
Facile room-temperature synthesis of layered transition metal phosphonates via hitherto unknown alkali metal tert-butyl phosphonates†
Anuj
Kumar
,
Aheli
Ghatak
and
Ramaswamy
Murugavel
 *
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400 076, India. E-mail: rmv@chem.iitb.ac.in
First published on 30th July 2024
Abstract
A facile room-temperature synthetic method is presented to produce alkali metal salts of tert-butyl phosphonic acid. The reaction between equimolar amounts of alkali metal carbonates and tert-butyl phosphonic acid in methanol results in the formation of [(tBuPO3H)Li(H2O)3·(H2O)] (1), [(tBuPO3)Na2(H2O)4]n (2), and [(tBuPO3H)K(H2O)]n (3). Solid-state structures of these compounds have been confirmed by single-crystal X-ray diffraction studies and further validated using numerous spectroscopic and analytical techniques. Compounds 1–3 are polymeric solids that are predominantly made up of a 1-D polymeric metal phosphonate chain. This synthetic approach leads to the formation of network structures/polymers in the solid state that otherwise are absent in solution due to the ionic nature of the interaction between the alkali metal ions and phosphonate anions. Apart from the multidentate nature of the phosphonate ligands, additional hydrogen bonding interactions involving water molecules, free P–OH groups, and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties allow these chains to be propagated into 2-D sheets. We have further utilized the completely metalated sodium phosphonate 2 to synthesize layered metal phosphonates [(tBuPO3)Ca(H2O)]n (4), [(tBuPO3)Mn(H2O)]n (5) and [(tBuPO3)Co(H2O)]n (6) via a simple metathesis reaction.
O moieties allow these chains to be propagated into 2-D sheets. We have further utilized the completely metalated sodium phosphonate 2 to synthesize layered metal phosphonates [(tBuPO3)Ca(H2O)]n (4), [(tBuPO3)Mn(H2O)]n (5) and [(tBuPO3)Co(H2O)]n (6) via a simple metathesis reaction.
Introduction
Metal phosphonates [RPO3M] have been studied for well over half a century owing to their utility in several fields such as adsorption and separation, sensing and laser technology, ion exchange materials, proton conductivity, nonlinear optical materials, water treatment, luminescent materials, catalysis, magnetism, and biochemical applications.1–11 While the focus of these studies has been mainly concentrated around the transition metal ions,12–18 the chemistry of s- and p-block elements containing phosphonates has been only sparsely investigated.5,19–21 Though homometallic alkali metal phosphonates are fewer in number,22,23 several aluminum, gallium, and other metal-containing phosphonates that simultaneously incorporate alkali metal ions such as Na+ and K+ have been studied to understand the structural chemistry of zeolites and the crown-ether-like coordination of alkali metal ions.24–26 The fact that the most comprehensive book on metal phosphonate chemistry published in 2012 does not devote a chapter or section to the study of alkali metal phosphonates summarizes the paucity of work on alkali metal phosphonates.1 This could, however, be directly related to the predominately ionic type of interaction between phosphonate anions and alkali metal ions, often resulting in very high solubility in water, unlike transition metal phosphonates that form stable 2-D layered structures through an extensive network of covalent bonds.27–31 Although their high solubility may appear to be a disadvantage to prepare stable frameworks in the solid state, alkali metal phosphonates can be used as soluble sources for the preparation of transition metal phosphonates through metathesis reactions with appropriately chosen starting materials under ambient reaction conditions.Since the 1970s, most metal phosphonates have been synthesized from phenylphosphonic acid, which has been readily available in large quantities.32–42 This, however, considerably changed during the 1990s with the introduction of tert-butyl phosphonic acid [tBuP(O)(OH)2] as a ligand in metal phosphonate chemistry.43–52tert-Butyl derivatives not only provide better solubility in organic solvents (vis-à-vis phenyl phosphonic acid) but they are also capable of producing various well-defined molecular clusters that resemble several zeolite building blocks apart from producing layered structures.43,45,53 As a result, this ligand system has garnered substantial interest where the initial focus was mainly concentrated on p-block tert-butyl phosphonates.4,21,24,54–59 The majority of these compounds are discrete clusters, which are soluble in organic solvents facilitating solid-structure determination by single-crystal X-ray diffraction studies. Subsequently, several transition metal phosphonates incorporating paramagnetic metal ions have been synthesized and structurally characterized with the study of molecular magnetism being the main focus.51,60–62 In spite of the rich chemistry that has been developed with tert-butyl phosphonic acid during the last 25 years, no single alkali metal phosphonate has ever been prepared.
In order to bridge this missing link in metal phosphonate chemistry, we have synthesized alkali metal phosphonates using appropriate metal precursors and crystallized the products to obtain 1-D polymeric structures of [(tBuPO3H)Li(H2O)3·(H2O)] (1), [(tBuPO3)Na2(H2O)4]n (2), and [(tBuPO3H)K(H2O)]n (3). This unique ligand, featuring two acidic protons and one phosphoryl oxygen, exhibits the ability to coordinate with neighboring metal ions and form larger supramolecular aggregates. These complexes display exceptional solubility in organic solvents. Moreover, we have utilized compound 2 as a precursor to synthesize layered metal phosphonates [(tBuPO3)Ca(H2O)]n (4), [(tBuPO3)Mn(H2O)]n (5) and [(tBuPO3)Co(H2O)]n (6) via a simple metathesis reaction.
Results and discussion
Synthesis and spectral characterization of alkali metal phosphonates 1–3
Preparation of alkali metal salts of various main group acids normally involves the use of organolithium reagents (such as n-BuLi, LiNMe2, etc.) or alkali metals. However, attempts to prepare Li, Na, and K salts of tert-butyl phosphonic acid in organic solvents using these methods have produced mixtures that could not be separated. In contrast, a synthetic methodology involving a reaction between alkali metal carbonates or bicarbonates (Li2CO3, NaHCO3, or KHCO3) and tert-butyl phosphonic acid (tBuP(O)(OH)2) in a methanol–water solution readily produces lithium tert-butyl phosphonate [(tBuPO3H)Li(H2O)3·(H2O)] (1), sodium tert-butyl phosphonate [(tBuPO3)Na2(H2O)4]n (2), and potassium tert-butyl phosphonate [(tBuPO3H)K(H2O)]n (3) (Scheme 1). Compounds 1–3 have been obtained as colorless crystals from the reaction mixture by slow evaporation of the solvent at room temperature and subsequently characterized by using various spectroscopic and analytical methods. The molecular structures of all three compounds have further been elucidated through single-crystal X-ray diffraction studies. Owing to their ionic nature, these compounds are freely soluble in water. The as-synthesized alkali metal phosphonate complexes are ionic in nature having several coordinated and lattice water molecules. ICP-AES analysis has provided evidence for the alkali metal-to-phosphorus ratio being close to 1 in all three samples (Table S10†). The removal of crystals from solution resulted in loss of water and hence the elemental analyses differed slightly from the calculated values. | ||
| Scheme 1 Synthesis of alkali metal phosphonates 1–6. | ||
The infrared spectra of compounds 1–3 show broad peaks at 3366 cm−1 for 1, 3426 and 3518 cm−1 for 2, and 3524 cm−1 for 3, corresponding to the O–H stretching frequency of H2O moieties. The absorption bands detected within the 3000–2900 cm−1 range are attributed to the C–H stretching vibration of the alkyl groups. Specifically, the absorption bands observed at 1144, 1050, and 902 cm−1 for 1, 1133, 1071, and 1013 cm−1 for 2, and 1130, 1041, and 900 cm−1 for 3 are assigned to the stretching frequencies of PO and P–O–M (antisymmetric and symmetric) bonds, respectively (Fig. S1†).
Additional evidence for the existence of alkali metal phosphonate complexes 1–3 was obtained through the analysis of their 31P NMR spectra. In the solution state, all three compounds exhibit a similar single resonance spectrum with almost identical chemical shifts in D2O at δ 33.09 ppm for 1, δ 32.73 ppm for 2, and δ 32.96 ppm for 3. This indicates that in solution the phosphonate is in an ionized state with no P–O–M linkage. Hence, solid-state NMR measurements were conducted for complexes 1–3 where a clear downfield shift is observed, which is consistent with the nature of bonding between phosphonate oxygen and metal ions. Single-pulse solid-state 31P NMR spectra show resonances at δ 40.20 and δ 39.65 ppm for 1, δ 37.31 ppm for 2, and δ 24.45 ppm for 3 (Fig. 1). The appearance of two signals for 1 could be rationalized as being due to small differences in the phosphorus environment caused by the position of the proton of the un-ionized P–OH group.
 | ||
| Fig. 1 CP-MAS 31P NMR spectra of 1 and 2 (162 MHz), and 3 (202 MHz) (δ in ppm). | ||
All the compounds exhibit a similar CP-MAS 13C NMR spectrum where the more intense upfield signals observed at δ 25.36 ppm for 1, δ 31.51 ppm for 2 and δ 22.86 ppm for 3 correspond to the methyl carbon of the tert-butyl group. The downfield doublets observed at δ 30.06 ppm for 1, δ 35.08 ppm for 2 and δ 26.83 ppm for 3 arise from the tert-carbon of the butyl group with a 1JP–C coupling of δ 141, 145, and 138 Hz, respectively. Additionally, the 7Li CP-MAS NMR spectrum of 1 exhibits a single well-defined resonance at δ 1.70 ppm, indicating the presence of a single type of Li center in compound 1 (Fig. S3†).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O solution (30:5 mL) at room temperature over a period of two weeks. Single-crystal X-ray diffraction studies reveal that compound 1 crystallizes in the triclinic P
:H2O solution (30:5 mL) at room temperature over a period of two weeks. Single-crystal X-ray diffraction studies reveal that compound 1 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit of compound 1 contains one Li ion, one mono-deprotonated tert-butyl phosphonate, and four H2O molecules. The Li ion is coordinated to the phosphonate group, forming a monomeric lithium tert-butyl phosphonate structure as shown in Fig. 2a. The tetra-coordinated Li ion is surrounded by one oxygen ion of the phosphonate group and three H2O molecules adopting a tetrahedral geometry.
space group. The asymmetric unit of compound 1 contains one Li ion, one mono-deprotonated tert-butyl phosphonate, and four H2O molecules. The Li ion is coordinated to the phosphonate group, forming a monomeric lithium tert-butyl phosphonate structure as shown in Fig. 2a. The tetra-coordinated Li ion is surrounded by one oxygen ion of the phosphonate group and three H2O molecules adopting a tetrahedral geometry.
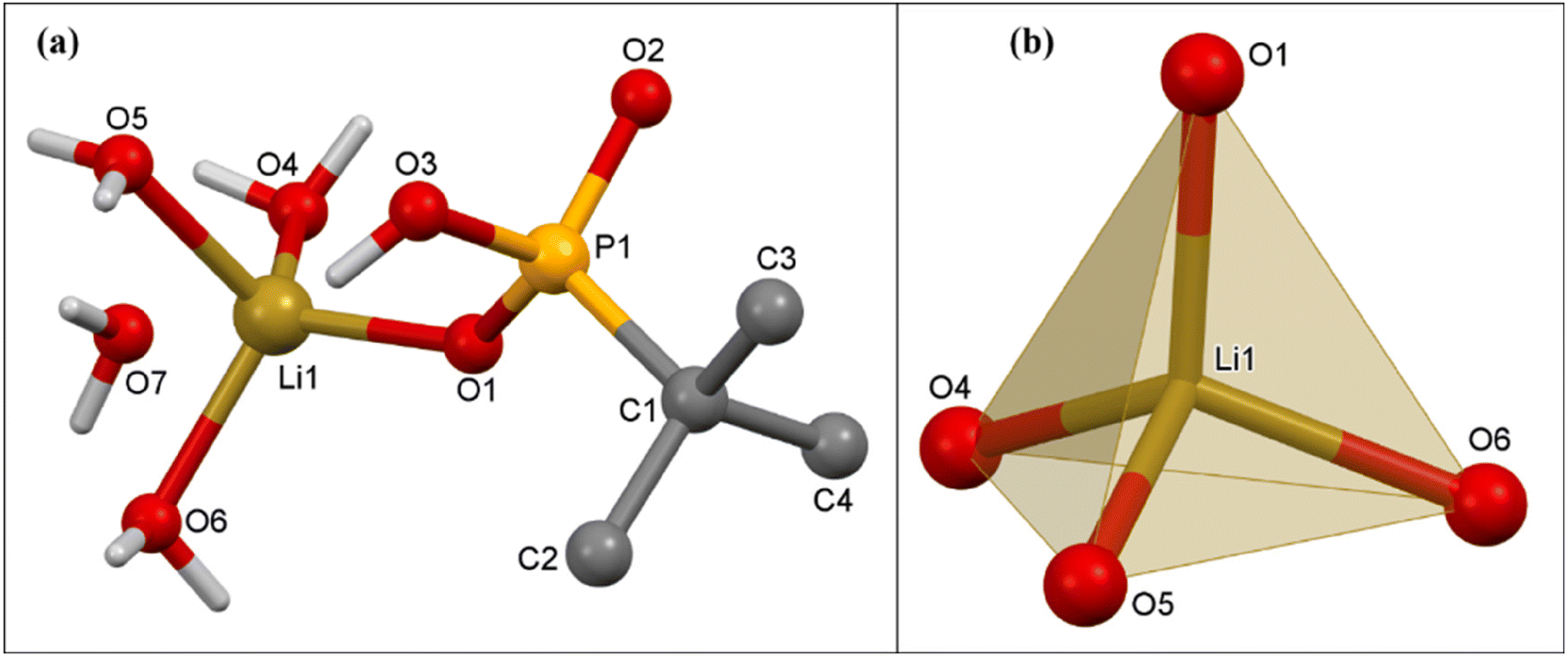 | ||
| Fig. 2 (a) Molecular structure of 1; (b) coordination polyhedron of a Li ion in 1. Selected bond lengths (Å) and bond angles (°): Li(1)–O(1) 1.906(5); Li(1)–O(4) 1.938(5); Li(1)–O(5) 1.975(5); Li(1)–O(6) 1.959(5); P(1)–O(1) 1.503(2); P(1)–O(2) 1.508(2); P(1)–O(3) 1.591(2); and P(1)–C(1) 1.815(3). O(1)–Li(1)–O(4) 115.6(3), O(1)–Li(1)–O(5) 109.7(2), O(1)–Li(1)–O(6) 111.9(2), O(4)–Li(1)–O(5) 103.9(2), O(4)–Li(1)–O(6) 109.3(2), and O(6)–Li(1)–O(5) 105.7(2). | ||
The Li–O distances are non-equal. The Li(1)–O(1) distance 1.906(5) Å is the shortest and corresponds to the lithium ion-to-phosphonate oxygen. The lithium-to-water oxygen bonds are comparatively longer (Li(1)–O(6) 1.959(5), Li(1)–O(5) 1.975(5), and Li(1)–O(4) 1.938(5) Å). The Li–O bond distances are comparable to those observed in similar Li phosphates recently reported by us.63 The observed P–O bond distances are P(1)–O(1) 1.503(1), P(1)–O(2) 1.508(2), and P(1)–O(3) 1.591(2) Å. The ∠O–Li–O angles vary over the range 103.9(2)° to 115.6(2)°. Intermolecular H-bonding between the uncoordinated P–OH group of one phosphonate and coordinated oxygen of a nearby water molecule in 1 leads to the formation of a H-bonded polymeric chain. This compound is also additionally stabilized by intermolecular hydrogen-bonding interactions between O(3)–H(3)⋯O(7) (2.585 Å, 153.9°), O(7)–H(7A)⋯O(1)#5 (2.730 Å, 170.4°), and O(4)–H(4A)⋯O(5)#1 (2.862 Å, 165.1°). (Fig. 3).
 | ||
| Fig. 3 (a) Intermolecular hydrogen bonding interactions in 1 between the hydrogen atoms of coordinated water molecules and O1, O4, and O5 of the phosphonate ligand, leading to (b) the formation of 2-D sheets, which are stacked one over the other. | ||
:H2O solution (30:5 mL) over a period of two weeks. Single-crystal X-ray diffraction studies revealed that compound 2 crystallizes in the monoclinic P21/c space group. The asymmetric part of the unit cell of 2 consists of two Na ions, one doubly deprotonated tert-butyl phosphonate, two terminal and two bridging H2O molecules. While one of the sodium ions, viz. the hexa-coordinated Na(1), is exclusively coordinated by water molecules, the second sodium ion, viz. tetra-coordinated Na(2), is coordinated by both the phosphonate ligand and water (Fig. 4).
 | ||
| Fig. 4 (a) A segment of the polymeric chain in 2; (b and c) coordination polyhedra of Na(1) and Na(2) ions in 2. Selected bond lengths (Å) and bond angles (°): Na(1)–O(4) 2.374(4); Na(1)–O(5) 2.523(4); Na(1)–O(6) 2.301(4); Na(1)–O(7) 2.558(4); Na(2)–O(1) 2.807(5); Na(2)–O(3)#4 2.676(5); Na(2)–O(4) 2.794(5); Na(2)–O(6) 2.713(5); P(1)–O(1) 1.596(3); P(1)–O(2) 1.502(4); P(1)–O(3) 1.514(3); and P(1)–C(1) 1.811(5). O(4)–Na(1)–O(4)#2 176.0(1); O(4)–Na(1)–O(5) 92.5(1); O(5)–Na(1)–O(7) 175.3(1); O(6)–Na(1)–O(6)#1 175.1(1); O(6)–Na(1)–O(4) 99.0(1); O(3)#4–Na(2)–O(1) 107.0(2); and O(6)#3–Na(2)–O(1) 137.4(2). #11 − X, −1/2 + Y, 1/2 − Z; #21 − X, 1/2 + Y, 1/2 − Z; #31 − X, 1 − Y, 1 − Z; #4+X, −1 + Y, +Z. | ||
In 2, the tBuPO32− ligands bridge the adjacent Na(2) ions along a polymeric chain through its deprotonated oxygen atoms O(1) and O(3). The Na(2) ions are additionally coordinated by bridging oxygen atoms of water O(4) and O(6), which emanate from an adjacent polymeric chain. This chain is made up of only Na(1) ions, which is coordinated with two terminal water molecules (O(5) and O(7)) apart from twice the number of O(4) and O(6). Overall, in the structure, alternating sodium phosphonate and hexa-aqua sodium polymers lead to the formation of two-dimensional structures (Fig. 5a), which are actually stacked one over the other to form a lamellar structure as shown in Fig. 5b. The adjacent layers are terminated by tert-butyl groups and are held together by van der Waals interactions. The Na–O(P), Na–O(water) and P–O distances listed in the caption of Fig. 4 are similar to those observed in the case of other ionic sodium phosphonates or phosphates reported earlier.38,63 The ∠O–Na–O angles vary over the range 83.1(1)° to 176.0(1)°. The geometry around Na(1) is nearly octahedral while the polyhedron around Na(2) can best be described as a trigonal pyramid (or vacant tbp), as evidenced by SHAPE analysis (Table S2†).
 | ||
| Fig. 5 (a) Ball and stick model of 2-D layered sheets of 2 (carbon and hydrogen atoms are omitted for clarity); (b) lamellar structure as viewed down the b-axis where the 2-D sheets stack one over the other separated by the tert-butyl groups. | ||
space group. The asymmetric part of the unit cell of 3 contains one hepta-coordinated K ion, one mono-deprotonated tert-butyl phosphonate, and one coordinated H2O molecule. In the polymeric structure, each K ion is surrounded by four phosphonate oxygen atoms emanating from three different tetradentate phosphonate ligands and three oxygen atoms of triply bridging water molecules (Fig. 6). Thus, each phosphonate in the polymeric structure is tetradentate and bridges three different adjacent potassium ions while each water molecule is tridentate and bridges three different metal ions. This multi-denticity along with the hydrophobicity of the tert-butyl phosphonate group, ensures this polymeric structure is organized into well-defined 2-D sheets, which stack one over the other to form lamellar (layered) structures. These 2-D sheets are held together with the aid of van der Waals interactions between the alkyl groups (Fig. 7). The K–O distances are non-equal with the values ranging from the shorter K(1)–O(1) at 2.712(4) Å to the longer K(1)–O(4) bond at 3.306(4) Å, which are comparable to those observed in similar K phosphates or phosphonates.23,63,64 The ∠O–K–O angles vary over the range of 51.8(1)° to 155.7(1)°. The observed P–O bond distances corresponding to the coordinated oxygen atoms are shorter (P(1)–O(1) 1.503(4) Å and P(1)–O(3) 1.508(4) Å) compared to the uncoordinated P(1)–O(2) bond distance (1.577(4) Å).
 | ||
| Fig. 6 (a) Contents of the asymmetric part of the unit of 3; (b) coordination polyhedron of K ion 3. Selected bond lengths (Å) and bond angles (°): K(1)–O(1)#1 2.835(4); K(1)–O(1) 2.712(4); K(1)–O(3)#1 2.944(4); K(1)–O(3)#3 2.742(4); K(1)–O(4) 3.306(4); K(1)–O(4)#4 2.836(4); K(1)–O(4)#5 2.857(4); P(1)–O(1) 1.503(4); P(1)–O(2) 1.577(4); P(1)–O(3) 1.508(4); and P(1)–C(1) 1.825(5). O(1)–K(1)–O(1)#1 84.7(1); O(1)–K(1)–O(3)#3 146.1(1); O(1)#1–K(1)–O(4)#5 155.7(1); O(3)#3–K(1)–O(1)#1 118.7(1); O(3)#3–K(1)–O(3)#1 77.8(8); O(3)#3–K(1)–O(4)#5 85.4(1); O(3)#3–K(1)–O(4) 96.5(1); O(4)#4–K(1)–O(3)#1 77.8(8); O(4)#4–K(1)–O(4) 154.8(7); and O(4)#5–K(1)–O(3)#1 145.8(1). #11 − X, 1 − Y, 1 − Z; #21 − X, 1 − Y, 2 − Z; #31 − X, 1/2 + Y, 3/2 − Z; #41 − X, −1/2 + Y, 3/2 − Z; #5+X, 3/2 − Y, 1/2 + Z. | ||
 | ||
| Fig. 7 (a) Ball and stick model of 2-D layered sheets of 3 (carbon and hydrogen atoms omitted for clarity); (b) lamellar structure as viewed down the c-axis where the 2-D sheets stack one over the other separated by the tert-butyl groups. | ||
Thus, it is interesting to note that although all three reactions were conducted under similar conditions, only in the case of sodium the phosphonic acid is completely neutralized by metal ion coordination. Moreover, the denticity of the phosphonate ligand also increases with the size of the alkali metal ion used (Fig. 8). For example, the mono-deprotonated ligand binds with only one lithium ion in the [1.100] mode, while in the case of the sodium compound the di-deprotonated ligand exhibits the [2.110] mode of coordination.65 The denticity, however, increases to [3.220] for complex 3 containing the largest of the cations.66 Consequently, compound 1 is a monomeric complex while 2 and 3 are 2-D polymeric layered solids, with higher framework density for 3 compared to 2.
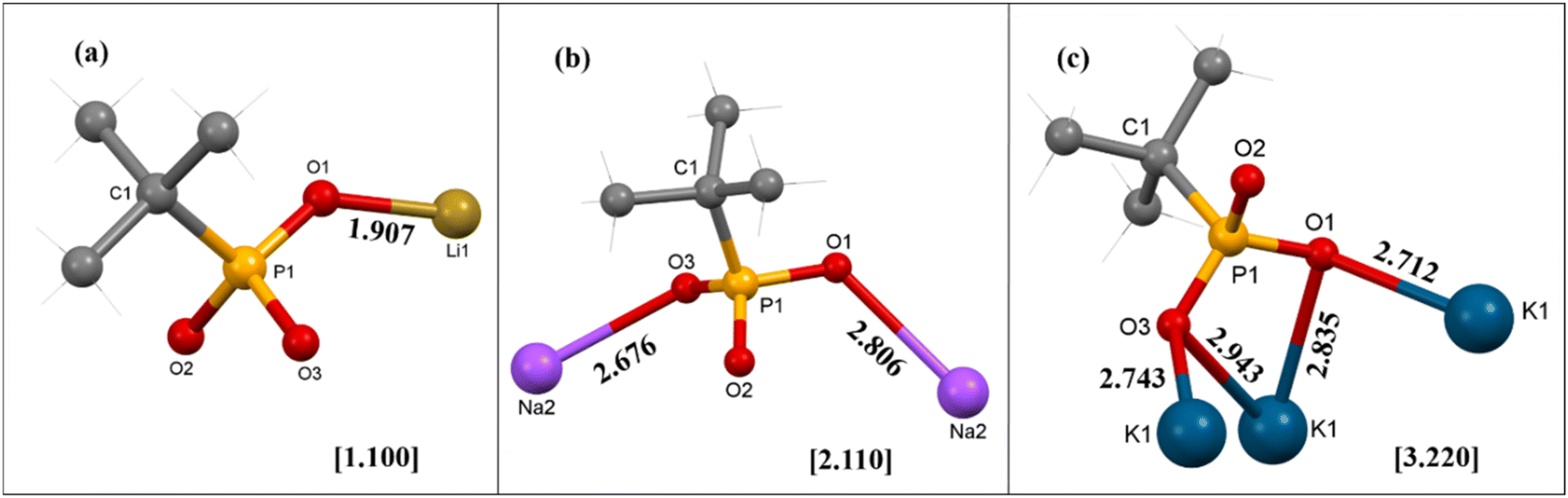 | ||
| Fig. 8 Binding modes of the tert-butyl phosphonate [tBuPO3]2− ligand and the corresponding Harris notations for complexes (a) 1, (b) 2, and (c) 3. | ||
Thermogravimetric analysis
In order to understand the stability of the obtained polymeric metal–organic phosphonate compounds, thermal analysis of 1–3 was conducted under a nitrogen atmosphere at a heating rate of 10 °C min−1. The thermal analysis of all three compounds shows a similar decomposition behavior with the initial loss of H2O molecules in the temperature range of 40–150 °C, and the second weight loss observed above 200–550 °C can be attributed to the degradation of the tert-butyl group of phosphonic acid (Fig. S7–S9†).Synthesis of layered phosphonates [(tBuPO3)M(H2O)]n (M = Ca (4), Mn (5), Co (6)) from 2
With all three ionic phosphonates being water soluble, we aimed at synthesizing layered metal phosphonates via metathesis by a simple salt removal reaction. Out of the three phosphonates synthesized, only sodium tert-butyl phosphonate (2) is completely metalated and hence it was chosen as the precursor for the metathesis reaction. Crystalline transition metal phosphonates are often synthesized from the reaction of aryl or alkyl phosphonic acids with suitable metal salt precursors at high temperature and autogenous pressure for prolonged periods.31,42,52 However, salt metathesis reactions can be carried out without employing any harsh reaction conditions. Sodium tert-butyl phosphonate (2) is prepared in situ in a mixture of water, methanol and half-equivalent of the corresponding metal chloride salt (CaCl2·4H2O for 4, MnCl2·4H2O for 5 and CoCl2·6H2O for 6) is added to it. The reactions are stirred overnight, and the products are obtained as insoluble precipitates. These products are centrifuged out of the solution and washed with copious amounts of water to remove the by-product NaCl.Compounds 4–6 give satisfactory elemental analyses consistent with the layered metal phosphonates reported in the literature52 with the general formula M(tBuPO3)·(H2O)n. The ICP-AES analysis for these insoluble materials provided evidence for the metal-to-phosphorus ratio being close to 1 in all three samples (Table S11†). The infrared spectra of compounds 4–6 show a broad O–H stretching frequency in the range of 3400–3430 cm−1 for the corresponding water molecule and the C–H stretching frequency of the tert-butyl groups can be observed at 2900 to 3000 cm−1 (Fig. S16†). The PO stretching of the parent compound 2 becomes shifted to lower frequency upon metathesis and can be seen as sharp peaks at 1050–1100 cm−1. More importantly, the powder X-ray diffraction of all the metal phosphonates produces a very similar pattern that is characteristic of layered metal phosphonates67 with low-angle peaks appearing at 2θ values of 7.17, 7.27, and 7.31° for compounds 4–6, respectively. These values correspond to d (interlayer spacing) values of 12.33 Å for 4, 12.16 Å for 5, and 12.09 Å for 6 (Fig. 9). These values are somewhat smaller than the value of 13.77 Å for the parent metal phosphonate 2. The PXRD pattern of 6 is also consistent with the single-crystal X-ray structure of the complex reported by Bujoli and co-workers, indicating phase purity of the bulk sample synthesized utilizing the current methodology (Fig. S17†).52 The layered morphology of the transition metal phosphonates 4–6 can be also confirmed via SEM images as shown in Fig. 10, and in Fig. S18, and S19.† Compound 4 is SEM-beam sensitive (Fig. S18†). Furthermore, SEM-EDX mapping confirmed that the respective elements are uniformly distributed throughout the layered structures of 4–6 indicating consistent composition and structure of these materials.
 | ||
| Fig. 9 PXRD patterns of 2 and 4–6. | ||
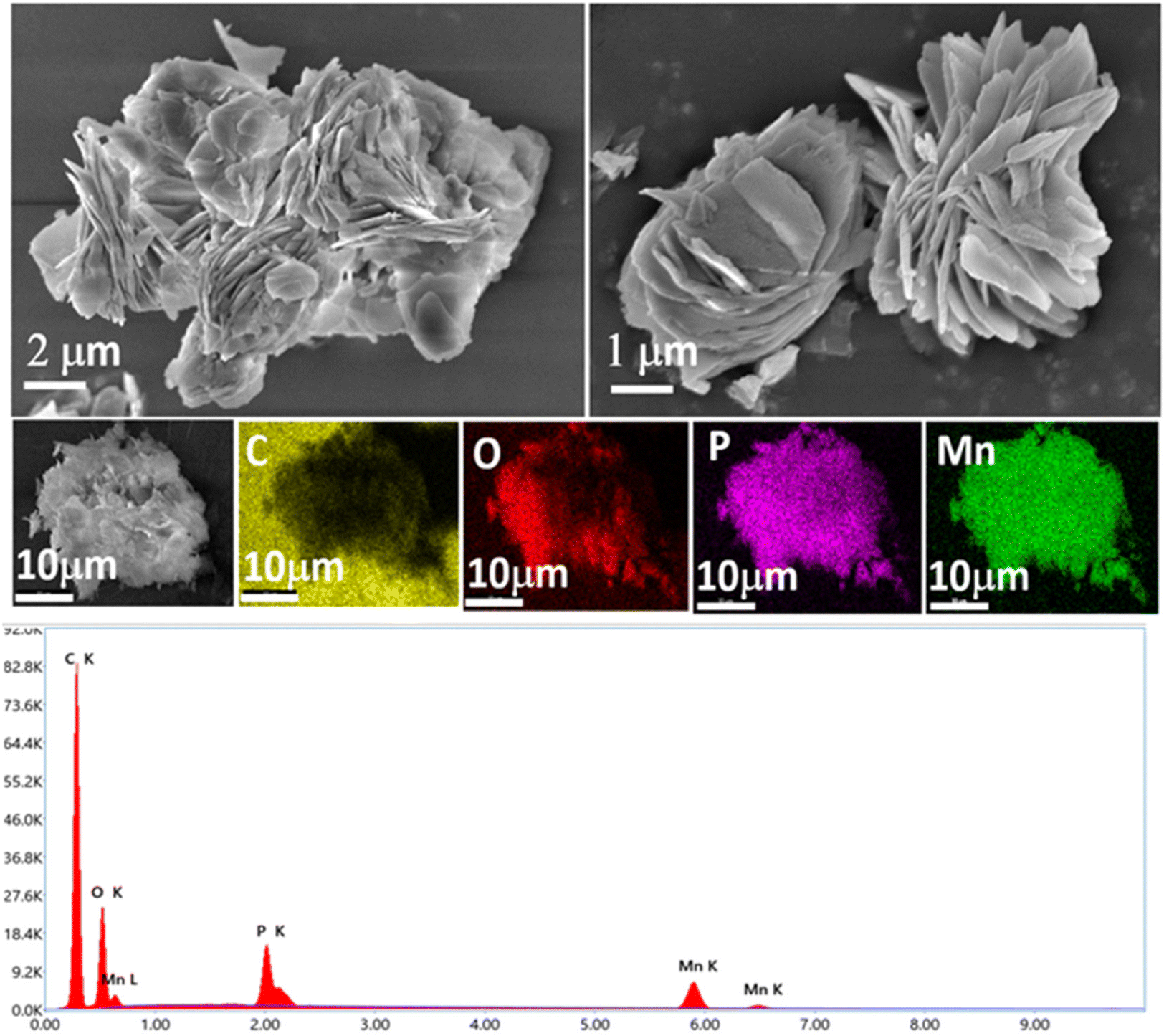 | ||
| Fig. 10 SEM images of compound 5 showing layered structures and EDX spectrum of 5 with elemental mapping. | ||
The DR-UV spectra of 4 and 5 display two intense absorption bands centered at around 250 and 300 nm, which are assignable to intra-ligand transitions originating from π → π* and n → π* of phosphonic acid (Fig. S20 and S21†). Additionally, for 6, intense absorption bands can be seen at 526, 603, and 643 nm due to d–d transitions originating from its Co(II) center (Fig. S22†).
Thermogravimetric analysis of 4–6 was carried out under a flow of N2 at a heating rate of 10 °C min−1. Compounds 4 and 5 undergo a two-stage thermal decomposition as shown in Fig. S23 and S24.† The first weight loss occurs below 200 °C corresponding to the loss of water molecules (∼8%), which is followed by the elimination of all organic moieties at 550–600 °C (∼29%) (Fig. S23–S25†).68 For compound 6, continuous weight loss can be observed until 400 °C, followed by 42% weight loss at 450 °C and another 10% weight loss at 550 °C through loss of water molecules due to the condensation of P–OH groups (Fig. S25†).
Conclusions
In summary, reactions of Li2CO3 and M(HCO3) (M = Na and K) with tert-butyl phosphonic acid (tBuP(O)(OH)2) result in the formation of polymeric chains of alkali metal di-tert-butyl phosphonates [(tBuPO3H)Li(H2O)3·(H2O)] (1), [(tBuPO3)Na2(H2O)4]n (2) and [(tBuPO3H)K(H2O)]n (3). These compounds are highly soluble in polar solvents, despite their extended structures in the solid state. The molecular structures have been determined through single-crystal X-ray diffraction measurements. These compounds have been fully characterized using various spectroscopic and analytical techniques. It is noteworthy that phosphonic acid utilized in the synthesis had not been fully neutralized. Consequently, the resulting framework structure contained a significant number of free P–OH groups. These free acid groups, along with the coordinated water molecules surrounding the metal ions, contributed to the formation of intriguing supramolecular structures through H-bonding interactions. Furthermore, compound 2 has been utilized to synthesize layered phosphonates [(tBuPO3)Ca(H2O)]n (4), [(tBuPO3)Mn(H2O)]n (5), and [(tBuPO3)Co(H2O)]n (6) via simple metathesis reactions. Thus, the reported methodology presents a simple procedure to synthesize layered metal phosphonates compared to the conventional solvothermal methods requiring drastic conditions.Experimental section
Methods and materials
Compounds 1–6 were synthesized at room temperature under ambient conditions. Solvents were purified according to standard procedures.69 Commercially available chemicals such as Li2CO3, NaHCO3 and KHCO3 (Spectrochem), and MCl2·nH2O [M = Ca, Mn, and Co (n = 2, 4, 6 respectively)] (Merck) were used as received. tert-Butyl phosphonic acid (tBuP(O)(OH)2) was synthesized following a reported procedure.70 The melting points were measured in glass capillaries and reported uncorrected. Infrared spectra were acquired using a PerkinElmer Spectrum One FT-IR spectrometer with KBr diluted discs. Elemental analyses were carried out using a VarioMicro Cube (Elementar Analysen Systeme GmbH) microanalyzer. Solution and solid-state NMR spectroscopic measurements were conducted using Bruker Avance DPX-400, Bruker Avance DPX-500 MHz and JEOL ECZR 600 MHz spectrometers. Thermogravimetric analyses (TGA) were carried out under a stream of dinitrogen gas at a heating rate of 10 °C min−1 using a PerkinElmer Pyris thermal analysis system for 1–3 and a Rigaku STA8122 Thermo plus EVO2 thermal analysis system for 4–6. The morphologies of the products were examined on a JEOL model JSM-7600F FEG-SEM operating at an accelerating voltage of 0.1 to 30 kV. The samples were prepared by smearing the powdered samples onto carbon tape and sputtering them with gold prior to imaging. The solid-state absorption spectra of 4–6 were measured in diffuse reflectance mode on an MPC-3100 Shimadzu spectrometer using an absolute specular reflectance measurement attachment. Powder X-ray diffraction studies were conducted on a Rigaku SmartLab SE powder X-ray diffractometer using Cu-Kα radiation (λ = 1.54190 Å).X-ray structure analysis
For unit cell determination and diffraction intensity data collection, a suitable crystal of each compound was mounted on a Bruker D8 QUEST single-crystal diffractometer equipped with a Mo-Kα radiation source (λ = 0.71073 Å). The Rigaku CrysAlisPro software package was used for data integration and indexing.71 Using Olex2,72 the structure was solved employing the ShelXT73 structure solution program utilizing intrinsic phasing. The structures were refined by ShelXL74 using least squares minimization. Anisotropic refinement was used for all non-hydrogen atoms. In their idealized places, the hydrogen atoms were refined isotropically as rigid atoms. Details of data collection, unit cell dimensions, refinement, and final residual values are listed in Table 1. All figures were created with either Mercury75 or Diamond 3.2 software.76 CCDC reference numbers 2330476, 2330477, and 2330478 contain supplementary crystallographic data for 1–3, respectively.† Metal coordination geometry analyses were performed using SHAPE2.1.77| Compounds | 1 | 2 | 3 |
|---|---|---|---|
| Identification code | RM-AK-251 | RM-AK-242 | RM-AK-252 |
| Empirical formula | C4H18LiO7P | C4H17Na2O7P | C4H12KO4P |
| Formula weight | 216 | 254 | 194 |
| Temperature/K | 150 | 100 | 100 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group |
P |
P21/c | P21/c |
| a/Å | 6.2774(3) | 14.5571(5) | 13.6615(6) |
| b/Å | 6.5742(4) | 6.7923(6) | 9.2070(4) |
| c/Å | 15.5423(8) | 13.4207(3) | 7.0197(3) |
| α/° | 79.599(4) | 90 | 90 |
| β/° | 88.942(4) | 104.188(3) | 101.967(4) |
| γ/° | 63.141(5) | 90 | 90 |
| Volume/Å3 | 561.36(6) | 1286.53(7) | 863.76(7) |
| Z | 2 | 4 | 4 |
| ρ calc/g cm−3 | 1.278 | 1.312 | 1.493 |
| μ/mm−1 | 0.248 | 0.288 | 0.761 |
| F(000) | 232.0 | 536.0 | 408.0 |
| Crystal size/mm3 | 0.3 × 0.3 × 0.1 | 0.8 × 0.3 × 0.15 | 0.19 × 0.16 × 0.16 |
| Radiation/Å | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 5.344 to 49.990 | 5.774 to 49.998 | 5.372 to 49.984 |
| Index ranges | −7 ≤ h ≤ 7 | −17 ≤ h ≤ 17 | −16 ≤ h ≤ 16 |
| −7 ≤ k ≤ 7 | −8 ≤ k ≤ 8 | −10 ≤ k ≤ 10 | |
| −18 ≤ l ≤ 18 | −15 ≤ l ≤ 15 | −8 ≤ l ≤ 8 | |
| Reflections collected | 12680 |
22368 |
14934 |
| Independent reflections | 1969 [Rint = 0.0924, Rsigma = 0.0460] | 2250 [Rint = 0.0771, Rsigma = 0.0370] | 1520 [Rint = 0.0647, Rsigma = 0.0288] |
| Data/restraints/parameters | 1969/0/136 | 2250/1/136 | 1520/1/95 |
| Goodness-of-fit on F2 | 1.066 | 1.110 | 1.115 |
| Final R [I ≥ 2σ(I)] | R 1 = 0.0632 | R 1 = 0.0953 | R 1 = 0.0815 |
| wR2 = 0.1661 | wR2 = 0.2483 | wR2 = 0.2322 | |
| Final R [all data] | R 1 = 0.0705 | R 1 = 0.1029 | R 1 = 0.0841 |
| wR2 = 0.1742 | wR2 = 0.2563 | wR2 = 0.2350 | |
| Largest diff. peak/hole/e Å−3 | 0.87/−0.37 | 0.77/−0.76 | 1.37/−0.48 |
Synthesis and characterization of [(tBuPO3H)Li(H2O)3·(H2O)] (1)
A solution of Li2CO3 (1 mmol, 74 mg) in MeOH (25 mL) was added to a methanolic solution (10 mL) of tBuP(O)(OH)2 (1 mmol, 138 mg) and the resulting mixture was stirred for 30 minutes at room temperature. Subsequently, H2O (5 mL) was added dropwise to the solution and the resulting mixture was stirred for 24 h at room temperature. After filtration, analytically pure 1 was obtained as colourless block-shaped crystals by leaving the reaction mixture for 2 weeks under ambient conditions. Yield 114 mg (53%), Mp >200 °C. Anal. (calcd) (%) for C4H18LiO7P (Mr = 216.09), C, 23.89 (22.23); H, 7.34 (8.40). 31P NMR (161 MHz, D2O) δ: 33.09 ppm. CP-MAS 7Li NMR (233 MHz) δ: 1.70 ppm, CP-MAS 31P NMR (242 MHz, ppm) δ: 40.20, 39.65, CP-MAS 13C NMR (150 MHz, ppm): δ 30.47, 29.65, 25.36. IR (as KBr pellet, cm−1): 3366 (br), 2975 (s), 1645 (s), 1481 (s), 1144 (s), 1050 (s), 902 (m), 661 (s), 517 (m), 502 (s). Temp. range °C (% Weight loss): 40–140 (33%, loss of coordinated and lattice H2O); 200–580 (26%, loss of organic moiety).Synthesis and characterization of [(tBuPO3)Na2(H2O)4]n (2)
A solution of NaHCO3 (1 mmol, 84 mg) in MeOH (25 mL) was added directly to a methanolic solution (10 mL) of (tBuP(O)(OH)2) (1 mmol, 138 mg) and the mixture was stirred for 2 h at room temperature. Then H2O (5 mL) was added dropwise to the solution, and the resulting mixture was warmed for 10 minutes on a water bath and then stirred for 24 h at room temperature. After filtration, analytically pure compound 2 was obtained as colourless plate-shaped crystals by leaving the reaction mixture for 2 weeks on the benchtop. Yield 162 mg (64%), Mp >200 °C. Anal. (calcd) (%) for C4H17Na2O7P (Mr = 253.12), C, 20.18 (19.0) H, 7.41 (6.74). 31P NMR (161 MHz, D2O) δ: 32.73 ppm. CP-MAS 31P NMR (162 MHz) δ: 37.31 ppm, CP-MAS 13C NMR (100 MHz, ppm): δ 35.81, 34.35, 31.51. IR (as KBr pellet, cm−1): 3426 (br), 2971 (s), 2909 (s), 1645 (s), 1477 (s), 1133 (s), 1071 (s), 1071 (s), 1013 (s), 893 (s), 654 (s), 502 (s), 425 (s). Temp. range °C (% Weight loss): 40–102 (28%, loss of coordinated H2O); 300–590 (22%, loss of organic moiety).Synthesis and characterization of [(tBuPO3H)K(H2O)]n (3)
A solution of KHCO3 (1 mmol, 100 mg) in MeOH (25 mL) was added directly to a methanolic solution (10 mL) of (tBuP(O)(OH)2) (1 mmol, 138 mg) and the mixture was stirred for 24 h at room temperature. After filtration, analytically pure 3 was obtained as colourless plate-shaped crystals by leaving the reaction mixture for 2–3 weeks on the benchtop. Yield 136 mg (71%), Mp >200 °C. Anal. (calcd) (%) for C4H12KO4P (Mr = 192.21), C, 24.72 (24.74); H, 5.44 (6.23). 31P NMR (202 MHz, D2O) δ: 32.96 ppm. CP-MAS 31P NMR (202 MHz) δ: 24.45 ppm, CP-MAS 13C NMR (125 MHz, ppm): δ 27.37, 26.29, 22.86. IR (as KBr pellet, cm−1): 3524 (br), 3243 (s), 2976 (m), 2869 (s), 1663 (s), 1130 (s), 1041 (s), 900 (s), 836 (s) 665 (s), 586 (s), 510 (s), 498 (s). Temp. range °C (% Weight loss): 40–120 (9%, loss of coordinated H2O); 200–550 (29%, loss of organic moiety).Synthesis and characterization of [(tBuPO3)Ca(H2O)]n (4)
To a solution of NaHCO3 (84 mg, 1 mmol) in methanol (10 mL), a methanolic solution (5 mL) of tBuPO(OH)2 (69 mg, 0.5 mmol) was added. The reaction mixture was sonicated for 35 min. 10 mL of water was added to the portion to solubilize the undissolved NaHCO3. After obtaining a clear solution, a solution of CaCl2·4H2O (55.5 mg, 0.5 mmol) in methanol (10 mL) was added to it. A white precipitate was obtained instantly. The reaction mixture was stirred overnight and the solid product was centrifuged out. The insoluble white precipitate was further washed with copious amounts of water to remove the by-product NaCl from the solution. Then it was air-dried to obtain a white solid powder. Yield 21 mg (22%), Mp >200 °C. Anal. (calcd) (%) for C4H11CaO4P (Mr = 194) C: 25.23 (24.74) H: 5.55 (5.71). DR-UV absorbance (λ, nm): 238, 308, 405. 31P NMR (162 MHz) δ: 37.75 ppm. CP-MAS 13C NMR (125 MHz, ppm): δ 38.04, 36.57, 32.82. IR (as KBr diluted disc, cm−1): 3424 (br), 2989 (m), 2056 (s), 1379 (m), 1092 (m), 1063 (s), 989 (s), 669 (m), 638 (br), 523 (m). Temp. range °C (% weight loss): 180–210 (8%, loss of coordinated H2O); 566–606 (29%, loss of organic moiety).Synthesis and characterization of [(tBuPO3)Mn(H2O)]n (5)
The same procedure as for 4 was used but instead of CaCl2·4H2O, MnCl2·4H2O (98.5 mg, 0.5 mmol) was added. A pinkish white precipitate was obtained instantly. The reaction mixture was stirred overnight and the solid product was centrifuged out. The insoluble pinkish white precipitate was further washed with copious amounts of water to remove the by-product NaCl from the solution. Then it was air-dried to obtain a powder. Yield 26 mg (24.9%), Mp >200 °C. Anal. (calcd) (%) for C4H11MnO4P (Mr = 209) C: 22.96 (22.98) H: 5.27 (5.30). DR-UV absorbance (λ, nm): 260, 293, 503. IR (as KBr diluted disc, cm−1): 3424 (br), 2951 (m), 2053 (s), 1640 (br), 1377 (m), 1063 (s), 1048 (s), 671 (m), 660 (br), 520 (m). Temp. range °C (% weight loss): 75–130 (8%, loss of coordinated H2O); 545–585 (29%, loss of organic moiety).Synthesis and characterization of [(tBuPO3)Co(H2O)]n (6)
The same procedure as for 4 was used but instead of CaCl2·4H2O, in this case, CoCl2·6H2O (118 mg, 0.5 mmol) was added. A purple precipitate forms instantly which turned dark violet after stirring for 45 min. The reaction mixture was stirred overnight and the solid product was centrifuged out. It was further washed with copious amounts of water to remove the by-product NaCl from the solution. Then it was air-dried to obtain a purple powder. Yield: 36.8 mg (34%), Mp >200 °C. Anal. (calcd) (%) C4H11CoO4P (Mr = 213) C: 22.66 (22.55) H: 5.29 (5.20). DR-UV absorbance (λ, nm): 526, 603, 643. IR (as KBr diluted disc, cm−1): 3430 (br), 2955 (m), 2868 (m), 2327 (m), 2070 (s), 1624 (br), 1487 (m), 1384 (s), 1142 (m), 1066 (s), 1010 (s), 938 (s), 834 (m), 667 (m), 513 (m). Temp. range °C (% weight loss): 65–406 (8%, loss of coordinated H2O); 421–485 (42%, loss of organic moiety); 555–578 (8%, loss of H2O).Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1–3 have been deposited at the CCDC under deposition numbers 2330476–2330478.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. M. acknowledges financial support from the SERB, New Delhi, through CRG/2022/002406 and SB/S2/JCB-85/2014 and from the MoE (SPARC/2019-2020/P1677/SL). A. K. thanks the CSIR, New Delhi and A. G. thanks the Prime Minister's Research Fellowship (PMRF) for a research fellowship. The authors thank the MoE IoE-funded central facilities and SAIF, IIT Bombay for characterization data. The authors thank the SPARC project partner, Prof. Suresh Valiyaveettil, NUS, Singapore, for useful discussions.References
- A. Clearfield and K. Demadis, Metal phosphonate chemistry: From synthesis to applications, RSC, Cambridge, 2011 Search PubMed.
- M. G. Walawalkar, H. W. Roesky and R. Murugavel, Acc. Chem. Res., 1999, 32, 117–126 CrossRef CAS.
- K. Rathore and R. Jangir, Inorg. Chim. Acta, 2024, 559, 121804 CrossRef CAS.
- J. Goura and V. Chandrasekhar, Chem. Rev., 2015, 115, 6854–6965 CrossRef CAS PubMed.
- K. D. Demadis and M. Papadaki, ACS Appl. Mater. Interfaces, 2010, 2, 1814–1816 CrossRef CAS.
- C. Queffelec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef CAS PubMed.
- Y.-P. Zhu, Z.-Y. Yuan and H. N. Alshareef, ACS Mater. Lett., 2020, 2, 582–594 CrossRef CAS.
- M. Mehring, M. Schürmann and R. Ludwig, Chem. – Eur. J., 2003, 9, 837–849 CrossRef CAS PubMed.
- S.-S. Bao, G. K. H. Shimizu and L.-M. Zheng, Coord. Chem. Rev., 2019, 378, 577–594 CrossRef CAS.
- L.-M. Zheng and Y. Duan, Metal Phosphonate Chemistry: From Synthesis to Applications, The Royal Society of Chemistry, 2012, pp. 235–278 Search PubMed.
- K. Gopal, S. Ali and R. E. Winpenny, Metal Phosphonate Chemistry, Royal Society of Chemistry, Cambridge, UK, 2011, pp. 364–419 Search PubMed.
- S. Langley, M. Helliwell, R. Sessoli, S. J. Teat and R. E. Winpenny, Dalton Trans., 2009, 3102–3110 RSC.
- M. Mehring, G. Guerrero, F. Dahan, P. H. Mutin and A. Vioux, Inorg. Chem., 2000, 39, 3325–3332 CrossRef CAS PubMed.
- M. G. Walawalkar, S. Horchler, S. Dietrich, D. Chakraborty, H. W. Roesky, M. Schäfer, H.-G. Schmidt, G. M. Sheldrick and R. Murugavel, Organometallics, 1998, 17, 2865–2868 CrossRef CAS.
- H. Xu, H. Zhou, L. Feng, Q. Wang, R. Chen, W. Huang and X. Wu, Dalton Trans., 2018, 47, 11226–11238 RSC.
- G. Anantharaman, M. G. Walawalkar, R. Murugavel, B. Gabor, R. Herbst-Irmer, M. Baldus, B. Angerstein and H. W. Roesky, Angew. Chem., Int. Ed., 2003, 42, 4482–4485 CrossRef CAS PubMed.
- J. K. Jabor, R. Stösser, M. Feist, P. Neubauer and M. Meisel, Inorg. Chem., 2008, 47, 9293–9302 CrossRef CAS PubMed.
- M. B. Dines and P. M. DiGiacomo, Inorg. Chem., 1981, 20, 92–97 CrossRef CAS.
- M. G. Walawalkar, R. Murugavel, H. W. Roesky and H.-G. Schmidt, Inorg. Chem., 1997, 36, 4202–4207 CrossRef CAS.
- Y. Yang, H.-G. Schmidt, M. Noltemeyer, J. Pinkas and H. W. Roesky, J. Chem. Soc., Dalton Trans., 1996, 17, 3609–3610 RSC.
- M. G. Walawalkar, R. Murugavel, H. W. Roesky and H.-G. Schmidt, Organometallics, 1997, 16, 516–518 CrossRef CAS.
- M. Srinu, N. B. Padalwar and K. Vidyasagar, Z. Anorg. Allg. Chem., 2015, 641, 2429–2434 CrossRef CAS.
- N. B. Padalwar, C. Pandu and K. Vidyasagar, J. Solid State Chem., 2013, 203, 321–325 CrossRef.
- M. G. Walawalkar, R. Murugavel, A. Voigt, H. W. Roesky and H.-G. Schmidt, J. Am. Chem. Soc., 1997, 119, 4656–4661 CrossRef CAS.
- M. G. Walawalkar, R. Murugavel, H. W. Roesky, I. Usón and R. Kraetzner, Inorg. Chem., 1998, 37, 473–478 CrossRef CAS PubMed.
- Y. Yang, J. Pinkas, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 1998, 37, 6404–6405 CrossRef CAS PubMed.
- M. M. Gómez-Alcántara, A. Cabeza, M. A. Aranda, A. Guagliardi, J. G. Mao and A. Clearfield, Solid State Sci., 2004, 6, 479–487 CrossRef.
- X.-Y. Yi, L.-M. Zheng, W. Xu and S. Feng, Inorg. Chem., 2003, 42, 2827–2829 CrossRef CAS PubMed.
- J.-G. Mao, Z. Wang and A. Clearfield, Inorg. Chem., 2002, 41, 2334–2340 CrossRef CAS PubMed.
- M. Pica, A. Donnadio, R. D'Amato, D. Capitani, M. Taddei and M. Casciola, Inorg. Chem., 2014, 53, 2222–2229 CrossRef CAS PubMed.
- B. Bujoli, O. Pena, P. Palvadeau, J. Le Bideau, C. Payen and J. Rouxel, Chem. Mater., 1993, 5, 583–587 CrossRef CAS.
- D. Grohol and A. Clearfield, J. Am. Chem. Soc., 1997, 119, 4662–4668 CrossRef CAS.
- M. Sunjuk, B. El-Eswed, J. N. Dawoud, A. Shtaiwi, M. Khanfar and M. El-Khateeb, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 558–575 CrossRef CAS.
- P. Tholen, L. Wagner, J. G. A. Ruthes, K. Siemensmeyer, T. H. Y. Beglau, D. Muth, Y. Zorlu, M. Okutan, J. C. Goldschmidt, C. Janiak, V. Presser, O. Yavuzcetin and G. Yucesan, Small, 2023, 19, e2304057 CrossRef PubMed.
- D. E. Bugaris and J. A. Ibers, Dalton Trans., 2010, 39, 5949–5964 RSC.
- G. Yucesan, W. Ouellette, V. Golub, C. J. O'Connor and J. Zubieta, Solid State Sci., 2005, 7, 445–458 CrossRef CAS.
- B. A. Breeze, M. Shanmugam, F. Tuna and R. E. Winpenny, Chem. Commun., 2007, 48, 5185–5187 RSC.
- K. P. Rao and K. Vidyasagar, Eur. J. Inorg. Chem., 2005, 24, 4936–4943 CrossRef.
- W. C. Grinonneau, P. L. Chapman, A. G. Menke and F. Walmsley, J. Inorg. Nucl. Chem., 1971, 33, 3011–3017 CrossRef CAS.
- G. Alberti, U. Costantino, S. Allulli and N. Tomassini, J. Inorg. Nucl. Chem., 1978, 40, 1113–1117 CrossRef CAS.
- D. Cunningham, P. J. Hennelly and T. Deeney, Inorg. Chim. Acta, 1979, 37, 95–102 CrossRef CAS.
- K. J. Martin, P. J. Squattrito and A. Clearfield, Inorg. Chim. Acta, 1989, 155, 7–9 CrossRef CAS.
- M. Mehring and M. Schurmann, Chem. Commun., 2001, 22, 2354–2355 RSC.
- V. Chandrasekhar, J. Goura and E. C. Sanudo, Inorg. Chem., 2012, 51, 8479–8487 CrossRef CAS PubMed.
- V. Chandrasekhar, L. Nagarajan, S. Hossain, K. Gopal, S. Ghosh and S. Verma, Inorg. Chem., 2012, 51, 5605–5616 CrossRef CAS PubMed.
- L. Zhang, R. Clerac, P. Heijboer and W. Schmitt, Angew. Chem., Int. Ed., 2012, 51, 3007–3011 CrossRef CAS PubMed.
- Y. Z. Zheng, M. Evangelisti, F. Tuna and R. E. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057–1065 CrossRef CAS PubMed.
- L. Zhang, B. Marzec, R. Clerac, Y. Chen, H. Zhang and W. Schmitt, Chem. Commun., 2013, 49, 66–68 RSC.
- J. Goura, P. Bag, V. Mereacre, A. K. Powell and V. Chandrasekhar, Inorg. Chem., 2014, 53, 8147–8154 CrossRef CAS PubMed.
- A. R. Patterson, W. Schmitt and R. C. Evans, J. Phys. Chem. C, 2014, 118, 10291–10301 CrossRef CAS.
- K. H. Zangana, E. M. Pineda and R. E. Winpenny, Dalton Trans., 2014, 43, 17101–17107 RSC.
- J. L. Bideau, A. Jouanneaux, C. Payen and B. Bujoli, J. Mater. Chem., 1994, 4, 1319–1323 RSC.
- S. Khanra, M. Kloth, H. Mansaray, C. A. Muryn, F. Tuna, E. C. Sanudo, M. Helliwell, E. J. McInnes and R. E. Winpenny, Angew. Chem., Int. Ed., 2007, 46, 5568–5571 CrossRef CAS PubMed.
- M. R. Mason, J. Cluster Sci., 1998, 9, 1–23 CrossRef CAS.
- Y. Yang, M. G. Walawalkar, J. Pinkas, H. W. Roesky and H. G. Schmidt, Angew. Chem., Int. Ed., 1998, 37, 96–98 CrossRef CAS.
- V. Chandrasekhar, J. Goura and A. Duthie, Inorg. Chem., 2013, 52, 4819–4824 CrossRef CAS PubMed.
- K. C. K. Swamy, C. G. Schmid, R. O. Day and R. R. Holmes, J. Am. Chem. Soc., 1990, 112, 223–228 CrossRef CAS.
- R. Murugavel and S. Shanmugan, Chem. Commun., 2007, 12, 1257–1259 RSC.
- R. Murugavel and S. Shanmugan, Dalton Trans., 2008, 5358–5367 RSC.
- V. Chandrasekhar, A. Dey, T. Senapati and E. C. Sañudo, Dalton Trans., 2012, 41, 799–803 RSC.
- R. Murugavel, N. Gogoi and R. Clerac, Inorg. Chem., 2009, 48, 646–651 CrossRef CAS PubMed.
- S. Langley, M. Helliwell, R. Sessoli, S. J. Teat and R. E. Winpenny, Inorg. Chem., 2008, 47, 497–507 CrossRef CAS PubMed.
- S. Verma and R. Murugavel, Inorg. Chem., 2022, 61, 6807–6818 CrossRef CAS PubMed.
- I. R. Salcedo, R. M. P. Colodrero, M. Bazaga-García, A. Vasileiou, M. Papadaki, P. Olivera-Pastor, A. Infantes-Molina, E. R. Losilla, G. Mezei, A. Cabeza and K. D. Demadis, CrystEngComm, 2018, 20, 7648–7658 RSC.
- D. Sahoo, R. Suriyanarayanan and V. Chandrasekhar, Dalton Trans., 2014, 43, 10898–10909 RSC.
- J.-y. Liu, C.-b. Ma, H. Chen and C.-n. Chen, CrystEngComm, 2015, 17, 8736–8745 RSC.
- B. Zhang, D. M. Poojary and A. Clearfield, Inorg. Chem., 1998, 37, 1844–1852 CrossRef CAS.
- J. E. Haky, J. B. Brady, N. Dando and D. Weaver, Mater. Res. Bull., 1997, 32, 297–303 CrossRef CAS.
- D. D. Perrin, W. L. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, 1988, Butterworth-Heinemann, Oxford, 4th edn, 1996 Search PubMed.
- L. D. Freedman and G. Doak, Chem. Rev., 1957, 57, 479–523 CrossRef CAS.
- CrysAlisPRO, Rigaku Oxford Diffraction Ltd, Yarnton, Oxfordshire, England Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- Diamond – Crystal and Molecular Structure Visualization, Crystal Impact – Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 2008, 2102, 53227 Bonn, Germany.
- M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE (Version 2.1), 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details, spectral characterization and additional figures and tables. CCDC 2330476 (1), 2330477 (2) and 2330478 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01668h |
| This journal is © The Royal Society of Chemistry 2024 |