
A double-chain based metallomicellar catalyst for aerobic oxidative synthesis of benzimidazoles in water†‡
Pragyansmruti
Sunani
 ,
Prabaharan
Thiruvengetam
and
Dillip Kumar
Chand
*
,
Prabaharan
Thiruvengetam
and
Dillip Kumar
Chand
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: dillip@zmail.iitm.ac.in
First published on 9th January 2025
Abstract
The oxomolybdenum complexes Mo1, Mo2 and Mo3, which share a common ONO donor ligand backbone but differ in their peripheral substituents, were explored to study their reactivity in organic transformations in water. The ligand backbones of Mo1 and Mo2 were covalently linked to a methyl group and a single hydrophobic n-hexadecyl chain via an ether linkage, respectively. The complex Mo3 was found to possess two n-hexadecyl chains attached to the ligand backbone via a common amine-N. Complexes Mo2 and Mo3 formed metallomicelle when dispersed in water due to the surfactant presence in their structures, enabling them to uptake organic substrates. The catalytic potential of the complexes was evaluated for the oxidative coupling of benzylamine with 1,2-diaminobenzene to synthesize benzimidazole in neat water using open air as the sole oxidant. The double-chain surfactant-type catalyst Mo3 displayed superior activity compared to the single-chain surfactant-type complex, Mo2. A wide variety of benzimidazoles were synthesized in good to excellent yields under environmentally benign conditions using Mo3 as the catalyst. The practical utility of the process was validated through multi-gram scale-up reactions and recyclability experiments. A plausible mechanism was proposed based on several controlled experiments and literature support.
Introduction
Surfactants are widely used as catalysts or as additives with suitable transition metal-based catalysts to promote organic transformations in water.1–4 In a metallosurfactant, the surfactant is integrated with the metal catalyst as a single molecule, thus offering greater advantages for organic transformations.5–13 Due to their amphiphilic behavior, metallosurfactants undergo spontaneous aggregation to form metallomicelle, which occurs above the critical micelle concentration (CMC). These metallomicelles enrich organic substrates within their hydrophobic core, bringing them closer to the catalytically active species.14–17 This enhanced interaction between the substrate and catalytic center significantly accelerates reaction rates. Our research focuses on developing diverse metallomicellar systems and exploring their application in various organic transformations in water.10–13In general, metallosurfactants are designed so that the ligand backbone is connected to a hydrophobic chain, either through electrostatic interactions10–12,18 or covalent bonding.13,19–22 Among these, covalently bonded metallomicellar systems have been proven to be more efficient catalysts compared to electrostatically linked systems, likely due to their enhanced catalytic stability and the fixed position of the metal units within the micellar aggregation.13 The success of metallomicellar-catalyzed organic reactions in water depends not only on the catalyst center but also on the structure and aggregation behaviour of the metallosurfactants.
Oxomolybdenum complexes are well-explored as efficient catalysts for various oxidation reactions, however, they often involve harsh reaction conditions, hazardous oxidants, additives, and/or environmentally unfriendly organic solvents.23–25 We have reported a non-surfactant-based molybdenum catalyst MoO2(L1)(OH2), Mo1, and a metallosurfactant, MoO2(L2)(OH2), Mo2, in which the ligand backbone is covalently bonded to one hydrophobic alkyl chain (Fig. 1). The metallosurfactant Mo2 could efficiently catalyzed the oxidation of alcohols and lignin mimics into their corresponding carbonyls in a water medium, using open air as the oxidant.13 We intended to prepare MoO2(L3)(OH2), Mo3 (Fig. 1), with two alkyl chains connected to the ligand backbone, expecting higher activity,21 and compare its reactivity with Mo2 and Mo1.
 | ||
| Fig. 1 Structures of oxomolybdenum complexes Mo1, Mo2, and Mo3. | ||
Herein we report the synthesis of double-chain based catalyst Mo3 and its use for oxidative coupling of benzylamine with 1,2-diaminobenzene to synthesize pharmaceutically important benzimidazoles26–29 in water. The advantage of a double-chain catalyst over its single-chain complex, Mo2, under comparable conditions is also established in terms of lower reaction temperature and lower catalyst loading. The newly developed double-chain connected metallomicellar catalyst, Mo3 produces the desired benzimidazoles in good yields under an aqueous environment, using open air as the sole oxidant, without involving any base/additives in contrast to the known methods that involve co-catalyst, additives, pressurized oxygen, and/or environmentally unfriendly organic solvents.30–35 Our catalyst is found to be compatible with synthesizing a wide range of benzimidazoles comprising various functional groups. Further, the practical utility of the process is validated by performing multi-gram scale synthesis and reusability experiments of the catalyst.
Results and discussion
The catalyst
Synthesis of the molybdenum complex, Mo3 is shown in Scheme 1. The precursors (P3, A3), Schiff base ligand (L3(H)2) and Mo3 are all new compounds but they were synthesized using methods developed for related compounds. The precursor P3 was synthesized36 using 3-aminophenol and 1-bromohexadecane as starting materials whereas A3 was synthesized by the Vilsmeier–Haack method.37 The Schiff base ligand L3(H)2 was prepared by treating A3 with ethanolamine in methanol under reflux conditions for 3 hours.38 The complex Mo3 was prepared13 by refluxing a mixture of the ligand L3(H)2 and MoO2(acac)2 in methanol for 3 hours followed by filtration to isolate a solid; the isolated solid was suspended in water and stirred for 1 hour to obtain the targeted complex. The obtained complex was thoroughly characterized by FT-IR, NMR spectroscopy, and ESI-MS spectrometry. The critical micellar concentration was determined using the conductivity method. The micellar aggregation behaviour in water was confirmed by high-resolution transmission electron microscopy (HR-TEM). | ||
| Scheme 1 Synthesis of the double-chain based oxomolybdenum complex Mo3. | ||
The presence of a single species in the solution was confirmed from 1H and 13C NMR of the complex recorded in CDCl3 (ESI, Fig. S13 and S14‡). The 1H NMR shows significant changes in chemical shift values for the complex Mo3 compared to those for the free ligand. The complex was further characterized by FT-IR spectroscopy. Two strong bands in the region of 897 and 931 cm−1 can be assigned as symmetric and asymmetric stretching frequencies, respectively, due to cis-dioxo moieties (cis-MoO2).39 The hydrogen-bonded OH present in the free ligand shows a stretching frequency near 2700 cm−1, which is absent in the spectrum recorded for complex Mo3. The presence of Mo–O bond with organic ligand was confirmed from the peaks appearing in the region of 636 cm−1.39 The bands at 1604 and 1523 cm−1 are assigned to the stretching frequency of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–O functional groups (ESI, Fig. S16‡). The ESI-MS data of the complex was recorded in positive ion mode in chloroform solvent. The segment containing molybdenum provides the isotopic pattern that corresponds to the mononuclear complex. The isotopic peak pattern (m/z = 757.4768) matched well with the simulated pattern (m/z = 757.4786) (Fig. 2). The crystallization of Mo3 was tried, however, a suitable crystal was not obtained for mounting. Hence, the energy-minimized structures of Mo3 are provided in lieu of the crystal structure (ESI, Fig. S17‡).
N and C–O functional groups (ESI, Fig. S16‡). The ESI-MS data of the complex was recorded in positive ion mode in chloroform solvent. The segment containing molybdenum provides the isotopic pattern that corresponds to the mononuclear complex. The isotopic peak pattern (m/z = 757.4768) matched well with the simulated pattern (m/z = 757.4786) (Fig. 2). The crystallization of Mo3 was tried, however, a suitable crystal was not obtained for mounting. Hence, the energy-minimized structures of Mo3 are provided in lieu of the crystal structure (ESI, Fig. S17‡).
 | ||
| Fig. 2 Partial ESI-MS spectrum of complex Mo3 in chloroform. | ||
The critical micelle concentration (CMC) of the metallosurfactant, Mo3 was determined from the conductivity measurement method and found to be 0.625 mM. It was observed that the double chain-based complex Mo3 self-assembled to form aggregates in lower concentration compared to the single chain molybdenum complex, Mo2, whose CMC was determined to be 0.996 mM (Fig. 3). The micellar aggregation of the complex Mo3 above CMC was further confirmed by HR-TEM analysis (Fig. 4).
 | ||
| Fig. 3 Determination of critical micelle concentration (CMC) of Mo2 and Mo3. | ||
 | ||
| Fig. 4 HR-TEM image of metallomicellar complex Mo3. | ||
Scope of the catalyst
After successful preparation and characterization of the metallomicellar complex Mo3, we intended to study the effect of double chain over single chain in reactivity of an organic transformation under comparable reaction conditions. In this regard, oxidative coupling of benzylamine with 1,2-diaminobenzene to generate 2-substituted benzimidazole in a water medium was chosen as the model reaction. The reaction was chosen with the purpose of delivering an efficient, cost-effective, and environmentally friendly route to synthesize N-heterocyclic compounds such as benzimidazoles from primary amine, unlike the reported methods that largely rely on the organic solvents for the required transformation.30–35The reaction conditions for the targeted organic reaction were optimized by taking benzylamine, 1a and 1,2-diaminobenzene, 1b as model substrates, varying the catalysts, catalyst loading, oxidant, temperature, and time. The acquired results are summarized in Table S1 (ESI‡).
We began our investigation by treating a slight excess of benzylamine, 1a (0.9 mmol) with 1,2-diaminobenzene, 1b (0.75 mmol) in 0.5 mL water, in the absence of a catalyst, at room temperature for 24 hours, under open air conditions. No product formation was observed (Table S1,‡ entry 1). However, when the reaction temperature was raised to 50 °C, a small amount of 2-phenyl-1H-benzimidazole, 1c (<05% yield) was observed (Table S1,‡ entry 2). Based on this observation, we introduced 1.0 mol% of non-micellar catalyst, Mo1, to the reaction mixture, and heated it at 50 °C for 24 hours, resulting in a slight increment of the product yield to 11% (Table S1,‡ entry 3). Notably, the micellar catalysts with a single chain, Mo2 (1.0 mol%), and double chain, Mo3 (1.0 mol%) in the ligand backbone were employed under similar conditions, and the yield of 1c increased significantly to 35% and 46%, respectively (Table S1,‡ entries 4 and 5). These results underscore the crucial role of the surfactant entity within the metal complex, which forms a metallomicelle during the reaction process, facilitating the reaction in a water medium. After proving the superior reactivity of the double chain-based catalyst, Mo3, over Mo2 and non-micellar catalyst, Mo1, further optimization was conducted using Mo3.
To achieve a better yield of the desired product, the reaction temperature was increased to 80 °C, and 1c was isolated in 83% yield (Table S1,‡ entry 6). A further increase in the reaction temperature to 100 °C did not result in a significant change in the product yield, where 1c was isolated in 87% (Table S1,‡ entry 7). Notably, when the catalyst loading was increased to 1.5 mol% and the reaction was conducted at 80 °C for 24 hours, complete conversion was achieved, and product 1c was isolated in 94% yield (Table S1,‡ entry 8). Reducing the reaction time to 18 and 12 hours led to isolated yields of 92 and 79%, respectively (Table S1,‡ entries 9 and 10). Thus, entry 9 in Table S1‡ is considered as the optimized reaction condition for the oxidative coupling of benzylamine, 1a with 1,2-diaminobenzene, 1b to form 2-substituted benzimidazole, 1c catalyzed by Mo3 (Scheme 2).
 | ||
| Scheme 2 Oxidative coupling of benzylamine, 1a and 1,2-diaminobenzene, 1b under optimized conditions using Mo3, Mo2, Mo1 and in the absence of a catalyst. | ||
Under these reaction conditions, the catalytic ability of Mo2 was tested, resulting in only 73% of benzimidazole 1c (Table S1,‡ entry 11). When the catalyst loading of Mo2 was increased to 2.5 mol%, 1c was isolated in 84% yield.
Only 28% and 11% yield of benzimidazole 1c were isolated when the reactions were carried out using Mo1 and in the absence of a catalyst under optimized reaction conditions (Scheme 2).
The effect of temperature on reaction progress using catalysts Mo3 and Mo2 was evaluated, by varying the reaction temperature while keeping other optimized conditions the same. The obtained results are plotted in Fig. 5. The yield of 1c increased with the reaction temperature and complete conversion was observed at 80 °C using the catalyst Mo3, whereas Mo2 requires a higher temperature (100 °C) for efficient conversion. Likewise, the effect of the catalyst concentration on the reaction rate was monitored using the more reactive catalyst Mo3. During the optimization of reaction conditions, the maximum yield of product 1c was obtained when the catalyst concentration was maintained at 22.5 mM. Hence, keeping the optimized reaction conditions, the concentration of the catalyst was lowered up to 3.75 mM and the product formation was evaluated (Fig. 6). The observed results signify the efficient product formation at higher catalytic concentrations.
 | ||
| Fig. 5 Yield of 1c catalyzed by Mo3 and Mo2 under optimized conditions as a function of temperature. | ||
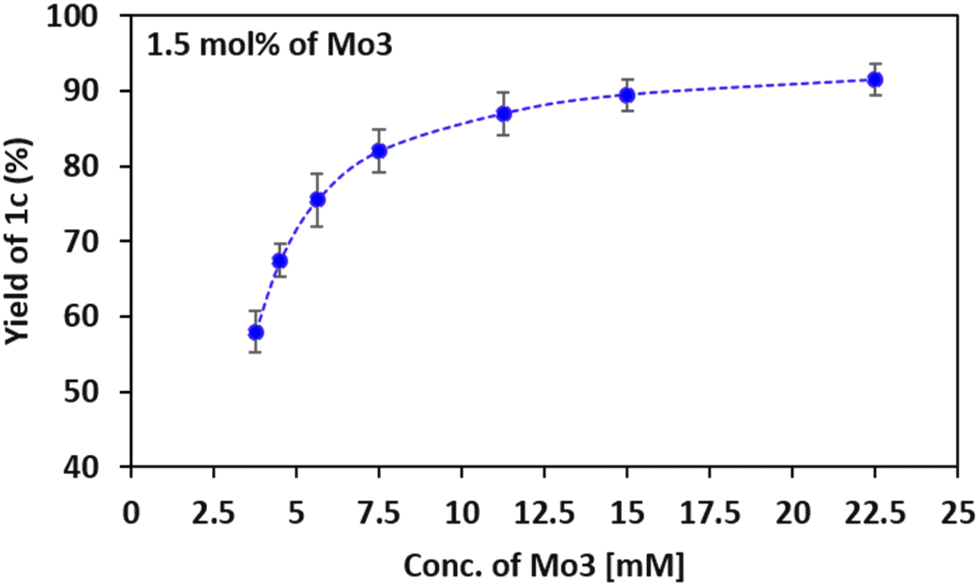 | ||
| Fig. 6 Yield of 1c under optimized conditions as a function of concentration of Mo3. | ||
For further understanding, we performed the reaction kinetics for the oxidative coupling of benzylamine, 1a with 1,2-diaminobenzene, 1b to form benzimidazole, 1c catalyzed by Mo2 and Mo3 under comparable reaction conditions. Conversion of 1,2-diaminobenzene, 1b to benzimidazole 1c was determined at specific time intervals by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard (Fig. 7a). The rates of the oxidative coupling reactions catalyzed by Mo2 and Mo3 were calculated from the −ln(1 − XA) vs. time plot (Fig. 7b); the slope (k′) values were obtained from the −ln(1 − XA) vs. time graph and found to be 0.083 and 0.166 (h−1), respectively. The second-order rate constants were calculated as 3.68 and 7.38 M−1 h−1 for Mo2 and Mo3, respectively, indicating the high reactivity of double chain-based catalyst, Mo3 compared to the single chain-based catalyst Mo2 under standard conditions.
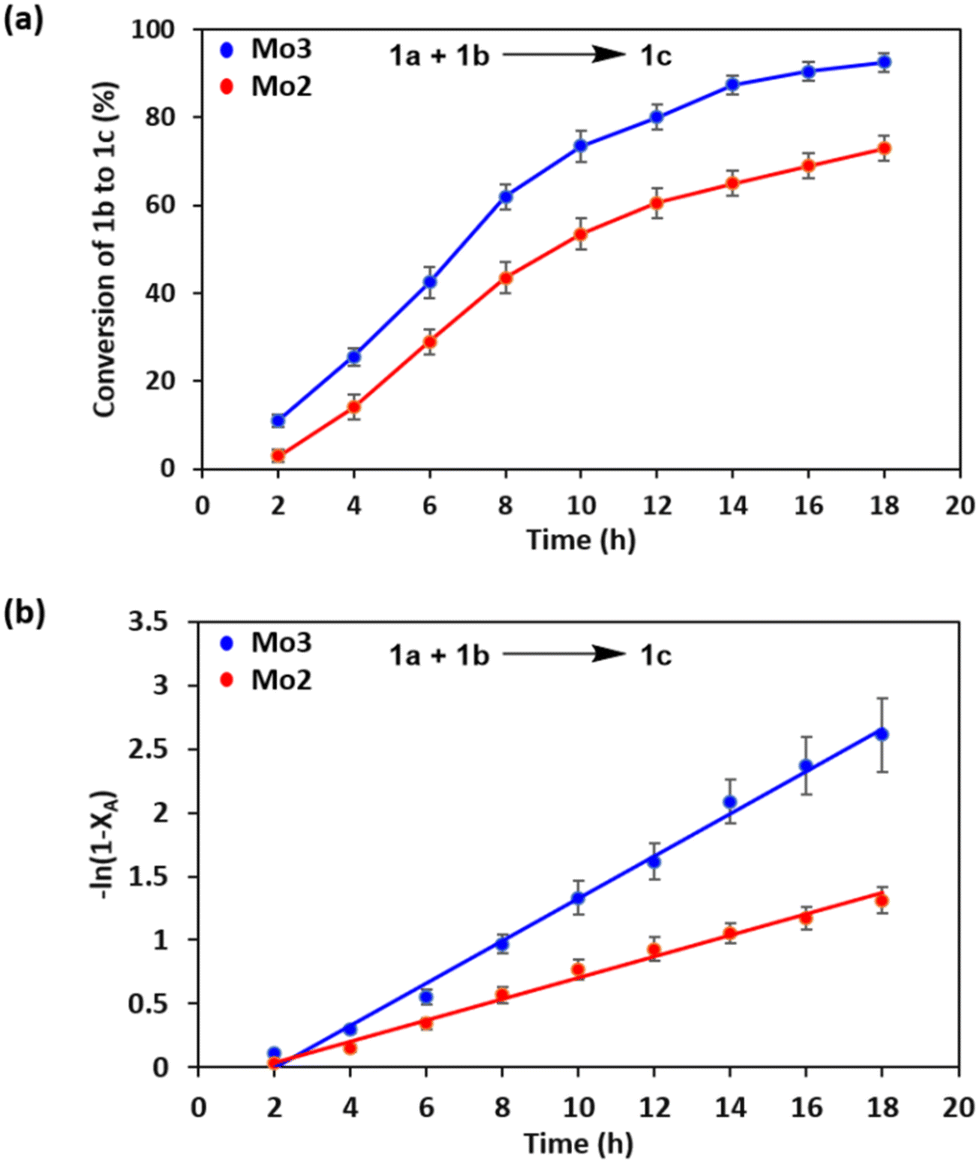 | ||
| Fig. 7 Mo2 and Mo3 catalyzed (a) conversion of 1b to 1c at 2 h time intervals; (b) plot of −ln(1 − XA) vs. time for the rate constant determination. | ||
The efficient and environmentally friendly approach for the synthesis of benzimidazoles from benzylamine using a highly reactive and cost-effective catalyst Mo3 inspired us to examine the versatility of the catalyst system towards a variety of substrates with different functional groups under the optimized reaction conditions and the results are summarized in Table 1. Interestingly, there is no apparent change in the product yield was observed despite the variations in the electronic nature of the benzene ring, indicating that the catalyst system operates effectively across a range of different substituents. Specifically, benzylamines substituted with typical electron-donating groups such as methoxy, methyl, isopropyl, and t-butyl groups (2a–6a) were efficiently converted to the corresponding benzimidazoles with good to excellent yields (2c–6c). The methoxy groups substituted at para position (2a) provided better yield compared to the ortho position (3a). For benzylamine substituted with t-butyl group (6a), the yield of the corresponding benzimidazole (7c) was slightly lower at 79%. This decrease in the yield is likely attributed to steric hindrance caused by the bulky t-butyl group.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a

Likewise, benzylamine substituted with electron-withdrawing groups such as fluoro, bromo, chloro, cyano, and trifluoromethyl groups (7a–11a) underwent oxidative coupling with 1,2-diaminobenzene, 1b and generated corresponding benzimidazoles in good yields (7c–11c). Benzylamines substituted with some other functional groups such as 4-trifluoromethoxy (12a), and naphthalen-1-ylmethanamine (13a) afforded corresponding benzimidazoles in good yields (12c–13c). The acetal group, a relatively weaker functionality, was tolerated during the reaction process and the corresponding benzimidazole 14c was isolated in 86% yield. Further, hetero-atom-containing amines such as furfuryl amine (15a), 2-thiophene-methylamine (16a), and 3-pyridine methylamine (17a) were evaluated under the standard reaction conditions and moderate to good yields of benzimidazoles were obtained (15c–17c).
The catalytic activity of Mo3 was also examined with methyl-substituted 1,2-diaminobenzene and reactions proceeded smoothly with a good yield of the desired products (18c–20c).
The excellent results from the substrate scope evaluation confirmed that the metallomicellar catalytic system is highly efficient in producing a wide range of benzimidazole derivatives with good to excellent yields in water using open air as the sole oxidant. This demonstrates the robustness, efficiency, and environmental sustainability of the catalyst system, as it operates under mild conditions without the need for external oxidants, making it a promising approach for green organic synthesis.
Gram-scale synthesis and recyclability experiments
To demonstrate the real-life applicability and synthetic validity of the current approach, gram-scale synthesis and recyclability experiments were performed under the optimized reaction conditions. We choose to synthesize 2-phenyl-1H-benzimidazole (1c) and a drug molecule, fuberidazole (17c) in a gram-scale for the scale-up reaction. In this line, 1.0 gram of benzylamine, 1a was reacted with 0.84 gram of 1,2-diaminobenzene, 1b with 1.5 mol% of the catalyst Mo3 maintaining the optimal conditions, and 85% of benzimidazole, 1c was obtained (Scheme 3a). Similarly, 1.0 and 2.0 grams of furfuryl amine, 17a were subjected to 0.92 and 1.8 grams of 1,2-diaminobenzene, 1b under standard conditions, and the desired product fuberidazole, 17c was obtained in 80 and 73% yield, respectively (Scheme 3b). The lowered product yield during the gram-scale synthesis is likely due to potential mass transfer challenges caused by the slurry formation. | ||
| Scheme 3 Multi-gram scale synthesis of benzimidazole using Mo3. | ||
The catalyst recyclability experiment was performed to evaluate the long-term constancy of the catalyst. A series of reactions were kept taking benzylamine, 1a (1.8 mmol) and 1,2-diamobenzene, 1b (1.5 mmol) with 1.5 mol% of catalyst Mo3 under optimized circumstances. The reusability and sustainability of the catalyst were examined by stopping the reaction after 18 h and using the same catalyst for the next run with the addition of fresh substrate. The organic compounds were separated from the aqueous layer by adding ethyl acetate to the reaction mixture. The organic layer was collected for the calculation of the yield of the desired product whereas the catalyst containing aqueous layer was used for the next cycle. The process was repeated for up to 8 cycles, and the yields were observed in the range of 92%–86% without any significant loss in the product yield (Scheme 4, blue color). The catalytic recyclability experiment was also performed under a kinetically controlled regime to evaluate the intrinsic ability of the catalyst throughout the reaction process. The reactions were stopped after 6 h and the organic compounds were extracted from the aqueous layer by phase separation adding ethyl acetate. The process was repeated for 8 cycles and the yield of benzimidazole 1c was isolated in the range of 46–41% (Scheme 4, orange color). These recyclability experiments prove the possibility of using the micellar catalyst Mo3 for practical applications in a water medium under open-air conditions.
 | ||
| Scheme 4 Recyclability experiments of catalyst Mo3. | ||
Benzaldehyde (oxidation product of excess benzylamine) was detected through a TLC experiment in the organic layer when the reaction was carried out for 18 h in a cycle. However, benzylamine (no benzaldehyde) could be detected when the reaction was stopped after 6 h. The presence of benzylamine in the water layer is inevitable in either case but such leftover benzylamine has no influence in the next cycle. This was confirmed by taking 1.5 equivalent of benzylamine in a separate experiment for synthesizing 1c and found that the time taken and yields are not changed as compared to the use of 1.2 equivalents of benzylamine.
Reaction mechanism
On the basis of the experimental study and literature support, we proposed the plausible mechanism for the oxidative coupling of benzylamine and 1,2-diaminobenzene catalyzed by Mo3. In order to determine the reaction followed by ionic or radical pathway, a few controlled experiments were performed. In this line, catalyst Mo3 was treated with 3 mol% of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT), both known as a radical inhibitor, under optimized reaction condition and no significant change in the yield of the product formation was observed (Scheme 5a and b). To obtain an insight into the mechanism and explore the formation of the oxoperoxo-molybdenum complex as the potential reaction intermediates, the catalyst Mo3 was treated in water under molecular oxygen (through bladder) at 80 °C temperature and was stopped after 10 hours (Scheme 5c). The sample was analyzed by ESI-MS spectrometry and we were unsuccessful in locating any oxodiperoxo-molybdenum complex intermediate even after several trials. Based on the evidence and lack of support for a radical mechanism, an ionic mechanism was proposed instead of a radical pathway. | ||
| Scheme 5 (a) and (b) Oxidative coupling reactions in water catalyzed by Mo3 in the presence of radical inhibitors; (c) trial conversion of molybdenum complex Mo3 into peroxo complex (not feasible under employed conditions). | ||
To determine the possible intermediates during the reaction for the formation of 2-substituted benzimidazole, a reaction was carried out in the absence of 1,2-diaminobenzene taking benzylamine as the starting material under standard conditions. We observed the complete conversion of benzylamine to benzaldehyde, 2a′ (Scheme 6a). However, when the reaction was carried out in a toluene medium (instead of H2O) under standard conditions, it generated the homo-coupled aldimine product N-benzylidene-1-phenylmethanamine, 3a′ (Scheme 6b). It is proposed that 1a is oxidized to phenylmethanimine, 1a′, which reacted with another unit of 1a to produce the homo-coupled product, 3a′. However, in the presence of water at 80 °C, phenylmethanimine, 1a′ is hydrolysed to provide benzaldehyde, 2a′ in the absence of 1,2-diamine. During the reaction starting with benzylamine, 1a and 1,2-diaminobenzene, 1b for synthesizing benzimidazole, aldehyde/imine was not traced. This signifies the generated imine from benzylamine immediately coupled with 1b to provide the desired product 1c. However, the complete or partial formation of aldehyde, 2a′ from imine, 1a′, through hydrolysis with water at 80 °C cannot be entirely ruled out. To verify this, benzaldehyde, 2a′ was reacted with 1,2-diaminobenzene, 1b and 1,2-disubstituted product, 1-benzyl-2-phenyl-1H-benzimidazole, 1c′ was found as a by-product, both, with or without the catalyst under optimized conditions. In contrast, the by-product 1c′ was not detected when the reaction started with benzylamine, 1a and 1,2-diaminobenzene, 1b. Hence, the slow production of imine from benzylamine using the catalyst Mo3 helps in the selective formation of 2-substituted benzimidazole, 1c. Further, to understand the role of oxygen, a separate reaction was carried out under an argon atmosphere and only 42% yield of 1c was obtained, which clearly indicates that the reaction proceeded through aerobic oxidation where oxygen plays a crucial role in the high yield of the desired product (Scheme 6c).
 | ||
| Scheme 6 Controlled experiments. | ||
To understand the fate of the catalyst Mo3 during the catalysis reaction, we stopped the catalysed synthesis of 1c after 10 h and separated the metal complex from the reaction mixture. The organic components were extracted from the aqueous reaction mixture using ethyl acetate. The metal-containing species were isolated by centrifugation of an aqueous layer and analysed by 1H NMR, HRMS and FT-IR techniques. While the evidence for the existence of Mo3 could be obtained, we do not have evidence to support the existence of metal–hydroxo species that are the likely intermediates.
Maeda et al. proposed a reaction mechanism for the oxidation of alcohol using an oxovanadium complex where the oxo–metal compound was converted to a hydroxo–metal intermediate.40 From all the above experimental studies and literature reports, we propose an ionic pathway for the oxidation of benzylamine to respective imine, 1a′ catalyzed by Mo3 as shown in Scheme 7. The amine 1a interacted with the molybdenum center of catalyst Mo3 replacing weakly coordinated water molecules with the formation of hydroxo–metal complex Mo3-I intermediate. Mo3-I undergoes β-hydrogen elimination from the methylene group of benzylamine to the unutilized MoO group or Mo–OH group, generating the intermediates Mo3-II or Mo3-II′, respectively. In this step, the formation of imine, 1a′ occurs which is weakly bound with the metal center. The intermediate complex Mo3-II or Mo3-II′ release the imine producing respective intermediates Mo3-III or Mo3-III′. In the final step, the intermediate complexes Mo3-III or Mo3-III′ react with ½O2 to produce the original catalyst Mo3. The generated imine coupled with 1,2-diaminobenzene to produce the desired compound 2-substituted benzimidazole, 1c.
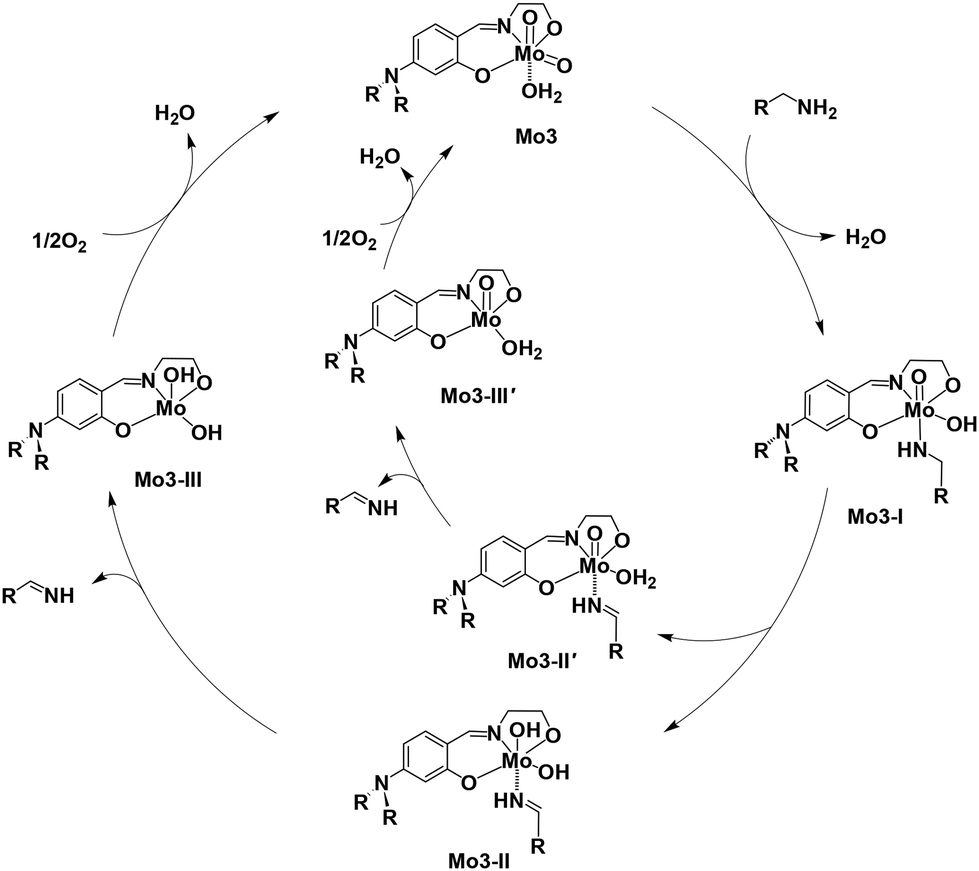 | ||
| Scheme 7 Proposed mechanism for the oxidation of benzylamine to phenylmethanimine using Mo3. | ||
Conclusions
In summary, a new oxomolybdenum-based metallomicellar catalyst, Mo3 was synthesized, where the coordination entity was connected with two long alkyl chains via covalent bonds. The complex Mo3 was thoroughly characterized using FT-IR, NMR spectroscopy and ESI-MS spectrometry techniques. The micellar aggregation of Mo3 in water was confirmed through CMC determination and HR-TEM analysis. The double-chain-based catalyst Mo3 demonstrated superior efficiency in the oxidative synthesis of 2-substituted benzimidazoles from benzylamine in water compared to a similar oxomolybdenum complex with a single alkyl chain in the ligand backbone, Mo2. Using the newly developed catalyst Mo3, a range of benzimidazole derivatives with different functional groups were synthesized in good to excellent yields in water, utilizing open air as the only oxidant without involving any base or additives. The practical applicability of the developed catalyst was probed by multi-gram scale synthesis and recyclability experiments. Finally, a plausible mechanism was proposed based on some controlled experiments and supporting literature.Experimental section
Materials and methods
3-Aminophenol, bis(acetylacetonato)dioxomolybdenum(VI), 1-bromohexadecane, bromomethane, 2,4-dihydroxybenzaldehyde were purchased from Sigma-Aldrich. Ethanolamine, POCl3, iPrOH, and ethyl acetate were purchased from SISCO Research Laboratories Pvt. Ltd, India. The benzylamines 1a–3a, 6a–8a, 11a, 13a, 15a and the deuterated solvents were purchased from Sigma-Aldrich, whereas 4a, 5a and 16a, 17a were purchased from TCI Chemicals (India Pvt. Ltd). Benzylamines 9a, 10a, 12a, 14a and 19a were purchased from BLD Chemicals and the 1,2-diaminobenzenes 1b and 2b were purchased from Sigma-Aldrich. All the aforementioned purchased chemicals from different sources were used without any further purification.The NMR spectra were recorded on Bruker Avance-400 and 500 instruments. Fourier Transform Infrared (FT-IR) spectra of the complexes were measured on a model Nexus 670 (FTIR), Centaurms 10× (Microscope) having spectral Range 4000 to 400 cm−1 with an MCT-B detector. The ESI-Mass spectrum of the complex was recorded on an Agilent Q-TOF spectrometer in a positive ion mode. HR-TEM images were obtained on HR-TEM, (TecnaiTM G2 TF20) working at an accelerating voltage of 200 kV. The wavelength of the laser used was 632.8 nm with a scattering angle of 90°. LMCM-20 conductivity meter from LABMAN scientific instruments was used for the determination of critical micellar concentration of the complexes. DFT study was performed using the Gaussian 09 software package. The B3LYP (Becke's three-parameter hybrid functional using the LYP correlation) functional was used for geometry optimizations and frequencies with LANL2DZ for the Mo atom, and the 6-31G* basis set for carbon, nitrogen, oxygen and hydrogen. Frequency calculations were performed for the optimized structures to confirm the absence of any imaginary frequencies.
Synthesis of precursor, 3-(dihexadecyl)aminophenol (P3)
The precursor 3-(dihexadecyl)aminophenol was synthesized following the literature report36 with necessary modification taking 3-aminophenol and 1-bromohexadecane as starting material. 3-Aminophenol (1.0 g, 9.16 mmol), 1-bromohexadecane (5.59 g, 18.32 mmol) and Na2CO3 (1.94 g, 18.32 mmol) were taken in H2O (10 mL) and iPrOH (10 mL) solvent mixture. The reaction mixture was allowed to stir for 12 hours under reflux conditions. After completion of the reaction, the solvent was removed under reduced pressure. The obtained crude product was purified by column chromatography using hexane and ethyl acetate as the eluent.1H NMR (400 MHz, CDCl3, 24 °C): δ 7.05–7.01 (m, 1H), 6.24–6.22 (m, 1H), 6.13–6.09 (m, 2H), 3.23–3.19 (m, 4H), 1.56 (m, 4H), 1.30–126 (m, 52H), 0.90–0.87 (m, 6H) ppm.
13C{1H} NMR (126 MHz, CDCl3, 24 °C): δ 156.85, 149.91, 130.16, 104.84, 102.29, 98.73, 51.31, 32.08, 29.85, 29.78, 29.71, 29.52, 27.40, 27.34, 22.84, 14.27 ppm.
HRMS (ESI) m/z: calculated for C38H71NO + H: 558.5534; found: 558.5609.
Synthesis of precursor, A3
Aldehyde, A3 was prepared by the Vilsmeier–Haack method following a literature protocol.37 Initially, 5.0 mL of dry DMF was cooled to 0–5 °C in an ice bath and POCl3 (0.34 g, 2.24 mmol) was added dropwise into it. The solution was heated at 40 °C for half an hour and a yellow-colored Vilsmeier reagent appeared. The solution was again cooled to 0–5 °C and 3-(hexadecyl)aminophenol (0.5 g, 0.89 mmol) was added dropwise with continuous stirring. The reaction mixture was allowed to stir for 15–20 minutes and heated at 70 °C for 6 hours. After completion of the reaction, the ice-cooled distilled water was added to the reaction mixture. The organic layer was separated from the aqueous layer by phase separation by adding DCM. The organic solvent was evaporated under reduced pressure and the desired product A3 was purified by column chromatography using hexane and ethyl acetate as eluent.1H NMR (500 MHz, CDCl3, 24 °C): δ 11.64 (s, 1H), 9.48 (s, 1H), 7.25–7.21 (m, 1H), 6.23–6.21 (m, 1H), 6.03 (s, 1H), 3.31–3.28 (m, 4H), 1.61–1.60 (m, 4H), 1.31–126 (m, 52H), 0.89–0.86 (m, 6H) ppm.
13C{1H} NMR (126 MHz, CDCl3, 24 °C): δ 191.95, 164.44, 154.68, 135.38, 111.51, 104.69, 96.99, 51.38, 32.07, 29.84, 29.80, 29.78, 29.74, 29.71, 29.58, 29.50, 27.43, 27.16, 22.83, 14.25 ppm.
HRMS (ESI) m/z: calculated for C39H71NO2 + H: 586.5485; found: 586.5553.
Synthesis and characterization of ligand, L3(H)2
The double chain integrated Schiff base ligand L3(H)2 was synthesized by adopting a literature protocol with necessary modifications.38 The precursor A3 (1.0 g, 1.7 mmol) was taken in 15 mL methanol and ethanolamine (0.1 g, 1.7 mmol) was added to the reaction mixture followed by a few drops of acetic acid as a catalyst. The reaction mixture was stirred for 3 h under reflux conditions. After completion of the reaction the obtained clear solution was allowed to stand overnight to obtain a yellow crystalline solid Schiff base compound.1H NMR (400 MHz, CDCl3, 24 °C): δ 8.05 (s, 1H), 6.99–6.97 (m, 1H), 6.13–6.11 (m, 1H), 6.05 (s, 1H), 3.86–3.84 (m, 2H), 3.64–3.63 (m, 2H), 3.27–3.24 (m, 4H), 1.58 (m, 4H), 1.30–126 (m, 52H), 0.89–0.86 (m, 6H) ppm.
13C{1H} NMR (126 MHz, CDCl3, 24 °C): δ 167.67, 164.48, 152.74, 133.36, 108.23, 103.59, 98.70, 62.44, 59.02, 51.19, 32.07, 29.84, 29.80, 29.75, 29.66, 29.50, 27.60, 27.25, 22.83, 14.26 ppm.
HRMS (ESI) m/z: calculated for C41H76N2O2 + H: 629.5907; found: 629.5974.
Synthesis of double chain based oxomolybdenum complex Mo3
The oxomolybdenum complex [MoO2(L3)(H2O)], Mo3 was synthesized following a general method demonstrated in our earlier report.13 The ligand L3(H)2 (0.5 g, 0.8 mmol) and bis(acetylacetonato)dioxomolybdenum(VI) (0.26 g, 0.8 mmol) were dissolved in 15 mL methanol and stirred for 3 h under reflux conditions. After completion of the reaction the obtained yellow colored solution was allowed to stand overnight. A yellow-colored semi-crystalline was observed, which was filtered and dried under vacuum. The obtained yellow solid was taken in 15 mL water and stirred for 1 hour at room temperature. The precipitate was further filtered and dried under a vacuum, providing the desired compound Mo3.1H NMR (500 MHz, CDCl3, 24 °C): δ 8.31 (s, 1H), 7.16–7.14 (m, 1H), 6.31–6.29 (m, 1H), 6.19 (s, 1H), 4.63–4.60 (m, 2H), 4.16–4.13 (m, 2H), 3.31–3.28 (m, 4H), 1.31–1.26 (m, 56H), 0.89–0.86 (m, 6H) ppm.
13C{1H} NMR (126 MHz, CDCl3, 24 °C): δ 164.09, 163.48, 155.45, 134.97, 109.82, 106.58, 99.88, 71.24, 63.72, 51.50, 32.07, 29.89, 29.85, 29.81, 29.74, 29.61, 29.50, 27.54, 27.20, 22.83, 14.25 ppm.
IR (KBr pellet) ν = 2917, 2856, 1607, 1523, 1461, 1233, 931, 897, 636 cm−1.
MS (ESI) m/z: calculated for [(Mo3 + H) − H2O]+: 757.4786; found: 757.4768.
General procedure for the oxidative coupling reactions
A mixture of benzylamine (0.9 mmol), 1,2-diaminobenzene (0.75 mmol) and Mo3 (1.5 mol%) was taken in 0.5 mL water in a ∼12.0 mL reaction tube (∼5.0 × 2.0 cm of height × width) fitted with a reflux condenser. The reaction mixture was stirred for 18 h at 80 °C temperature under open air atmosphere. The progress of the reaction was monitored by TLC. After completion of the reaction, 0.5 mL water was added to the reaction mixture, and the aqueous layer was extracted with ethyl acetate (3 × 1 mL). The organic layer containing benzimidazole was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The desired product was purified by column chromatography (silica gel) using hexane–ethyl acetate as an eluent. The purified product was characterized by 1H and 13C NMR spectroscopy.Author contributions
D. K. C., P. S. and P. T. designed the project. P. S. and P. T. were involved in the synthesis of surfactant-based ligands, molybdenum complexes, and their characterization. P. S. carried out the experiments related to catalysis. P. T. was involved in scientific discussions with P. S. on a regular basis. The manuscript was written by all authors. D. K. C. is the principal investigator and managed the project.Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. K. C. acknowledges financial support provided by IIT Madras under an Exploratory Research Project (RF20210651CYRFER008112). We thank the Department of Chemistry, IIT Madras for the instrumentation facilities.References
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174–7199 CrossRef CAS PubMed.
- C.-J. Li and L. Chen, Chem. Soc. Rev., 2006, 35, 68–82 RSC.
- G. L. Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS PubMed.
- J. Zhang, X. G. Meng, X. C. Zeng and X. Q. Yu, Coord. Chem. Rev., 2009, 253, 2166–2177 CrossRef CAS.
- J. Li, Y. Tang, Q. Wang, X. Li, L. Cun, X. Zhang, J. Zhu, L. Li and J. Deng, J. Am. Chem. Soc., 2012, 134, 18522–18525 CrossRef CAS PubMed.
- J. E. W. Cull, A. Richard and J. Scott, Green Chem., 2013, 15, 362–364 RSC.
- X. Liang, Y. Gui, K. Li, J. Li, Z. Zha, L. Shi and Z. Wang, Chem. Commun., 2020, 56, 11118–11121 RSC.
- R. D. Chakravarthy, V. Ramkumar and D. K. Chand, Green Chem., 2014, 16, 2190–2196 RSC.
- P. Thiruvengetam, R. D. Chakravarthy and D. K. Chand, J. Catal., 2019, 376, 123–133 CrossRef CAS.
- P. Thiruvengetam and D. K. Chand, J. Org. Chem., 2022, 87, 4061–4077 CrossRef CAS PubMed.
- P. Thiruvengetam, P. Sunani and D. K. Chand, ChemSusChem, 2024, 17, e202301754 CrossRef CAS PubMed.
- J. K. Virdi, A. Dusunge and S. Handa, JACS Au, 2024, 4, 301–317 CrossRef CAS PubMed.
- T. Lorenzetto, D. Frigatti, F. Fabris and A. Scarso, Adv. Synth. Catal., 2022, 364, 1776–1797 CrossRef CAS.
- L. Onel and N. J. Buurma, Annu. Rep. Prog. Chem., Sect. B, 2010, 106, 344–375 RSC.
- P. C. Griffiths, I. A. Fallis, T. Chuenpratoom and R. Watanesk, Adv. Colloid Interface Sci., 2006, 122, 107–117 CrossRef CAS PubMed.
- Q. Gao, Y. Liu, S.-M. Lu, J. Li and C. Li, Green Chem., 2011, 13, 1983–1985 RSC.
- T. Rispens and J. B. F. N. Engberts, Org. Lett., 2001, 3, 941–943 CrossRef CAS PubMed.
- A. P. H. J. Schenning, J. H. Lutje Spelberg, D. H. W. Hubert, M. C. Feiters and R. J. M. Nolte, Chem. – Eur. J., 1998, 4, 871–880 CrossRef CAS.
- J. Li, Z. Lin, Q. Huang, Q. Wang, L. Tang, J. Zhu and J. Deng, Green Chem., 2017, 19, 5367–5370 RSC.
- T. Lorenzetto, F. Fabris and A. Scarso, Curr. Opin. Colloid Interface Sci., 2023, 64, 101689 CrossRef CAS.
- M. Amini, M. M. Haghdoost and M. Bagherzadeh, Coord. Chem. Rev., 2013, 257, 1093–1121 CrossRef CAS.
- S. C. A. Sousa and A. C. Fernandes, Coord. Chem. Rev., 2015, 284, 67–92 CrossRef CAS.
- P. Thiruvengetam and D. K. Chand, J. Indian Chem. Soc., 2018, 95, 781–788 CAS.
- O. O. Ajani, D. V. Aderohunmu, S. J. Olorunshola, C. O. Ikpo and I. O. Olanrewaju, Orient. J. Chem., 2016, 32, 109–120 CrossRef CAS.
- Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208–6236 CrossRef CAS PubMed.
- D. I. Shah, M. Sharma, Y. Bansal, G. Bansal and M. Singh, Eur. J. Med. Chem., 2008, 43, 1808–1812 CrossRef CAS PubMed.
- S. A. Galal, K. H. Hegab, A. S. Kassab, M. L. Rodriguez, S. M. Kerwin, A.-M. A. El-Khamry and H. I. El Diwani, Eur. J. Med. Chem., 2009, 44, 1500–1508 CrossRef CAS PubMed.
- K. M. H. Nguyen and M. Largeron, Chem. – Eur. J., 2015, 21, 12606–12610 CrossRef CAS PubMed.
- K. Gopalaiah and S. N. Chandrudu, RSC Adv., 2015, 5, 5015–5023 RSC.
- K. M. H. Nguyen and M. Largeron, Eur. J. Org. Chem., 2016, 1025–1032 CrossRef CAS.
- C. Weerakkody, D. Rathnayake, J. He, B. Dutta, P. Kerns, L. Achola and S. L. Suib, ChemCatChem, 2019, 11, 528–537 CrossRef CAS.
- K. Bano, D. A. Kisan and T. K. Panda, Eur. J. Inorg. Chem., 2022, e202200023 CrossRef CAS.
- M. Kocsis, M. Szabados, S. B. Ötvös, G. F. Samu, Z. Fogarassy, B. Pécz, Á. Kukovecz, Z. Kónya, P. Sipos, I. Pálinkó and G. Varga, J. Catal., 2022, 414, 163–178 CrossRef CAS.
- J. F. Ge, C. Arai, M. Kaiser, S. Wittlin, R. Brun and M. Ihara, J. Med. Chem., 2008, 51, 3654–3658 CrossRef CAS PubMed.
- R. Nazir, P. Danilevicius, A. I. Ciuciu, M. Chatzinikolaidou, D. Gray, L. Flamigni, M. Farsari and D. T. Gryko, Chem. Mater., 2014, 26, 3175–3184 CrossRef CAS.
- C. R. Bhattacharjee, C. Datta, G. Das and P. Mondal, Liq. Cryst., 2012, 39, 639–646 CrossRef CAS.
- O. A. Rajan and A. Chakravorty, Inorg. Chem., 1981, 20, 660–664 CrossRef CAS.
- Y. Maeda, N. Kakiuchi, S. Matsumura, T. Nishimura, T. Kawamura and S. Uemura, J. Org. Chem., 2002, 67, 6718–6724 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Vadapalli Chandrasekhar on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization of ligands, Mo-complexes, metallomicellar systems, catalysis protocols, reaction kinetics and characterization of catalytic products. See DOI: https://doi.org/10.1039/d4dt03406f |
| This journal is © The Royal Society of Chemistry 2025 |