DOI:
10.1039/D4DT03382E
(Paper)
Dalton Trans., 2025,
54, 4518-4529
Synthesis and characterization of In(III) S-thiobenzoylthioglycolate complexes and their catalytic applications in CO2 fixation and multicomponent synthetic reactions†
Received
5th December 2024
, Accepted 28th January 2025
First published on 29th January 2025
Abstract
Indium(III) complexes of S-thiobenzoylthioglycolate (Stbtg) with nitrogen-donor ligands, such as 2,2′-bipyridyl and 1,10-phenanthroline, have been synthesized. The complexes [In(1,10-phen)(Stbtg)3] (1), [In(2,2′-bipy)(Stbtg)3] (2), and a thioglycolate salt, Na[In(2,2′-bipy)(SCH2COO)2]·H2O (3), obtained by the decomposition of thiobenzoylthioglycolate complexes, were fully characterized using NMR and IR spectroscopy and single-crystal X-ray diffraction. The catalytic activities of these complexes were evaluated for two significant types of reactions. Complex 1 demonstrated exceptional catalytic efficiency in the Knoevenagel condensation and Knoevenagel-initiated multicomponent reactions (MCRs) for the synthesis of 2-amino-4H-chromene derivatives, including cyclohexane-1,3-dione, 5,5-dimethylcyclohexane-1,3-dione, 4-hydroxycoumarin, barbituric acid, and 2-aminobenzimidazole. Additionally, the complexes were found to be highly effective in CO2 fixation reactions, with complex 1 exhibiting the highest activity, followed by 2 and 3. These results highlight the potential of In(III) complexes as catalysts in a variety of applications, such as organic synthesis and environmentally significant CO2 fixation, demonstrating the broad applicability of non-transition metal complexes in sustainable chemical processes.
Introduction
The unique qualities of indium and its derivatives make their use increasingly common in chemical synthesis.1,2 In organic synthesis, Lewis acids like indium(III) can assist in a number of processes, including Aldol condensations3–5 and Friedel–Crafts acylations.3 These catalysts are particularly useful under mild reaction conditions, which prevent the degradation of sensitive functional groups and minimize unintended side reactions. These are, therefore, employed in the synthesis of natural products and pharmaceutical intermediates as well as in asymmetric synthesis with excellent enantioselectivity.6 In cyclic compounds, In(III) reagents stimulate ring-opening events, resulting in complex structures.7–9 They also provide effective pathways to functionalize organic compounds in C–C and C–N bond-forming processes.10,11 Reductive couplings12–15 between distinct functional groups are facilitated by indium metal, which is also employed as a mediator in cross-coupling reactions16 to create carbon–carbon bonds between organic fragments.10,14 However, organic synthesis is a dynamic science and reagents may become less or more popular over time. Many recent academic researchers have examined the use of indium salts,1 organoindium compounds, and indium metal–organic frameworks (MOFs) in heterogeneous and homogeneous catalysis16–21 such as the classical Ugi reaction,22–24 the epoxide-based Passerini reaction,17,18 the multiple isocyanide insertion reaction,25 the A3-coupling reaction,26 the Knoevenagel-initiated MCRs,27–32 the synthesis of Hantzsch-type dihydropyridines and pyrroles,33–35 as well as CO2 conversion reaction.36–38 (Notably, several transition metal complexes of V,39 Ni,40 Zn,41 Cu,42 Au,43 Ag,44 and Fe45 also demonstrated their suitability as catalysts in MCRs.) The conversion of CO2 is attracting more attention as atmospheric CO2 levels surpassed 420 ppm in 2023,46 compounded by the high costs associated with CO2 capture, transport, and purification,47 posing a challenge before the scientific society. One promising solution is the transformation of CO2 into valuable products, such as cyclic carbonates.36,48 These carbonates have low vapor pressure, low toxicity, and a mild odor, making them applicable in various industries,49 including those associated with lithium-ion batteries,50 cosmetics,51,52 and the production of complex molecule precursors.52,53 Researchers have explored the use of some In(III) salts and complexes as catalysts for this process.54 However, there is still a need for simpler and more efficient catalysts that can operate at atmospheric pressure given the intricate ligand structures of the existing catalysts. There is an urgent need for a straightforward, scalable catalytic system to convert diluted CO2 from sub-atmospheric waste streams.55–57 The wide range of applications for pyran-annulated heterocyclic compounds in biology,58 medicine, and pharmacology have motivated chemists to devise new synthetic routes.59 Although a number of strategies have been previously proposed, the most efficient and straightforward strategies involve utilizing various homogeneous/heterogeneous catalysts in a one-pot, three-component reaction involving aryl aldehyde, malononitrile, and C–H-activated compounds (such as cyclohexane-1,3-dione, 5,5-dimethylcyclohexane-1,3-dione, 4-hydroxycoumarin, and barbituric acid) and imidazopyrimidine. While there are benefits to the catalytic multicomponent synthesis of pyrans documented in the literature, there are also some disadvantages, such as prolonged reaction times, unfavorable reaction conditions, high catalyst requirements, high costs of catalysts, and the formation of side products. Therefore, there is a need for a high-yield, environmentally friendly, and cost-effective method that requires low-catalyst loading. The use of indium complexes in the catalysis of the CO2 conversion reaction and the multicomponent synthesis of 2-amino-4H-chromene derivatives has been documented in a very small number of investigations.27,60–62 In view of these facts we report herein the synthesis and structures of three In(III) complexes and their catalytic efficiencies for two different types of important reactions: (a) fixation of CO2 and (b) multicomponent synthesis of 2-amino-4H-chromene derivatives. The significance of various reaction conditions has been examined. The Kneovenagel condensation reaction intermediates were judiciously isolated and characterized for multicomponent synthesis.
Experimental
Synthesis of [In(1,10-phen)(Stbtg)3] (1)
S-Thiobenzoylthioglycolic acid (0.212 g, 1 mmol) was added to a methanolic solution (5 mL) of sodium methoxide (0.023 g, 1 mmol) in an ice bath. The resulting solution was added to a stirred suspension of indium trichloride (0.073 g, 0.33 mmol) and 1,10-phenanthroline (0.060 g, 0.33 mmol) in methanol (10 mL) at room temperature. After stirring for approximately 2 h, a light pink precipitate formed which was filtered, washed successively with methanol and diethyl ether, and dried under vacuum. Light pink block-shaped crystals suitable for analysis were obtained by recrystallization from a chloroform/toluene mixture (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), yield: (0.770 g, 83%), m.p. 201 °C. Anal. calc. for C39H29InN2O6S6: C, 50.43; H, 3.15; N, 3.02; O, 10.33% found: C, 49.85; H, 3.25; N, 3.11; O, 9.97%, IR data (KBr, cm−1): 1559 (OCO), 1416 (OCO), 1234 (C
:1), yield: (0.770 g, 83%), m.p. 201 °C. Anal. calc. for C39H29InN2O6S6: C, 50.43; H, 3.15; N, 3.02; O, 10.33% found: C, 49.85; H, 3.25; N, 3.11; O, 9.97%, IR data (KBr, cm−1): 1559 (OCO), 1416 (OCO), 1234 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) 1046 (C–O), 765 (C–S), 1H NMR (500 MHz, CDCl3, δ ppm): 4.16 (6H, –CH2– of Stbtg), 7.31 (t, J = 7 Hz, 8H, Stbtg & 1.10-phen.), 7.47 (t, J = 7.5 Hz, 3H, Stbtg), 7.88 (d, J = 7 Hz, 6H, Stbtg), 8.0 (s, 2H, 1.10-phen), 8.54 (s, J = 7.5 Hz, 2H, 1.10-phen), 9.41 (d, J = 5 Hz, 2H, 1.10-phen). 13C NMR (125 MHz, CDCl3, δ ppm): 35.72 (CH2) 118.46–145.68 (aromatic), 176.61 (COO), 222.22 (CSS).
S) 1046 (C–O), 765 (C–S), 1H NMR (500 MHz, CDCl3, δ ppm): 4.16 (6H, –CH2– of Stbtg), 7.31 (t, J = 7 Hz, 8H, Stbtg & 1.10-phen.), 7.47 (t, J = 7.5 Hz, 3H, Stbtg), 7.88 (d, J = 7 Hz, 6H, Stbtg), 8.0 (s, 2H, 1.10-phen), 8.54 (s, J = 7.5 Hz, 2H, 1.10-phen), 9.41 (d, J = 5 Hz, 2H, 1.10-phen). 13C NMR (125 MHz, CDCl3, δ ppm): 35.72 (CH2) 118.46–145.68 (aromatic), 176.61 (COO), 222.22 (CSS).
Synthesis of [In(2,2′-bipy)(Stbtg)3] (2)
S-Thiobenzoylthioglycolic acid (0.212 g, 1 mmol) was added to a methanolic solution (5 mL) of sodium methoxide (0.023 g, 1 mmol) in an ice bath. The resulting solution was then added to a suspension of indium trichloride (0.073 g, 0.33 mmol) and 2,2′-bipyridine (0.052 g, 0.33 mmol) in methanol (10 mL) at room temperature. After stirring the reaction mixture for about 3 h, the red-orange precipitate formed was filtered, washed successively with methanol and diethyl ether, and then dried under vacuum. A crop of red-orange block-shaped crystals was obtained upon recrystallization from chloroform/acetonitrile (5:1). Yield: (0.768 g, 85%), m.p. 180 °C, anal. calc. for C37H29InN2O6S6: C, 49.12; H, 3.23; N, 3.10; O,10.61%; found: C, 49.00; H, 2.26; N, 3.07; O,10.70%; IR data (KBr, cm−1): 1598 (OCO), 1442 (OCO), 1241 (CS) 1045 (C–O), 764 (C–S); 1H NMR (500 MHz, CDCl3, δ ppm): 4.18 (6H, –CH2–), 7.33 (t, J = 7.5, 6H, Stbtg), 7.49 (t, J = 7.5, 3H, Stbtg), 7.63 (t, J = 6, 2H, 2,2-bipy), 7.92 (d, J = 7.5, 6H, Stbtg), 8.11 (t, J = 7.5 Hz, 2H, 2,2-bipy), 8.21 (t, J = 8 Hz, 2H, 2,2-bipy.), 9.12 (d, J = 4.5 Hz, 2H, 2,2-bipy.); 13C NMR (125 MHz, CDCl3, δ ppm): 39.94 (CH2) 121.49–150.03 (aromatic), 176.09 (COO), 227.15 (CSS).
Synthesis of Na[In(2,2′-bipy)(SCH2COO)2]·(H2O) (3)
S-Thiobenzoylethioglycolic acid (0.212 g, 1 mmol) was added to a methanolic solution (5 mL) of sodium methoxide (0.023 g, 1 mmol) in an ice bath. The resulting solution was then added to a suspension of indium trichloride (0.073 g, 0.33 mmol) and 2,2′-bipyridine (0.052 g, 0.33 mmol) in methanol (10 mL). The reaction mixture was refluxed for approximately 12 h. The solvent was reduced to dryness under vacuum using a rotary evaporator, and the residue was extracted with chloroform. The extract was left to crystallize, yielding block-shaped, diffraction-quality brownish-yellow crystals from chloroform. Yield: (0.380 g, 77%) m.p. 300 °C (dec), anal. calc. for C14H14InN2NaO5S2: C, 35.52; H, 3.38; N, 5.52; O, 16.25% found: C, 34.96; H, 3.29; N, 5.40; O, 16.10; % IR data (KBr, cm−1): 1552 (OCO), 1398–1441 (OCO), 1023 (C–O), 770 (C–S); 1H NMR (500 MHz, DMSO-d6, δ ppm): 2.08 (2H, CH2), 3.85 (1H, CH2), 3.43 (1H, CH2), 7.07 (t, J = 8.5 Hz, 2H, 2,2-bipy), 7.53 (t, J = 8 Hz, 2H, 2,2-bipy.), 7.97 (s, 2H, 2,2-bipy.), 8.27 (s, 2H, 2,2-bipy.); 13C NMR (125 MHz, DMSO-d6δ ppm): 39.94 (CH2) 123.71–148.27 (aromatic), 174.45 (COO), 212.47 (–SCS).
General procedure for the Knoevenagel condensation reactions
The previously documented techniques were used to carry out the Knoevenagel condensation reactions.27,28 The following ingredients were added to a round bottomed flask containing acetonitrile and toluene (1:1 ratio, 6 mL) in a prototype reaction: 4-methyl benzaldehyde (0.118 g, 1 mmol), malononitrile (0.079 g, 1.2 mmol), triethylamine (140 μL, 1 mmol), and catalyst 1 (0.93 mg, 0.1 mol%). The mixture was then stirred for 10 min at room temperature. On completion of the reaction (monitored by TLC), solvent from the reaction mixture was evaporated on a rotary evaporator and the product was purified using column chromatography (5% ethyl acetate/n-hexane mixture). The ESI includes the products’ yields and NMR spectra in data S4 (Fig. S10–S16†).
One-pot multicomponent reactions
Synthesis of 2-amino-4H-chromene derivatives of cyclohexane 1,3-dione/5,5′ dimethyl cyclohexane 1,3-dione.
The following ingredients were added to a round-bottomed flask: 4-methyl benzaldehyde (145 μL, 1.2 mmol), malononitrile (0.079 g, 1.2 mmol), cyclohexane 1,3-dione (0.112 g, 1 mmol) or 5,5′ dimethyl cyclohexane 1,3-dione (0.140 g, 1 mmol), triethyl amine (140 μL, 1 mmol), catalyst 1 (0.93 mg 0.1 mol%), and acetonitrile + toluene (1:1 ratio, 4 mL). The reaction mixture was then swirled at room temperature for 10–15 minutes. Column chromatography using ethyl acetate/n-hexane (1:4 ratio) was used to purify the product. The yields and NMR spectra of 2-amino-4H-chromene derivatives of cyclohexane 1,3-dione/5,5′ dimethyl cyclohexane 1,3-dione are shown in ESI data S5–S6 (Fig. S16–S28†).
Synthesis of 2-amino-4H-chromene derivatives of 4-hydroxycoumarin.
A mixture of the desired aromatic aldehyde (1.2 mmol), 4-hydroxycoumarin (1.0 mmol), and malononitrile (1.2 mmol) was combined with triethylamine (1.0 mmol) in a solvent mixture of acetonitrile and toluene (1:1). Complex 1 (0.93 mg 0.1 mol%) was used as a catalyst at room temperature, and the reaction was completed within 10 minutes, as monitored by TLC. After completion, the solvent was removed using a rotary evaporator, and the residue was purified by column chromatography using an ethyl acetate/n-hexane mixture (1:4 ratio). Data-S7 (Fig. S28–S33) of the ESI† contains the yields and NMR spectra of the representative compound and its derivatives.
Synthesis of 2-amino-4H-chromene derivatives of barbituric acid.
A mixture of 4-methyl aromatic aldehyde (1.2 mmol), barbituric acid (1.0 mmol), and malononitrile (1.2 mmol) was prepared in the presence of triethylamine (1.0 mmol) and a solvent mixture of MCN and toluene (1:1). Complex 1 (0.93 mg, 0.1 mol%) was used as a catalyst at room temperature. Upon completion of the reaction, yielding over 94% of the product within 5 minutes, the precipitate was filtered and purified by repeated washing with dilute HCl and water to obtain the pure product. Data S8 (Fig. S33–S38) of the ESI† contains the yields and NMR spectra of the representative compound and its derivatives.
Synthesis of imidazopyrimidine derivatives.
In a 10 mL round-bottomed flask containing MeCN and toluene (1 mL each), a combination of benzaldehyde (122 μL, 1.2 mmol), malononitrile (0.079 g, 1.2 mmol), 2-aminobenzimidazole (0.133 g, 1 mmol), triethyl amine (140 μL, 1.0 mmol), and catalyst 1 (0.93 mg, 0.1 mol%) was stirred at room temperature for 1 h. TLC was used to monitor the reaction's completion. The white solid product was isolated by filtration and washed with ethanol two times. Data S9 (Fig. S38–S42) of the ESI† contains the yields and NMR spectra of the representative compound and its derivatives.
CO2 conversion reaction
In a 25 mL Schlenk tube, 1 mmol of epichlorohydrin, 0.01 mmol of Bu4NBr, and 0.001 mmol of catalyst 1 were combined. The setup was purged with dry nitrogen gas, and carbon dioxide was supplied using CO2 balloons. The reaction mixture was then heated to reflux at 100 °C for 5 h with continuous stirring. After completion, the crude reaction mixture was collected and analyzed using NMR spectroscopy. The conversion of epoxide to carbonate was determined through analysis of the 1H NMR spectra. Data S10 (Fig. S44–S49) of the ESI† contains the yields and NMR spectra of the representative compounds.
Results and discussion
Synthesis and spectral characterization of the complexes
The complexes 1 and 2 of S-thiobenzoylthioglycolate were synthesized by reacting indium(III) chloride with sodium S-thiobenzoylethioglycolate and an auxiliary ligand (1,10-phenanthroline for complex 1 and 2,2′-bipyridyl for complex 2) in stoichiometric proportions (1:3:1, respectively) at room temperature. Complex 3 was synthesized in refluxing MeOH using the same reactants as in the case of complex 2 (as shown in Scheme 1A). In Scheme 1b, the reaction mechanism for the formation of complex 3 has been illustrated, showing the stepwise transformation of InCl3 into the final complex 3. All the complexes were fully characterized by IR and NMR spectral analyses and single-crystal X-ray diffraction techniques. The crystallographic and structure refinement data of the complexes are shown in Table S1.†
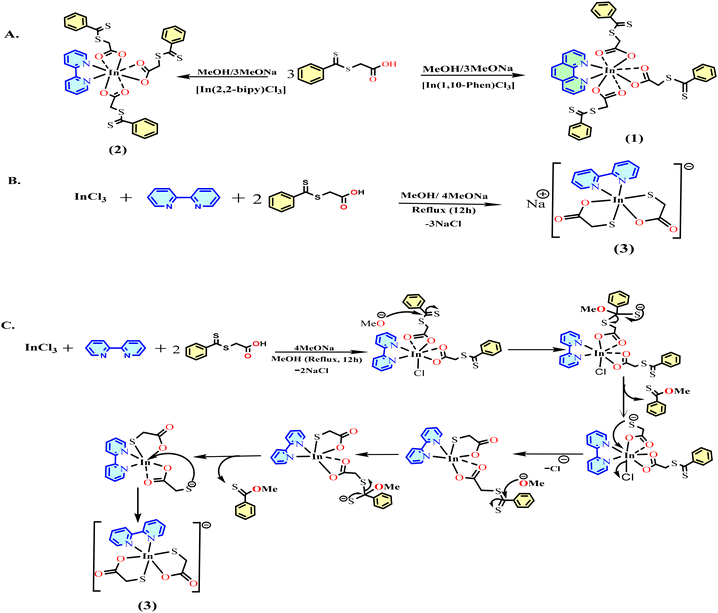 |
| | Scheme 1 (A) Reaction scheme for the synthesis of complexes 1 and 2. (B) Reaction scheme for the synthesis of complex 3 and (C) plausible mechanism for the formation of complex 3. | |
Crystal structure description
The structures of complexes 1 and 2 are depicted in Fig. 1 and 2, respectively, along with the selected bond lengths and bond angles. The complex 1 was crystallized in a monoclinic system with the space group Pn, whereas molecule 2 was in an orthorhombic system with the space group Pca21. Both molecules show a bicapped octahedral geometry around In(III). Coordination number 8 around the In(III) center though very uncommon has been reported earlier.63–65 In complex 1, the atoms N1, N2, O5 and O6 constitute the approximate square plane from which O6 shows a maximum deviation of 0.218 Å, O1 and O4 lie almost perpendicular to this plane and subtend an angle of 167.85°. The other two oxygen atoms O2 and O3 cap the triangular faces O4, O5, N2 and O1, O5, N2, respectively. Notably, the differences in In–O bond lengths are rather small (2.268–2.330 Å).
 |
| | Fig. 1 (a) Thermal ellipsoid plot (at 50% probability) of complex 1, (b) the bicapped octahedral geometry at the molecular core. Selected bond lengths (Å): In1–O1 2.309(6), In1–O2 2.331(5), In1–O3 2.272(6), In1–O4 2.333(4), In1–O5 2.316(4), In1–O6 2.268(5), In1–N1 2.288(4), In1–N2 2.291(6). Selected bond angles (°): O1–In–O4 123.67(19), O1–In1–O2 55.10(19), O1–In–O5 112.93(19), O2–In1–O4 76.28(17), O3–In–O4 56.66(17), O3–In1–N2 94.6(2), O3–In1–O2 86.3(2), O3–In1–O1 90.7(2), O3–In1–N1 80.9(2), O3–In–O5 135.45(19), O5–In1–O4 79.01(16), O5–In1–O2 78.63(17), O6–In1–O1 82.6(2), O6–In1–O2 97.9(2), O6–In1–O3 167.92(15), O6–In1–O4 135.32(18), O6–In1–O5 56.62(18), N1–In1–O4 128.1(2), N1–In1–N2 72.0(3), N1–In1–O2 133.6(2), N1–In1–O1 80.5(3), N1–In1–O5 137.9(2), N2–In1–O1 150.8(2), N2–In1–O2 153.88(19), N2–In1–O4 82.44(18), N2–In1–O5 −82.6(2), O6–In1–N1 88.1(2), O6–In1–N2 86.6(2). | |
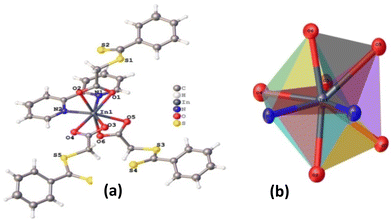 |
| | Fig. 2 (a) Thermal ellipsoid plot of complex 2, (b) 3D representation of the molecular core. Selected bond lengths (Å): In1–O1 2.421(7), In1–O2 2.217(7), In1–O3 2.257(10), In1–O4 2.327(9), In1–O5 2.325(7), In1–O6, 2.270(7), In1–N1 2.290(12), In1–N2 2.314(10), selected bond angles (°): O2–In1–O4 82.6(3), O2–In1–O5 136.0(3), O2–In1–O3 88.5(3), O2–In1–O1 56.6(3), O2–In1–O6 166.6(3), O2–In1–N1 93.2(3), O2–In1–N2 87.3(3), O4–In1–O1 119.0(3), O5–In1–O4 124.0(3), O5–In1–O1 79.4(3), O3–In1–O4 57.0(3), O3–In1–O5 81.2(3), O3–In1–O1 76.9(3), O3–In1–O6 95.4(3), O3–In1–N1 150.9(3), O3–In1–N2 138.2(4), O6–In1–O4 88.8(3), O6–In1–O5 57.4(3), O6–In1–O1 136.9(2), O6–In1–N1 89.5(3), O6–In1–N2 81.2(3), N1–In1–O4 152.0(4), N1–In1–O5, 77.5(3), N1–In1–O1 79.9(4), N1–In1–N2 70.9(4), N2–In1–O4 81.2(4), N2–In1–O5 127.5(3), N2–In1–O1 131.9(3). | |
In complex 2, there are two molecules present in one unit cell. The bicapped octahedral geometry surrounds the In(III) center. The atom O3 exhibits a maximum deviation of 0.268 Å from the approximate square plane formed by N1, N2, O4, and O3 atoms. Atoms O2 and O6 lie nearly perpendicular to this plane and subtend an angle of 166.40° at In(III). The triangular faces O6, O3, N1 and O2, O3, N1 are capped by the remaining two oxygen atoms, O5 and O1. In this case, however, the variations in In–O bond lengths are significant (2.214–2.421 Å).
The structure of complex 3 is depicted in Fig. 3, along with the selected angles and bond lengths. This is an ionic complex, crystallized in a monoclinic system with the space group P21/c. In an asymmetric unit, the indium(III) metal shows six coordination in which two coordination sites are occupied by two nitrogen atoms of the 2,2-bipyridyl ligand, and the other four sites are occupied by two sulfur atoms (S1 and S2) and two oxygen atoms (O3 and O4); the two S atoms are cis to each other. The octahedral geometry around In(III) is warped. The approximate square planar base is defined by the atoms N1, N2, S2, and O3, with the largest deviation observed at 0.283 Å for atom N1. S1 and O4 subtend an angle of 167.25° and are nearly perpendicular to this plane. The results of the DFT computation also support the type of bonding. According to the analysis of the Fock matrix using second-order perturbation theory in the natural bond orbital calculation, the total energy change resulting from the electron transfers in S1–In and S2–In bonds is approximately equal (184 kcal mol−1), while the same is significantly less (94 kcal mol−1) in O4–In and O3–In bonds. The atom O2 is connected with Na1 which is also associated with the O1 (H2O) molecule; these sodium metals have a distorted octahedral geometry in the ionic lattice (as shown in Fig. 3c).
 |
| | Fig. 3 (a) Asymmetric unit of complex 3 plotted at a 50% probability level. (b) Arrangement of complex anions and Na+ cations along the b axis, (c) octahedral geometry about the In(III) center. Selected bond lengths (Å): In1–S1 2.4903(7), In1–S2 2.4613(8), N1–In1 2.349(2), In1–N2 2.312(2), In1–O4 2.2276(18), In1–O3 2.1790(17), Na1–O2 2.279(2), O1–Na1 2.356(3) Na1–O44 2.457(2), Na1–O33 2.4154(19), Na1–O23 2.495(2), Na1–O54 2.563(2). Selected bond angles (°): S2–In1–S1 104.79(3), O4–In1–S1 167.25(5), N1–In1–S1 93.75(6), N1–In1–S2 95.07(6), O4–In1–S2 81.00(5), O4–In1–N2 80.45(7), O3–In1–N1 156.80(8), O3–In1–S1 82.01(5), O3–In1–S2 108.09(5), O3–In1–O4 85.40(7), O3–In1–N2 88.13(7), In1–O4–Na12 140.64(9), N2–In1–N1 69.68(8), N2–In1–S1 97.11(6), N2–In1–S2 154.19(6). | |
Catalytic properties
The synthesized In(III) complexes have been employed in the Knoevenagel-initiated one-pot multicomponent reactions (MCR) for the synthesis of 2-amino-4H-chromene derivatives. Additionally, these complexes have demonstrated catalytic activity in CO2 conversion reactions, highlighting the potential of non-transition metal complexes in diverse catalytic processes. To evaluate their efficiency in the Knoevenagel condensation, a model reaction was conducted using 4-methylbenzaldehyde (1.0 mmol), malononitrile (1.0 mmol), triethylamine (1.0 mmol), and catalyst (0.1 mol%), in a solvent mixture of acetonitrile and toluene (1:1, 4 mL) at room temperature. Complex 1 achieved 95% aldehyde conversion within 10 minutes. In contrast, complexes 2 and 3 required significantly longer reaction times (see in Table 1), notably under the same conditions, even after 6 h. Table 2 summarizes the reaction parameters, including catalyst concentration and solvent type that were evaluated during the study. Recent studies emphasized the role of indium in MOF/CP structures for catalytic applications. In this study, In(III) complexes demonstrated exceptional catalytic efficiency, achieving high product yields (up to 95%) with minimal catalyst loading and rapid reaction times. Scheme 2 illustrates their broad applicability across a range of substrates. The plausible mechanism for the reaction has been given in Scheme S1 (ESI†). Product yields were observed to improve when aldehydes contained electron-donating groups, while electron-withdrawing groups led to slightly reduced yields.
 |
| | Scheme 2 Product yields and reaction times for Knoevenagel condensation using catalyst 1. | |
Table 1 Screening of catalysts 1–3 for the Knoevenagel condensation of 4-methyl benzaldehyde with malononitrile
Table 2 Screening of catalyst 1 for the Knoevenagel condensation of 4-methyl benzaldehyde with malononitrile
Among the three complexes, the catalytic efficiency in the presence or absence of a base followed the order: 1 > 2 > 3 (see Table 2). This highlights the superior performance of complex 1 under mild conditions.
Multicomponent reactions
Multicomponent reactions involving aromatic aldehydes, malononitrile, and various carbonyl compounds, including cyclohexane-1,3-dione, 5,5-dimethylcyclohexane-1,3-dione, 4-hydroxycoumarin, barbituric acid, and 2-aminobenzimidazole, were studied. Optimal results with catalyst 1 (0.1 mol%) were achieved with a 96% yield within 10 minutes at room temperature using a solvent mixture of acetonitrile and toluene (1:1) in the presence of a base (Et3N). Under identical reaction conditions, In(III) acetate, when used as a catalyst yielded only 70% of the product. It is worth mentioning here that the reaction does not proceed with In(OAc)3 in the absence of a base. To evaluate the performance of three In(III) complexes as catalysts, it was necessary to identify the most favorable reaction conditions. Systematic adjustments were made to catalyst loading, solvent choice, and reaction time. The ideal reaction conditions are summarized in Table 3. Complex 1 was selected as the representative catalyst due to its superior performance. Remarkably, reducing the catalyst loading from 0.1 mol% to 0.01 mol% in the optimized solvent mixture of acetonitrile and toluene (1:1) did not significantly impact the yield, which remained nearly the same within the same reaction time. The turnover number (TON) for the synthesis of a 4-methyl derivative of cyclohexane-1,3-dione and 5,5-dimethylcyclohexa-1,3-dione was 9500, while the same for the synthesis of a 4-methoxy derivative of 4-hydroxycoumarin was 9400.
Table 3 Screening of catalysts for the synthesis of 2-amino-4H-chromene derivatives
Syntheses of 2-amino-4H-chromene derivatives of cyclohexane-1,3-dione/5,5-dimethylcyclohexane-1,3-dione.
The reaction was tested across various solvents, including acetonitrile, toluene, ethanol, and a mixture of acetonitrile and toluene, with and without the base (Et3N). Among these, the acetonitrile and toluene (1:1) mixture in the presence of a base consistently demonstrated the best results, achieving up to 94% yield within 10 minutes with a catalyst loading of 0.1 mol%. With complex 1 under the optimized conditions, one-pot multicomponent reactions of various dicarbonyl compounds, malononitrile, and aromatic aldehydes were performed. Aldehydes with electron-donating substituents showed superior conversion rates (90–96%) compared to benzaldehyde, while aldehydes with electron-withdrawing groups exhibited similar or slightly lower yields. For comparison, indium chloride was also tested under similar conditions. Indium chloride, without the base, achieved only a 40% yield, while the yield increased to a maximum of 55% in the presence of Et3N. However, we have reported earlier that in the presence of the base alone, the yield is only 32% while in the absence of both the catalyst and base, the reaction does not occur. These findings highlight the efficiency and versatility of complex 1 in facilitating multicomponent reactions with high yields and rapid reaction times under mild conditions. The one-pot reaction involving malononitrile, an aromatic aldehyde, and an active methylene diketone was facilitated by the use of complex 1 as a catalyst. The reaction was carried out at room temperature with a catalyst loading of 0.93 mg (0.1 mol%) in the presence of a base (triethylamine) and a solvent mixture of MeCN and toluene (1:1, 4 mL). Remarkably, the reaction was completed within 10 minutes, yielding 95% of the desired product. In contrast, when the reaction was conducted without the catalyst, only 40–60% of the product was obtained. Table 3 summarizes the substrates and their corresponding catalytic products, demonstrating the effectiveness of the catalyst in accelerating the reaction and improving the yield.
Syntheses of a 2-amino-4H-chromene derivative of 4-hydroxycoumarin.
Substituents on the aromatic aldehyde play a crucial role in the synthesis 2-amino-4H-chromene derivatives of 4-hydroxycoumarin catalysed by complex 1. High-purity derivatives are obtained through a one-pot process involving malononitrile, 4-hydroxycoumarin, and aryl aldehydes with weak electron-withdrawing or electron-donating groups. Notably, the yield decreases when a weak electron-withdrawing substituent is present. Scheme 3 illustrates all the 2-amino-4H-chromene derivatives of 4-hydroxycoumarin synthesized under these conditions.
 |
| | Scheme 3 Syntheses of 2-amino-4H-chromene derivatives of dicarbonyl compounds viz. (5) cyclohexane-1,3-dione, (6) 5,5-dimethylcyclohexane-1,3-dione, (7) 4-hydroxycoumarin and (8) barbituric acid, using different aromatic aldehydes. | |
Syntheses of 2-amino-4H-chromene derivatives of barbituric acid.
The model reaction involved barbituric acid reacting with malononitrile and 4-methylbenzaldehyde in the presence of a base (triethylamine) and a solvent mixture of acetonitrile and toluene (1:1, 6 mL). Complex 1 (0.1 mol%) served as the catalyst at room temperature, achieving a 95% product yield within 10 minutes. The nature of substituents on the aromatic aldehyde significantly impacted the synthesis of 2-amino-4H-chromene derivatives of barbituric acid. Aldehydes with electron-donating groups increased the yield to over 90%, while electron-withdrawing groups led to reduced yields. Scheme 3 highlights various substrates, their corresponding products, and the resulting catalytic yields.
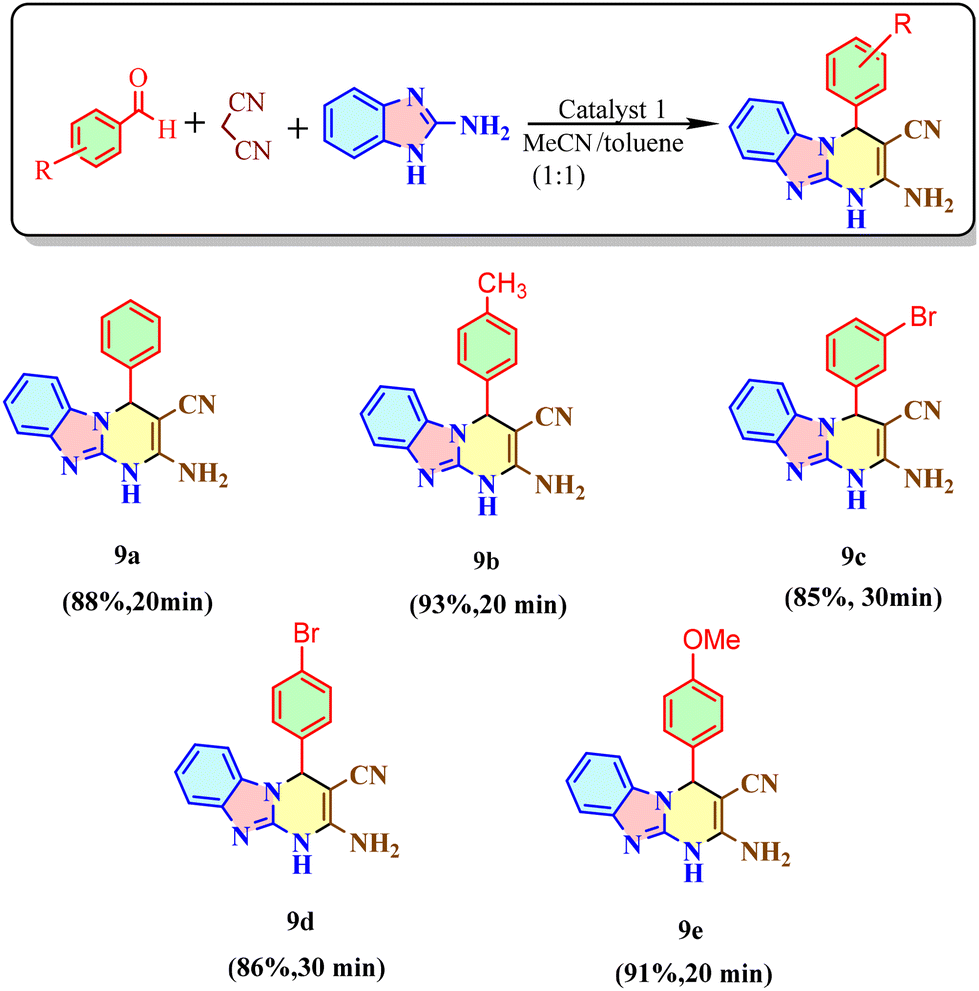
Syntheses of imidazopyrimidine derivatives
We employed complex 1 extensively for the one-pot synthesis of imidazopyrimidine derivatives due to its exceptional catalytic efficiency. In the model reaction, 2-aminobenzimidazole was reacted with malononitrile and benzaldehyde in the presence of a base (triethylamine) and a solvent mixture (MeCN:toluene 1:1 6 ml), using complex 1 (0.1 mol%) as the catalyst. The reaction proceeded at room temperature and afforded a 93% yield of the product within 20 minutes. Aldehydes with weak electron-donating groups significantly enhanced the yield to over 90%, whereas electron-withdrawing groups resulted in lower yields. Scheme 4 provides a detailed overview of various substrates, their corresponding products, and the associated catalytic yields and reaction times. Additionally, at a catalyst loading of 0.01%, the turnover number (TON) for the 4-methyl derivative of 2-aminobenzimidazole was calculated to be 9300.
 |
| | Scheme 4 Syntheses of derivatives of 2-aminobenzimidazole using different aromatic aldehydes via catalyst 1. | |
Plausible reaction mechanism for the In(III)-catalyzed one-pot multicomponent reaction
Multicomponent one-pot reactions and Knoevenagel condensation are well-known, and in recent years, some research on the catalytic routes has been published.27,40,66–68 The electrophilic In(III) core of the catalyst activates the carbonyl carbon of benzaldehyde as the initial step in the Knoevenagel condensation reaction. A C–C bond is formed in the next step as a result of the base abstracting a proton from malononitrile. Scheme S1(A)† illustrates the tenable mechanism.69 As seen in Scheme S1(B),† In(III) facilitates the enolization of the cyclohexanedione during the synthesis of 2-amino-4H-chromene derivatives. It then proceeds via Michael addition70 of malanobenzylidene, tautomerization, and cyclization. Likewise, in Scheme S1(C),† the C–N bond is created by the Michael addition of the Knoevenagel product and 2-aminobenzaimidazole, which are then tautomerized and cyclized one after the other.
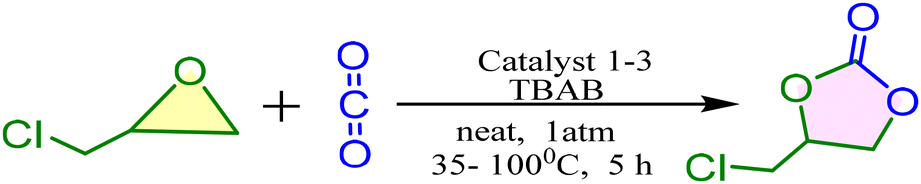
Catalytic activity for CO2 fixation
The complexes were tested for their efficiency as catalysts in the cycloaddition reaction of epoxides and CO2 to form cyclic carbonates. The catalytic cycloaddition of CO2 and epichlorohydrin (ECH) was performed as a model reaction in the presence of n-Bu4NBr (TBAB) (as a co-catalyst) at 100 °C and 1 atm CO2 pressure (Scheme 5). Key results are summarized in Table 4 and the plausible mechanism for the reaction has been given in Scheme S2 (ESI†).
 |
| | Scheme 5 Cycloaddition reaction of CO2 with different epoxides to form cyclic carbonates by using catalysts 1–3. | |
Table 4 Catalyst screening for the cycloaddition of epichlorohydrin and CO2a
|

|
| Entry |
Catalyst (mol%) |
Co catalyst (mol%) |
Temp. (°C) |
Conversionb (%) |
Yield (%) |
|
All reactions were carried out under solvent-free conditions: 1 mmol of epichlorohydrin, indium complex and TBAB was used as a co-catalyst.
Conversion was determined by 1H NMR.
|
| 1 |
1 (0.1) |
1
|
100 |
96 |
92 |
| 2 |
2 (0.1) |
1
|
100 |
82 |
80 |
| 3 |
3 (0.1) |
1
|
100 |
75 |
71 |
| 4 |
1 (0.1) |
1
|
40 |
35 |
33 |
| 5 |
1 (0.1) |
1
|
60 |
37 |
34 |
| 6 |
— |
1
|
20 |
Trace36 |
— |
| 7 |
1 (0.1) |
— |
20 |
Trace36 |
— |
A catalyst loading of 0.1 mol% was initially used to explore the relationship between coupling activity and the ratio of 1.0 mol% co-catalyst. We observed yields of 97%, 82% and 75% with catalysts 1, 2 and 3 respectively (Table 4, entries 1–3). The effect of reaction temperature with catalyst 1 on the cycloaddition of CO2 and epichlorohydrin to form chloropropene carbonate was studied across a temperature range of 40 and 60 °C (Table 4, entries 4 and 5). Under identical conditions (catalyst loading at 0.1 mol%, TBAB loading at 1 mol%, CO2 pressure at 1 atm, and 5 h reaction time), the yield of chloropropene carbonate marginally increased as the temperature rose from 40 °C to 60 °C. Experimental results showed that the cycloaddition of CO2 and ECH, co-catalyzed by 1.0 mol% n-Bu4NBr, was effective in producing chloropropene carbonate (CPC) with good ECH conversion rates. We also performed the reaction by time variation (12 h) at 60 °C and obtained 60% conversion (57% yield). However, in the absence of a catalyst, the ECH conversion was negligible, indicating that the indium catalyst provides active site(s) that significantly enhance the activation of CO2 or ECH in this catalytic process (Table 4, entry 6). Similarly, the CO2 fixation products could not be isolated in the absence of TBAB. These results indicate that both the In(III) complex catalyst and TBAB are essential for the catalytic conversion of CO2 into carbonates (Table 4, entry 7). The metal center acts as a Lewis acid, and the Br− anion serves as a Lewis base or nucleophile. Together, they work cooperatively to open the epoxide ring. The scope of the catalysis was expanded to include cyclohexene oxide (CHO) (aliphatic epoxide) and styrene epoxide (SO) (aromatic epoxide). These reactions were carried out under optimized conditions using catalysts 1, 2, and 3. We observed that the % conversion rates with epoxy hexane epoxides were 91% (yield 85%), 90% (yield 85%), and 73% (yield 70%) for catalysts 1, 2, and 3, respectively. In contrast, for styrene epoxide, the yields were significantly lower: 49% (yield 45%), 58% (yield 54%), and 25% (yield 22%) for catalysts 1, 2, and 3, respectively. Catalyst 1 demonstrated greater catalytic activity compared to catalysts 2 and 3. Notably, the catalytic yields of aliphatic epoxides were higher than those for similarly sized aromatic epoxides in forming the corresponding cyclic carbonates.
Plausible reaction mechanism for the conversion of CO2 into a cyclic carbonate
Based on the experimental results and previously discussed mechanisms in the literature,71–75 a possible co-catalytic mechanism for the cycloaddition reaction is proposed in Scheme S2.† In this cycle, the epoxide is first activated through coordination with the Lewis acidic In(III) metal center, which facilitates ring-opening by activating the C–O bond. Next, the Br− ion of TBAB attacks the less hindered side of the epoxide, leading to a metal alkoxide intermediate through ring-opening. This is followed by CO2 insertion and intra-molecular ring closure to form the cyclic carbonate, regenerating the catalyst.
Applicability of the catalyst on a larger scale
Using a similar reaction protocol, we also carried out a model gram-scale reaction with four methyl benzaldehyde (1.20 g, 10 mmol), triethyl amine (1.4 mL, 10 mmol), malononitrile (760 μL, 12 mmol), and catalyst 1 (0.1 mol%) in acetonitrile and toluene (1:1 ratio, 20 mL) to confirm the catalyst's suitability on a larger scale. A good yield of the product was observed (1.15 g, 75%). Similarly, a gram-scale multicomponent synthesis of 2-amino-4H-chromene using 4-methoxy benzaldehyde (1.21 mL, 10 mmol), 5,5-dimethyl cyclohexane-1,3-dione (1.40 g, 10 mmol), and malononitrile (760 μL, 12 mmol) in acetonitrile and toluene (1:1, 20 mL) yielded the desired product in 80% yield (2.59 g). We also tested the catalyst's applicability on a larger scale for the CO2 conversion reaction, achieving a 40% conversion and a 38% yield. The reaction conditions included a catalyst 1 loading of 0.1 mol%, a TBAB loading of 1 mol%, a CO2 pressure of 1atm, and a reaction time of 5 h.
Conclusion
Three indium(III) complexes have been synthesized and structurally characterized using NMR, IR, and single-crystal X-ray diffraction analyses. The discrete molecules of complexes 1 and 2 exhibited a bicapped octahedral In(III) core while the complex anion 3 showed an octahedral geometry. The complexes, particularly complex 1, showed excellent catalytic efficiency for the Knoevenagel-initiated multicomponent reactions. Their versatile catalytic potential was further demonstrated in producing cyclic carbonates from CO2 and epoxides, achieving high yields and short reaction times under mild conditions. Among the complexes, Complex 1 exhibited the highest activity, with a catalytic efficiency trend of 1 > 2 > 3. The findings underscore the dual utility of these In(III) complexes in both organic synthesis and environmental applications, suggesting a promising direction for the further development of non-transition metal catalysts in advanced catalytic processes.
Author contributions
RP prepared the complexes, characterized them, and along with RKS carried out the catalytic synthesis of Knoevenagel condensation products. TM and HST performed the CO2 fixation reactions. SB planned the project. MDP and SB contributed resources, conceptualization, supervision, reviewing, and editing of the manuscript draft. The manuscript was drafted by RP and TM. All the authors have given their consent to publish the paper.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgements
Financial support (faculty incentive grant of IoE to MDP and SB and a fellowship to RP) from Banaras Hindu University, Varanasi, is gratefully acknowledged. TM is thankful to UGC-CSIR for a fellowship.
References
- J. S. Yadav, A. Antony, J. George and B. V. Subba Reddy, Eur. J. Org. Chem., 2010, 591–605 CrossRef CAS.
- M. E. Desat and R. Kretschmer, Dalton Trans., 2019, 48, 17718–17722 RSC.
- K. K. Chauhan and C. G. Frost, J. Chem. Soc., Perkin Trans. 1, 2000, 3015–3019 RSC.
- C. G. Frost and J. P. Hartley, Mini-Rev. Org. Chem., 2004, 1, 1–7 CrossRef CAS.
- A. Kawata, K. Takata, Y. Kuninobu and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 7793–7795 CrossRef CAS PubMed.
- K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS.
- M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- T. Iwase, A. Habibzadeh, C. Goonesinghe, K. Nyamayaro, J. Chang, J. W.-L. Poon, S. H. Rushdy, J. Koh, B. O. Patrick and P. Mehrkhodavandi, Dalton Trans., 2024, 53, 19337–19341 RSC.
- U. Schneider and S. Kobayashi, Acc. Chem. Res., 2012, 45, 1331–1344 CrossRef CAS.
- L. Vanoye, A. Hammoud, H. Gérard, A. Barnes, R. Philippe, P. Fongarland, C. De Bellefon and A. Favre-Réguillon, ACS Catal., 2019, 9, 9705–9714 CrossRef CAS.
- S. Ohtaka, K. Mori and S. Uemura, Heteroat. Chem., 2001, 12, 309–316 CrossRef CAS.
- D. Xu, M. Y. Jin, Y. Chen, D. Han, L. Tao and X. Xing, ACS Catal., 2024, 14, 3241–3247 CrossRef CAS.
- M. R. Pitts, J. R. Harrison and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 2001, 955–977 RSC.
- G. S. Kumar, K. Bano, P. Biswal, S. Dey, R. Kumar, A. Sau, V. Chandrasekhar and T. K. Panda, Dalton Trans., 2025, 54, 1552–1559 RSC.
- I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155–4160 CrossRef.
- L. M. Aguirre-Díaz, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, CrystEngComm, 2013, 15, 9562–9571 RSC.
- A. Corma, H. García and F. X. Llabrés I Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- N. Bayati and S. Dehghanpour, Mater. Adv., 2023, 4, 4921–4928 RSC.
- K. M. Osten, D. C. Aluthge and P. Mehrkhodavandi, Dalton Trans., 2015, 44, 6126–6139 RSC.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- K. Lingaswamy, P. R. Krishna and Y. L. Prapurna, Synth. Commun., 2016, 46, 1275–1282 CrossRef CAS.
- Z.-Q. Liu, Curr. Org. Chem., 2014, 18, 719–739 CrossRef CAS.
- Y. Tian, L. Tian, X. He, C. Li, X. Jia and J. Li, Org. Lett., 2015, 17, 4874–4877 CrossRef CAS PubMed.
- T. L. da Silva, R. S. Rambo, D. da Silveira Rampon, C. S. Radatz, E. V. Benvenutti, D. Russowsky and P. H. Schneider, J. Mol. Catal. A:Chem., 2015, 399, 71–78 CrossRef.
- K. Kumar, R. K. Sahani, S. Garai and S. Bhattacharya, Dalton Trans., 2023, 52, 17499–17513 RSC.
- Y. Ogiwara, K. Takahashi, T. Kitazawa and N. Sakai, J. Org. Chem., 2015, 80, 3101–3110 CrossRef CAS.
- N. Lakshmi, S. Kiruthika and P. Perumal, Synlett, 2011, 1389–1394 CAS.
- N. V. Lakshmi, S. E. Kiruthika and P. T. Perumal, Can. J. Chem., 2013, 91, 479–485 CrossRef CAS.
- M. Ali, S. Shamim, U. Salar, P. Taslimi, S. M. Saad, T. Taskin-Tok, M. Taha and K. M. Khan, J. Mol. Struct., 2025, 1321, 139863 CrossRef CAS.
- E. Kamali, F. Dreekvandy, A. Mohammadkhani and A. Heydari, BMC Chem., 2024, 18, 78 CrossRef CAS PubMed.
- V. K. Sharma and S. K. Singh, RSC Adv., 2017, 7, 2682–2732 RSC.
- M. M. Khan, S. Khan and S. Iqbal, RSC Adv., 2016, 6, 42045–42061 RSC.
-
X. Bugaut, T. Constantieux, Y. Coquerel and J. Rodriguez, in Multicomponent Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M. Wang, Wiley, 1st edn, 2014, pp. 109–158 Search PubMed.
- H. A. Baalbaki, H. Roshandel, J. E. Hein and P. Mehrkhodavandi, Catal. Sci. Technol., 2021, 11, 2119–2129 RSC.
- C.-Y. Chou and R. F. Lobo, Appl. Catal., A, 2019, 583, 117144 CrossRef CAS.
- F. Esteve, R. Porcar, M. Bolte, B. Altava, S. V. Luis and E. García-Verdugo, Chem. Catal., 2023, 3, 100482 CrossRef CAS.
- M. R. Maurya, S. K. Maurya, N. Kumar and F. Avecilla, J. Org. Chem., 2024, 89, 12143–12158 CrossRef CAS PubMed.
- S. A. Patra, M. Nandi, M. R. Maurya, G. Sahu, D. Mohapatra, H. Reuter and R. Dinda, ACS Omega, 2024, 9, 31910–31924 CrossRef CAS.
- M. Neetha, K. R. Rohit, S. Saranya and G. Anilkumar, ChemistrySelect, 2020, 5, 1054–1070 CrossRef CAS.
- F. Peringer, J. E. R. Do Nascimento, P. B. Abib, T. Barcellos, E. V. Van Der Eycken, G. Perin, R. G. Jacob and D. Alves, Eur. J. Org. Chem., 2017, 2579–2586 CrossRef CAS.
- R. Visbal, S. Graus, R. P. Herrera and M. C. Gimeno, Molecules, 2018, 23, 2255 CrossRef PubMed.
- G. Abbiati and E. Rossi, Beilstein J. Org. Chem., 2014, 10, 481–513 CrossRef PubMed.
- S. Saranya, T. Aneeja, M. Neetha and G. Anilkumar, Appl. Organomet. Chem., 2020, 34, e5991 CrossRef CAS.
- S. Basu, D. F. Baker, F. Chevallier, P. K. Patra, J. Liu and J. B. Miller, Atmos. Chem. Phys., 2018, 18, 7189–7215 CrossRef CAS.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. Van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS.
- W. Rong, M. Ding, Y. Wang, S. Kong and J. Yao, Sep. Purif. Technol., 2025, 353, 128427 CrossRef CAS.
- N. von der Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 RSC.
- H. Zhao, S.-J. Park, F. Shi, Y. Fu, V. Battaglia, P. N. Ross and G. Liu, J. Electrochem. Soc., 2013, 161, A194 CrossRef.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- P. Rollin, L. K. Soares, A. M. Barcellos, D. R. Araujo, E. J. Lenardão, R. G. Jacob and G. Perin, Appl. Sci., 2021, 11, 5024 CrossRef CAS.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- C. Calabrese, F. Giacalone and C. Aprile, Catalysts, 2019, 9, 325 CrossRef.
- D. N. Gorbunov, M. V. Nenasheva, M. V. Terenina, Yu. S. Kardasheva, S. V. Kardashev, E. R. Naranov, A. L. Bugaev, A. V. Soldatov, A. L. Maximov and E. A. Karakhanov, Pet. Chem., 2022, 62, 1–39 CrossRef CAS.
- R. Cauwenbergh, V. Goyal, R. Maiti, K. Natte and S. Das, Chem. Soc. Rev., 2022, 51, 9371–9423 RSC.
- E. A. Couladouros and A. T. Strongilos, Angew. Chem., Int. Ed., 2002, 41, 3677–3680 CrossRef CAS.
- E. C. Witte, P. Neubert and A. Roesch, Ger Offen DE, 1986, 3427985 Search PubMed.
- I. Shibata, I. Mitani, A. Imakuni and A. Baba, Tetrahedron Lett., 2011, 52, 721–723 CrossRef CAS.
- L. Xu, M.-K. Zhai, X.-C. Lu and H.-B. Du, Dalton Trans., 2016, 45, 18730–18736 RSC.
- D. J. Cabrera, R. D. Lewis, C. Díez-Poza, L. Álvarez-Miguel, M. E. Mosquera, A. Hamilton and C. J. Whiteoak, Dalton Trans., 2023, 52, 5882–5894 RSC.
- M. A. Malyarick, A. B. Ilyuhin and S. P. Petrosyants, Main Group Met. Chem., 1994, 17, 707–718 CAS.
- H. R. Maecke, A. Riesen and W. Ritter, J. Nucl. Med., 1989, 30, 1235–1239 CAS.
- B. Zhang, W. Wang, B. Liu and L. Hou, Dalton Trans., 2021, 50, 5713–5723 RSC.
- B. Parmar, P. Patel, V. Murali, Y. Rachuri, R. I. Kureshy, H. K. Noor-ul and E. Suresh, Inorg. Chem. Front., 2018, 5, 2630–2640 RSC.
- G. B. B. Varadwaj, S. Rana and K. M. Parida, Dalton Trans., 2013, 42, 5122–5129 RSC.
- M. R. Maurya, N. Kumar and F. Avecilla, Inorg. Chem., 2024, 63, 2505–2524 CrossRef CAS.
- K. Madasamy, S. Kumaraguru, V. Sankar, S. Mannathan and M. Kathiresan, New J. Chem., 2019, 43, 3793–3800 RSC.
- P. Sharma, M. Gupta, R. Kant and V. K. Gupta, RSC Adv., 2016, 6, 32052–32059 RSC.
- G.-M. Huang, L.-X. Pan, S.-M. Li, M.-X. Ma, L.-C. Gui and Q.-L. Ni, J. Solid State Chem., 2023, 320, 123849 CrossRef CAS.
- B. N. Cabral, J. L. S. Milani, Á. F. A. da Mata, G. F. S. Andrade, H. B. da Silva and R. P. das Chagas, Inorg. Chem. Commun., 2023, 156, 111315 CrossRef CAS.
- Y. Liu, D. Hou and G. Wang, Chem. Phys. Lett., 2003, 379, 67–73 CrossRef CAS.
- Y. Xu, N. Al-Salim, C. W. Bumby and R. D. Tilley, J. Am. Chem. Soc., 2009, 131, 15990–15991 CrossRef CAS PubMed.
- M. Alberti, M. Gianelli, N. Panza, D. Zákutná, I. Matulková and A. Caselli, Eur. J. Inorg. Chem., 2024, 27, e202400494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra, selected computational results, characterization data, and plausible mechanisms of reactions. CCDC 2334707, 2405392 and 2405398 for the complexes 1, 2, and 3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03382e |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Subrato
Bhattacharya
* and
Subrato
Bhattacharya