
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gigawatt-hour to terawatt-hour salt cavern supercapacitors and supercapatteries†
Tingting Jiang *a,
Jingjie Lia and
George Z. Chen*b
*a,
Jingjie Lia and
George Z. Chen*b
aThe State Key Laboratory of Refractories and Metallurgy, Faculty of Materials, Wuhan University of Science and Technology, Wuhan 430081, P. R. China. E-mail: ttjiang@wust.edu.cn
bDepartment of Chemical and Environmental Engineering, Faculty of Engineering, University of Nottingham, Nottingham NG2 7RD, UK. E-mail: george.chen@nottingham.ac.uk
First published on 28th May 2025
Abstract
The advancement of energy technology has led to a notable increase in the contribution from renewable energy sources to the global energy supply and consumption landscape. Nevertheless, although inexhaustible and clean, the intermittency and instability of these energy sources present significant challenges to their wider deployment, necessitating the development of robust energy storage systems. Also, it is historical that the demand for power supply also varies significantly between day and night, and between different time zones, requesting large scale storage capacity for not only load levelling but also power supply security. In this article, salt caverns, which offer a sealable and unmatched large space and are currently employed for storage of compressed energy gases, are proposed for construction of giga- to tera-watt-hour scale supercapacitors and supercapatteries as an effective storage solution to renewable energy farms and national and international power grids. Following an introduction to salt caverns and their uses for storage of compressed air, natural gas, hydrogen and carbon dioxide, the potential is explored for construction of supercapacitors and supercapatteries in salt caverns. The discussion is specially focused on aqueous electrolytes that can be formed by utilising the salty water or brine from the construction of the salt cavern, and the respective electrode materials suitable for such aqueous electrolytes. Furthermore, calculations and analyses are given on the prospects of construction and application of giga- to tera-watt-hour supercapacitors and supercapatteries in salt caverns. Last, but not least, foreseeable challenges of such unprecedented ultra-large scale electrochemical energy storage devices are discussed with possible solutions.
1. Introduction: energy storage in salt caverns
1.1. Basics of salt caverns
As society and the economy develop and progress, so do the demands on energy supply and storage. At the same time, there is an urgent need to mitigate the environmental impact caused by the use of fossil fuels. Many countries have set targets to reduce carbon emissions and achieve carbon neutrality.1 The supply and use of renewable energy from, for example, wind and sunlight, have seen rapid development and application in recent years, and the share of renewable energy in the total energy market is increasing and will continue to increase significantly. For example, electricity generation from renewables in the UK was 33 terawatt-hour (TW h) in the second quarter of 2024, accounting for 51.6% of the total electricity generation in the same period and 19.0% increase from the second quarter of the previous year.2However, both wind and solar power have the limitations of intermittency and instability, which would hinder the direct power supply to the grid. Similarly, the demand for energy supply also varies significantly with time and regions. Such mismatches between the supply and user sides make it imperative and necessary to match the large storage capacity of energy with the peak and valley shifting of renewable resources and market variation.3 Currently, energy storage technologies include pumped hydro, flywheel,4 hydrogen, compressed gas (air),5,6 and (flow) batteries.7,8 As the earliest used and most mature energy storage system, pumped hydro energy storage has a long history, but is limited by geology and water resources.9 Hydrogen storage has attracted much attention in recent decades due to its high energy storage density (compared with other gases), long-term storage, and its ease of converting into different forms of energy.10,11 However, there are some unresolved issues regarding production costs, efficiency and safety. Compressed air storage has the advantages of large scale, fast response and low cost and is believed to be a promising candidate.12,13 In addition, there are emerging novel energy storage technologies.14,15 For example, batteries and their derivations with or without redox active electrolytes that may also be flowing can offer electricity storage with high energy efficiency, great power capability, and wide scalability because of their modular nature. On the other hand, salt caverns, a type of cave or space formed in the underground salt rock, can be widely used in a variety of energy storage fields.16,17
Salt rocks belong to the class of sedimentary rocks, which often consist of NaCl as the main mineral component, and polyhalite and argillaceous rock as secondary mineral components. Salt rock is widely distributed on the Earth, including two types: the lacustrine and marine sedimentaries. These two types of salt rock can be formed into (1) the fault basin structure, which usually consists of various organic impurities with many thin or interlayered salt layers, and (2) the bottom-splitting salt dome structure, which has large layer thickness, high salt rock purity and few interlayered parts.18 Both types of salt rock reserve rich oil and gas resources, especially the salt dome from marine sedimentary.
Based on the salt rock, the construction of salt caverns can lead to a large underground space for various storage applications. For example, in the salt dome structure, a salt cavern of 1 mile in diameter and 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 feet in height (≈1.83 × 1010 m3) can be formed. However, the fault basins or salt beds have usually wide, shallow, thin geological features with the salt layer thickness being less than 1000 ft (<300 m). Such horizontally extended salt rock layers are generally considered unsuitable for construction of salt caverns for storage of pressurised gases in comparison with vertically structured caverns.19
000 feet in height (≈1.83 × 1010 m3) can be formed. However, the fault basins or salt beds have usually wide, shallow, thin geological features with the salt layer thickness being less than 1000 ft (<300 m). Such horizontally extended salt rock layers are generally considered unsuitable for construction of salt caverns for storage of pressurised gases in comparison with vertically structured caverns.19
Although with different thicknesses, the walls of salt caverns always have excellent plasticity, good mechanical stability and self-healing capability under complex conditions, which are all desirable for construction. More importantly, the walls have low porosity and therefore low permeability which translates into high air tightness compared with other underground spaces, and low chemical reaction activity with other substances.20,21 Based on these characteristics, salt caverns are ideally suitable for various applications in the field of energy storage.
1.2. Formation of salt caverns
In general, there are three types of existing salt caverns: natural formations resulting from the dissolution of salt rock and the flow of groundwater; abandoned cavities after oil and gas extraction, and artificial constructions using a series of techniques, which will be discussed below. The depleted oil and gas reservoirs usually show the porous storage form, while artificially created caverns are commonly a single space with gas tight walls for storage of energy gases.22Leaching, or solution mining, is the primary construction technique for most salt caverns. The salt rock, which consists mainly of NaCl, is highly soluble in water. By using the water leaching method, the underground salt rock can be easily removed without damaging the surrounding cavern rock. Generally, there are several steps involved in creating a salt cavern by water leaching. First, boreholes are designed and drilled in the location surfaces where salt rocks are present, and a pipe string system is built. High-pressure water is then injected to fracture the salt rock and dissolve some of the salt, creating a cavity. The brine formed in the course of drilling is then pumped up to the surface though pipes. By repeating these processes, as the water is injected and the salt rock dissolves continuously, a cavern of a designated shape and volume is created, which becomes the desired salt cavern. The rate of dissolution is the main parameter influencing the construction of the salt cavern, which is different in the top, lateral, and bottom layers of the salt rock. The introduction of low-density media, such as diesel oil or nitrogen gas could help to prevent the over-dissolution of the upper layer of the salt cavern and control the final cavern to be in the desired geometry.22–24 The discharged brine can be treated or reused in three ways:
Pumping into the sea, salt production, and as a raw material for the chemical industry.27
To date, most of the construction of salt caverns around the world has been based on the single-well cavern construction method.22 There is only one group of concentric inner, intermediate, and outer pipes. The injection of fresh water, the extraction of brine and the introduction of protective media can all be achieved though these concentric pipes. The salt cavern created by this method is usually cylindrical and elongated vertically. However, there are some drawbacks to this method, including high cost, long time for the cavern formation, and inappropriateness for complex geological environments such as the lacustrine sedimentary.
Therefore, the horizontal multi-stage leaching method to form a horizontal cavern with two vertical wells has been developed and demonstrated.25,28 The construction process differs from that of the single well cavern. First, two vertical wells are drilled and then a horizontal tunnel is drilled to be connected to one of the vertical wells (the first) at the bottom of the target salt rock layer. Pipes with or without the concentric structure are then installed in both the vertical wells and the connected horizontal tunnel through which fresh water is injected to dissolve the salt rock, i.e. solution mining. The resulting brine is extracted from the second vertical well, which means the solution mining can be operated continuously. Obviously, if concentric pipes are used, both vertical wells can be used for water injection and brine extraction. The cavern construction is completed by repeating or continuing these processes until the salt cavern is formed with a designated shape, e.g. similar cross-sectional dimensions in the horizontal direction. In addition, diesel (or pressurised gas, e.g. nitrogen) is injected from the annulus between the outer and intermediate casing to form an oil blanket. This isolation layer suppresses roof dissolution and controls lateral leaching for controlled cavern geometry.11 A schematic illustration of the horizontal cavern with two vertical wells is shown in Fig. 1a for the purpose of oil storage. The two-well horizontal cavern formation method has the advantage of quicker injection and extraction to form the salt cavern with larger sizes. In addition, the shape of the salt cavern can be controlled more flexibly. This method is more suitable for bedded salt rocks with a stratified structure, for example in China and the UK.
 | ||
| Fig. 1 Schematic illustrations of (a) two well horizontal salt cavern constructions, reproduced from ref. 25 with permission from Taylor & Francis, copyright 2020 and (b) compressed air energy storage in salt caverns, reproduced from ref. 26 with permission from Elsevier, copyright 2021. | ||
1.3. Fluids storage in salt caverns for energy purposes
Due to their distinctive characteristics such as self-healing, high tightness and hence low permeability, salt caverns have great significance in large-scale energy storage technologies that involve a fluid such as energy gases and liquid fuels, which are different from those energy stores relying on solid materials, e.g. flywheel and springs. In addition, because of their vast storage capacity, salt cavern energy storage can serve as an integral component of larger energy regulators for renewable energy plants and the power grid. In commercial practices, salt caverns have already been utilised for the storage of oil, natural gas, hydrogen, compassed air, as well as CO2 for the reduction of carbon emissions. The principle, dis-/advantages, and applications of storage of fluids in salt caverns are summarized in Table 1 and more details can be found in Section S1 of the ESI.†Fig. 1b illustrates compressed air energy storage (CAES) in salt caverns. As one of the most established technologies, CAES uses off-peak electricity to compress air in caverns and releases it to drive the turbine for electricity generation during peak demand, enhancing grid stability and integrating with renewables. CAES in caverns has been commercialized in Germany, the USA and China. Natural gas storage, widely deployed, offers strategic peak-shaving capacity but conflicts with decarbonization goals due to its fossil fuel nature. Hydrogen storage exhibits high potentials, but faces barriers such as high electrolysis costs and hydrogen embrittlement risks. CO2 sequestration contributes to emission reductions through supercritical state storage but capture costs are high. Generally, salt caverns must balance technological maturity (gas/oil), sustainability (H2/CO2), and economics to help the energy transition.
Currently, salt caverns in China are distributed in more than 150 mining areas with depths of 100–4000 metres, divided into the shallow, medium, deep and ultra-deep categories. Typical examples include Yunying in Hubei (shallow, natural gas storage), Jintan in Jiangsu (medium depth, CAES) and Chuzhou in Jiangsu (deep, natural gas storage).22 UK salt caverns include Teesside (hydrogen storage), East Yorkshire (>1600 m, natural gas storage), Cheshire Basin and Wessex Basin.29 Based on the geological exploration information, modelling simulations can be developed to derive information on the construction process, energy storage performances, and long-term stability of fluid filled salt caverns (Table 1).21,30–32
| Energy form | Principle | Energy density | Advantages | Disadvantages | Technical challenges | Geographical constraints | Case studies | Ref. |
|---|---|---|---|---|---|---|---|---|
| CAES | Store compressed air in salt caverns for electricity generation when needed | ∼2.9 kW h m−3 | Large-scale, long duration, low cost | Fossil fuel dependency, high initial cost | Cyclic pressure stability | Thick salt layers or salt dome | Huntorf (Germany), McIntosh (USA), Jintan (China) | 3, 16, 17 and 33 |
| Natural gas | Inject high-pressure gas into caverns for seasonal/emergency supply | ∼1000 kW h m−3 | Rapid response (minutes), mature technology, stable long-term | Deformation risk under pressure swings | Thin-bedded salt morphology control with high permeability interlayers | High-purity salt, salt domes or bedded salt | Gulf Coast (USA) | 3, 16, 17 and 34 |
| H2 | Electrolyze water to produce H2, store in caverns for fuel cell use | ∼300 kW h m−3 | High energy density, zero emissions, renewable integration | Hydrogen embrittlement, high permeability risk | Diffusion control, corrosion-resistant material | Deep caverns (800–1500 m) for sealing | Spindletop (USA, 2014) | 3, 10, 11, 16, 17 and 35–37 |
| CO2 | Inject supercritical CO2 for sequestration or resource utilization | ∼0.77 t m−3 (supercritical) | High sealing, potential resource cycling, long-term stability | Limited per-cavern capacity, requires carbon capture techniques | Chemical reactions with interlayer | Thick salt layers or domes | 3, 16, 17 and 38 | |
| Others | Oil: inject oil displacing brine; waste: utilize low permeability for containment | Oil: low cost; waste: Reliable long-term containment | High construction cost, chemical compatibility | Frequency-dependent fatigue effects | 16 and 17 |
Although salt caverns are already playing an important role in the field of energy storage and utilisation, there are additional potential applications that warrant further investigation. As discussed later, we propose to use salt caverns for construction of large scale aqueous supercapacitors and supercapatteries with the salty water or brine from solution mining as the electrolyte with or without modification. The current status of salt cavern energy storage indicates some obstacles to be overcome in terms of its promotion and application.
First, from the perspective of salt cavern construction, while techniques such as the two-well horizontal cavern formation method have been gradually developed, there remain technical challenges in the construction of salt caverns that are fast, efficient, and safe. The construction of salt caverns, especially those of a large scale, should overcome the variable and complex geological conditions that they are built in. Previous multiple theoretical analyses and simulations may provide a basis for the construction process.
Second, reliable supporting facilities are required to align with the salt cavern energy storage systems during the utilisation process. These include gas injection, extraction, and transportation methods and equipment.
Third, it is of great importance to pay close attention to the safety and stability aspects when storing certain gases, such as natural gas and hydrogen. Meanwhile, contamination and purification of these gases represent a significant challenge that has not yet been fully addressed in the context of more widely used compressed air salt cavern storage systems.
Fourth, despite the high air tightness of salt caverns, there is a residual risk of leakage over extended periods of storage. The potential environmental impact and hazards associated with such leakage should be rigorously analysed, monitored and controlled.
Finally, it is notable that the cost of salt cavern energy storage, particularly in relation to hydrogen storage, is considerably less than that of alternative storage solutions. However, construction and operational costs can vary significantly across different geographical regions, which also needs careful and thorough considerations.
1.4. Potential of electrochemical energy storage in salt caverns
The aforementioned energy storage applications of salt caverns are primarily utilised as a means of storing gases of which the so-called green hydrogen gas has been considered widely as the promising option because it can be produced by water electrolysis utilising renewable energy. There are four commercial or commercial-ready options, namely alkaline water electrolysis (industrial), polymer electrolyte membrane (PEM) electrolysis (commercial), alkaline electrolyte membrane (AEM) electrolysis (commercial-ready) and solid oxide membrane (SOM) electrolysis (laboratory, 700–850 °C).39,40 Of these, AEM electrolysis is perhaps the most cost-effective approach because it requires no or less precious metal catalysts, uses cheaper electrolyte membranes and offers higher energy efficiency and longer service life.40 It is worth noting that the catalysts used in these conventional methods may be incorporated into the recently proposed half-electrolysis41,42 to further improve process efficiency.Fig. S1a and b (ESI†) show schematically a single cell AEM electrolyser which is structurally similar to a single cell PEM electrolyser, and the bipolarly connected stack of multiple AEM cells in a representive workflow diagram. In alkaline electrolysis, two moles of water are reduced at the cathode, producing one mole of hydrogen gas and two moles of hydroxide ions (H2O + 2e ⇌ H2 + 2OH−). The evolved H2 diffuses from the cathode surface, while the remaining OH− ions migrate through a porous separator to the anode under the applied electrical potential. At the anode, these OH− ions are subsequently oxidised, generating 0.5 moles of oxygen gas and one mole of water (2OH− ⇌ 1/2O2 + H2O + 2e). The multiple AEM system consists of the water/gas loop systems, heating system, and control system.43 The working cell voltage of AEM electrolysis ranges from 1.4 to 2.0 V (lower than 1.6 to 2.2 V for PEM electrolysis),39 which means the voltage efficiency (equivalent to the energy efficiency at the same current density) is 70 to 21%. Energy output from the hydrogen gas is more efficient using the H2–O2 fuel cells whose optimal efficiency for hydrogen to power conversion is between 60 to 40%. Therefore, the overall energy efficiency from water electrolysis to power generation is between 42 and 8%. In comparison, the energy efficiencies of Li-ion batteries and supercapacitors are typically higher than 70% and 90%, respectively. Such large differences in energy efficiency make green hydrogen production and storage far less favourable than direct electrochemical energy storage.
Based on a salt cavern volume of 105 m3 proposed by the EWE company in 2016, a novel concept involving the construction of salt caverns as containers for the fabrication of large-scale redox flow batteries (RFBs) has been put forth.44 As a typical example of electrochemical energy storage techniques, RFBs can offer the advantages of long cycle life, and high safety.45 These attributes have led to the consideration of their potential for use in large-scale energy storage. The feasibility of this approach has been demonstrated in laboratory settings, with specific electrolytes and electrodes validated for use in large-scale salt caverns.
In recent years, electrochemical energy storage (EES) has attracted considerable attention, in particular metal ion batteries and supercapacitors, which have emerged as promising energy storage technologies due to their high energy or power densities and favourable cycle stabilities. The advantages of salt caverns, including their vast underground space and stable mechanical properties, make them ideal for use as electrolyte and electrode containers for metal ion batteries or supercapacitors, and offers a promising strategy for developing large-scale salt cavern EES batteries.
Another unique but rarely explored prospect is to use the brine from solution mining of salt caverns as the electrolyte for EES devices, which should offer a great economic advantage. This prospect coincides well with past and ongoing research of the authors in terms of electrode materials and device design strategies particularly in relation to aqueous electrolytes. Whilst more discussion is given later, it is worth mentioning here that one of the authors (G. Z. C.) consulted on the technical feasibility of developing giant batteries in deep salt caverns between 2009 and 2010.46 In the view of the authors, the potential application of combining salt caverns with EES techniques represents an important direction for large-scale power plant development at the GW to TW levels and will be the primary focus of the following discussions.
2. Electrode materials and aqueous electrolytes
In recent years, the objective of reducing carbon emissions and the policies of many countries have driven the rapid development of EES. As the technologies mature and the costs decrease, its applications and potential applications are expanding, which has been utilised or is predicted to be utilised in electronic devices, electric vehicles, and power stations. Materials and technologies based on electrochemical reactions, which are typically represented by metal-ion batteries with high energy density and supercapacitors with high power performances, excellent energy efficiencies and long cycle lives, are receiving more attention and recognition.Nevertheless, the deployment of EES in the field of large-scale energy storage is still facing improvement demands. For example, lithium-ion batteries (LIBs) are the commercial champions for EES purposes but their large scale applications are not yet ideal because of two primary factors: the voltage loss due to internal factors such as the inefficient electron and ion transfer and transport, and safety concerns associated with the flammability and toxicity of the raw materials, particularly the organic and flammable electrolytes.47,48 These two unfavourable factors are basically minimal in supercapacitors (and some rechargeable batteries) with aqueous electrolytes. Furthermore, the considerable potentials of supercapacitors, in conjunction with those that integrate rechargeable batteries and supercapacitors, e.g. supercapatteries, to supplement or even replace batteries in the future have been widely investigated. In the course of their development, supercapacitors and supercapatteries have shown the predicted capability to deliver high power output in a safe and stable manner with an extended operational lifespan. Although their demonstrated energy densities are still lower than those of LIBs, supercapacitors and supercapatteries can be shown to possess a much greater potential for combination with salt caverns, representing great promise for future large-scale EES plants.
2.1. Basic principles
Fig. 2a shows a schematic of the generic EES device configuration.49–54 Supercapacitors bridge the high power characteristics of conventional capacitors with the high energy capacity of rechargeable batteries, among which electrochemical (or electrical) double-layer (EDL) capacitors are most prevalent. Their operation relies on reversible electrostatic adsorption of electrolyte ions at the porous electrode–electrolyte interface (both the positive and negative electrodes (positrode and negatrode) for anions and cations, respectively).49 Because adsorption of anions occurs at a much more positive potential than that of cations, electrical energy is thus stored in the EDL capacitor.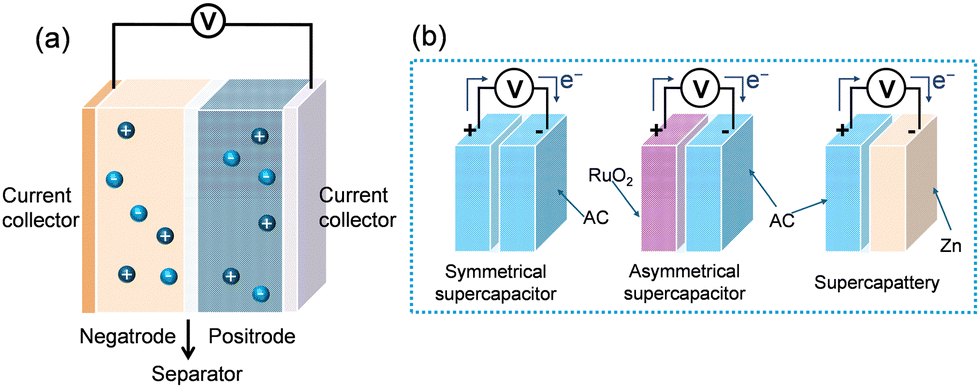 | ||
| Fig. 2 Schematic illustrations of (a) the structure of a generic EES device (cell) and (b) charging three different EES devices as indicated. | ||
While general views consider EDL capacitor energy storage to be physical in nature (ad-/desorption of ions without invoking chemical reactions),14 the de-/solvation of ions adsorbed on the surface induces chemical bonding changes, indicating a non-exclusively physical process.50
The classical Helmholtz bilayer model was initially employed to account for the EDL capacitor, which was later extended by the diffuse bilayer model incorporating an ion distribution gradient into the electrolyte. Currently, EDLs actually possess multilayered structures with compact and diffused layers.50 Despite limited specific capacity, EDL capacitors enable rapid dis-/charging. In general, the larger the specific surface area and the higher the porosity of the electrode material, the greater the number of sites that can undergo bilayer adsorption, and the higher the amount of charges that can be stored in the corresponding material. Activated carbon is the most common EDL active material.51,52 The dis-/charging processes of batteries, supercapacitors and supercapatteries (cf. Fig. 2) are characterised by different features on cyclic voltammograms (CVs) as well as linear galvanostatic (constant-current) charging and discharging curves (GCDs), as shown in Fig. 3.
 | ||
| Fig. 3 Illustration of CVs of electrodes and GCD profiles of two-electrode cells of a battery, supercapacitor and supercapattery (redrawn from ref. 50). | ||
Unlike EDL capacitance, charge storage can also be achieved via reduction and oxidation (redox) reactions in the electrode that break and reform chemical bonds, leading to chemical energy storage. Such reactions always involve the transfer of electrons across an interface such as that between electrode and electrolyte (solid|liquid), or between the current collector and active material (solid|solid) and are called Faradaic processes.50 It is proposed that, depending on the chemical bonding, the Faradaic process can proceed in two different ways.
The first is Nernstian. Consider the oxidation of a species in the reduced state, Re, to its oxidised state, Ox, as shown in reaction (1), on a positrode upon the application of a potential, E+.
| Re ⇌ Ox + ne | (1) |
If reaction (1) obeys the following Nernst eqn (2) which includes an equilibrium potential, E+0, that is both theoretically and experimentally characteristic of reaction (1),
 | (2) |
The above discussion refers to both Re and Ox being in the liquid electrolyte. This means that no direct interactions occur between the same or different types of redox active species. If they are anchored on the electrode and do not interfere with each other, the Nernst equation can still be valid. Mainly because of E+0 or E−0, a Nernstian process exhibits distinct peak-shaped cyclic voltammograms (CVs) and non-linear galvanostatic charging–discharging plots (GCDs) often with a potential plateau. The Nernstian process can also cause phase transition in the electrode material. The combination of a Nernstian positrode and a Nernstian negatrode is responsible for the charge storage in conventional rechargeable batteries with E+0−E−0 being the theoretical cell voltage.
It should be pointed out that in an electrochemical cell, the electrode with a reduction reaction is named as the cathode, while that with an oxidation reaction as the anode. In an electrolysis cell, the cathode is the negatrode and the anode the positrode. However, in a rechargeable battery, charging is the same as electrolysis, but discharging will have oxidation on the negatrode and reduction on the positrode. Therefore, the terms anode and cathode should not be used to name the electrodes in any rechargeable electrochemical cell, such as battery, redox flow battery, supercapacitor or supercapattery.
Another Faradaic process is widely known as pseudocapacitance.45,46 It may result from reaction (2) if Ox and Re are both on the electrode and these redox active species can electronically interact with each other. This situation is commonplace amongst semiconducting redox active materials, such as RuO2 and MnO2 and also polypyrrole, in which the electrons or more accurately valence electrons are delocalised to a certain zone or range in the electrode. It is worth mentioning that, unfortunately, pseudocapacitance was used for at least two different electrochemical processes in the literature, resulting in confusions and misleading claims.50,55–57 To avoid confusion, Faradaic capacitance or linear pseudocapacitance has been proposed.53 In the following discussion, Faradaic capacitance is used in place of pseudocapacitance that was used broadly and also confusingly in the respective literatures.
The phenomenon of Faradaic capacitance was first observed in RuO2.54 Other transition metal oxides (TMOs) and intrinsically and electronically conducting polymers have also been shown to store charge in a capacitive way, which is in accordance with the concept of Faradaic capacitance.58,59 Nevertheless, this category of capacitive energy storage processes, despite following the Faraday reaction rule, do not align with the Nernst equation.60 This is because, experimentally, rectangular CVs and linear GCDs are also commonly observed in materials with Faradaic capacitance. In comparison with materials of EDL capacitance, Faradaic capacitive materials can offer higher specific capacitance, but slower kinetics because of the relatively slow transport of charges (mainly ions) that is also well known in Nernstian or battery materials. It is worth noting that it is impossible to differentiate between EDL and Faradaic capacitances by electrochemical means only, but the electron transfer reactions of Faradaic capacitance can often be detected by spectroscopic analyses, such as electron spin resonance (ESR) spectroscopy.53
Accordingly, in terms of electrode materials, rechargeable batteries use Nernstian materials, whilst supercapacitors have their two electrodes being either EDL or Faradaic capacitive (symmetrical) or both (asymmetrical). Symmetrical supercapacitors are devices comprising identical positive and negative electrode materials, with the same charge storage mechanism but the opposite ion movements or reactions in, for example, the carbon-based EDL capacitor. Asymmetrical supercapacitors are those with different positrode and negatrode materials but both are capable of capacitive charge storage, or with different positrode and negatrode energy storage mechanisms, or with the same materials in both electrodes but different mass loadings. A third configuration, known as a supercapattery (= supercapacitor + battery) comes naturally from combing a Nernstian (or battery-type) electrode with either an EDL or Faradaic capacitive electrode. Electrochemical characteristics of these three configurations are illustrated in Fig. 3.
Terminology-wise, a supercapattery is an asymmetrical or hybrid device, but it should not be called a “hybrid supercapacitor” (which is in fact the same as an asymmetrical supercapacitor). In terms of energy storage, supercapatteries are expected to combine the merits of EDL or Faradaic capacitive electrode materials and battery-type or Nernstian materials to provide both high specific capacity and high multiplicity performance, although it may not always be so in reality, making it crucially important to pair the electrode materials with matching properties and to design the electrodes and cell for optimal performances.
The construction of large EES devices including supercapacitors and supercapatteries utilising salt caverns necessitates the use of stable electrode materials and electrolytes. Electrode materials and aqueous electrolytes for salt cavern energy storage will be discussed in the following sub-sections, respectively.
2.2. Electrode materials
The electrochemical performance, cycling and storage stabilities, and manufacturing costs of supercapacitors are all significantly influenced by the choice of electrode materials. In recent decades, a variety of electrode materials, including carbon-based materials,61,62 transition metal oxides,54,58,63 hydroxides,62 sulfides, phosphides, and nitrides,64 and various electronically conducting polymers65,66 have been extensively investigated and employed in EES technologies. Typically, electrode materials for supercapacitors should offer high specific capacitance, a broad potential window, good conductivity and high electrochemical stability, and a large specific surface area, while also low resource and manufacturing costs.Carbon-based materials have been the subject of extensive investigation and concern in the field of supercapacitors due to their highly controllable porosity, good electrical conductivity and chemical stability, rich resources, and relatively low or affordable costs. Over the past decades, a range of carbon materials have been prepared and utilised as electrode materials, including activated carbon (AC), carbon nanotubes (CNTs), graphene, and carbon nano fibres (CNFs).52 In particular, they work very well for capacitive charge storage in various aqueous electrolytes with or without additives that, for example, improve water wettability on carbon surfaces.67 These attributes, particularly the affordable cost and high stability, offer promising potential for their application in salt cavern EES.
Among the various carbon-based charge storage materials, AC is the most widely used due to its favourable materials and performance characteristics as mentioned above. Limited by specific surface area, and electrolyte selection, the real capacitance can only reach 200–300 F g−1 due to the low efficiency of ions accessing the available active sites in the porous structure, which is mainly composed of microporous regions. The main focus of AC research is on the rational design of porous structures to achieve a higher specific surface area, whilst in most studies and capacitive storage applications, AC based symmetrical or asymmetrical supercapacitors use aqueous electrolytes.
It is well known that electric conductivity varies inversely against, but specific capacitance increases with, material porosity. In order to address the issues of lower electrical conductivity and specific capacitance associated with AC, a number of potential solutions have been put forth, including the utilisation of carbon materials with enhanced electrical conductivity or distinctive structural characteristics. Promising candidates include carbon nanotubes and graphene. The special hollow structure of CNTs could help the creation of a large number of charge transfer pathways, which would result in an enhanced energy or power density.68 Graphene, a representative 2D material, is known to be a very promising charge storage material in supercapacitors due to its excellent conductive and mechanical properties, and high specific surface area.62,69 Graphene could exhibit a very high specific capacitance because of its intrinsic double layer capacitive nature, and long cycling stability which may be attributed to its high flexibility. After modifying graphene by doping with other atoms such as boron, nitrogen and phosphorus, the charge storage performance of graphene can be improved because of the introduction of additional active reaction sites.
In addition to their use as EDL electrode materials, carbon-based materials have been widely used as a support to form composites with other electrochemically active materials. This approach can deliver significantly enhanced energy storage capacities and power capability due to the synergistic effect of, for example, structure and redox activity changes.70,71
Various candidates from the TMO group have been applied as the Faradaic capacitive electrodes in supercapacitors or as Nernstian (or battery type) electrodes in rechargeable batteries. As one of the earliest discovered metal oxides with excellent capacitive properties, RuO2 has been shown to produce rectangular CVs with typical Faradaic capacitive characteristics due to its stable structure, high conductivity, large specific capacitance and great reversibility during the dis-/charging processes.54 The unique nanostructures and redox activity of RuO2 enable highly reversible dis-/charging processes with a large specific capacitance in acidic aqueous electrolytes. Also, nanostructured RuO2 offers large specific surface areas and high conductivity to both electrons and protons. The measured high capacitance must have both Faradaic and EDL contributions. However, the construction of EES devices in salt caverns would be technically meaningful and more economical if neutral aqueous electrolytes are employed. In addition, the cost of RuO2 electrodes is too high for large scale applications.
The relatively inexpensive metal oxide, MnO2, has also attracted much attention due to its high theoretical specific capacitance (= nF/(ΔEM) = 1 × 96485/(1 × 87) = 1109 F g−1 where M is the formular mass), wide electrochemical window (ΔE = 1.0 V), and morphology tunability.72 MnO2 exhibits Faradaic capacitance with a rectangular CV due to reversible changes between the valances of Mn(IV) and Mn(III). The reduction of Mn(III) to Mn(II) is only partially reversible and should be avoided in capacitive charge storage. However, the inherently low to medium conductivity and the dis-/charging induced agglomeration of nanoparticles may impede the fast charge storage and stability of MnO2 as an electrode material.73 Recently, by growing MnO2 nanoparticles on mesoporous bowl-like carbon (MnO2/MBC),74 additional electroactive sites were introduced and the mechanical strength of MnO2 particles was enhanced. The asymmetrical supercapacitor assembled with a MnO2/MBC positrode and an AC negatrode in a water-in-salt (WIS) electrolyte could deliver a fairly high specific energy and power of 70.2 W h kg−1 and 700 W kg−1 with a high operational cell voltage of 2.8 V.
In addition, considerable research has been conducted into other metal oxides. For example, V2O5,75 which exhibits multiple oxidation valences (V, IV, III and II) with a layered crystal structure, could deliver optimal energy storage performance in aqueous electrolytes by constructing a unique structure to overcome the instability and low electron conductivity associated with conventional materials. Another TMO example is the low cost Fe2O3,76 which has a theoretical specific capacitance of 1208 F g−1 (= 2 × 96485/(1 × 159.7)) for the reduction of Fe(III) to Fe(II) between 0 and −1.0 V vs. Ag/AgCl. It is a suitable negatrode material in supercapacitors after nanostructure design and compositing with conductive additives to achieve a high capacitance and long cycle life. In the literature, reported specific capacitances ranged from 100 to over 1000 F g−1 depending on structures.
In addition to oxides, metal sulphides, phosphides and nitrides are also being investigated as supercapacitor electrode materials because of their metal-like electrical conductivity and interesting redox properties.77–80 It is worth pointing out that not all redox active metal compounds, particularly oxides and sulphides, can offer Faradaic capacitance, but studies on such materials often report misleadingly high specific capacitances. For example, a high value of 1095 F g at 3 A g−1 was claimed for a composite of Fe2O3/graphene, but the electrochemical features as reported were all Nernstian in nature, i.e. current peaks on CVs and potential plateaus on GCDs.81
Electronically conducting polymers (ECPs) are another important category of Faradaic capacitive materials, offering enhanced suitability for next generation flexible and wearable energy storage devices.66,82 For instance, polyaniline (PANi), polypyrrole (PPy), polythiophene (PTh), and their derivatives have been investigated as charge storage materials through Faradaic capacitance and de-/doping of ions. For capacitive storage, ECPs and their composites with EDL or other Faradaic capacitive materials can offer multiple advantages, including low density, good processability, low cost, and large charge capacity via reversible redox reactions. ECPs can be doped by oxidation or reduction processes, allowing for the control of conductivity through the manipulation of doping levels. Representative ECPs such as PANi and PPy, are capable of operating in aqueous electrolytes and exhibit high specific capacitances (200–500 F g−1).83,84 Additionally, ECPs have been composited with other polymers or inorganic materials to improve their charge storage performance.82
It is interesting to note that the concept of and commercial interests in supercapacitors appeared earlier in the literature than lithium-ion batteries (LIBs) but the latter gained more research attention and commercial development in the past three decades.85,86 The reasons are mainly due to LIBs having a much greater energy density or specific energy which is determined by two main factors: cell voltage and electrode charge capacity. In theory, the cell voltage of supercapacitors can be the same as the electrochemical stability windows (ESWs) of the electrolytes. However, the cell voltages of LIBs are limited by the potential differences between the positive and negative electrodes which are usually narrower than the ESWs of the electrolytes. This understanding has invoked early research efforts in searching for supercapacitor electrode materials with large specific charge capacities. Fast growing research followed particularly the discovery of RuO2 being capable of dis-/charging in a capacitive manner,54 leading to the development of a large family of unique redox active materials, i.e. Faradaic capacitive materials such as TMOs and ECPs. Such materials offer reversible electrochemical capacitive performances, differing from that of battery electrodes in accordance with Nernst's law.
It should be pointed out that the specific charge capacities of both Faradaic capacitive and Nernstian materials are very much comparable because of their redox origins and are much higher than those of carbonaceous materials that store charges in their EDLs. However, when used alone at a relatively high loading (e.g. >10 mg cm−2), redox active materials become more resistive to both the electron and ion conductions. Particularly, the ingression or egression of ions in the redox material become diffusion controlled, limiting the dis-/charging rates and hence the power capability.
A pioneering effort to enhance electron and ion conduction inside electrode materials is to distribute the redox active materials as a thin coating on individual carbon nanotubes (CNTs).70,71,87–89 As shown in Fig. 4a–c, individually coated CNTs with polypyrrole (PPy) or MnO2 were successfully prepared. These CNTs not only functioned as pathways for electron conduction, but also enabled the formation of micro and macro pores for ion transport. Electrochemical analyses in aqueous chloride electrolytes revealed significantly enhanced charge storage capacities of these composites in comparison with their parent components as exemplified in Fig. 4d. Furthermore, these CNT based composites present consistent dis-/charging performances at increasing areal loadings beyond 30 mg cm−2 without invoking diffusion control as evidenced in Fig. 4e and f.70 A record areal capacitance (>5 F cm−2) was claimed for the CNT-65 wt% MnO2 composite in aqueous KCl (2 mol L−1), whilst the loss in capacitance after 1000 dis-/charging cycles was less than 5%.
 | ||
| Fig. 4 TEM images of carbon nanotubes (CNTs) coated with (a) polypyrrole (CNT-PPy), reproduced from ref. 71 with permission from John Wiley and Sons, copyright 2000, and (b) manganese dioxide (CNT-MnO2), reproduced from ref. 70 with permission from John Wiley and Sons, copyright 2007. The HRTEM image in (c)71 confirms the enclosure of individual CNTs by uniform PPy coatings. Cyclic voltammograms were obtained from (d) pure PPy and CNT-PPy of the same deposition charge in aqueous 0.5 mol L−1 KCl,88 and (e) CNT-MnO2 in aqueous 2.0 mol L−1 KCl at different overall loadings which are translated to the respective areal loadings in (f).70 Reproduced from ref. 71 with permission from John Wiley and Sons, copyright 2000, from ref. 88 with permission from Elsevier, copyright 2007, and from ref. 70 with permission from John Wiley and Sons, copyright 2007. | ||
A key step in making composites with well dispersed CNTs is to firstly disperse the highly entangled CNTs (commercial products from the chemical vapour deposition method) in aqueous solutions. It was most effectively achieved in the laboratory via partial oxidation of the commercial CNTs in strong sulfonitric acids (mixture of concentrated H2SO4 and HNO3).71,87,88 Partial oxidation of CNTs results in the formation of oxygen containing groups (OCGs) on the surface of CNTs that become anionic in neutral aqueous solutions. Therefore, such a suspension of CNTs was directly used in electro-deposition of ECPs without any additional electrolyte salt. However, this sulfonitric process emits nitrogen oxides (NOx) and is not suitable for commercial scale-up. In a recent study, by replacing nitric acid with hydrogen peroxide (H2O2), a novel mechano-Fenton-Piranha oxidation process was demonstrated, capable of dispersing commercial CNTs in water without any emission of NOx. Such produced CNTs were dispersed well in water for making CNT-PPy composites without compromising the desirable specific capacitance.90
Not all monomers can dissolve in water to a sufficiently high concentration for polymerisation. A good example is poly[3,4-ethylene-dioxythiophene] (PEDOT) whose monomer can be electro-polymerised into stable films of high performance on various electrode surfaces, e.g. graphite and platinum, in an acetonitrile solution of 0.5 mol L−1 LiClO4, for example. An attempt91 to mix the acetonitrile solution of the monomer with an aqueous suspension of well dispersed CNTs led to an organoaqueous emulsion that remained stable for hours. This metastability enabled successful electro-deposition of the CNT-PEDOT composites whose surface morphology showed interesting crater-like features as shown in Fig. 5a–c, apparently resulting from the drying of the emulsion. Fig. 5c further reveals that the CNTs were assembled in the banks of craters, implying their function as the stabiliser (similar to surfactant) of the emulsion at the interfaces between the organic and aqueous phases. Again, at the same deposition charge, the CNT-PEDOT outperformed the PEDOT in charge storage capacity, although both exhibited comparable cycling stabilities.88
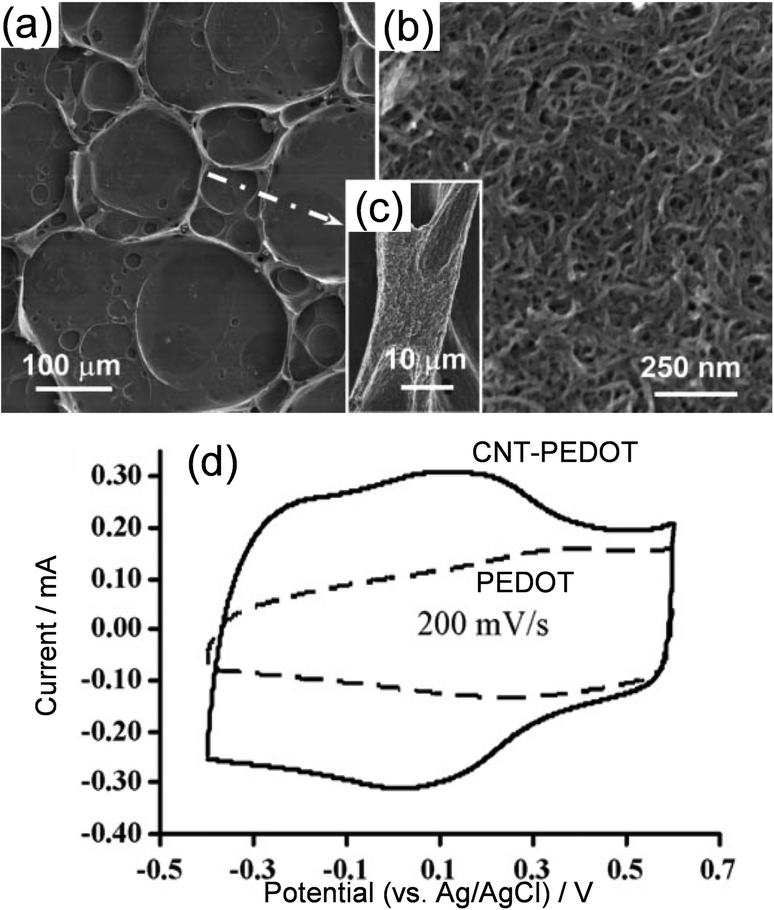 | ||
| Fig. 5 SEM images of electro-deposited CNT-PEDOT films from a CNT-assisted metastable emulsion, showing (a) interconnected craters of various sizes with enlarged views of (b) the base and (c) the bank of the crater. Reproduced from ref. 91 with permission from Royal Society of Chemistry, copyright 2006. (d) Cyclic voltammograms of pure PEDOT and CNT-PEODY of the same deposition charge in aqueous 0.5 mol L−1 KCl. Reproduced from ref. 88 with permission from Elsevier, copyright 2007. | ||
The electro-co-deposition method discussed above is effective in operation and control of product quality, but it requires the dispersion of the CNTs in the electrolyte which is not aways convenient and cost effective. Alternatively, it is possible to firstly anchor the CNTs or other high surface area and conducting materials such as activated carbon and graphenes that are capable of forming a suitable porous structure on the electrode surface. Then, the redox material is electro-deposited onto the modified electrode surface.92–95 Fig. 6 shows an example of direct electro-deposition of PPy onto an array of vertically aligned CNTs which were grown onto a quartz plate via chemical vapour deposition and then transferred to the surface of gold-coated Mylar foil with the aid of silver paint.94 It can be seen that the PPy coating was uniform on individual CNTs, as evidenced by the insert showing a naked portion of the CNT. Clearly, both SEM (inset in Fig. 6a) and TEM (Fig. 6b) studies demonstrated the PPy coating was much thicker than that revealed in Fig. 4c. This is not surprising because the direct electro-deposition on the vertically aligned CNTs proceeded until a deposition charge of 40 C cm−2. Under similar conditions, directly deposited pure PPy on the electrode showed a capacitance plateau at deposition charges larger than 10 C cm−2, suggesting the deposit was becoming resistive to conduction of either or both ions and electrons. However, when the deposition charge was further increased beyond 40 C cm−2, the channels between the CNTs were blocked by the overgrown PPy.
 | ||
| Fig. 6 (a) SEM images of vertically aligned CNTs coated with PPy via direct electro-deposition (deposition charge: 40 C cm−2). The inset is an enlarged view showing the uncoated portions of the CNTs. (b) A single CNT with the left portion being coated with PPy and the right portion being naked. (c) Areal capacitance of PPy coated vertically aligned CNTs (triangles) and that of pure PPy (diamonds) as a function of deposition charge. Reproduced from ref. 92 with permission from John Wiley and Sons, copyright 2002. | ||
It is worth mentioning that the examples discussed above have been selected for their excellent performances in aqueous electrolytes. Accordingly, in the construction of EES in salt caverns, it is possible to select conventional carbon-based materials or metal oxides or conductive polymers as electrode materials. However, it is essential to give more comprehensive consideration to the electrolyte and the cost implications.
2.3. Electrolytes
It is widely overinterpreted that the thermodynamic decomposition voltage of water (1.23 V at room temperature) is a limiting factor of the cell voltage of aqueous EES devices and hence results in a low energy density. An exemplar fact against this overinterpretation is that the very common lead-acid battery operates at a cell voltage over 2.1 V without invoking any problematic water decomposition. In more general practices, there are several strategies that can be employed to expand the electrochemical window of aqueous electrolytes. Firstly, an extra overpotential is required on the electrodes (e.g. carbon and lead) against the oxygen/hydrogen evolution reactions. Secondly, the interaction between the solvent and solute ions can also be enhanced, such as in the case of water-in-salt, so that there is little or no free water molecules in the electrolyte to undertake electrode reactions, impeding effectively water decomposition. Last, but not least, it has been demonstrated that with an ion conducting membrane (separator), the electrolyte on the negatrode side can be made highly alkaline to minimise hydrogen evolution, whilst using a strong acidic electrolyte on the positrode side to push the oxygen reaction to more positive potentials. Consequently, by using electrodes with high overpotentials for the HER and OER, separating the HER and OER with an ion conducting membrane, and regulating the aqueous electrolyte on concentration, pH value, or additives, high voltage (≥2.0 V) aqueous EES devices can be achieved to assist applications in salt caverns.
Another thermodynamic prediction of water electrolysis is that variation of the pH of an aqueous electrolyte does not affect the water decomposition voltage. In accordance with the Nernst equation, the pH value of the electrolyte exerts a modulating influence on the potentials of the negatrode for the hydrogen evolution reaction (HER) and positrode for the oxygen evolution reaction (OER). The thermodynamic potentials of the OER and HER can be calculated according to eqn (3) and (4).102–104 Because the decomposition (or cell) voltage is the difference between the OER potential and HER potential, the pH influence is cancelled.
| φOER = 1.23 − 0.0591 pH | (3) |
| φHER = −0.0591 pH | (4) |
However, the kinetics for the HER and OER are very different, which can be further distorted by using electrode materials that favour differently towards the HER and OER at different pH values, leading to kinetically increased water decomposition voltage.
Indeed, it has been demonstrated that a considerable number of electrochemical reactions or processes in aqueous electrolytes are markedly affected by the pH value of the electrolyte, including water electrolysis.102,105 The stable potential window of an EDL capacitive carbon-based electrode, for example, is usually 0.8 V in acidic or alkaline electrolytes, while it can be extended to more than 1.4 V in neutral electrolytes.106
An interesting and useful observation of water reduction on an AC electrode is that when the potential reaches at or slightly beyond the overpotential for the HER, e.g. −1.23 V vs. H+/H2, the local water molecules at the carbon|electrolyte interface undergo partial reduction and form nascent hydrogen atoms which are then trapped inside the micropores of the AC material, partly assisted by electrochemical adsorption. If there was sufficient activation energy, the adsorbed hydrogen atoms would further react to form hydrogen molecules. Because the trapped hydrogen atoms can be re-oxidised at more positive potentials, by controlling the potential, it is possible to utilise the nascent hydrogen atoms for additional charge storage.
Given that HER and OER processes in neutral electrolyte result in low concentrations of H+ and OH−, the local reduction of water molecules to free OH− causes a local pH increase, thereby shifting the hydrogen generation potential further in the negative direction. Consequently, a higher applied voltage is required to drive the HER, and this difference is referred to as the overpotential. As a result, the generated hydrogen binds more readily to the porous carbon surface rather than hydrogen evolution. Therefore, the electrochemical window of the aqueous electrolyte is expanded.
Abbas et al.106 investigated the HER response of AC electrodes by studying the HER in aqueous electrolytes of 1 M Li2SO4 (pH = 6.5) and 1 M BeSO4 (pH = 2.1), as illustrated in Fig. 7a. It was found that in the neutral electrolyte, due to water reduction, a small amount of OH− was produced, and thus a local pH value increase resulted in a higher overpotential for dihydrogen formation. Subsequently, the current oscillation initiated by hydrogen evolution started from −0.8 V vs. SHE. In contrast, the process had no such impact in the acidic electrolyte, where the hydrogen generation occurred at −0.3 V vs. SHE, which is close to the thermodynamic reduction potential of water. Consequently, there is a notable distinction in the overpotentials in acidic and neutral aqueous electrolytes.
 | ||
| Fig. 7 (a) CVs of AC at 2 mV s−1 in 1 M Li2SO4 with pH = 6.5 (left) and in 1 M BeSO4 with pH = 2.1 (right). Reproduced from ref. 106 with permission from IOP Publishing, copyright 2015. (b) The HER and OER potentials and electrochemical window of WIS electrolytes. (c) Illustration of the Li+ solvation sheath in diluted and water-in-salt solutions. Reproduced from ref. 108 with permission from Springer Nature, copyright 2013. (d) Schematic of the capacitive and Faradaic charge storage processes with the redox couple in the electrolyte. (e) Reduction potentials of the indicated redox couples (BV, benzyl viologen; EV, ethyl viologen; HV, heptyl viologen; MV, methyl viologen). Reproduced from ref. 114 with permission from Springer Nature, copyright 2015. | ||
Furthermore, the utilisation of a cation exchange membrane serves to segregate the positrode and negatrode, thereby extending the operational voltage range.107 However, this approach is more complex. In the context of salt caverns, neutral electrolytes are more likely to exist stably, thereby enabling the achievement of an extended working voltage. On the one hand, the comprehensive regulation of the electrolyte concentration and composition is essential to ensure the stability of the electrochemical energy storage process and more importantly to prevent the secondary dissolution of the rock salt on the walls of the salt cavern if it is used directly to contain the aqueous electrolyte. The latter will be further discussed in later sections.
In addition, the ultra-high Li+ ion concentration leads to the formation of an anion-containing solvation sheath, which results in strong coordination between water molecules and Li+ ions (see Fig. 7c). Consequently, the majority of water molecules in the electrolyte are constrained around the Li+ ion solvent layer, and thereby are markedly reduced in their electrochemical activity. Furthermore, the reduction potential of TFSI− is more positive than that of hydrogen generation, according to the density functional theory. Therefore, TFSI− will be reduced prior to the occurrence of hydrogen evolution, resulting in the formation of a LiF passivation layer on the electrode surface. This phenomenon leads to a more negative potential for HER. Subsequently, this electrolyte was employed to construct an aqueous lithium-ion full battery, in conjunction with Mo6S8 as the negatrode and LiMn2O4 as the positrode, which demonstrated a voltage of 2.3 V and exhibited a stable capacity for 1000 cycles.109
Subsequently, WIS electrolytes were also employed extensively in supercapacitors, which could enhance the voltage and energy density of the devices. For example, symmetrical supercapacitors comprising AC electrodes and LiTFSI as the electrolyte offered a wide voltage window of 2.4 V and a high specific energy of 24 W h kg−1.110 In light of the considerably expensive LiTFSI salt, water-in-salt electrolytes with alternative salts also begin to emerge. For instance, Bu et al. developed a water-in-salt electrolyte of 17 M NaClO4 and designed supercapacitors capable of operating at 2.3 V and delivering a specific energy of 23.7 W h kg−1, which further reduced the cost of the water-in-salt electrolyte.111 Subsequently, the research group also proposed NaNO3 as the salt with a concentration of 12 M to widen the electrochemical window to 2.56 V, demonstrating improved energy storage characteristics in symmetrical supercapacitors based on commercial AC.112
In order to further reduce the free water content in WIS, researchers have proposed the utilisation of water-in-salt electrolytes with bimetallic salts to broaden the electrochemical window. For example, a new ultra-high concentration water-in-salt electrolyte consisting of LiTFSI (21 M) and additional lithium trifluoromethane sulfonate (LiTOf, 7 M) demonstrated an electrochemical window of up to 3.1 V as a result of the further reduction of reaction activity of water and a more compact and efficient LiF layer.113
However, water-in-salt electrolytes also possess certain intrinsic drawbacks, including high viscosity and low conductivity, due to the strong coordination between anions and cations. These characteristics affect the electrochemical stability and multiplicity performance of supercapacitors. In addition, solute precipitation occurs at low temperatures, which is a significant concern when the electrolyte is applied in salt caverns, where temperature varies in the huge space and the concentration of the salt in WIS may be challenging to stabilise over time.
Researchers discovered that by incorporating a modest quantity of organic solvents into the WIS electrolyte as a co-solvent, they could leverage the characteristics of organic solvents such as a low freezing point and low viscosity, to improve the conductivity and low-temperature stability, and further broaden the electrochemical window of the mixed electrolyte.115,116 One example is the addition of acetonitrile to the WIS solution of LiTFSI (21 M), which can reduce the interactions between anions and cations and promote the ion transport.116 The hybrid WIS electrolyte could improve the cycling performance of the supercapacitor to 14000 cycles at the operation volage of 2.2 V. Furthermore, the electrochemical window of acetonitrile containing WIS of NaClO4 (8 M) can be extended to 3.16 V.117
Although water-in-salt electrolytes can significantly address the issue of the limited potential window of water-based electrolytes, their high costs, low temperature instability, and low conductivity present significant challenges which must be overcome to further develop their application in the construction of large-scale EES devices in salt caverns. As ion transport efficiency is an important factor influencing the energy storage characteristics of large-scale EES devices, any measure that can increase ion transport efficiency is worth investigation. If the electrolyte may underperform against design and expectation, further consideration of device structure optimisation is necessary. Additionally, the potential impact of filling up salt caverns with the WIS electrolyte on the environment requires further evaluation.
The introduction of redox-active additives into the electrolyte enables the generation of supplement Faradaic capacitance or capacity and an enhancement of the charge storage capacity of the supercapacitor.114,118–120 In addition, the selection of additives with redox potentials in proximity to the HER and OER potentials can also effectively protect water from decomposition, while simultaneously widen the potential window to a certain extent. The selected redox-active additives are typically highly soluble, ensuring the Faradaic contributions. They can undergo oxidation and reduction at the positrode and negatrode, respectively. It is also important to ensure that the respective redox potentials should reach the limit of the OER and HER in order to circumvent the shuttle effect and self-discharge, as shown in Fig. 7d. Reduction potentials of various redox couples considered relative to the thermodynamic stability window of water at neutral pH can be seen in Fig. 7e. For example, by adding 0.1 M Fe(CN)63−/Fe(CN)64− to a 1 M Na2SO4 electrolyte, a wide operating potential of 2 V was achieved for symmetrical supercapacitors based on AC.121
In conclusion, although there is a vast array of potential electrolytes, aqueous electrolytes are most suitable for large-scale EES in salt caverns. Furthermore, a neutral electrolyte is preferable as it can remain stable when in contact with the salt-cavern walls. In order to achieve higher operating voltages and hence energy densities of EES, it is necessary to modulate and modify the aqueous electrolyte by utilising WIS, tuning the pH value, and using redox-active additives in conjunction with electrode materials with high overpotentials for the HER and OER.
3. Perspective: salt cavern supercapacitors and supercapatteries
3.1. Advantages of supercapacitors and supercapatteries in salt caverns
It is anticipated that salt caverns can be employed as liquid electrolyte containers for supercapacitors and supercapatteries, given their distinctive physicochemical attributes. These include favourable rheological properties, low porosity, low permeability, self-healing capability and plasticity. Salt caverns are typically of considerable magnitude in terms of size, with depths extending from hundreds to thousands of metres and volumes reaching beyond 1 × 1013 L (10 tera-litres).19 This huge volume can be translated to an overall energy capacity of 1014 W h = 100 TW h (terawatt-hour) and power capability of 1014 kW = 100 PW (petawatt) based on the very moderate energy density (ca. 10 W h L−1) and power density (ca. 10 kW L−1) of AC (activated carbon) based symmetrical supercapacitors with aqueous electrolytes (AC‖AC).127A drawback of AC‖AC supercapacitors is their vulnerability to self-discharging that impacts the shelf-life of the stored charge (or charge retention capability). This problem can be much improved in supercapatteries. For example, a supercapattery consisting of a zinc metal negatrode and an AC positrode (Zn‖AC), which is also known as a zinc ion capacitor,128 was tested in different aqueous electrolytes. The specific energy and power were around 100 W h kg−1 and 1000 W kg−1.129 These values correspond to energy and power densities of 120 W h L−1 and 1200 W L−1, respectively, assuming an apparent density of 1.2 kg L−1 for the aqueous device. More importantly, the charge retention was highly satisfactory in a test time of 500 h. Therefore, using this Zn‖AC supercapattery can lead to at least a ten-fold increase in energy density without compromising the power density too much.
Although the AC‖AC supercapacitor may remain favourable, considering that repeated electro-deposition and -dissolution of Zn metal still suffer from dendrite formation,130 there has been continuous research studies looking for solutions to self-discharging in supercapacitors such as using an ion conducting but solvent impermeable membrane to separate the positrode and negatrode.131 The impact of the dendritic deposition of Zn may also be effectively minimised by using, for example, a properly structured porous carbon negatrode substrate.130
Nevertheless, the above simple but promising estimations and the technical issues with EES in salt caverns warrant further considerations, such as materials and engineering, before more efforts toward feasibility verification and demonstration, and also commercial and environmental gains. Also bearing in mind that because of their ultra large volumes, salt caverns are well-suited to either accommodate substantial quantities of electrolyte and/or facilitate the construction of large-scale electrochemical cells that can be stacked via bipolar plates.132,133 Furthermore, the confinement and structural stability of salt caverns provide the foundation for their long-term reliability for EES. The extensive geographic distribution of salt caverns enables the construction of salt caverns for EES in proximity to large-scale renewable energy power plants, such as on and off shore photovoltaic or wind power farms, for the storage of the energy generated from these intermittent renewable sources and to balance the power grid at peak and valley times.
In the following sections, two potential scenarios will be described, analysed and discussed: (1) utilisation of the brine water from solution mining, after necessary processing (cleaning), as the electrolyte, and (2) using electrolytes that have zero or insignificant interaction with the wall of the salt cavern to build the EES devices inside the salt cavern. Apparently, scenario (1) would be low cost but also have a low energy density, and be suitable for large scale energy storage at GW h to TW h levels, whilst scenario (2) would be more expensive with high energy density and could be applied to geological conditions that do not allow for the construction of stable and ultra large salt caverns, but the high energy density should still enable energy storage to the MWh levels.
3.2. Electrolyte selection
Preliminary experimental studies that have been conducted thus far have focused on the design and construction of redox flow batteries (RFBs) connected to salt caverns.134 Similarly, the construction of supercapacitors and supercapatteries in salt caverns can be carried out in a series of device designs by combining the special properties of salt caverns.It is recommended that an aqueous electrolyte should be used to construct supercapacitors and supercapatteries in salt caverns. Utilisation of the brine from solution mining as the electrolyte is an attractive low cost and environment benign approach. The composition of salt rocks is dominated by NaCl (>95 wt%) resulting originally from natural drying of seawater, including that from inland salty lakes. The other ionic impurities include Mg2+, Ca2+, K+ and Li+ (cations) and SO42− and Br− (anions). These ions, except for Br−, are electrochemically inert in water and hence should benefit EDL capacitive charge storage. The Br− anion can undergo reversible oxidation to form the tribromide ion, Br3−. Both Br− and Br3− can be electro-adsorbed onto the internal walls of a porous AC positrode, and hence contribute to both capacitive and Faradaic charge storage.135 Unfortunately, because the content of Br− is about 3 orders of magnitude smaller than that of Cl−, its influence is likely negligible.136 Another point about the extracted brine is its concentration. It is unlikely that the brine as extracted is always saturated and hence should not be used directly as the electrolyte can attack (dissolve) the walls of salt cavern. Thus, further processing of the brine, such as removal of unwanted components and additions of NaCl to the saturation concentration and other functional agent, is necessary before injection to the supercapacitor or supercapattery.
The solubility of NaCl in water is about 5.41 M (360 g L−1) at room temperature which is not high enough to form a WIS solution, but is sufficiently high for supercapacitor application. It was reported that AC electrodes behaved in a highly capacitive way in neutral aqueous electrolytes of alkali chlorides (3 M), giving rise to a potential window wider than 1.8 V.127 Interestingly, such a wide potential window was not achieved in conventional symmetrical supercapacitors, but when the positrode to negatrode capacitance ratio was increased, the cell voltage could reach 1.8 V and beyond.61,137
In order to obtain a wider voltage window, apart from organic electrolytes and ionic liquids, WIS represents a favourable alternative. The construction of symmetrical supercapacitors utilising WIS as the electrolyte in AC‖AC supercapacitors offers a straightforward and convenient approach to the development of salt cavern supercapacitors. As mentioned above, because NaCl is unable to form a WIS solution, another salt has to be used, such as LiTFSI. Although WIS solutions have ultrahigh concentrations, in the absence of Na+ and Cl− ions, they could highly likely interact with the walls of the salt cavern until they become saturated with NaCl. Such interactions may be avoided or minimised if the WIS solution is pre-saturated with NaCl before injection into the supercapacitor. This strategy may be applied to other non-aqueous electrolytes. Therefore, the saturated NaCl solution with other conceivable additives could be applied in salt caverns for long time running. Besides, periodic addition of NaCl to the electrolyte may also be effective in extending the stability and service life of salt caverns. Combining stability and cost considerations, saturated NaCl solution is the most suitable electrolyte for salt caverns. Through co-salt and/or co-solvent strategies, low-cost NaCl-based electrolytes could be designed with widened voltage windows and high stability in the salt cavern environment.
Considering the elevated cost of most WIS electrolytes, the selection of low-cost salts, such as sodium acetate (CH3COONa), sodium nitrate (NaNO3) and sodium perchlorate (NaClO4),111,117,138,139 can be considered as electrolyte salts for WIS. In addition, the temperature in the salt caverns is typically different from the room temperature, and the potential negative effects of high viscosity and low conductivity of WIS should be taken into account. Similar considerations may be applied to those non-aqueous electrolytes. It should also be pointed out that using WIS and non-aqueous electrolytes requires careful and effective measures on all potential hazards against health, safety and environment.
3.3. Device design
Unlike the storage of gases or liquids, the construction of a large scale supercapacitor or other EES devices in a salt cavern likely requires machines and even humans to work inside the cavern and also the necessary transport of materials and engineering parts for assembly and installation purposes. This means at least one of the vertical wells used for solution mining needs to be widened for such access. Because salt cavern construction is beyond the scope of this article, the following discussion will be focused on the electrochemical aspects.A basic and common technical feature of supercapacitors and supercapatteries is that they can be fully discharged to zero volt, at least for analytical purposes. This would be destructive to rechargeable batteries whose minimum working voltage is determined by the Nernstian reactions on the two electrodes. This is largely because capacitive electrodes, either EDL or Faradaic, are capable of storing charge in a much wider potential range than a Nernstian electrode. Furthermore, the capacitive potential range (CPR) of the positrode can overlap, at least partly, with that of the negatrode and vice versa. In principle, the amount of charge (and also the current) must be the same between the positrode and negatrode in any electrochemical cell. For a capacitive electrode whose capacitance is C, its charge capacity, Q, is proportional to the potential range, U, i.e. Q = CU (U ≤ CPR).137,140,141
Thus, supercapacitors and supercapatteries can maximise their cell voltage via unequalisation of the capacitance or mass of positrode to negatrode. Combining with the overpotentials of the hydrogen evolution reaction (HER) and/or oxygen evolution reaction (OER) on some electrode materials, such as carbon, lead and zinc, the cell voltage of an aqueous supercapacitor and supercapattery can be extended well beyond the thermodynamic decomposition voltage of water.
Obviously, this approach should also increase the energy capacity of the cell. For example, as shown in Fig. 8, application of this unequalisation principle to an aqueous supercapacitor of “(+) polyaniline/carbon nanotubes‖carbon (−)” led to an over 80% increase in specific energy.137 Also, at the positrode to negatrode capacitance (mass) ratio of 4:3, the voltage of a “carbon‖carbon” supercapacitor was extended beyond 1.9 V in aqueous K2SO4, accompanied by an increase of 38% in energy capacity.61 It is worth noting that this unequalisation principle requires no change in materials selection and hence incurs zero or low additional cost, which should be applicable to the construction of both aqueous and non-aqueous supercapacitors and supercapatteries. Because Nernstian reactions occur in relatively narrow potential ranges, this unequalisation principle should have a little or limited influence on rechargeable battery.
 | ||
| Fig. 8 (a) CVs and (b) the specific energy of the cell of “polyaniline/carbon nanotubes|1.0 M HCl|carbon” at the indicated positrode to negatrode capacitance ratios. Reproduced from ref. 137 with permission from RSC Publishing, copyright 2010. | ||
Although salt caverns have been proposed for EES with the RFB design, the caverns are only used as containers for separate storage of the charged and discharged redox electrolytes. The electrochemical cells are built above the ground. Salt cavern EES is in principle capable of approaching the GW h to TW h levels, and the working voltage of individual electrochemical cells is still too small for electrical engineering. The conventional solution to serially stack multiple cells is to achieve a high overall voltage. While external serial connection of individual cells is widely used in the range from kW h to MW h, internal stacking via bipolar plates (electrodes) can be advantageous. For example, when stacking 1000 cells, for external connection, the number of electrodes is 2000, but for bipolar connection, only 1001 electrodes are needed. Furthermore, the external connection requires additional connectors (cables) whose total weight and resistance are not negligible, in addition to the cost of connectors and assembling operation.
Laboratory attempts were successful in stacking asymmetrical supercapacitor cells of “AC|3 M KCl|PPy-CNTs” with titanium bipolar plates.133 For building stacked supercapacitors with bipolar plates and cleaned brine as the electrolyte, it is necessary to utilise the walls of the salt cavern as the container to seal every individual cell serially connected via the bipolar plates.
Fig. 9 shows schematically the design of a laboratory simulation of the salt cavern supercapattery, including five bipolarly connected (−) Zn|brine + ZnCl2|AC (+) cells, utilising the brine from solution mining of the salt cavern as the electrolyte. The brine as produced may need necessary cleaning to remove the undesirable components. The positrode is made from AC with necessary additives such as binder and conductive agents. The negatrode could use appropriately structured cloth (e.g. woven carbon fibres) to offer a sufficiently high specific area that is completely or mostly accessible for electro-deposition of Zn so that dendrite formation is avoided. The separator can be a porous membrane such as a glass-fibre felt or a Celgard product,142 or an ion conducting membrane such as one of those Na+ ion conducting Nafion materials.143
 | ||
| Fig. 9 Laboratory simulated salt cavern supercapattery stack with bipolarly connected 5 Zn‖AC cells. (a) Cross section view of the repeating bipolar plate (electrode) assembly in the bipolar stack. (b) Cross section and (c) top views of the bipolar stack (HDPE: High density polyethylene). | ||
In Fig. 9, the design sketches of a simulated salt cavern are presented. In the laboratory, this can be prepared by melting and solidifying NaCl in a stainless steel container with or without an alumina liner (coating) on the internal walls of the container, and then using a water jet to make the central space. Also, holes and spaces are provided above and asides the stacked cells for electrolyte spill or overflow because charging and discharging of supercapacitors and supercapatteries involve ingression and egression of hydrated ions, leading to variation in electrolyte volume. The vertical pipes for brine addition and extraction are for regular electrolyte cleaning and replacement. The bottom plate is used to hold the bipolar assembly in place during installation, and also to ensure good physical contacts between neighbouring bipolar assemblies in operation.
Assuming the working voltage range of each cell to be 0.5 to 1.5 V, to output a voltage of, for example, 115 kV for connection to the grid, 77000 cells are needed (or slightly more to counterbalance the voltage loss due to internal resistance, i.e. the equivalent serial resistance (ESR). Obviously, for supply security against accidentally malfunctioning individual cells, it is unwise to connect these many cells directly via bipolar plates. Instead, properly designed external serial and parallel connections of a certain number of smaller stacks or modules would be a better engineering and more secured option.
Obviously, it is also possible to use a power electronic converter to interface between the low DC voltage of the salt cavern supercapattery and the ultrahigh AC voltage of the power grid. Thus, internal parallel connection of individual cells remains an option which is also easier for engineering and installation because there is no need to seal individual cells from each other. Fig. 10 shows a laboratory simulation of a salt cavern supercapattery stack of five cells in parallel connection. Both bipolar and parallel stacks of the same number of cells should in principle offer the same energy storage capacity, but there are three main differences between them.
 | ||
| Fig. 10 Laboratory simulated salt cavern supercapattery stack with 5 Zn‖AC cells in parallel connection. (a) Cross section view of the repeating parallel plates (electrodes) assembly. (b) Cross section and (c) top views of the parallel stack. HDPE: High density polyethylene. | ||
Firstly, the maximum working voltage of the bipolar stack is five times that of the parallel stack, whilst the latter can output a maximum current five times greater than the former. Also, all individual cells must be physically sealed from each other in the bipolar stack, but the cells are all open and share the same electrolyte in the parallel stack. Last, but may not be the least, installation of the parallel stack needs minimal work on the walls of the salt cavern, whilst the bipolar stack as shown in Fig. 9 requires careful engineering procedures to ensure the close match between the bipolar plates and the salt cavern walls.
To obtain an overall working voltage reasonably higher than that of a single cell, e.g. 110 V vs. 1.8 V, it is necessary to apply serial connections externally to multiple parallel stacks. One option is to construct 65 (instead of 62 considering voltage loss through external connections) small salt caverns with each accommodating one 1.8 V parallel stack. Alternatively, the 65 parallel stacks can be all installed in a large salt cavern in which 65 pools are formed in the floor to immerse a parallel stack in each pool. Obviously, the latter option requires humans working in the large salt cavern in the course of construction and installation. This may be advantageous for maintenance and repair.
3.4. Other considerations
The construction of asymmetrical supercapacitors, such as EDL combined with Faradaic capacitive electrodes, can optimise the positrode and negatrode matching, thereby effectively improving the specific capacity of supercapacitors. Alternatively, capacitive electrodes can be paired with battery electrodes to construct a hybrid energy storage device, namely supercapattery, which can demonstrate both the improved power capability and energy capacity. Nevertheless, when selecting electrodes, particular attention needs to be paid to matching the potential ranges of the positrode with those of the negatrode, and the capacitance or weight ratio of the two electrodes. Additionally, due to the large-scale characteristics of salt cavern energy storage, it is also necessary to consider the cost of electrode materials, and the issues associated with the preparation of large-scale electrode materials.A further crucial aspect of the construction of supercapacitors and supercapatteries utilising salt caverns is the optimisation of electrode material design and the matching of these materials with electrolytes. The structural design of electrode materials, including nano and porous materials can enhance the number of active sites and improve charge storage efficiency. Additionally, interface engineering and elemental doping can further optimise the charge storage properties of electrode materials. It is recommended that different optimisation designs or composite designs be carried out for different types of electrode materials. For example, carbon-based materials have a relatively wide range of electrolyte matching, whereas battery-type electrode materials have more specific requirements for electrolytes. This necessitates a comprehensive evaluation of several aspects, including the choice of a wide voltage window and electrode material matching. In line with this consideration, it is worth considering using organic electrolytes and ionic liquids in salt cavern EES devices, such as that shown in Fig. 9. This thought is derived from the fact that, as mentioned at the end of Section 1.3.5, salt caverns are already used for storage of crude oil and petroleum, which means a great stability of the salt cavern walls towards the interactions with hydrocarbons. It is acknowledged that organic solvent molecules for electrolyte making are mostly polar to assist dissolution of salts. Although the solubility of NaCl in such solvents is expected to be insignificant, it is still worth saturating the organic electrolytes with NaCl before injecting them into the salt cavern supercapattery or supercapacitor.
The volume of the salt cavern is considerably greater than that of the supercapacitor and supercapattery in the conventional sense. Consequently, the adaption of either a bipolar or parallel stack in a single large cavern or multiple smaller ones needs great efforts in planning and designing. The optimal configuration and parameters must be selected and adjusted according to the specific site conditions. Additionally, given the need for a separator membrane between the positrode and negatrode, the incorporation of some functionalities into the separator may also be a viable option, which could enable electrode reactions that may not occur directly in aqueous electrolytes, and further expand the voltage window of the electrolyte and enhance the energy storage capacity.
3.5. Construction process
In the construction of supercapacitors and supercapatteries within salt caverns, the initial and crucial step is the selection of an appropriate site. In view of the uniformity of the electrolyte, the charge transfer efficiency of the electrode materials and the difficulty of fabrication, it is recommended that salt caverns of small to medium sizes should be selected, and that the nearby geological conditions should be subjected to close examination. In contrast to storing compressed air or natural gas in salt caverns, the liquid electrolyte may interact with and erode the salt cavern walls. Consequently, there are specific requirements for mechanical properties of the salt cavern. Furthermore, as previously discussed, large-scale electrochemical storage in salt caverns is more suitable for low population densities and closer proximity to other new energy power plants.In light of the structural characteristics of supercapacitors and supercapatteries, the two-well horizontal cavern method is the more optimal approach for the construction of salt caverns for EES. This method allows for the creation of larger internal dimensions within the resulting salt caverns, facilitating the installation of the positrode and negatrode at the position of two wells. Additionally, it enables the establishment of a connection to the external power supply upon the completion of the well construction. According to the current technology of salt cavern construction, the assembly of EES devices may be achieved in two ways. After precise exploration of the shape and size of the salt cavern, the design of the EES module is confirmed. Then, the module can be assembled overground before being transported and placed inside the salt cavern, followed by injection of the electrolyte into the salt cavern via pipes. This method requires a sufficiently large size of the channel connecting the salt cavern to the ground. The second plan is to assemble the EES devices underground in the salt cavern. Unit cells can be transported underground through the salt cavern mining channel and then assembled in the salt cavern. To realize this approach, engineering technology and personal experience in underground coal mining can be used. Alternatively, it can be considered to combine with an intelligent robot to complete the operation.
In addition, there are specific requirements for sealing the gaps at different connections. In the bipolar plate connected cells in the stack of Fig. 9, the individual cells should be separated from each other without electrolyte flowing between them. The separation can be realized by conventional rubber models, or by the salt wall itself which can be shaped during the salt cave manufacturing process. It is thought that in the saturated electrolyte, the possibly present gaps between the bipolar plates and the salt cavern walls should make it difficult or inefficient for local convection, which may promote salt crystallisation or deposition in and hence help seal the gap.
3.6. Challenges
Salt cavern EES has the potential to become a highly promising large-scale energy storage technology. It offers advantages in terms of scalability, storage capacity, and power performance, which could make it a valuable support for intermittent renewable energy power stations in the future. However, there are still challenges to be addressed before this technology can be realised.The current salt cavern energy storage technologies primarily utilise the large volume capacity to store energy gas or compressed air, thereby fulfilling the function of energy reserves and peak shifting. If the salt caverns are to be used as a supercapacitor and supercapattery container, they must be injected with electrolyte and accommodate electrodes to form the electrochemical energy storage devices. This would introduce new requirements with regard to the stability and safety of the salt caverns. Therefore, the location and construction of salt caverns should be reconsidered in terms of mechanical stability and sealing, especially in the case of long-term filling of the liquid electrolyte. The potential for environmental hazards to be introduced to the surrounding environment, both under and above ground, must be weighed against the benefits of the proposed energy storage solution.
The issue of cost is a significant factor to be considered. In addition to the common cost components, i.e. the manufacturing cost of the salt caverns, it is also necessary to involve the cost of the supercapacitor or supercapattery compared to gas storage in salt caverns in terms of materials, manufacture, underground installation, maintenance and repair, and decommission. Specifically, the costs of electrode materials and electrolytes are likely the highest in terms of investment. Therefore, the application of brine can significantly reduce electrolyte costs while being compatible with current aqueous supercapacitors and supercapatteries. Furthermore, the brine electrolyte should be highly conductive and tuneable. The cost assessment should therefore be based on the generic rule that the larger the amounts of energy input and output, the greater the quantity of electrode materials and the more optimised the electrode properties. The current supercapacitor and supercapattery technologies are based on the modular construction strategy, and are mainly to suit utilisation in portable electric devices or electric vehicles. For salt cavern applications, it is worth exploring the possibility of scaling up the individual modular supercapacitors and supercapatteries.
Due to the large scale of EES in salt caverns, the overall cost for design, materials, manufacturing and installation can reach a very high level. Although a comprehensive analysis is not yet possible, it is still interesting to have an estimate according to the cost of the Zn‖AC supercapattery designed in Fig. 9. Currently, the cost of commercial lithium-ion batteries is about $100–150 per kW h, mainly due to the high costs of lithium metal or compounds (lithium carbonate: $14000 per ton)144 and organic electrolytes ($1000–7000 per ton, depending on the type of LIBs).145 In comparison, zinc metal ($2800 per ton)146 as well as aqueous electrolytes (e.g. the cleaned brine from salt caverns) are much cheaper. Because the specific energy of Zn-based supercapatteries is approaching the level of LIBs, e.g. 269 W h kg−1,146 the cost of the Zn‖AC supercapattery may be about or less than $50 per kW h. Thus, the cost of a 500 GW h EES in the salt cavern can be up to $25 billion.
The preparation of electrode materials suitable for large supercapacitors and supercapatteries remains an unresolved issue. The comprehensive cost of aqueous electrolyte is contingent upon the electrolyte salt. In order to widen the voltage window and enhance the stability, the electrolyte stored in the salt cavern should be of higher concentrations, comprising a greater proportion of electrolyte salt. The incorporation of a substantial quantity of electrolyte, coupled with the inclusion of supplementary additives or stabilisers in the electrolyte, will inevitably result in an increase in cost.
Operating temperatures of EES in salt caverns also represent a significant challenge. The majority of EESs currently in use operate at ambient or atmospheric temperatures. The stability of electrolyte and electrode materials and the charge transfer efficiency which may influence the cycling and rate performance of EES devices all rely on temperature. However, the temperature within salt caverns can vary considerably, depending on depth. Furthermore, the temperature gradient increases with depth, where the temperature rises as the distance from the surface increases. Therefore, it is essential to select salt caverns with appropriate geological situations, for example, locations within 1000 metres in depth, to maintain the temperature to be near room temperature.
To ensure highly efficient and uniform charge transfer processes in large-scale EES cells is expected to be a significant challenge. The volume of a single salt cavern can range from 104 to 106 m3, depending on the geological conditions and formation method. Similarly, the apparent overall working area of a single electrode within the cavern may vary from 10 to 103 m2. To make a single electrode (plate) of such a large surface area is not impossible, but it would be more realistic to use the parallel plates design as shown in Fig. 10. Verification of such designs should begin with computer modelling before experimental testing at the pilot scale.
Long stability of EES in salt cavern is a very important parameter for real applications. Due to the inherent plasticity and self-healing capability of salt cavern walls (i.e. salt rock), macroscopic cracking is almost unlikely to happen, but there is the possibility of long-term surface changes when the wall is in contact with the saturated electrolyte. For example, there are small but expected differences in temperature, solubility, and convection current in different regions due to the huge span of the salt cavern, leading to a slow transfer of mass (mostly salt). Therefore, the salt cavern EES works in a long-term dynamic equilibrium and the following aspects should be considered to maintain long-term stability. Firstly, enough thickness of the salt cavern wall should be reserved to minimise the impact from possible mass movement on the wall due to the dynamic nature of the equilibrium between the wall and the saturated electrolyte. The design and manufacturing process should be carried out after precise calculation in view of various perturbation factors to ensure the stability of the salt cavern. Secondly, the concentration changes of electrolyte during the dis-/charging process should be controlled to a minimal range (e.g. ±0.1%) through the power design of EES. Last, the internal convection current of the electrolyte in the salt cavern can be reduced through rational design of suitable barriers between supercapattery stacks and salt cavern walls. These strategies are expected to help maintain the stability of the salt cavern walls over a longer period (e.g. 10–20 years) before periodic evacuation and cleaning of the electrolyte, inspection and check, maintenance, repair and replacement.
4. End remarks
Salt caverns, particularly manmade, are based on a rich natural underground resource of deposited thick, wide and dense layers of salt rock, and offer great realities and perspectives for energy storage technologies. They possess ideal physical and mechanical properties, such as non-permeability and self-healing capability, and chemical inertness towards organic substances, and are traditionally utilised for safe and stable storage of fossil gases and oils, particularly natural gas, crude oil and petroleum. The capacity of energy storage is proportional to the volume of the salt cavern, ranging typically from gigawatt-hour to terawatt-hour levels. With the development of more advanced and cleaner energy technologies, salt caverns are being considered and tested for storage of the hydrogen gas and compressed air in relation with renewable energy sources.The utilisation of salt caverns for electrochemical energy storage is rarely reported and a recent proposal was to use salt caverns as the separate stores for the charged and discharged electrolytes of redox flow batteries. This approach is in fact similar to hydrogen and compressed air storage in that the salt caverns are used in a passive manner with the actual power generation occurring aboveground. The active use of salt caverns for large scale electrochemical energy storage is proposed in this report based on the conventional developments of electrode materials and electrolytes for supercapacitors and supercapatteries. The unique prospect is to reuse the cleaned brine from the solution mining of the salt caverns as the electrolytes in the supercapacitor or supercapattery.
A basic estimation has shown that salt cavern supercapacitors and supercapatteries with aqueous electrolytes can offer energy storage capacities to a comparable level as energy gases storage, i.e. up to terawatt-hours. On the technical aspects, aqueous electrolytes, particularly the cleaned brine from solution mining of the salt cavern are recommended for low cost and large scale installation, whilst the more expensive water in salt and organic electrolytes are also considered for higher voltage storage in smaller carvens. Electrode materials are considered on their charge storage mechanisms, namely Nernstian (battery-like), electric double layer and Faradaic capacitive dis-/charging processes. The well established zinc‖carbon supercapattery (i.e. zinc ion capacitor) is used as an exemplar single cell for the design and analysis of stacks of multiple cells connected in series via bipolar plates (high voltage) or in parallel using parallel plates (high current). The analysis suggests a better match between the bipolar plates stack and aqueous electrolytes, whilst the parallel plate stack can be coupled with WIS or organic electrolytes. Although there are still various challenges on the engineering aspects, salt cavern supercapacitors and supercapatteries are promising and highly feasible large scale energy storage approaches for mitigating the impacts of the intermittency of renewable energy sources and the peak-valley nature of power grids.
Author contributions
TTJ: conceptualization; writing – original draft; visualisation. JJL: data and reference collection; formal analysis. GZC: conceptualization; writing – review & editing; visualisation; supervision; funding acquisition; project administration.Data availability
No original research results, software, or codes have been included in this feature article which contains all data and information needed for analysis, derivation and comparison.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge past and ongoing financial support from the NSFC (No. 51602234), the Natural Science Foundation of Hubei Province (2021CFB434), the Ningbo Municipal Bureau of Science and Technology (3315 Plan and 2014A35001-1), EPSRC (EP/J000582/1, GR/R68078), Innovate UK (Smart Grants, 10017140), and E.ON AG (Energy Storage Award, 2007) and other industrial sponsors, and families of our postgraduate students to our research on electrochemical technologies and liquid salts for materials, energy and environment innovations.References
- C. Duffy, R. Prudhomme, B. Duffy, J. Gibbons, P. P. M. Iannetta, C. O’Donoghue, M. Ryan and D. Styles, Nat. Sustainability, 2022, 5, 973–980 CrossRef.
- Energy Trends, Department for Energy Security and Net Zero, https://www.gov.uk/government/collections/energy-trends.
- H. Blanco and A. Faaij, Renewable Sustainable Energy Rev., 2018, 81, 1049–1086 Search PubMed.
- M. E. Amiryar and K. R. Pullen, Appl. Sci., 2017, 7, 286 CrossRef.
- S. Koohi-Fayegh and M. A. Rosen, J. Energy Storage, 2020, 27, 101047 CrossRef.
- M. Mahmoud, M. Ramadan, A. G. Olabi, K. Pullen and S. Naher, Energy Convers. Manage., 2020, 210, 112670 CrossRef.
- M. L. Perry and A. Z. Weber, J. Electrochem. Soc., 2016, 163, A5064–A5067 CrossRef CAS.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2017, 2, 16080 CrossRef CAS.
- S. Rehman, L. M. Al-Hadhrami and M. M. Alam, Renewable Sustainable Energy Rev., 2015, 44, 586–598 CrossRef.
- N. Ma, W. Zhao, W. Wang, X. Li and H. Zhou, Int. J. Hydrogen Energy, 2024, 50, 379–396 CrossRef CAS.
- T. Peng, J. Wan, W. Liu, J. Li, Y. Xia, G. Yuan, M. J. Jurado, P. Fu, Y. He and H. Liu, J. Energy Storage, 2023, 60, 106489 CrossRef.
- A. G. Olabi, T. Wilberforce, M. Ramadan, M. A. Abdelkareem and A. H. Alami, J. Energy Storage, 2021, 34, 102000 CrossRef.
- M. Budt, D. Wolf, R. Span and J. Y. Yan, Appl. Energy, 2016, 170, 250–268 CrossRef.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 Search PubMed.
- T. S. Mathis, N. Kurra, X. H. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS.
- B. Hou, S. Shangguan, Y. Niu, Y. Su, C. Yu, X. Liu, Z. Li, J. Li, X. Liu and K. Zhao, Energy Sources, Part A, 2023, 46, 621–635 Search PubMed.
- W. Liu, Q. Li, C. Yang, X. Shi, J. Wan, M. J. Jurado, Y. Li, D. Jiang, J. Chen, W. Qiao, X. Zhang, J. Fan, T. Peng and Y. He, Energy Storage Mater., 2023, 63, 103045 CrossRef.
- G. Zhang, Z. Wang, J. Liu, Y. Li, Z. Cui, H. Zhang, L. Wang and L. Sui, Bull. Eng. Geol. Environ., 2020, 79, 4205–4219 Search PubMed.
- J. G. Speight, in Natural Gas, ed. J. G. Speight, Gulf Professional Publishing, Boston, 2nd edn, 2019, pp. 149–186 DOI:10.1016/B978-0-12-809570-6.00005-9.
- T. Wang, L. Ao, B. Wang, S. Ding, K. Wang, F. Yao and J. J. K. Daemen, Energy, 2022, 238, 121906 CrossRef.
- Z. Zhang, W. Liu, Q. Guo, X. Duan, Y. Li and T. Wang, J. Energy Storage, 2022, 50, 104454 CrossRef.
- Z. Ding, T. Wang, T. He, D. Xie, Y. Liao, J. Chen, J. Li and L. Chen, J. Energy Storage, 2024, 78, 110080 Search PubMed.
- X. Wei, X. Shi, Y. Li, P. Li, S. Ban, T. Xue, S. Zhu, H. Liu and C. Yang, Rock Mech., 2023, 57, 287–305 Search PubMed.
- X. Zhang, W. Liu, D. Jiang, W. Qiao, E. Liu, N. Zhang and J. Fan, Energy, 2021, 231, 120968 CrossRef.
- F. Ban, G. Yuan, J. Wan and T. Peng, Energy Sources, Part A, 2020, 43, 3082–3100 CrossRef.
- A. G. Olabi, T. Wilberforce, M. Ramadan, M. A. Abdelkareem and A. H. Alami, J. Energy Storage, 2021, 34, 102000 CrossRef.
- Y. Zheng, Y. Zhao, G. Ding, Z. Wu, S. Lu, X. Lai, X. Qiu, D. Yang, B. Han and L. Wang, Pet. Explor. Dev., 2017, 44, 139–145 CrossRef.
- W. Liu, Z. Zhang, J. Chen, J. Fan, D. Jiang, D. Jjk and Y. Li, Energy, 2019, 185, 682–694 CrossRef.
- J. D. O. Williams, J. P. Williamson, D. Parkes, D. J. Evans, K. L. Kirk, N. Sunny, E. Hough, H. Vosper and M. C. Akhurst, J. Energy Storage, 2022, 53, 105109 CrossRef.
- J. Wang, Z. Wang, Q. Zeng, G. Ding, K. Li, Q. Wanyan and Y. Wang, J. Energy Resour. Technol., 2023, 145, 022001 CrossRef.
- X. Wang, J. Wang, Q. Zhang, Z. Song, X. Liu and S. Feng, J. Energy Storage, 2022, 55, 105843 CrossRef.
- T. Wang, Z. Ding, T. He, D. Xie, Y. Liao, J. Chen and K. Zhu, J. Energy Storage, 2024, 84, 110817 CrossRef.
- T. Wang, C. Yang, H. Wang, S. Ding and J. J. K. Daemen, Energy, 2018, 147, 464–476 CrossRef.
- C. Yang, T. Wang and H. Chen, Engineering, 2023, 25, 168–181 CrossRef.
- S. R. Thiyagarajan, H. Emadi, A. Hussain, P. Patange and M. Watson, J. Energy Storage, 2022, 51, 104490 CrossRef.
- M. AbuAisha and J. Billiotte, J. Energy Storage, 2021, 38, 102589 Search PubMed.
- A. I. Osman, N. Mehta, A. M. Elgarahy, M. Hefny, A. Al-Hinai, A. A. H. Al-Muhtaseb and D. W. Rooney, Environ. Chem. Lett., 2021, 20, 153–188 CrossRef.
- X. Wei, S. Ban, X. Shi, P. Li, Y. Li, S. Zhu, K. Yang, W. Bai and C. Yang, Energy, 2023, 272, 127120 CrossRef CAS.
- X. Guo, H. Zhu and S. Zhang, Int. J. Hydrogen Energy, 2024, 49, 1048–1059 CrossRef CAS.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- Y. Chen and G. Z. Chen, ACS Appl. Energy Mater., 2023, 6, 6104–6110 CrossRef CAS PubMed.
- Y. Chen and G. Z. Chen, Next Sustainability, 2024, 3, 100029 CrossRef.
- Q. Xu, L. Zhang, J. Zhang, J. Wang, Y. Hu, H. Jiang and C. Li, Energy Chem., 2022, 4, 100087 CrossRef CAS.
- H. Wang, W. Ke, Z. Wei, D. Li and L. Chen, Chem. Lett., 2021, 50, 389–391 CrossRef CAS.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- N. U. Consultants, Exchange (University of Nottingham), May, 2010, p. 13.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- G. Z. Chen, Int. Mater. Rev., 2017, 62, 173–202 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- G. Z. Chen, Prog. Nat. Sci.: Mater. Int., 2021, 31, 792–800 CrossRef CAS.
- S. Trasatti and G. Buzzanca, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29, A1–A5 CrossRef.
- B. Akinwolemiwa, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2015, 162, A5054–A5059 CrossRef CAS.
- L. Guan, L. Yu and G. Z. Chen, Electrochim. Acta, 2016, 206, 464–478 CrossRef CAS.
- G. Z. Chen, Curr. Opin. Electrochem., 2020, 21, 358–367 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Li, J. O’Shea, X. Hou and G. Z. Chen, Chem. Commun., 2017, 53, 10414–10417 RSC.
- J. H. Chae and G. Z. Chen, Electrochim. Acta, 2012, 86, 248–254 Search PubMed.
- P. Hao, Z. H. Zhao, Y. H. Leng, J. Tian, Y. H. Sang, R. I. Boughton, C. P. Wong, H. Liu and B. Yang, Nano Energy, 2015, 15, 9–23 CrossRef CAS.
- L. Demarconnay, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2011, 196, 580–586 CrossRef CAS.
- T. C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, J. Electrochem. Soc., 1998, 145, 1882–1888 CrossRef CAS.
- K. K. Patel, T. Singhal, V. Pandey, T. P. Sumangala and M. S. Sreekanth, J. Energy Storage, 2021, 44, 103366 Search PubMed.
- N. N. Loganathan, V. Perumal, B. R. Pandian, R. Atchudan, T. N. J. I. Edison and M. Ovinis, J. Energy Storage, 2022, 49, 104149 Search PubMed.
- D. G. Gromadskyi, J. H. Chae, S. A. Norman and G. Z. Chen, Appl. Energy, 2015, 159, 39–50 Search PubMed.
- B. Kim, H. Chung and W. Kim, Nanotechnology, 2012, 23, 155401 CrossRef PubMed.
- S. Korkmaz and I. A. Kariper, J. Energy Storage, 2020, 27, 101038 Search PubMed.
- X. Jin, W. Zhou, S. Zhang and G. Z. Chen, Small, 2007, 3, 1513–1517 CrossRef CAS PubMed.
- G. Z. Chen, M. S. P. Shaffer, D. Coleby, G. Dixon, W. Zhou, D. J. Fray and A. H. Windle, Adv. Mater., 2000, 12, 522–526 CrossRef CAS.
- Q. Z. Zhang, D. Zhang, Z. C. Miao, X. L. Zhang and S. L. Chou, Small, 2018, 14, 1702883 CrossRef PubMed.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- Y. Qian, Z. Zhou, Q. Zhang, H. Zhao, H. Chen, J. Han, H. Wan, H. Jin, S. Wang and Y. Lei, Small, 2024, 20, e2310037 CrossRef PubMed.
- Q. Qu, Y. Zhu, X. Gao and Y. Wu, Adv. Energy Mater., 2012, 2, 950–955 Search PubMed.
- Z. Zhou, Q. Zhang, J. Sun, B. He, J. Guo, Q. Li, C. Li, L. Xie and Y. Yao, ACS Nano, 2018, 12, 9333–9341 Search PubMed.
- J. Xiao, L. Wan, S. Yang, F. Xiao and S. Wang, Nano Lett., 2014, 14, 831–838 Search PubMed.
- W. Wen, J. Yao, H. Tan and J.-M. Wu, J. Mater. Chem. A, 2019, 7, 21378–21385 RSC.
- S. Surendran, S. Shanmugapriya, A. Sivanantham, S. Shanmugam and R. Kalai Selvan, Adv. Energy Mater., 2018, 8, 1800555 CrossRef.
- T. Jiang, Y. Wang and G. Z. Chen, Small Methods, 2023, 7, e2201724 CrossRef PubMed.
- Y. Zeng, M. Yu, Y. Meng, P. Fang, X. Lu and Y. Tong, Adv. Energy Mater., 2016, 6, 1601053 CrossRef.
- S. Banerjee and K. K. Kar, in Handbook of Nanocomposite Supercapacitor Materials II: Performance, ed. K. K. Kar, Springer International Publishing, Cham, 2020, pp. 333–352 DOI:10.1007/978-3-030-52359-6_13.
- A. Eftekhari, L. Li and Y. Yang, J. Power Sources, 2017, 347, 86–107 CrossRef CAS.
- R. Z. Xing, Y. Q. Xia, R. L. Huang, W. Qi, R. X. Su and Z. M. He, Chem. Commun., 2020, 56, 3115–3118 RSC.
- WiKipedia, Lithium-ion_battery, https://en.wikipedia.org/wiki/Lithium-ion_battery.
- WiKipedia, Supercapacitor, https://en.wikipedia.org/wiki/Supercapacitor.
- M. Hughes, G. Z. Chen, M. S. P. Shaffer, D. J. Fray and A. H. Windle, Chem. Mater., 2002, 14, 1610–1613 CrossRef CAS.
- C. Peng, J. Jin and G. Z. Chen, Electrochim. Acta, 2007, 53, 525–537 CrossRef CAS.
- M. Wu, G. A. Snook, G. Z. Chen and D. J. Fray, Electrochem. Commun., 2004, 6, 499–504 CrossRef CAS.
- C. Wei, B. Akinwolemiwa, Q. Wang, L. Guan, L. Xia, D. Hu, B. Tang, L. Yu and G. Z. Chen, Adv. Sustainable Syst., 2019, 3, 1900065 CrossRef CAS.
- C. Peng, G. A. Snook, D. J. Fray, M. S. P. Shaffer and G. Z. Chen, Chem. Commun., 2006, 4629–4631, 10.1039/B609293D.
- M. Hughes, M. S. P. Shaffer, A. C. Renouf, C. Singh, G. Z. Chen, D. J. Fray and A. H. Windle, Adv. Mater., 2002, 14, 382–385 CrossRef CAS.
- C. Downs, J. Nugent, P. M. Ajayan, D. J. Duquette and K. S. V. Santhanam, Adv. Mater., 1999, 11, 1028–1031 Search PubMed.
- M. Gao, S. Huang, L. Dai, G. Wallace, R. Gao and Z. Wang, Angew. Chem., Int. Ed., 2000, 39, 3664–3667 CrossRef CAS PubMed.
- M. J. Bleda-Martínez, C. Peng, S. G. Zhang, G. Z. Chen, E. Morallón and D. Cazorla-Amorós, J. Electrochem. Soc., 2008, 155, A672–A678 CrossRef.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- C. Y. Foo, A. Sumboja, D. J. H. Tan, J. Wang and P. S. Lee, Adv. Energy Mater., 2014, 4, 1400236 CrossRef.
- Z. Lin, D. Barbara, P.-L. Taberna, K. L. Van Aken, B. Anasori, Y. Gogotsi and P. Simon, J. Power Sources, 2016, 326, 575–579 CrossRef CAS.
- X. Peng, H. Liu, Q. Yin, J. Wu, P. Chen, G. Zhang, G. Liu, C. Wu and Y. Xie, Nat. Commun., 2016, 7, 11782 CrossRef CAS PubMed.
- M. Ishikawa, K. Dokko, H. Teng, S. Lindberg, J. Ajuria, A. Balducci and E. Frackowiak, Electrochemistry, 2024, 92, 074003 CrossRef CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 RSC.
- Q. Gou, S. Zhao, J. Wang, M. Li and J. Xue, Nano-Micro Lett., 2020, 12, 98 CrossRef CAS PubMed.
- A. Slesinski, C. Matei-Ghimbeu, K. Fic, F. Béguin and E. Frackowiak, Carbon, 2018, 129, 758–765 Search PubMed.
- N. Zhao, H. Fan, M. Zhang, J. Ma, W. Zhang, C. Wang, H. Li, X. Jiang and X. Cao, Electrochim. Acta, 2019, 321, 134681 CrossRef CAS.
- Q. Abbas, P. Ratajczak, P. Babuchowska, A. L. Comte, D. Bélanger, T. Brousse and F. Béguin, J. Electrochem. Soc., 2015, 162, A5148–A5157 Search PubMed.
- P. Ratajczak and F. Béguin, ChemElectroChem, 2018, 5, 2518–2521 CrossRef CAS.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, J. Mater. Chem. A, 2016, 4, 6639–6644 RSC.
- P. Lannelongue, R. Bouchal, E. Mourad, C. Bodin, M. Olarte, S. le Vot, F. Favier and O. Fontaine, J. Electrochem. Soc., 2018, 165, A657 CrossRef CAS.
- X. Bu, L. Su, Q. Dou, S. Lei and X. Yan, J. Mater. Chem. A, 2019, 7, 7541–7547 RSC.
- J. H. Guo, Y. L. Ma, K. Zhao, Y. Wang, B. P. Yang, J. F. Cui and X. B. Yan, ChemElectroChem, 2019, 6, 5433–5438 CrossRef CAS.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- S.-E. Chun, B. Evanko, X. Wang, D. Vonlanthen, X. Ji, G. D. Stucky and S. W. Boettcher, Nat. Commun., 2015, 6, 7818 CrossRef CAS PubMed.
- D. Xiao, Q. Dou, L. Zhang, Y. Ma, S. Shi, S. Lei, H. Yu and X. Yan, Adv. Funct. Mater., 2019, 29, 1904136 CrossRef CAS.
- Q. Dou, S. Lei, D.-W. Wang, Q. Zhang, D. Xiao, H. Guo, A. Wang, H. Yang, Y. Li, S. Shi and X. Yan, Energy Environ. Sci., 2018, 11, 3212–3219 RSC.
- Q. Dou, Y. Lu, L. Su, X. Zhang, S. Lei, X. Bu, L. Liu, D. Xiao, J. Chen, S. Shi and X. Yan, Energy Storage Mater., 2019, 23, 603–609 CrossRef.
- L.-Q. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y.-L. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923 CrossRef PubMed.
- E. Frackowiak, K. Fic, M. Meller and G. Lota, ChemSusChem, 2012, 5, 1181–1185 Search PubMed.
- J. Lee, B. Krüner, A. Tolosa, S. Sathyamoorthi, D. Kim, S. Choudhury, K.-H. Seo and V. Presser, Energy Environ. Sci., 2016, 9, 3392–3398 Search PubMed.
- J. Y. Hwang, M. Li, M. F. El-Kady and R. B. Kaner, Adv. Funct. Mater., 2017, 27, 1605745 Search PubMed.
- L. Xia, L. Yu, D. Hu and G. Z. Chen, Mater. Chem. Front., 2017, 1, 584–618 RSC.
- L. Xia, J. Zhu and G. Z. Chen, in Encyclopedia of Electrochemical Power Sources, ed. J. Garche, Elsevier, Oxford, 2nd edn, 2025, pp. 467–479 DOI:10.1016/B978-0-323-96022-9.00258-9.
- Y. L. Xia Lan, H. Di and C. Z. George, Acta Chim. Sinica, 2017, 75, 1183–1195 CrossRef.
- N. Zhang, X. Gao, B. Yan, Y. Zhang, S. Ji and X. Shi, Processes, 2024, 12, 1709 Search PubMed.
- J. Tillerson, Geomechanics investigations of SPR crude oil storage caverns, Sandia Labs, Albuquerque, NM (USA), 1979 Search PubMed.
- J. H. Chae and G. Z. Chen, Particuology, 2014, 15, 9–17 CrossRef CAS.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Adv. Energy Mater., 2021, 11, 2100201 Search PubMed.
- Z. Huang, T. Wang, H. Song, X. Li, G. Liang, D. Wang, Q. Yang, Z. Chen, L. Ma, Z. Liu, B. Gao, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2021, 60, 1011–1021 CrossRef CAS PubMed.
- C. Mao, Y. Chang, X. Zhao, X. Dong, Y. Geng, N. Zhang, L. Dai, X. Wu, L. Wang and Z. He, J. Energy Chem., 2022, 75, 135–153 CrossRef CAS.
- W. Shang, W. Yu, X. Xiao, Y. Ma, Y. He, Z. Zhao and P. Tan, Adv. Powder Mater., 2023, 2, 100075 CrossRef.
- K. C. Ng, S. Zhang, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2009, 156, A846 CrossRef CAS.
- X. Zhou, C. Peng and G. Z. Chen, AIChE J., 2012, 58, 974–983 CrossRef CAS.
- H. Guodong, C. Jie, L. Wei and C. He, IOP Conf. Ser. Earth Environ. Sci., 2020, 570, 062020 CrossRef.
- B. Akinwolemiwa, C. Wei, Q. Yang, L. Yu, L. Xia, D. Hu, C. Peng and G. Z. Chen, J. Electrochem. Soc., 2018, 165, A4067 CrossRef CAS.
- V. Vandeginste, Y. Ji, F. Buysschaert and G. Anoyatis, Deep Undergr. Sci. Eng., 2023, 2, 129–147 CrossRef.
- C. Peng, S. Zhang, X. Zhou and G. Z. Chen, Energy Environ. Sci., 2010, 3, 1499–1502 RSC.
- X. Bu, Y. Zhang, L. Su, Q. Dou, Y. Xue and X. Lu, Ionics, 2019, 25, 6007–6015 CrossRef CAS.
- J. Guo, Y. Ma, K. Zhao, Y. Wang, B. Yang, J. Cui and X. Yan, ChemElectroChem, 2019, 6, 5433–5438 CrossRef CAS.
- C. Peng, D. Hu and G. Z. Chen, Chem. Commun., 2011, 47, 4105–4107 RSC.
- Z. Dai, C. Peng, J. H. Chae, K. C. Ng and G. Z. Chen, Sci. Rep., 2015, 5, 9854 CrossRef CAS PubMed.
- Celgard, Celgard Products Data Sheets, https://www.celgard.com/product-data.
- Nafion, Ion Exchange Membranes Product Overview, https://www.nafion.com/ Search PubMed.
- B. W. Jaskula, U.S. Geological Survey, Mineral Commodity Summaries, U.S., 2025.
- Shanghai Metals Market, Electrolyte prices, https://www.metal.com/price/New-Energy/Electrolyte.
- W. Zhang, X. Gao, X. Yang, T. Zhang, Y. Li and J. Zhang, Chem. Eng. J., 2023, 460, 141824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06169a |
| This journal is © The Royal Society of Chemistry 2025 |