMed.
- X. Luo, S. Li, Y. Tian, Y. Tian, L. Gao, Q. Wang and Y. Zheng, Eur. J. Org. Chem., 2024, e202400254 CrossRef CAS.
- D. Bhattacharjee, B. Kshiar and B. Myrboh, RSC Adv., 2016, 6, 95944–95950 RSC.
- A. G. Woldegiorgis, Z. Han and X. Lin, Org. Lett., 2022, 24, 4058–4063 CrossRef CAS PubMed.
- G. Nie, J. Sun, C. Mou, K. Tang, Y. R. Chi and T. Li, Org. Lett., 2023, 25, 134–139 CrossRef CAS PubMed.
- Y. Li, X. Huang, J. He, S. Peng, J. Wang and M. Lang, Adv. Synth. Catal., 2023, 365, 490–495 CrossRef CAS.
- Q. Wu, J. Han, J. Huang, H. Zhang, M. Ren, X. Zhang and Z. Fu, Org. Biomol. Chem., 2023, 21, 6898–6902 RSC.
- J. Li, X.-Y. Duan, X. Ren, Y. Li and J. Qi, J. Org. Chem., 2023, 88, 16621–16632 CrossRef CAS PubMed.
- S. Zhu, X. Tian, J. Liu, B. Dai and S.-W. Li, Org. Lett., 2024, 26, 3487–3492 CrossRef CAS PubMed.
- W. S. Hamama, M. E. Ibrahim and H. H. Zoorob, Synth. Commun., 2013, 43, 2393–2440 CrossRef CAS.
-
(a) K. Zhu, J. Mo, H. Z. Lin, Y. Chen and H. P. Sun, Bioorg. Med. Chem., 2018, 26, 3065–3075 CrossRef PubMed;
(b) A. Sysak and B. Obminska-Mrukowicz, Eur. J. Med. Chem., 2017, 137, 292–309 CrossRef CAS PubMed;
(c) M. A. Barmade, P. R. Murumkar, M. K. Sharma and M. R. Yadav, Curr. Top. Med. Chem., 2016, 16, 2863–2883 CrossRef CAS PubMed.
- H. Liu, Y. Yan, M. Li and X. Zhang, Org. Biomol. Chem., 2021, 19, 3820–3824 RSC.
- M. Li, Y. Chen, Y. Yan, M. Liu, M. Huang, W. Li, L. Cao and X. Zhang, Org. Biomol. Chem., 2022, 20, 8849–8854 RSC.
- S. Gao, X. Sun, S. Peng, Z. Zha, Q. Sun and Z. Wang, Org. Biomol. Chem., 2024, 22, 3391–3395 RSC.
- J. Jiaping, A. G. Woldegiorgis and X. Lin, RSC Adv., 2023, 13, 18964–18973 RSC.
- T. A. Schmidt, V. Hutskalova and C. Sparr, Nat. Rev. Chem., 2024, 8, 497–517 CrossRef PubMed.
- W. Luo, H. Guo, X. Qiu, M. Ming, L. Zhang, H. Zhu and J. Zhou, Org. Lett., 2024, 26, 2564–2568 CrossRef CAS PubMed.
- H. Liu, Y. Yan, J. Zhang, M. Liu, S. Cheng, Z. Wang and X. Zhang, Chem. Commun., 2020, 56, 13591–13594 RSC.
- W. Li, M. Gao, M. Li, S. Yang and X. Zhang, Asian J. Org. Chem., 2024, 13, e202400055 CrossRef CAS.
- K. Li, S. Gao, Z. Zha and Z. Wang, Org. Biomol. Chem., 2023, 21, 4404–4408 RSC.
- X. Huang, Y. Li, J. He, S. Peng, J. Wang and M. Lang, Org. Chem. Front., 2023, 10, 963–969 RSC.
-
(a) R. V. Kaberdina and V. I. Potkin, Russ. Chem. Rev., 2002, 71, 673–694 CrossRef;
(b) A. Alam, K. Shimada, W. Khan and D. Hossain, Med. Anal. Chem. Int. J., 2019, 3, 00137 Search PubMed;
(c) A. V. Kletskov, N. A. Bumagin, F. I. Zubkov, D. G. Grudinina and V. I. Potkin, Synthesis, 2020, 159–188 CAS.
- Y. Yan, M. Li, Q. Shi, M. Huang, W. Li, L. Cao and X. Zhang, Asian J. Org. Chem., 2023, 12, e202200578 CrossRef CAS.
-
(a) Y.-H. Miao, Y.-H. Hu, J. Yang, T. Liu, J. Sun and X.-J. Wang, RSC Adv., 2019, 9, 27510–27540 RSC;
(b) A. Mushtaq, A. F. Zahoor, S. Ahmad, M. J. Saif, A. ul Haq, S. G. Khan, A. A. Al-Mutairi, A. Irfan, S. A. Al-Hussain and M. E. A. Zaki, ACS Omega, 2024, 9, 20728–20752 CrossRef CAS PubMed;
(c) H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483–504 CrossRef CAS PubMed.
- Y. Wang, X. Huang, H. Ji, H. Zhou, H. Gao and J. Xu, J. Org. Chem., 2023, 88, 15486–15493 CrossRef CAS PubMed.
- H. Zhang, S. Li, Q. Kang and Y. Du, Org. Chem. Front., 2019, 6, 3683–3687 RSC.
- S. Barik, S. Shee, A. Ghosh and A. T. Biju, Org. Lett., 2020, 22, 3865–3869 CrossRef CAS PubMed.
- X.-F. Ding, R.-H. Su, W.-L. Yang and W.-P. Deng, Adv. Synth. Catal., 2018, 360, 4168–4177 CrossRef CAS.
- C. He, Y. Zhou, Z. Li, J. Xu and X. Chen, Org. Chem. Front., 2021, 8, 1569–1574 RSC.
- Z. Li, H. Zhou and J. Xu, Org. Lett., 2021, 23, 6391–6395 CrossRef CAS PubMed.
- L. Li, C. Li, S. Zhang, X. Wang, P. Fu and Y. Wang, J. Org. Chem., 2024, 89, 5170–5180 CrossRef CAS PubMed.
-
(a) R. S. Keri, K. Chand, S. Budagumpi, S. B. Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed;
(b) T. M. Dhanya, G. Anjali Krishna, D. P. Savitha, A. A. Shanty, K. M. Divya, S. K. Priya and P. V. Mohanan, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 198, 283–299 CrossRef.
- B. Deka, M. L. Deb and P. K. Baruah, Top. Curr. Chem., 2020, 378, 22 CrossRef CAS PubMed.
- S. Zhang, C. Lin, C. Liu and D. Du, J. Org. Chem., 2022, 87, 10441–10448 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Alicia
Monleón
,
Marc
Montesinos-Magraner
,
Amparo
Sanz-Marco
*,
Alicia
Monleón
,
Marc
Montesinos-Magraner
,
Amparo
Sanz-Marco