
Transforming delayed fluorescence into blue-shifted phosphorescence in aminoboranes via oxygen-to-sulfur substitution on the donor amine†
Akkarakkaran Thayyil
Muhammed Munthasir
 *,
Satyam
Jena
and
Pakkirisamy
Thilagar
*
*,
Satyam
Jena
and
Pakkirisamy
Thilagar
*
Department of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore, 560012, India. E-mail: thilagar@iisc.ac.in
First published on 23rd January 2025
Abstract
Thermally activated delayed fluorescence (TADF) and room-temperature phosphorescence (RTP) materials are found in diverse applications, from optoelectronic devices to time-gated bioimaging. Recently, aminoboranes with donor–acceptor structures have been identified as promising candidates due to their inherent capacity to harvest the triplet excitons by their unique orbital configurations (El Sayed rule). This work reports the delayed luminescence behaviors of two aminoboranes, BNO and BNS, featuring phenoxazine (PXZ) or phenothiazine (PTZ) donors coupled with a dixylylborane acceptor. BNO exhibits efficient TADF emission in aggregates, thin films, and solid states. In contrast, BNS shows delayed fluorescence (DF) in aggregate states and RTP in solid and thin-film states. Notably, BNS shows a rare blue-shifted phosphorescence relative to its prompt fluorescence, which has not been reported for aminoboranes. Photoluminescence studies and computational calculations reveal that ISC and rISC processes in these systems involve higher triplet states. The unprecedented blue-shifted phosphorescence in BNS is attributed to perturbations in energy levels, which are driven by unique quasi-axial and quasi-equatorial conformations and the stronger spin–orbit coupling of heavier S over O.
Introduction
Luminescence efficiency is a critical parameter that determines the viability of a material for practical applications.1–6 Contemporary research has primarily focused on developing innovative designs and strategies to achieve energy-efficient systems. Conventional fluorescence relies solely on singlet-state emission, prompting extensive research into harnessing the dark triplet state to enhance the luminescence efficiency.7–10 To date, several innovative approaches have been reported, including phosphorescence (PH),11–16 delayed fluorescence (DF),17–22 processes such as triplet–triplet annihilation (TTA),23–26 thermally activated delayed fluorescence (TADF),27–32 and, most recently, hybridized local charge transfer (HLCT),33–36 offering new avenues for improved performance. Among these, phosphorescence and TADF materials are found to be prominent and have found application in various fields such as organic light-emitting diodes (OLEDs),31,37–40 bioimaging and biosensing,41–43 security and anti-counterfeiting,44–46 sensing,47–49 solar cells, energy harvesting, etc.50–52Phosphorescence involves two consecutive spin-forbidden processes, intersystem crossing (ISC) (S1 → Tn) followed by radiative decay from Tn → S0.11–16 The weak spin–orbit coupling (SOC) in organic molecules limits the development of purely organic phosphorescence materials. However, this hurdle has been overcome by incorporating heteroatoms (N, P, O, S, etc.), and heavy atoms (Br, Cl, Se etc.), which enhances ISC rates and triplet state populations, consistent with El-Sayed's rule and the heavy atom effect.53–57 Meanwhile, TADF is an unimolecular process that utilizes reverse intersystem crossing (rISC) to convert triplet-state populations to singlet states, facilitated by thermal energy. The rate of this up-conversion process is inversely correlated with the singlet to triplet energy gap, ΔEST, governed by the equation

Aminoboranes are a new class of systems recently added to the family of delayed luminescent materials. Their unique molecular architecture and access to electronic states of different symmetry (π–π* and n–π*/CT and LE) between singlet and triplet leads to enhanced SOC, which enables efficient ISC and rISC for delayed emission.38–40,60–63 Moreover, aminoboranes are well known for their distinctive stimuli-responsive properties.64–66 Thus, aminoboranes find applications in various fields. For instance, in 2017, Yun Chi and co-workers reported the first TADF from an aminoborane system, using an acridine-decorated aminoborane to develop a green OLED.39 In 2018, Chen et al. developed boryl-substituted phenoxazine that exhibits efficient TADF capable of producing highly efficient orange OLEDs.38 In 2020 Yun Chi's group again developed an OLED with blue light emission from a methoxy-substituted carbazole-based aminoborane.40 He and co-workers demonstrated ultralong organic phosphorescence (UOP) from a carbazole-based aminoborane that same year.61 Our group has also been active in aminoborane research for the past decade. In 2017, Neena et al. showed aggregation-induced emission AIE and triboluminescence properties in diphenylamine and phenothiazine-decorated aminoboranes, respectively.64,67 In 2018, we explored a vinyl-pyridyl-decorated aminoborane for barrier-free DF.63 Recently, in 2023, we reported intense blue circularly polarized luminescence from a chiral aminoborane, and in the same year, we demonstrated persistent RTP with a 470 ms lifetime from molecularly dispersed aminoboranes.62,66
As part of our ongoing program, we intend to investigate the influence of the heteroatoms and the conformational dynamics of the nonplanar donor attached to an aryl borane acceptor on its singlet/triplet state energetics and delayed luminescence behavior. For this study, we have chosen the phenoxazine (PXZ) and phenothiazine (PTZ)-decorated aminoboranes BNO and BNS, respectively, as the lighter atom, O in phenoxazine, and heavier atom, S in phenothiazine, may impart different amounts of spin–orbit coupling in these molecules and endow them with unique delayed luminescence characterstics (Fig. 1). Furthermore, the nonplanar heterocycles PXZ and PTZ in these molecules can exhibit quasi-equatorial and quasi-axial conformational dynamics that are susceptible to the microenvironment, by this means modifying the energetics of the singlet and triplet states and, therefore, their emission properties.68–75 In light of these factors, we present a comprehensive photophysical and theoretical analysis of BNO and BNS. The results reveal that BNO exhibits efficient TADF emission in both dispersed and condensed states, while BNS demonstrates delayed fluorescence in aggregates and rare, blue-shifted phosphorescence in the solid state and as polymer-doped film. Our detailed study concludes that the heavy atom effect of sulfur over oxygen, coupled with the pronounced flip-flop motion of PTZ, is responsible for the distinct delayed emission features in BNS over BNO. These intriguing findings are discussed in this manuscript.
 | ||
| Fig. 1 Chemical structures of the aminoboranes under investigation. | ||
Results and discussion
Synthesis and structural studies
Both BNO and BNS were synthesized following the literature procedures (Schemes 1 and S1, S2†).38,64 The synthesis involves lithiation of phenoxazine or phenothiazine using n-butyllithium, followed by quenching of the lithiated intermediate with bis(2,6-dimethylphenyl)fluoroborane. Analytically pure BNO and BNS were obtained through column chromatography, which was followed by recrystallization. Both compounds were characterized using NMR spectroscopy (1H, 13C, 11B) and high-resolution mass spectrometry (HRMS) (Fig. S1–S8†). The molecular structure of BNO was confirmed by single-crystal X-ray diffraction (SCXRD) analysis (Fig. 2 and Table S1†). 1H NMR analysis indicated that BNO adopts C2 symmetry in CDCl3, similar to its mesityl analog (R1)38 and many reported aminoboranes.38–40,61–67 The 11B NMR resonance for BNO appears at 50.7 ppm, consistent with aminoborane systems exhibiting significant B–N π-bond character (Fig. S3†).38–40,61–67 The characterization data for BNS exactly matched the literature report, so the molecular structure previously reported by Neena et al. was used for further structural comparisons.64 | ||
| Scheme 1 Synthesis scheme for BNO and BNS. | ||
 | ||
| Fig. 2 (a) Molecular structure of BNO, and (b) supramolecular dimer chains of BNO. Cyan and yellow dashed lines represent the CH⋯π interactions. Atom colour code – carbon: grey, nitrogen: blue, oxygen: red, and hydrogen: off-white. | ||
Pale yellow block-shaped crystals of BNO suitable for single-crystal X-ray diffraction studies were obtained by slow evaporation of a DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane solution (3:1 ratio). BNO crystallizes in the triclinic crystal system with the P
:hexane solution (3:1 ratio). BNO crystallizes in the triclinic crystal system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 2a and Table S1†). Similar to its mesityl analog R1 (Fig. S13†), the B(1) center in BNO adopts trigonal planar geometry with a B–N bond length of 1.433 Å, comparable to that in R1 (1.438 Å), BNS (1.455 Å) and other aminoboranes (Table S2†).38–40 The dihedral angle between the C(28)N(1)C(C17) plane (PXZ) and the C(1)B(1)C(7) plane (boryl) is 29°, which is higher than the value noted for R1 (25°) and BNS (22°), suggesting better in-plane arrangement of donor–acceptor moieties and greater electronic communication in BNO than in the other two compounds (Fig. S9†).38,64 The dihedral angle between the aryl groups on boron is larger in BNO (86.80°) than in BNS (82.73°), allowing more spatial accommodation for PXZ and resulting in a slightly shorter B–N bond (Fig. S10†). Additionally, the twist angle between the two benzene rings of PXZ in BNO is 146.35°, similar to that in R1 (142.70°), indicating a saddle-like nonplanar geometry for PXZ (Fig. S11†). However, this twist angle is notably higher in the PTZ unit of BNS (134.08°), suggesting that PXZ connected to arylborane in BNO is more planar than PTZ in BNS. This planarity likely contributes to denser packing in BNO (ρcalcd = 1.215 g cm−3) than in BNS (ρcalcd = 1.174 g cm−3).
space group (Fig. 2a and Table S1†). Similar to its mesityl analog R1 (Fig. S13†), the B(1) center in BNO adopts trigonal planar geometry with a B–N bond length of 1.433 Å, comparable to that in R1 (1.438 Å), BNS (1.455 Å) and other aminoboranes (Table S2†).38–40 The dihedral angle between the C(28)N(1)C(C17) plane (PXZ) and the C(1)B(1)C(7) plane (boryl) is 29°, which is higher than the value noted for R1 (25°) and BNS (22°), suggesting better in-plane arrangement of donor–acceptor moieties and greater electronic communication in BNO than in the other two compounds (Fig. S9†).38,64 The dihedral angle between the aryl groups on boron is larger in BNO (86.80°) than in BNS (82.73°), allowing more spatial accommodation for PXZ and resulting in a slightly shorter B–N bond (Fig. S10†). Additionally, the twist angle between the two benzene rings of PXZ in BNO is 146.35°, similar to that in R1 (142.70°), indicating a saddle-like nonplanar geometry for PXZ (Fig. S11†). However, this twist angle is notably higher in the PTZ unit of BNS (134.08°), suggesting that PXZ connected to arylborane in BNO is more planar than PTZ in BNS. This planarity likely contributes to denser packing in BNO (ρcalcd = 1.215 g cm−3) than in BNS (ρcalcd = 1.174 g cm−3).
The intermolecular CH⋯π interaction (2.849 Å) between a C–H of a xylyl ring and the xylyl π-cloud of an adjacent molecule generates a supramolecular dimer (Fig. 2b). The neighboring dimers are stitched together by the CH⋯π interaction (3.031 Å) between the methyl proton (H16B) on the xylyl unit of one molecule and the π cloud of one of the benzo groups of phenoxazine on the adjacent dimer leading to the formation of the supramolecular 2D chain (Fig. S12†). In contrast, the more puckered nature of PTZ in BNS leads to a less dense packing arrangement, forming a “3D supramolecular structure with cylindrical columns of diameter 4.61 Å”, as reported by Neena et al.64 These structural and packing differences between BNO and BNS could have a significant impact on their optoelectronic properties.
Photophysical and theoretical studies
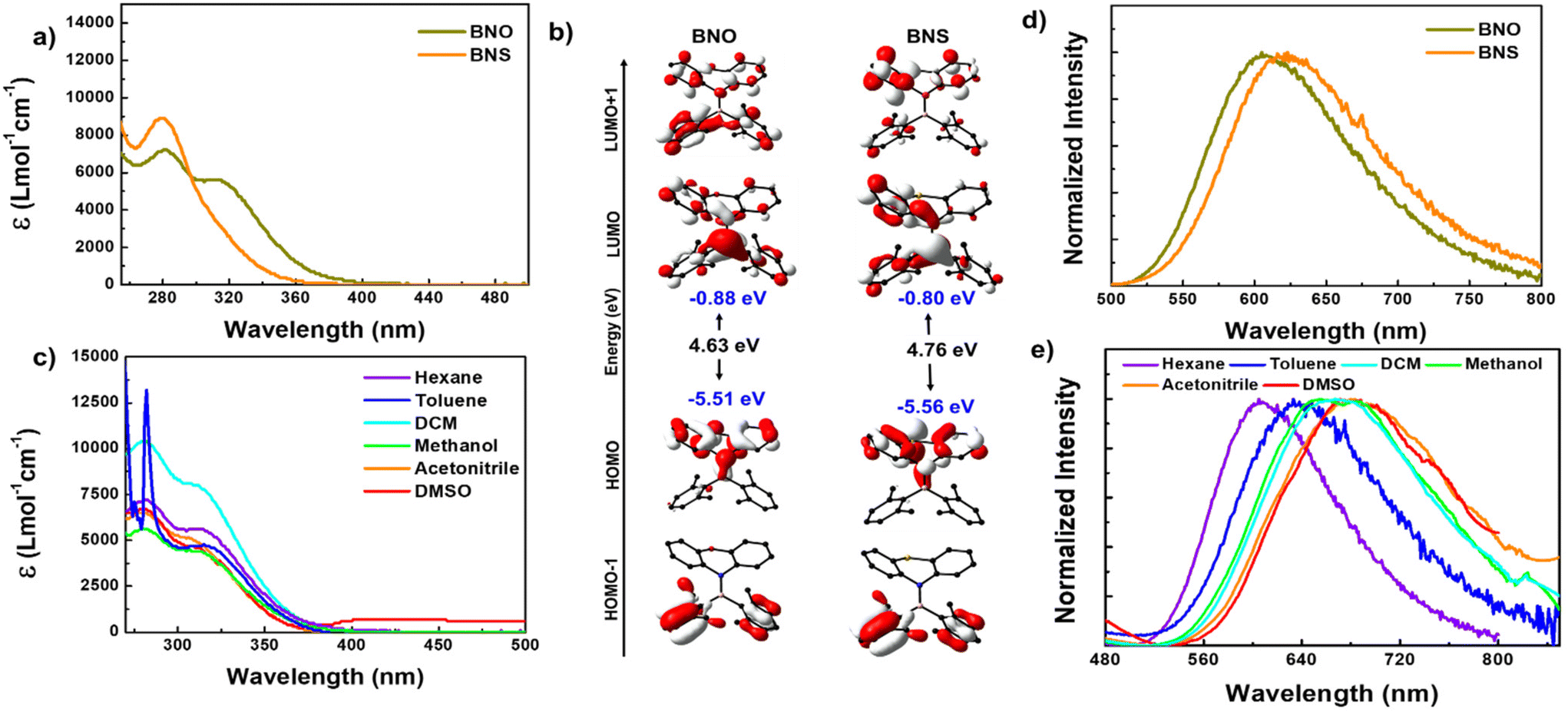 | ||
| Fig. 3 (a) Absorption spectra of BNO and BNS in hexane (conc. 10−5 M). (b) Frontier molecular orbitals for BNO and BNS obtained from the optimized geometry using DFT [the B3LYP/6-31G(d) level of theory]. (c) Absorption spectra of BNO in different solvents (conc. 10−5 M). (d) Steady-state PL spectra for BNO and BNS in hexane and (e) steady-state PL spectra of BNO in different solvents (conc. 10−5 M) at λex = 310 nm for BNO and 290 nm for BNS under ambient conditions. | ||
To understand the absorption features of BNO in detail, we optimized the ground state geometry for BNO, and singlet vertical transitions were calculated using density functional theory (DFT) and time-dependent DFT (TD-DFT), respectively (Fig. 3b and S14, S15†). The bond parameters of the optimized geometry are closely matched with those of the crystal structure, validating the choice of level of theory (Table S3†). The HOMO is localized over the phenoxazine moiety, whereas the LUMO is localized all over the molecule with a significant contribution from boron. Similar features were observed in BNS also (Fig. 3b). Upon changing PTZ to PXZ (in BNO), the HOMO of the molecule is slightly destabilized, while the LUMO is stabilized, resulting in a reduction in the band gap (ΔEg = 4.63 eV) compared to BNS (ΔEg = 4.76 eV), with the same reflected in their respective absorption spectra (Fig. 3a and c).
The vertical transition calculation revealed that the lower energy absorption band at 320 nm for BNO results from the S0 to S1 transition involving the HOMO and LUMO (f = 0.1589) (Fig. S14†). As discussed above, the HOMO and LUMO are localized on different parts of the molecule suggesting the lowest energy HOMO → LUMO transition is an intramolecular charge transfer (ICT) from the donor amine to the acceptor boryl moieties. On the other hand, the higher energy band is a collection of S0 to Sn (n = 2, 3 and 4) transitions involving electronic distribution within the PXZ ring or boryl units (Fig. S14 and Table S4†). A similar scenario is observed in the case of BNS; however, the transition probability for S0 to S1 (303.04 nm, f = 0.0094) is quite low, resulting in a weak CT band (Fig. S15 and Table S5†).
The photoluminescence (PL) spectra of BNO in hexane display a broad emission band ranging from 500 to 800 nm with a peak maximum at 605 nm. This emission band is 15 nm blue-shifted compared to the PL of BNS (Fig. 3d). Upon increasing solvent polarity, the emission maximum is red-shifted with a drastic decrease in the PL intensity, which is typical of D–A systems (Fig. 3e and S17c†). Similar to R1, the BNO also shows a very large Stokes shift (15217 cm−1) indicating that large structural reorganization occurred in the excited state.38 The TD-DFT-optimized S1 geometry indeed showed large structural variations; the nonplanar PXZ ring in S0 became completely planar along with the BN bond elongation, supporting the large Stokes shift (Fig. S16 and Table S3†). The degree of ring planarization in BNO is slightly lower than that in its mesityl analog R1; consequently, BNO exhibits a smaller Stokes shift than R1. The phenothiazine ring in BNS also undergoes planarization (to a greater extent than that in BNO) resulting in a large Stokes shift (Fig. S16 and S18†).38 Because of the large structural reorganization/flip-flop motion of the PXZ unit, BNO is weakly emissive in the dilute solution. Despite this weak emission, BNO shows a delayed fluorescence (DF) band, which overlaps smoothly with the fluorescence spectra, revealing the DF nature of the emission (Fig. S19a†). The lifetime measurement revealed that BNO exhibits both ns (13.60 ns) and μs (6.38 μs) for the same peak maximum further affirming the TADF characteristics (Fig. S19d and Table S7†). Under the oxygen environment, the PL intensity, lifetime, and photoluminescence quantum yield (PLQY) of BNO decreased considerably (Fig. S19 and Table S7†). No such DF is observed for BNS under similar conditions.
Furthermore, to understand the Lewis acidic nature of the boron center, these aminoboranes are titrated against pyridine/F− and the binding events are monitored using absorption and PL spectrometers. The result shows that pyridine does not bind to the Lewis acid boron; however, F− binds with these aminoboranes and causes significant spectral changes. Upon binding with fluoride (TBAF), the absorption bands show a bathochromic shift, while the PL bands exhibit a hypsochromic shift (Fig. S20 and S21 in the revised ESI†). These spectral changes can be attributed to the geometry changes at the boron center of trigonal planar to tetrahedral and the breaking of conjugation between the empty p orbital of boron and lone-pair electrons of nitrogen in the amine moiety. The calculated binding constants for BNO and BNS are 2.09 × 104 and 1.94 × 104, respectively. These values are significantly lower than the ones reported for triarylboranes elsewhere, indicating that the Lewis acidity of B is significantly reduced in aminoboranes due to the resonance bonding interactions between the empty p orbital of acceptor and lone-pair electrons of N in the donor moiety.76–78
 | ||
| Fig. 4 (a) Steady-state PL spectra of BNO in DMSO with different fractions of water [fw] at λex = 310 nm. (b) Prompt and time-gated/delayed [20 μs delay] PL spectra for BNO in degassed DMSO–water mixture [fw = 90%] at λex = 310 nm. (c) Steady-state and (d) time-gated/delayed [20 μs delay] PL spectra for BNO in DMSO–water mixture [fw = 90%] in a different environment at λex = 310 nm. (e) Fluorescence and (f) delayed fluorescence decay for BNO in DMSO–water mixture [fw = 90%] in a different environment at λex = 310 nm and λem = 580 nm. | ||
We further analysed the delayed emission behaviour of BNO aggregates at fw = 70% and fw = 90%. As anticipated, the aggregates exhibited more efficient DF than the dilute solution (Fig. 4b–f and S23†). The prompt and delayed emission spectra overlapped smoothly, and both the lifetime and quantum yield increased significantly compared to the solution (Fig. 4b–f, S23 and Table S8†). The lifetime (prompt and delayed) and PLQY of aggregates at fw = 70% (ΦTotal = 0.23) were found to be higher than those at fw = 90% (ΦTotal = 0.18) and showed greater sensitivity to oxygen (Fig. S23, and Table S8†). This difference can be attributed to the greater crystallinity of the particles formed at fw = 90% than at fw = 70% (Fig. 4c–f, S24, and Table S8†). These results suggest that both singlet and triplet manifolds are affected by the size, morphology, and crystallinity of the aggregates formed at different water fractions (fw).
Although the AIEE behavior of BNS was reported previously, its aggregate-state delayed luminescence features had not been explored.64 To further substantiate the DF phenomena at the aggregate level, we conducted similar experiments on BNS. This compound exhibited significant emission enhancement with a blue shift (580 nm) in a water–DMSO mixture at fw = 90%, consistent with observations reported elsewhere (Fig. S25a†). Time-gated spectra recorded for these aggregates revealed the DF nature of BNS with a smooth overlap of prompt and delayed spectra. BNS shows higher PLQY (0.32) than BNO (0.25), though with a DF lifetime value nearly half that of BNO (Fig. S25b–S25f and Table S9†). Detailed studies including lifetime and quantum yield and various radiative and non-radiative decay rates are given in Table 1 and S8–S10.† Furthermore, the aggregates formed from BNS are amorphous, hence, the molecules may not adopt a specific conformation such as quasi-axial or quasi-equatorial (Fig. S24†). In contrast, in solid or thin-film forms, the molecules exhibit high crystallinity and adopt a preferential conformation, which explains the observed differences in their emission properties. This is discussed in detail in the following section.
| λ ex (nm) | λ em (nm) | Fluorescence | Delayed fluorescence | Φ Total | |||
|---|---|---|---|---|---|---|---|
| τ 1 (A1) | τ 2 (A2) | τ 1 (A1) | τ 2 (A2) | ||||
| τ = lifetime, A is the amplitude of intensity, ΦTotal is total PLQY, and λex and λem are the excitation and emission wavelengths, respectively. | |||||||
| BNO | |||||||
| Hexane | 310 | 605 | 13.60 ns (100%) | 6.38 μs (100%) | 0.12 | ||
| DMSO–water mixture (fw = 70%) | 310 | 535 | 14.58 ns (100%) | — | 60.07 μs (6.71%) | 146.04 μs (93.29%) | 0.25 |
| DMSO–water mixture (fw = 90%) | 310 | 580 | 10.39 ns (14.93%) | 15.64 ns (85.07%) | 24.98 μs (78.17%) | 109.49 μs (21.83%) | 0.19 |
| BNS | |||||||
| Hexane | 280 | 620 | 11.18 ns (100%) | ||||
| DMSO–water mixture (fw = 90%) | 280 | 580 | 10.31 ns (100%) | — | 13.52 μs (66.58%) | 32.96 μs (33.42%) | 0.32 |
In short, BNO exhibits a weak fluorescence and DF in a molecularly dispersed solution due to active molecular motion, which increases non-radiative decay from the emissive state. However, at the aggregate state, both BNS and BNO demonstrate luminescence enhancement. Both BNO and BNS showed efficient delayed fluorescence in the aggregated state. Moreover, the PL features of BNO depend on the morphology and size of the aggregates. Though the phenomenon of aggregation-induced delayed fluorescence (AIDF) is well documented in the literature, such phenomena are rare in B–N systems, highlighting the significance of these findings.82–86
 | ||
| Fig. 5 (a) Steady-state PL (dashed lines) and excitation spectra for the solid samples of BNO and BNS (λex = 375 nm). (b) Prompt and time-gated [50 μs delay] PL spectra for solid samples of BNO and BNS (λex = 375 nm and 350 nm) at 298 K under ambient conditions. (c) Time-gated [50 μs delay] PL spectra and (d) delayed fluorescence decay (λem = 540 nm) for the solid sample of BNO. (e) Time-gated [50 μs delay] PL spectra and (f) phosphorescence decay (λem = 500 nm) for solid samples of BNS under vacuum (absence of oxygen) and ambient (in the presence of oxygen) conditions. | ||
| Compound | λ em (Fl) (nm) | PF lifetime | λ em (DF/PH) (nm) | PH/DF lifetime | Φ Total | k r [106] s−1 | kn r [107] s−1 |
|---|---|---|---|---|---|---|---|
| Fl is the fluorescence, τ = average lifetime, ΦTotal is the total PLQY, while kr (=Φ/τFl), knr (=1−Φ/τFl), are the radiative decay from S1, the non-radiative decay from S1, the λex, and λem, are the excitation wavelength and the emission wavelength, respectively. | |||||||
| BNO (solid) | 545 | 13.92 ns | 540 | 103.65 μs | 0.74 | 54.33 | 1.91 |
| BNO (Neat film) | 545 | 14.02 ns | 545 | 99.02 μs | 0.63 | 44.93 | 2.64 |
| BNO (1 wt%@ PMMA) | 568 | 15.92 ns | 568 | 41.63 μs | 0.3 | 19.62 | 4.57 |
| BNS (solid) | 520 | 9.87 ns | 500* | 91.03 μs | 0.2 | 20.26 | 8.10 |
| BNS (Neat film) | 500 | 8.71 ns | 535* | 73.30 μs | 0.6 | 68.88 | 4.59 |
| BNS (1 wt%@ PMMA) | 575 | 12.26 ns | 550* | 69.05 μs | 0.41 | 33.44 | 4.81 |
To further validate the nature of DF, the PL studies were conducted at various temperatures under vacuum (Fig. S28 and Table S11†). As the temperature is increased, the prompt PL intensity and lifetime increase significantly, indicating the thermally activated delayed fluorescence (TADF) process (Fig. S28a†). In contrast, the DF intensity decreased with increasing temperature, which can be attributed to the temperature-assisted deactivation of triplet states at a higher temperature state (Fig. S28c†). At lower temperatures, this compound showed a longer PL decay time than at higher temperatures (Fig. S28d and Table S11†). At 298 K, rISC predominates over radiative and non-radiative decay from the triplet state, resulting in TADF emission.
Steady-state PL and triboluminescence characteristics of BNS were demonstrated by Neena et al. in 2017.64 However, they did not explore the delayed luminescence features of this compound. Hence, we studied the delayed luminescence of this compound in detail and compared the results with those of BNO. BNS exhibits a broad emission like BNO with a peak maximum at 520 nm and lifetime of ∼9.87 ns, which are comparable to the reported data (Fig. 5a and Table 2). The time-gated PL spectrum of BNS appeared at ∼510 nm, which is 10 nm blue-shifted compared with the prompt fluorescence (PF) spectrum (Fig. 5a). This band (∼510 nm) exhibited a lifetime of 91.03 μs in contrast to the ns lifetime of PF (Fig. 5f and Table 2). The PL intensity and lifetime of delayed bands are sensitive to oxygen, and thus, this band has been ascribed to phosphorescence (PH) (Fig. 5e, f and S29†).
Furthermore, the PF and PH lifetimes of this compound were recorded at different temperatures (Fig. S30 and Table S11†). The intensity of both PF and PH bands increased steadily as the temperature decreased from 298 to 77 K. Furthermore, the peak maximum of the PH is ∼5 nm red-shifted at 77 K than at 298 K. However, such a spectral shift is absent for PF, indicating that temperature-dependent structural reorganization affects the triplet manifold, not the singlet (Fig. S30a and c†). Furthermore, the lifetime of PF showed marginal changes with respect to temperature; however, the PH lifetime steeply increased from microseconds to hundreds of milliseconds (Fig. S30d and Table S11†). These results indicate that BNS shows blue-shifted RTP, a phenomenon very rarely demonstrated in the literature.87–91 This blue-shifted RTP band can be attributed to PH from a higher energy triplet state (Tn), which is at slightly higher energy than S1 and has a significantly larger energy gap from the T1 state. Moreover, the inherent conformation-switching properties of phenothiazine between quasi-axial and quasi-equatorial at the excited state can also result in re-organizing electronic states. This hypothesis is well-supported by the computational studies (vide infra) detailed in the last sub-section.68–75
 | ||
| Fig. 6 Steady-state PL spectra for (a) BNO and (d) BNS doped into PMMA at different doping concentrations and neat film at λex = 330 nm under vacuum (in the absence of oxygen). Prompt and time-gated/delayed [30 μs delay] PL spectra of (b) BNO and (e) BNS doped into PMMA (1wt%) and neat film at λex = 330 nm under vacuum (in the absence of oxygen). (c) Delayed fluorescence decay for BNO doped into PMMA at different doping concentrations and neat film at λex = 330 nm under vacuum (in the absence of oxygen). (f) Phosphorescence decay for BNS doped into PMMA at different doping concentrations and neat film at λex = 330 nm under vacuum (in the absence of oxygen). | ||
The PF maxima of films doped with (1, 10, and 50 wt%) BNO (λem = 568 nm) and BNS (λem = 575 nm) show a bathochromic shift compared to their respective solids (Fig. 6a and d). However, the spectral shift is more significant in BNS (55 nm) than in BNO (23 nm). The PF band intensity in BNO increased when the doping concentration increased from 1 wt% to 10 wt%, while the PL intensity decreased for 50 wt% doping. In contrast, for BNS, the PL intensity gradually increased when increasing the doping concentration from 1 wt% to 50 wt% (Fig. 6a and d). The PF of neat films of both compounds show a hypsochromic shift (545 nm for BNO and 500 nm for BNS) compared to their doped films; however, BNS showed a stronger blue shift (75 nm) than BNO (23 nm) (Fig. 6b and e). Interestingly, the neat film of BNO showed brighter luminescence than doped films (ΦTotal = 0.3, 0.48, 0.5, and 0.63 for 1 wt%, 10 wt%, 50 wt%, and neat, respectively). On the other hand, the neat film of BNS was less luminescent (ΦTotal = 0.60) than 50 wt% (ΦTotal = 0.87) doped film and higher than 10 wt% (ΦTotal = 0.54) and 1 wt% doped film (ΦTotal = 0.41). Furthermore, the PF of the neat film of BNO matched exactly with the PF of the solid (545 nm). In contrast, the BNS (500 nm) showed a 20 nm blue shift compared to its solid PF (520 nm). The PF lifetime of these compounds falls within the ns region confirming the fluorescence nature of these bands. Moreover, the PF lifetime values follow the same trend observed for their respective PF intensity values (Tables S12 and S14†).
The DF intensity of doped (1, 10, and 50 wt%) and neat films of BNO, exhibited a similar trend that was observed for PF intensities. In contrast, the doped films (1, 10, and 50 wt%) of BNS show blue-shifted PH, and the peak intensity follows the same trend as that observed for PF (Fig. 6b and e). The extent of the hypsochromic spectral shift is susceptible to the doping concentrations; the maximum spectral shift was observed for 1 wt% doping (25 nm). Interestingly, the PH of the neat film of BNS (535 nm) shows a bathochromic shift compared to its PF (500 nm) and PH of crystals (500 nm). The DF and PH of neat and doped films of BNO and BNS exhibit PL lifetimes in the μs range; furthermore, these lifetimes decrease upon increasing the doping concentration (Fig. 6c and f). The PL lifetime of the neat film of both compounds was higher than that of doped films and lower than their respective solids.
The PF and DF (BNO) or PH (BNS) of these compounds were recorded at different temperatures, under ambient (in the presence of O2) and vacuum (in the absence of O2) conditions at RT (298 K) (Fig. S33–S35 and S38–40†). The PL intensity of BNO and BNS are sensitive to oxygen and showed stronger luminescence under vacuum. The PF and DF peak intensity of both BNO and BNS increased as the temperature decreased from 298 K to 77 K. However, in both cases, the change in peak intensity is more pronounced for DF/PH compared to their respective PF intensity.
The PH band of thin films (doped and neat films) of BNS shows a blue shift as the temperature decreases from 298 to 77 K. Unlike in the crystalline state, the PF of neat films of BNS also shows a hypsochromic shift at low temperatures. Such spectral shifts are not observed in thin films of BNO. The temperature-dependent PF and PH of BNS can be attributed to the temperature-dependent preferential stabilization of its conformer. Moreover, the unprecedented shift in the PF and PH band in BNS upon changing the doping concentration from 1 wt% to neat can be attributed to perturbations in the dynamics between different molecular conformers (quasi-axial and quasi-equatorial), which alter the energy of the singlet and triplet manifold.68–75 The PL lifetimes recorded for BNO and BNS at different temperatures show that both the PF and DF or PH are progressively increased upon lowering the temperature (Fig. S33f, S35f, S38f, and S40f†). However, the quantum of increment is considerably higher for the DF and PH than it is for their PF, which can be attributed to the blocking of non-radiative decay of triplet manifolds at lower temperatures. Furthermore, the DF in BNO and PH in BNS clearly suggest that the stabilization of the triplet manifold and the rate of the rISC process highly depend on the heteroatom attached to the cyclic amine donor.53–55,92 This conclusion is further augmented by computational results discussed vide-infra.
 | ||
| Fig. 7 Energy level diagrams showing plausible singlet to triplet transitions (ISC/rISC), the singlet–triplet energy gap, spin–orbit coupling constant (ξSOC), and geometry of the corresponding electronic states for (a) BNO and (b) BNS. FL = fluorescence, DF = delayed fluorescence, PHI = blue-shifted phosphorescence (doped film and solid), and PHII = red-shifted phosphorescence (neat film). Energy levels in the diagram are not to scale and obtained from the vertical transition calculations using TD-DFT and the B3LYP/6-31G(d) level of theory. Geometry optimization is done using DFT/TD-DFT at the same level of theory by taking coordinates from crystal data and after single-point energy calculations. Natural transition orbitals for (c) BNO and (d) BNS at the S1, T5, and T6 states. | ||
The energy levels of BNO and BNS, derived from singlet and triplet vertical transition calculations, are illustrated in Fig. 7a and b (Fig. S45 and Tables S18, S19†). It is quite evident from the diagram that the ΔES1–T1 in BNO (0.67 eV) and BNS (0.70 eV) is relatively large, making efficient intersystem crossing (ISC) challenging. However, higher triplet states (Tn), specifically T5 for BNO and T6 for BNS, are energetically closer to S1 (ΔES1–Tn < 150 meV), making them favorable for efficient ISC. The experimental ΔEST value determined from the spectral onset values further supports the claim (Table S17; see the ESI† for more details).
For BNO, T5 is energetically closer to S1 (ΔES1–T5 = 140 meV) and exhibits a significant spin–orbit coupling constant (ξSOC) of 0.1910 cm−1. The substantial energy difference between T5 and T4 favors efficient reverse intersystem crossing (rISC) rather than radiative or non-radiative decay from T5 and drives delayed fluorescence at room temperature (Fig. 7a, S45a and Table S18†). Moreover, the natural transition orbital (NTO) analysis shows that T5 has a pronounced locally excited (LE) character, whereas S1 has a pure charge-transfer (CT) character. This difference in symmetry further promotes ISC and rISC (El-Sayed's rule) (Fig. 7c and S43†).56,57
In BNS, T5 and T6 are energetically close to S1 through which efficient spin crossover is possible. Among these two states, T6 is slightly higher in energy than S1 (ΔES1–T6 = 10 meV) but has a considerably higher ξSOC (0.7679 cm−1) than T5 (0.4358 cm−1), and is about four times higher than that of BNO, leading to efficient ISC to T6 (Fig. 7b, S45b and Table S18†). This large difference in SOC can be attributed to the heavy atom effect of S over O. Additionally, the large energy gap between T5 and T4 reduces the chances of non-radiative decay, confining competing processes to rISC (T6 → S1) and radiative decay from T6 → S0 or T5 → S0. The ξSOC calculated for S0–T6 is nearly three times higher than that of S1–T6, resulting in a high-energy (blue-shifted) phosphorescence (PHI), especially in a rigid isolated environment. While in the neat film, non-radiative deactivation of the T6 → T5 state occurred due to molecular collision at high compound concentration, followed by emission from T5 (red-shifted phosphoresce – PHII) (Fig. 7b and S45b†). In addition, BNS in T5 and T6 adopted a quasi-axial conformation with a prominent LE character, which is different from the S1 geometry (quasi-equatorial with pure CT character) further facilitating ISC and phosphorescence (Fig. 7d and S44†).
In short, the large energy gap between the S1 and the T1 states in BNO and BNS results in the spin crossover (ISC and rISC) occurring via the energetically favorable T5 or T6 states. In BNO, efficient rISC occurs under favorable energy conditions at room temperature, leading to TADF. In contrast, for BNS, due to the large energy gap between T6 or T5 and T4 combined with the higher SOC between S0 and T6, a radiative emission from T6 is favored, producing blue-shifted phosphorescence in isolated rigid environments (solid and doped films). Additionally, the involvement of different conformations in excited states (especially in BNS) causes these states to vary their energetics with respect to the microenvironment and molecular interactions.68–75 This sensitivity may explain the anomalous shift in the phosphorescence band observed in thin films as the doping concentration increases from 1 wt% to 100 wt%, leading to unprecedented phosphorescent behaviour.
Conclusion
In conclusion, we investigated the delayed luminescence properties of aminoboranes BNO and BNS with phenoxazine (PXZ) or phenothiazine (PTZ) donors and dixylylborane acceptors. BNO and BNS exhibit broad emission in solution (450–800 nm) with large Stokes shifts and low PLQYs due to the excited state conformational changes of nonplanar cyclic amine donors. BNO shows TADF in the solution state; however, BNS does not show TADF, which can be attributed to the combined heavy atom (S) and the extensive flip-flop motion of phenothiazine-mediated non-radiative triplet deactivation in dilute solutions. Both the compounds exhibit AIEE and DF in aggregates due to the restriction of intramolecular motion. The aggregates of BNO demonstrate tunable emission from greenish-yellow to orange-red and efficient TADF in solid and thin films. In contrast, BNS shows rare, blue-shifted phosphorescence in solid and doped films. The extent of this blue shift in phosphorescence compared to the prompt fluorescence depends on the doping concentration, confirming the role of intermolecular interactions in stabilizing the triplet state. On the other hand, neat films of BNS exhibit PTZ-centered, red-shifted phosphorescence. Excited-state computational studies reveal that these compounds undergo significant conformational changes, alternating between quasi-axial and quasi-equatorial forms. These dynamic conformations perturb the energetics of triplet and singlet states, particularly in BNS, which leads to unusual phosphorescence shifts. Additionally, ISC and rISC were found to occur via higher triplet states (T5 or T6) in both compounds. In BNO, low ΔES1–T5 favors rISC for efficient TADF emission. In BNS, the substantial energy gap between T6 or T5 and T4, combined with strong spin–orbit coupling between T6 and S0, results in unique, blue-shifted phosphorescence, which has not been observed for aminoboranes.Data availability
Details on the synthesis and structural characterization, photophysical and DFT and TD-DFT results are available in the ESI, and Crystallographic data available in CCDC (CCDC 2402292 for BNO).The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank IISc, SERB for the financial support and Prof K.R. Prasad, Department of Organic Chemistry for the HRMS facility. M. M. A. T. thanks CSIR India for the research fellowship. S. J. thanks IISc for the research fellowship.References
- Y. Yang, F. Gao, Y. Wang, H. Li, J. Zhang, Z. Sun and Y. Jiang, Molecules, 2022, 27, 8421 CrossRef PubMed.
- H.-Z. Li, F.-M. Xie, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2023, 11, 6471–6511 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef.
- K. R. Naveen, H. I. Yang and J. H. Kwon, Commun. Chem., 2022, 5, 149 CrossRef PubMed.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 160544 Search PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef.
- T. Zhang, Y. Xiao, H. Wang, S. Kong, R. Huang, V. Ka-Man Au, T. Yu and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301896 CrossRef PubMed.
- H. E. Hackney and D. F. Perepichka, Aggregate, 2022, 3, e123 CrossRef.
- A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
- Z. Y. Liu, J. W. Hu, C. H. Huang, T. H. Huang, D. G. Chen, S. Y. Ho, K. Y. Chen, E. Y. Li and P. T. Chou, J. Am. Chem. Soc., 2019, 141, 9885–9894 CrossRef PubMed.
- Y. Tao, C. Liu, Y. Xiang, Z. Wang, X. Xue, P. Li, H. Li, G. Xie, W. Huang and R. Chen, J. Am. Chem. Soc., 2022, 144, 6946–6953 CrossRef PubMed.
- R. Wang, Y. Zhu, Z. Xia, K. Liang, L. Kong, J. Liu, W. Shi and C. Lu, J. Mater. Chem. C, 2022, 10, 17182–17189 RSC.
- Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Würthner, C. M. Marian, L. Ji and T. B. Marder, Angew. Chem., Int. Ed., 2020, 59, 17137–17144 CrossRef PubMed.
- Y. Huo, J. Lv, Y. Xie, L. Hua, Y. Liu, Z. Ren, T. Li, S. Ying and S. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 57092–57101 CrossRef PubMed.
- U. P. Pandey, R. P. Nandi and P. Thilagar, Front. Chem., 2020, 8, 541331 CrossRef PubMed.
- U. P. Pandey and P. Thilagar, Adv. Opt. Mater., 2020, 8, 1902145 CrossRef.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Inorg. Chem., 2020, 59, 3142–3151 CrossRef PubMed.
- S. Jena, P. Dhanalakshmi, G. Bano and P. Thilagar, J. Phys. Chem. B, 2020, 124, 5393–5406 CrossRef PubMed.
- S. K. Sarkar, M. Pegu, S. K. Behera, S. K. Narra and P. Thilagar, Chem. – Asian J., 2019, 14, 4588–4593 CrossRef PubMed.
- K. K. Neena, P. Sudhakar and P. Thilagar, Angew. Chem., 2018, 130, 17048–17052 CrossRef.
- S. R. Pristash, K. L. Corp, E. J. Rabe and C. W. Schlenker, ACS Appl. Energy Mater., 2020, 3, 19–28 CrossRef.
- T. Serevičius, R. Komskis, P. Adomènas, O. Adomènienè, G. Kreiza, V. Jankauskas, K. Kazlauskas, A. Miasojedovas, V. Jankus, A. Monkman and S. Juršènas, J. Phys. Chem. C, 2017, 121, 8515–8524 CrossRef.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef PubMed.
- D. Hertel, H. Bässler, R. Guentner and U. Schert, J. Chem. Phys., 2001, 115, 10007–10013 CrossRef.
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882–2886 CrossRef.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 10, 678–668 CrossRef.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef.
- D. G. Congrave, B. H. Drummond, P. J. Conaghan, H. Francis, S. T. E. Jones, C. P. Grey, N. C. Greenham, D. Credgington and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 18390–18394 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef.
- W. Chi, J. Chen, W. Liu, C. Wang, Q. Qi, Q. Qiao, T. M. Tan, K. Xiong, X. Liu, K. Kang, Y. T. Chang, Z. Xu and X. Liu, J. Am. Chem. Soc., 2020, 142, 6777–6785 CrossRef.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510–515 CrossRef.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef.
- S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef.
- D. G. Chen, T. C. Lin, C. L. Chen, Y. T. Chen, Y. A. Chen, G. H. Lee, P. T. Chou, C. W. Liao, P. C. Chiu, C. H. Chang, Y. J. Lien and Y. Chi, ACS Appl. Mater. Interfaces, 2018, 10, 12886–12896 CrossRef.
- Y. J. Lien, T. C. Lin, C. C. Yang, Y. C. Chiang, C. H. Chang, S. H. Liu, Y. T. Chen, G. H. Lee, P. T. Chou, C. W. Lu and Y. Chi, ACS Appl. Mater. Interfaces, 2017, 9, 27090–27101 CrossRef PubMed.
- P. Ganesan, D. G. Chen, W. C. Chen, P. Gnanasekaran, J. A. Lin, C. Y. Huang, M. C. Chen, C. S. Lee, P. T. Chou and Y. Chi, J. Mater. Chem. C, 2020, 8, 4780–4788 RSC.
- J. Zhi, Q. Zhou, H. Shi, Z. An and W. Huang, Chem. – Asian J., 2020, 15, 947–957 CrossRef.
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao and J. Zhang, Adv. Sci., 2021, 8(24), 2102970 CrossRef.
- J. R. Caine, P. Hu, A. T. Gogoulis and Z. M. Hudson, Acc. Mater. Res., 2023, 4, 879–891 CrossRef.
- J. You, X. Zhang, Q. Nan, K. Jin, J. Zhang, Y. Wang, C. Yin, Z. Yang and J. Zhang, Nat. Commun., 2023, 14, 4163 CrossRef.
- Y. Li and P. Gao, Chemosensors, 2023, 11, 489 CrossRef.
- R. Arumugam, A. T. M. Munthasir, R. Kannan, D. Banerjee, P. Sudhakar, V. R. Soma, P. Thilagar and V. Chandrasekhar, Chem. Sci., 2024, 15, 18364–18378 RSC.
- M. Ji and X. Ma, Ind. Chem. Mater., 2023, 1, 582–594 RSC.
- D. Li, J. Guo, L. Zhao, G. Zhang and G. Yan, RSC Adv., 2019, 9, 31953–31959 RSC.
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge and Z. M. Hudson, ACS Appl. Mater. Interfaces, 2020, 12, 20000–20011 CrossRef PubMed.
- F.-Y. Meng, I.-H. Chen, J.-Y. Shen, K.-H. Chang, T.-C. Chou, Y.-A. Chen, Y.-T. Chen, C.-L. Chen and P.-T. Chou, Nat. Commun., 2022, 13, 797 CrossRef PubMed.
- X. Chen, J. Zhang, C. Liu, Q. Lou, K. Zheng, X. Yin, L. Xie, P. Wen, C. Liu and Z. Ge, ACS Appl. Energy Mater., 2021, 4, 11112–11120 CrossRef.
- Y. Meng, J. Zhang, C. Liu, K. Zheng, L. Xie, S. Bu, B. Han, R. Cao, X. Yin, C. Liu and Z. Ge, Adv. Funct. Mater., 2023, 33, 2210600 CrossRef.
- D. R. Lee, K. H. Lee, W. Shao, C. L. Kim, J. Kim and J. Y. Lee, Chem. Mater., 2020, 32, 2583–2592 CrossRef.
- U. Balijapalli, M. Tanaka, M. Auffray, C. Y. Chan, Y. T. Lee, Y. Tsuchiya, H. Nakanotani and C. Adachi, ACS Appl. Mater. Interfaces, 2020, 12, 9498–9506 CrossRef PubMed.
- S. Jena, S. K. Behera, J. Eyyathiyil, M. Kitahara, Y. Imai and P. Thilagar, Adv. Opt. Mater., 2023, 11, 2300923 CrossRef.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef PubMed.
- J. Eng and T. J. Penfold, Chem. Rec., 2020, 20, 831–856 CrossRef PubMed.
- N. Aizawa, Y. Harabuchi, S. Maeda and Y. J. Pu, Nat. Commun., 2020, 1, 3909 CrossRef PubMed.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- L. Xu, K. Zhou, X. Qiu, B. Rao, D. Pei, A. Li, Z. An and G. He, J. Mater. Chem. C, 2020, 8, 14740–14747 RSC.
- R. P. Nandi, N. K. Kalluvettukuzhy, S. Pagidi and P. Thilagar, Inorg. Chem., 2023, 62, 1122–1134 CrossRef PubMed.
- K. K. Neena, P. Sudhakar and P. Thilagar, Angew. Chem., 2018, 130, 17048–17052 CrossRef.
- K. K. Neena, P. Sudhakar, K. Dipak and P. Thilagar, Chem. Commun., 2017, 53, 3641–3644 RSC.
- P. Sudhakar, K. K. Neena and P. Thilagar, J. Mater. Chem. C, 2017, 5, 6537–6546 RSC.
- N. K. Kalluvettukuzhy, P. Sudhakar, J. Eyyathiyil, N. Hara, Y. Imai and P. Thilagar, Org. Lett., 2023, 25, 6067–6071 CrossRef PubMed.
- K. K. Neena and P. Thilagar, J. Mater. Chem. C, 2016, 4, 11465–11473 RSC.
- R.-N. Su, Q.-Q. Pan, G.-Y. Ding, J. Sun, L.-L. Wen, K.-Z. Shao, S.-B. Wang, G.-G. Shan and Z.-M. Su, J. Mater. Chem. C, 2023, 11, 9908–9915 RSC.
- Y. Tian, J. Yang, A. Li, J. Ren, X. Li, M. Fang and Z. Li, ACS Mater. Lett., 2024, 6, 943–953 CrossRef.
- Q. Zhou, C. Yang and Y. Zhao, Chem, 2023, 9, 2446–2480 Search PubMed.
- F. Khan and R. Misra, J. Mater. Chem. C, 2023, 11, 2786–2825 RSC.
- M. Gao, Y. Tian, X. Li, Y. Gong, M. Fang, J. Yang and Z. Li, Angew. Chem., Int. Ed., 2023, 5, e202214908 Search PubMed.
- M. Gao, Y. Tian, J. Yang, X. Li, M. Fang and Z. Li, J. Mater. Chem. C, 2021, 9, 15375–15380 RSC.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 40, 2101719 CrossRef.
- T. Song, H. Liu, J. Ren and Z. Wang, Adv. Opt. Mater., 2024, 1, 2301215 CrossRef.
- C. A. Swami and P. Thilagar, Inorg. Chem., 2014, 53, 2776–2786 CrossRef PubMed.
- S. K. Sarkar, S. E. Rao and P. Thilagar, J. Phys. Chem. B, 2020, 124, 8896–8903 CrossRef PubMed.
- P. Thilagar, R. P. Nandi and S. Ghosh, Chem. – Eur. J., 2024, 30, e202400398 CrossRef PubMed.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Organometallics, 2018, 37, 1900–1909 CrossRef.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef.
- X. Lu, Y. Hu, H. Liu, J. Qi, X. Chen, Y. Sun and W. Hu, Adv. Opt. Mater., 2022, 20, 2201075 CrossRef.
- J. Xu, X. Zhu, J. Guo, J. Fan, J. Zeng, S. Chen, Z. Zhao and B. Z. Tang, ACS Mater. Lett., 2019, 1, 613–619 CrossRef.
- N. Aizawa, C.-J. Tsou, I. S. Park and T. Yasuda, Polym. J., 2017, 49, 197–202 CrossRef.
- D. Barman and P. K. Iyer, J. Phys. Chem. C, 2023, 127, 2694–2704 CrossRef.
- B. Zhang, Y. Kong, H. Liu, B. Chen, B. Zhao, Y. Luo, L. Chen, Y. Zhang, D. Han, Z. Zhao, B. Z. Tang and L. Niu, Chem. Sci., 2021, 12, 13283–13291 RSC.
- K. Veys and D. Escudero, Acc. Chem. Res., 2022, 55, 2698–2707 CrossRef.
- W. Xie, W. Huang, J. Li, Z. He, G. Huang, B. S. Li and B. Z. Tang, Nat. Commun., 2023, 14, 8098 CrossRef.
- S. Gao, J. Ding, S. Yu and F. Li, J. Mater. Chem. C, 2023, 11, 6400–6406 RSC.
- Z. Zhu, Z. Kuang, L. Shen, S. Wang, X. Ai, A. Abdurahman and Q. Peng, Angew. Chem., Int. Ed., 2024, e202410552 Search PubMed.
- B. H. Jhun, D. Y. Jeong, S. Nah, S. Y. Park and Y. You, J. Phys. Chem. C, 2021, 9, 7083–7093 Search PubMed.
- S. Jena, J. Eyyathiyil, S. K. Behera, M. Kitahara, Y. Imai and P. Thilagar, Chem. Sci., 2022, 13, 5893–5901 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2402292. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03200d |
| This journal is © The Royal Society of Chemistry 2025 |