
A hydrogen-bridged bis(silylene) complex as a platform for small molecule activation via adjacent metal–ligand cooperation†
Hisako
Hashimoto
 *,
Satomi
Matsuo
,
Yuto
Odagiri
and
Hiromi
Tobita
*,
Satomi
Matsuo
,
Yuto
Odagiri
and
Hiromi
Tobita
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: hisako.hashimoto.b7@tohoku.ac.jp
First published on 13th December 2024
Abstract
Effective activation of small molecules (alcohol, phenol, ketones, amine, etc.) by a hydrogen-bridged bis(silylene) complex, which has two adjacent Lewis acidic centers and a hydridic bridging hydrogen with weak Si⋯H bonds, is described, along with product characterization by NMR and X-ray diffraction studies.
Activation of small molecules is one of the leading topics in current chemistry. Recent extensive studies in this field have increasingly provided new strategies beyond the traditional activation that relies on coordinatively unsaturated transition-metal (TM) centres. Activation via frustrated Lewis acid–base pairs (FLP)1–3 and bifunctional low-valent main group element species4–8 and activation via metal–metal9,10 and metal–ligand cooperation11,12 have emerged as promising new strategies for molecular activation. In particular, the metal–ligand cooperative activation has been found in a variety of complexes in the past decade, from originally suggested complexes where a metal centre and the active moiety of the ligand are indirectly connected,11,12 to those that have a direct metal–ligand bond, e.g., metal–tetryl double13,14 and triple15–17 bonds between TM and heavier group 14 elements.
In this regard, we have previously reported the synthesis and characterization of hydrogen-bridged bis(silylene) complexes, which have novel four-centre bonding interaction and can be described as two canonical forms where the Si–H is activated at the adjacent silylene ligand by coordinating its σ-bonding electrons18,19 (Fig. 1). These complexes have two Lewis acidic silicon centres close to each other and a hydridic bridging hydrogen atom, as suggested by detailed DFT calculations, which inspired us to investigate the reactivity toward small molecules. Here we report the reactions of Cp*W(CO)2{Me2Si⋯H⋯Si(tBu)Me} (1), which was previously synthesised,18 with several small nucleophiles (alcohol, phenol, ketones, amine, etc.) and product characterization including single-crystal X-ray diffraction (SC-XRD) analysis of five complexes.
 | ||
| Fig. 1 Description of hydrogen-bridged bis(silylene) complexes: charge distribution and the Si–H bond activation form on the adjacent silylene ligand. R = alkyl groups. | ||
Treatment of 1 with 1 equiv. of phenol at room temperature almost instantaneously led to an air-sensitive phenoxy-bridged bis(silylene) complex Cp*W(CO)2{Me2Si⋯O(Ph)⋯Si(tBu)Me} (2) in 91% NMR yield, accompanied by evolution of H2 (4.46 ppm in C6D6) (eqn (1)). The use of p-cresol instead of phenol led to an analogous complex Cp*W(CO)2{Me2Si⋯O(p-tol)⋯Si(tBu)Me} (3). Complexes 2 and 3 were isolated in 68% and 65% yields, respectively, after recrystallisation and characterised by 1H, 13C and 29Si NMR, IR spectroscopy and elemental analysis. The appearance of moderately downfield-shifted 29Si NMR resonances (108.0 and 121.8 ppm) of 2 is indicative of the formation of base-stabilised silylene ligands,20–25 and these resonances are comparable to those of the methoxy-bridged bis(silylene) complex Cp*W(CO)2{Me2Si⋯O(Me)⋯Si(tBu)Me} (A) (97.5 ppm for SiMe2 and 111.4 ppm for SiMetBu).18 Thus, as expected, 1 reacted with weak protic acids at the bridging hydrogen moiety, in line with its hydridic nature previously reported, resulting in the replacement of the bridging hydrogen atom with the aryloxy group by releasing dihydrogen. Complex 1 also reacted with iPrOH just as MeOH18 despite being bulkier, to afford a similar complex Cp*W(CO)2{Me2Si⋯O(iPr)⋯Si(tBu)Me} (4). The extremely easy and clean occurrence of the reaction and concomitant dihydrogen evolution can be notable features.
 | (1) |
Remarkably, the exactly same complex 4 was formed in an excellent yield of 75% by the treatment of 1 with acetone at room temperature (Scheme 1). This reaction is viewed as one-pot hydro-bis(silylation) of the carbonyl compound, which is unprecedented, to the best of our knowledge. This reaction can also be viewed as the insertion of the carbonyl compound into the Si–H–Si moiety in the WSiHSi ring, demonstrating a new pattern of insertion: the two ligand moieties located next to each other acted as a cooperative multi-site activation platform toward the substrate, enabling the one-pot hydro-bis(silylation) reaction. During the reaction, the bridging hydrogen atom of 1 was efficiently and rapidly transferred onto the carbonyl carbon atom, implying a concerted process. It should be noted here that there are several examples of insertion of olefins26–28 and carbonyl compounds29,30 into the Si–H bond of hydrosilylene complexes with a Si(sp2)–H bond. In the former cases, a concerted mechanism based on direct addition of the olefin C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to the Si(sp2)–H bond is proposed, while several mechanisms are discussed for the latter cases.
C bond to the Si(sp2)–H bond is proposed, while several mechanisms are discussed for the latter cases.
 | ||
| Scheme 1 Reactions of 1 with ketones. | ||
A similar reaction occurred on using adamantanone with the quantitative formation of Cp*W(CO)2{Me2Si⋯O(Ad)-⋯Si(tBu)Me} (5) (Ad = adamantyl). Complexes 4 and 5 were characterized based on multiple spectroscopic data and the structure of 5 was confirmed by SC-XRD analysis. The molecular structure of 5 is illustrated in Fig. 2, which clearly reveals the WSiOSi four-membered-ring structure with the O(Ad) group bridging over the two silicon moieties. The W–Si bond lengths [2.535(3), 2.500(3) Å] are slightly longer than those of a previously reported methoxy-bridged tungsten complex CpW(CO)2{Me2Si⋯O(Me)⋯SiMe2} [2.490(3), 2.490(3) Å],31 but are very close to those of complex A [2.537(3), 2.509(3) Å] (see Fig. S26 in the ESI†), which has exactly the same bis(silylene) moieties as those of 5. The Si–O bond lengths [1.871(8), 1.816(8) Å] at the bridging moiety of 5 are significantly longer than those of usual Si–O single bonds (1.63–1.65 Å),32 evidently indicating their coordination character. In the 29Si NMR spectrum of 5, two resonances were observed at 99.3 and 110.2 ppm, accompanied by clear satellite JWSi couplings of 83 and 117 Hz, respectively (Table 1). The chemical shift values of 5 are close to those of A and 2. The spin–spin coupling constants JWSi, which reflect the s-character of the nuclei involved in the bond, are nearly intermediate between those of the silyl (sp3 Si) and silylene (sp2 Si) ligands: for instance, 30.5 Hz for WSiMe3 and 154.9 Hz for W = SiMes2 in Cp*W(CO)2(SiMes2)(SiMe3).33 All these features are characteristics of base-stabilised bis(silylene) complexes.24
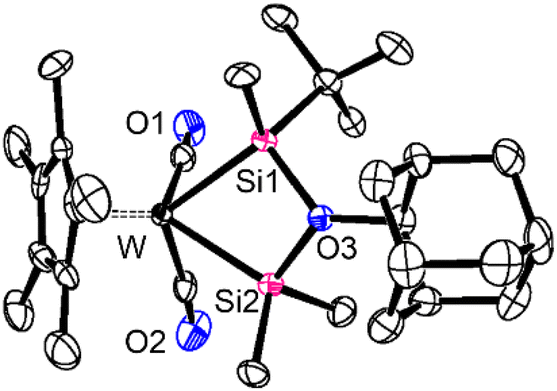 | ||
| Fig. 2 Crystal structure of Cp*W(CO)2{Me2Si⋯O(Ad)⋯Si(tBu)Me} (5). Selected interatomic distances (Å): W–Si1 2.535(3), W–Si2 2.500(3), Si1–O3 1.871(8), Si2–O3 1.817(8). Hydrogen atoms have been omitted for clarity. | ||
| Complex | 29Si[1H] NMR (JWSi)b | Complex | 29Si[1H] NMR (JWSi)b |
|---|---|---|---|
| a From ref. 18. b ppm (Hz). c Not obtained due to thermal instability. | |||
A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
97.5 (87.1) SiMe2, 111.4 (94.1) SiMetBu | 6 | 102.6, 115.3 |
| 2 | 108.0, 121.8 | 7 | —c |
| 3 | 106.9 (93), 126.4 (116) | 8 | 100.8, 117.7 |
| 4 | 99.3, 110.2 | 9 | 22.9 (118) SiHMetBu, 101.4 SiMe2 |
| 5 | 99.1 (82.9), 120.8 (116.9) | 10 | 26.9 (70.4) SiHMetBu, 86.7 (114.5) SiMe2←DMAP |
To further examine the reactivity, we also tested the reactions of 1 with carbonyl compounds bearing functional groups, i.e. a vinyl group or an ethynyl group. Although there are two possible products that can be formed via 1,2-addition or 1,4-addition, both reactions of 1 with methyl vinyl ketone and ethyl vinyl ketone selectively gave rise to Cp*W(CO)2{Me2Si⋯O[CHR′(CHCH2)]⋯Si(tBu)Me} (6: R′ = Et, 7: R′ = Me) in nearly quantitative yields. Thus, the 1,2-addition type of reaction solely occurred. In the case of 6, there were two diastereomeric isomers in the solution, and one of them was crystallised and structurally confirmed by SC-XRD analysis. The molecular structure of the crystal is depicted in Fig. 3(a), which clearly reveals its alkoxy-bridged bis(silylene) backbone with a four-membered ring. The main bond distances and angles quite resemble those of 5 (Table S2 in the ESI†). The presence of a vinyl group on the alkoxy bridging ligand is evidenced by the C21–C22 bond distance of 1.292(8) Å, which is considerably shorter than the normal C–C single bond distance of the ethyl group [C23–C24 [1.435(8) Å]]. In the 1H NMR spectrum, two sets of resonances for the major and minor diastereomers appeared. For instance, two resonances of Cp* were observed at 1.93 and 1.94 ppm with a 5:3 intensity. The Ha proton of the vinyl group (–CHaCHbHc) appeared with a pattern of ddd (doublet of doublet of doublet) at 5.66 ppm (major) and at 5.66 ppm (minor) with a good separation (see Fig. S13-2†). The other signals were rationally assigned using multiple techniques such as 1H–1H COSY and 1H–29Si{1H} COSY measurements (Fig. S14 and S16†). Complex 7 was extremely thermally unstable to decompose but its structure was supported by HRMS (high-resolution mass spectrum) and the similarity of the resonances in the 1H NMR (Fig. S18†) and IR (vCO) bands of 7 to those of 6 (see the ESI†).
 | ||
Fig. 3 Crystal structures of (a) Cp*W(CO)2{Me2Si⋯O[CHEt(CHCH2)]⋯Si(tBu)Me} (6) and (b) Cp*W(CO)2{Me2Si⋯O[CHMe(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)]⋯Si(tBu)Me} (8). Selected interatomic distances (Å): W–Si1 2.5188(9), W–Si2 2.5037(9), Si1–O3 1.828(3), Si2–O3 1.813(3), C21–C22 1.292(8), C23–C24 1.435(8) for 6; W–Si1 2.5236(13), W–Si2 2.5049(13), Si1–O3 1.829(4), Si2–O3 1.818(4), C11–C12 1.174(8) for 8. Hydrogen atoms have been omitted for clarity. CH)]⋯Si(tBu)Me} (8). Selected interatomic distances (Å): W–Si1 2.5188(9), W–Si2 2.5037(9), Si1–O3 1.828(3), Si2–O3 1.813(3), C21–C22 1.292(8), C23–C24 1.435(8) for 6; W–Si1 2.5236(13), W–Si2 2.5049(13), Si1–O3 1.829(4), Si2–O3 1.818(4), C11–C12 1.174(8) for 8. Hydrogen atoms have been omitted for clarity. | ||
Previously, Sakaba et al. demonstrated that an alkynyl moiety can be coordinated to the silylene ligand by its CC triple bond in the formation of Cp*(OC)2W(SiPh2)(CCtBu).34 If a similar coordination of the alkynyl moiety of 3-butyn-2-one to a silylene ligand is possible, we could expect the formation of a cyclic base-stabilized silyl(silylene) complex a (Scheme 2) by the reaction with 1. However, instead of the expected complex a, exclusive 1,2-addition at the CO bond occurred to form the four-membered-ring complex Cp*W(CO)2{Me2Si⋯O[CHMe(CCH)]–⋯Si(tBu)Me} (8). SC-XRD analysis [Fig. 3(b)] confirmed the structure of 8 possessing an ethynyl group on the alkoxy bridging ligand, as indicated by the very short C11–C12 distance [1.174(8) Å]. The 29Si NMR resonances that resemble those of 2–6 are consistent with its structure (Table 1).
 | ||
| Scheme 2 Reactions of 1 with conjugated molecules. | ||
Furthermore, we tested the reaction with a secondary amine to clarify whether the N–H bond can be activated or not in this system. No N–H bond activation of the amine occurred in the reaction with 1 unlike the O–H bond of alcohol and phenol. Instead, coordination of the N lone pair to the silylene moiety led to a base-stabilized silyl(silylene) complex without a ring structure, Cp*(OC)2W(SiHMetBu)-(SiMe2←NHEt2) (9). A similar simple coordination occurred on using a more basic substrate, dimethyl aminopyridine (DMAP), to afford Cp*(OC)2W(SiHMetBu)(SiMe2←DMAP) (10), whose structure was also confirmed by SC-XRD (Fig. S27 and Table S2†). Both complexes exhibited clearly two 29Si NMR resonances in very different fields at around 20–30 and 86–100 ppm for silyl and base-coordinated silylene ligands, respectively (Table 1). These examples indicate the convertibility of the bridging form of the SiHSi moiety of 1 into the open form, i.e. the hydrosilyl–silylene complex (Scheme 3).
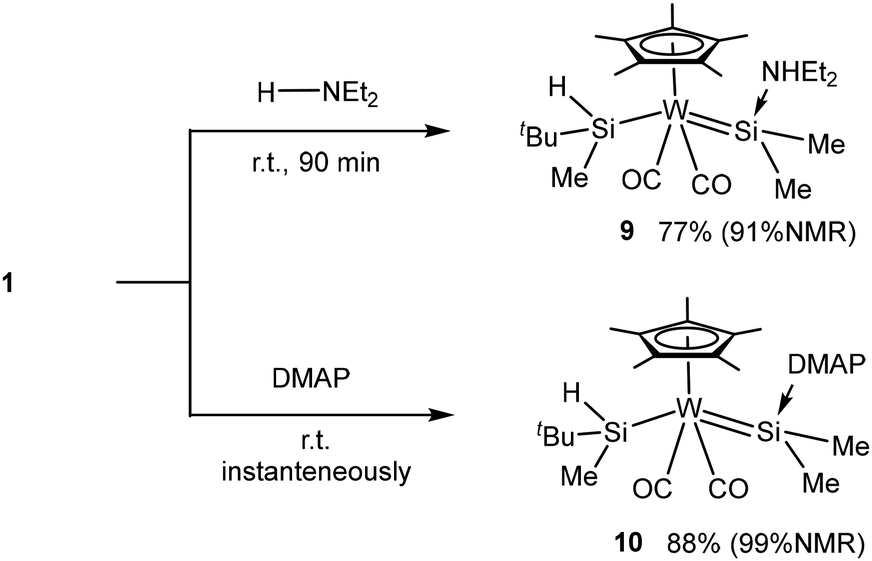 | ||
| Scheme 3 Reactions of 1 with diethylamine and DMAP. | ||
The mechanisms for these unique reactions of 1 with ROH and OCR2 are not clear yet at present but the main route can be considered as follows: it starts from the nucleophilic approach of the substrates to the silicon centre through their oxygen atoms. Then, the O–H bond of ROH or the OC bond of OCR2 interacts with the activated Si⋯H bond, followed by evolution of H2 (for the former case) or H-migration to the carbonyl carbon (for the latter case). As another possible H-migration process for carbonyl compounds, the approach of the substrate to the silicon centre gives rise to a hydrosilyl(R2CO→silylene) complex, from which H-migration occurs through a 6-membered-ring transition state to form an (alkoxysilyl)silylene complex. Finally, the alkoxyl group on the silyl ligand bridges over the silylene ligand to afford the four-membered-ring products. During the processes, it seems that the two adjacent Lewis acidic unsaturated silicon centres and a hydridic hydrogen atom function cooperatively in transforming the substrates.
In summary, complex 1 bearing a hydrogen-bridged bis(silylene) moiety displayed remarkable reactivity toward various small molecules (phenol, alcohol, carbonyl compounds, amine, DMAP). All reactions proceeded almost instantaneously and quantitatively at room temperature. Carbonyl compounds were hydro-bis-silylated selectively at the CO bond. These results demonstrate that the hydrogen-bridged bis(silylene) complex can provide a new platform for the efficient activation of small molecules via a new type of adjacent multi-site metal–ligand cooperation. Further investigations on the detailed reaction mechanism and dynamic behaviour of 1 by theoretical calculations are in progress.
Data availability
Electronic supplementary information (ESI) available: synthetic and experimental procedures, characterisation data, details of X-ray crystallographic analysis and NMR spectra. CCDC 2403405 (for A), 2403406 (for 5), 2403407 (for 6), 2403408 (for 8) and 2403409 (for 10) can be obtained from CCDC (https://www.ccdc.cam.ac.uk/structures/).The data supporting this article have been included in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grants JP19K05490, JP22K05123 and JP22H02088 from the Japan Society for the Promotion of Science (JSPS). We are also grateful to the Research and Analytical Centre for Giant Molecules (Tohoku University) for mass and elemental analyses.References
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- D. W. Stephane and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592 RSC.
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- D. Martin, D. M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389 RSC.
- K. Oberdorf and C. Lichtenberg, Chem. Commun., 2023, 59, 8043 RSC.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS.
- L. Wang, Y. Li, Z. Li and M. Kira, Coord. Chem. Rev., 2022, 457, 214413 CrossRef CAS.
- Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839 RSC.
- R. I. Petrikat, S. T. Steiger, E. Barani, P. J. Boden, M. E. Huber, M. R. Ringenberg, G. Niedner-Schatteburg, K. Fink and S. Becker, Chem. – Eur. J., 2023, 29, e202203438 CrossRef CAS PubMed.
- T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393 CrossRef CAS PubMed.
- W. A. Nugent and J. M. Mayer, in Metal-Ligand Multiple Bonds, Wiley, New York, 1988, p. 334 Search PubMed.
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 3488 CrossRef CAS PubMed.
- J. Schoening, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2023, 26, e202200638 CrossRef CAS.
- H. Hashimoto and H. Tobita, Coord. Chem. Rev., 2018, 355, 362 CrossRef CAS.
- S. Saini, A. Agarwal and S. K. Bose, Dalton Trans., 2020, 49, 17055 RSC.
- H. Hashimoto and K. Nagata, Chem. Lett., 2021, 50, 778 CrossRef CAS.
- H. Hashimoto, Y. Odagiri, Y. Yamada, N. Takagi, S. Sakaki and H. Tobita, J. Am. Chem. Soc., 2015, 137, 158 CrossRef CAS PubMed.
- H. Hashimoto, K. Komura, T. Ishizaki, Y. Odagiri and H. Tobita, Dalton Trans., 2017, 46, 8701 RSC.
- K. Ueno, H. Tobita, M. Shimoi and H. Ogino, J. Am. Chem. Soc., 1988, 110, 4092 CrossRef CAS.
- H. Tobita, K. Ueno, M. Shimoi and H. Ogino, J. Am. Chem. Soc., 1990, 112, 3415 CrossRef CAS.
- M. D. Curtis and P. S. Epstein, Adv. Organomet. Chem., 1981, 19, 213 CrossRef CAS.
- H. K. Sharma and K. H. Pannell, Chem. Rev., 1995, 95, 1351 CrossRef CAS.
- H. Ogino, Chem. Rec., 2002, 2, 291 CrossRef CAS PubMed.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2003, 493 RSC.
- P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640 CrossRef CAS PubMed.
- P. G. Hayes, C. Beddie, M. B. Hall, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 428 CrossRef CAS PubMed.
- E. Calimano and T. Don Tilley, J. Am. Chem. Soc., 2009, 131, 11161 CrossRef CAS PubMed.
- M. Ochiai, H. Hashimoto and H. Tobita, Dalton Trans., 2009, 1812 RSC.
- E. Calimano and T. Don Tilley, Organometallics, 2010, 29, 1680 CrossRef CAS.
- K. Ueno, A. Masuko and H. Ogino, Organometallics, 1997, 16, 5023 CrossRef CAS.
- W. S. Sheldrick, in The Chemistry of Organic Silicon Compounds, ed. S. Patai and Z. Rappoport, John Wiley & Sons, New York, 1989, ch. 3, vol. 2 Search PubMed.
- K. Ueno, S. Asami, N. Watanabe and H. Ogino, Organometallics, 2002, 21, 1326 CrossRef CAS.
- H. Sakaba, M. Yoshida and C. Kabuto, J. Am. Chem. Soc., 2005, 127, 7276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures, characterisation data, details of X-ray crystallographic analysis and NMR spectra. CCDC 2403405 (for A), 2403406 (for 5), 2403407 (for 6), 2403408 (for 8) and 2403409 (for 10). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03226h |
| This journal is © The Royal Society of Chemistry 2025 |