
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral Si(IV) complex bearing a 1,2,4-triazole-2,2′-diphenol ligand: synthesis, (chiro-)optical properties and computational investigation†
Valerio
Giuso‡
 a,
Thibault
Thierry‡
a,
Christophe
Gourlaouen
b,
Pierluigi
Mercandelli
c,
Nicolas
Vanthuyne
d,
Matteo
Mauro
*a and
Stéphane
Bellemin-Laponnaz
*a
a,
Thibault
Thierry‡
a,
Christophe
Gourlaouen
b,
Pierluigi
Mercandelli
c,
Nicolas
Vanthuyne
d,
Matteo
Mauro
*a and
Stéphane
Bellemin-Laponnaz
*a
aInstitut de Physique et Chimie des Matériaux de Strasbourg UMR 7504 - Université de Strasbourg & CNRS, 23 rue du Loess, 67034 Strasbourg, France. E-mail: bellemin@unistra.fr; mauro@unistra.fr
bLaboratoire de Modélisation et Simulations Moléculaires, UMR7140-Chimie de la Matière Complexe, Institut Le Bel – 4, rue Blaise Pascal, CS 90032, FR-67081 Strasbourg Cedex, France
cUniversità degli Studi di Milano, Dipartimento di Chimica, 20133 Milan, Italy
dAix-Marseille Université, CNRS Centrale Marseille, iSm2, 13284 Marseille, France
First published on 17th March 2025
Abstract
The reactivity of bis-3,5-phenol 1-phenyl-1,2,4-triazole, a non-symmetrical tridentate O^N^O proligand derived from Deferasirox, towards a Si(IV) precursor is herein studied. The reaction of the proligand (O1NO2)H2 with SiCl4 afforded a highly stable homoleptic hexacoordinate complex, namely Si(O1NO2)2, in high yield. While the emission profile of the proligand – arising from an excited-state intramolecular proton transfer (ESIPT) mechanism – appears featureless and broad, the Si(IV) complex exhibits enhanced photoluminescence in the violet-to-deep-blue region, with a quantum yield of up to 32% in spin-coated thin films. A comprehensive study, combining photophysical methods and (time-dependent) density functional theory (TD-DFT) calculations, has rationalized the emissive behavior of this complex. Due to the non-symmetrical nature of the ligand, the homoleptic Si complex is obtained as a racemate of two Δ/Λ enantiomers that were separated by chiral chromatography and characterized by electronic circular dichroism (ECD). Subsequent TD-DFT calculations enabled the modelling of the ECD spectra and the assignment of the absolute configuration of the resolved enantiomers.
Introduction
Organosilicon compounds are currently of great importance in the development of new chemical reagents and functional materials, such as polymers, glasses and ceramics.1 Organosilicon derivatives offer great molecular diversity, also because, unlike carbon, silicon allows coordination greater than four.2 Some of the major achievements in organosilicon chemistry over the last few decades have been related to hypercoordinated intermediates.3,4 This coordination diversity has been studied to yield interesting tetra-, penta- or hexa-coordinated compounds with attractive applications in various fields, such as electroluminescent and electron transport materials as well as photodynamic therapy agents.5 For example, hexacoordinated N–Si complexes containing two 2,6-bis(benzimidazol-2-yl)pyridine pincer-type ligands have demonstrated excellent optoelectronic properties and exceptional thermal stability suitable for applications in optoelectronics, such as electroactive materials in organic light-emitting diodes (OLEDs) and photovoltaic devices (OPVs).6,7 Very recently, we have reported the synthesis and photo-/electroluminescence properties of a neutral homoleptic hexacoordinate Si(IV) complex based on two pincer-type tridentate O^C^O N-heterocyclic carbene ligands (Scheme 1). This complex, which shows remarkable saturated true-blue emission, was then employed as an emitter in a proof-of-concept OLED.8 This result led us to investigate other tridentate ligands for the synthesis of homoleptic Si(IV) complexes, and among the available options, we focused on the molecular structure of Deferasirox, a tridentate O^N^O iron chelator used for the treatment of iron overload that has also recently been used in chemotherapy and as an antifungal and antimicrobial agent.9–14 Additionally, Deferasirox derivatives have shown interesting photophysical properties for practical applications as sensors and detectors in physiological environments15,16 and have been shown to yield phosphorescent Pt(II) complexes with excited-state lifetimes on the order of microseconds and photoluminescence quantum yields (PLQYs) of up to 17%.17 | ||
| Scheme 1 Structures of Deferasirox (top, left), the previously studied NHC pincer-type O^C^O proligand (OCO)H3 (top, right) and the synthetic strategy for obtaining the Si(IV) complex of the current work (bottom). | ||
Herein, we report the easy access of a chiral, homoleptic, neutral and hexacoordinate luminescent complex, namely, Si(O1NO2)2, bearing the asymmetric bis-aryloxide triazole ligand (O1NO2)H2 along with its (chiro-)optical and computational investigation.
Results and discussion
It is important to note that due to the presence of the phenyl substituent on the triazole ring, the two phenoxy moieties are chemically different. As a consequence, the tridentate proligand, named (O1NO2)H2 for the sake of further considerations, yields a racemic octahedral complex with chiral meridional C2 symmetry and the general structure rac-[Si(O1NO2)2] upon coordination to the Si(IV) centre. Therefore, the Si(O1NO2)2 complex exists in two possible absolute configurations, namely, and
and  , owing to the two possible mutual orientations in space of the tridentate O1NO2 scaffolds around the Si(IV) atom (see Fig. S7† for details on the absolute configuration).
, owing to the two possible mutual orientations in space of the tridentate O1NO2 scaffolds around the Si(IV) atom (see Fig. S7† for details on the absolute configuration).
As shown in Scheme 1, the bis-tridentate, homoleptic Si(IV) complex was obtained by the direct reaction of SiCl4 with the 3,5-bisphenol 1-phenyl-1,2,4-triazole proligand (O1NO2)H2 and was easily purified by column chromatography.
1H NMR spectroscopy at room temperature exhibits a highly symmetric resonance pattern, indicative of fluxional behaviour around the Si(IV) centre and coherent with the small energetic barrier associated with the ring puckering motion of the coordinated tridentate O1NO2 ligands that deviate from planarity. The fast exchange between the different possible ligand conformations gives rise to the different helical twists that each of the two ligands may independently adopt, namely (δ,δ), (δ,λ), (λ,δ), and (λ,λ). Similarly, 29Si NMR spectroscopy confirms the hexacoordinate nature of silicon with a chemical shift of δ = −194.1 ppm, that falls in the range typically observed for other hexacoordinated complexes.6,8 NMR spectra are reported in Fig. S1–S6 of the ESI.†
Single crystals of rac-Si(O1NO2)2 suitable for X-ray crystallographic analysis were obtained by vapour diffusion of Et2O into a CH2Cl2 solution of the racemate and the molecular structure of the Δ isomer is shown in Fig. 1. The complex crystallised in the P21/c monoclinic space group with an octahedral geometry around the Si centre, showing O–Si–O angles in the range of 87.92–91.86° and 178.63–179.27°, O–Si–N angles in the ranges 89.46–91.20° and an N–Si–N angle of 177.79°. The octahedral Si centre bridges two tridentate ligands in a mer-O1NO2 coordination motif, with an average length of the Si–O bonds of 1.756 Å and more elongated Si–N bonds, averaging 1.873 Å. Although the environment around silicon is perfectly octahedral, the chelate ligands adopt an arrangement that positions them at an average angle of ca. 50° to each other with respect to the N1–N4 axes (Fig. 1b displays a view along the N1–N4 axes). Interestingly, the complex has a very high thermal stability as shown by thermogravimetric analysis (TGA), with degradation starting above 300 °C and a 5% weight loss temperature T5% being as high as 366 °C (Fig. S8†).
 | ||
| Fig. 1 (a) Molecular structure of Δ-Si(O1NO2)2. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Si(1)–O(2), 1.7468(16); Si(1)–O(3), 1.7546(15); Si(1)–O(4), 1.7583(15); Si(1)–O(1), 1.7642(15); Si(1)–N(4), 1.8721(18); Si(1)–N(1), 1.8740(18); O(2)–Si(1)–O(3), 89.45(7); O(2)–Si(1)–O(4), 91.85(7); O(3)–Si(1)–O(4), 178.62(8); O(2)–Si(1)–O(1), 179.26(8); N(2)–N(3)–N(6)–N(5), −49.83(18). (b) View of Δ-Si(O1NO2)2 along the N1–N4-axes. Hydrogen atoms and phenyl moieties are omitted for clarity (CCDC 2422992†). | ||
The two enantiomers were separated under standard conditions using a Chiralpak® IB-N5 chiral stationary phase with a heptane/ethanol/CH2Cl2 mixture (60/20/20) as the eluent with an enantiomeric excess (ee) of over 99.5%. The successful chiral resolution of the enantiomers was confirmed by analytical chiral HPLC characterization and electronic circular dichroism (ECD) spectra. Lastly, the high configurational stability of the complexes was confirmed by ECD measurements carried out on enantiopure samples before and after thermal treatment in refluxing toluene for 24 hours (see the ESI† for further details).
Photophysical characterization
First, the electronic absorption of the ligand (O1NO2)H2 and rac-Si(O1NO2)2 was investigated in dilute CH2Cl2. The electronic absorption spectra are displayed in Fig. 2 and the corresponding data are listed in Table 1. | ||
| Fig. 2 UV-vis absorption and photoluminescence emission in dilute CH2Cl2 (2 × 10−5 M) for the complex Si(O1NO2)2 (solid traces) and the proligand (O1NO2)H2 (dashed traces). Emission spectra were recorded upon excitation at λexc = 320 nm. | ||
| λ max (ε) [nm, (103 M−1 cm−1)] | λ em [nm] | PLQY (%) | τ obs [ns] |
|
k r [107 s−1] | k nr [108 s−1] | |
|---|---|---|---|---|---|---|---|
| CH2Cl2, 2 × 10−5 M, air-equilibrated | |||||||
| sh denotes a shoulder. | |||||||
| (O1NO2)H2 | 298sh (12.81), 308 (14.60), 323sh (6.99) | 519 | 1 | 2.49 (65%), 11.34 (35%) | 8.76 | 0.1 | 1.13 |
| Si(O1NO2)2 | 322 (21.52), 345sh (13.60) | 414 | 12 | 3.26 | — | 3.7 | 2.70 |

In the 300–350 nm region, the electronic absorption spectrum of the proligand (O1NO2)H2 exhibits a slightly structured profile with moderate intensity (ε = 1.46 × 104 M−1 cm−1) at the relative maximum at λabs = 308 nm. Upon complexation onto the Si(IV) centre, the absorption maximum in Si(O1NO2)2 is bathochromically shifted by ca. 1400 cm−1 at λabs = 322 nm with ε values of 2.15 × 104 M−1 cm−1 and 1.36 × 104 M−1 cm−1 at the shoulder at λabs = 345 nm. The concomitant increase in band intensity is related to the presence of two chromophoric ligands in the Si(O1NO2)2 complex.
Upon increasing the solvent polarity, a steady hypsochromic shift of the absorption can be observed along the series toluene → THF → CH2Cl2 → CH3CN → MeOH, which is indicative of a negative solvatochromic effect (see Fig. S9 and Table S1†). Comparison with the UV-vis spectrum of the ligand (O1NO2)H2 in CH2Cl2 allows us to associate the high-energy flank at λabs = 250–270 nm of the absorption profile of the complex Si(O1NO2)2 with an electronic transition of mainly singlet ligand-centred (1LC) nature. Lastly, the broad absorption bands at λabs = 320–350 nm of Si(O1NO2)2 can be overall ascribed to overlapping electronic transitions with mixed singlet-manifold intraligand charge transfer (1ILCT) and 1LC with πPhO → π*Trz and πO1NO2 → π*O1NO2 character, respectively.
As far as the photoluminescence properties of the (O1NO2)H2 proligand in CH2Cl2 are concerned, the emission profile appears featureless and broad with a maximum at λem = 519 nm and low intensity, with PLQY being as low as 1% (see Table 1). Interestingly, its spectrum is characterized by a very large Stokes’ shift, as large as 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 cm−1. This emission is attributed with confidence to the excited-state keto form (K*), resulting from the photoinduced keto-enol tautomerisation process between the acidic phenolic proton and the H-accepting N-atoms of the substituted 1,2,4-triazole ring, after the excited-state intramolecular proton transfer (ESIPT) process, in agreement with recently reported findings.18
200 cm−1. This emission is attributed with confidence to the excited-state keto form (K*), resulting from the photoinduced keto-enol tautomerisation process between the acidic phenolic proton and the H-accepting N-atoms of the substituted 1,2,4-triazole ring, after the excited-state intramolecular proton transfer (ESIPT) process, in agreement with recently reported findings.18
In sharp contrast, upon excitation at λexc = 320 nm, a dilute CH2Cl2 sample of Si(O1NO2)2 shows a featureless photoluminescence profile in the violet-to-deep-blue region with a maximum centred at λem = 414 nm and higher intensity with a PLQY of 12%. The emission maximum of Si(O1NO2)2 is slightly affected by the polarity of the solvent, yet in an opposite way compared to the absorption spectra, with the peak shifting from λem = 414 to 410 nm upon decreasing the solvent polarity. This spectral shift is accompanied by a remarkable increase in the PLQY value from 2% in MeOH and CH3CN to 21% in toluene.
As far as the fluorescence decay of Si(O1NO2)2 is concerned, time-resolved spectroscopy yields data that can be nicely fitted with a mono-exponential decay model for the three most apolar solvents, namely, toluene, THF and CH2Cl2, while the CH3CN and MeOH samples yield kinetic data that require a two-exponential decay model. As listed in Table 1 and Table S1,† the observed lifetimes are in the range of a few nanoseconds, the longest being τ = 3.30 ns in toluene and τ = 3.27 ns in CH2Cl2 and the shortest being the two-exponential τ1 = 1.44 ns (29%) and τ2 = 0.88 ns (69%) in MeOH, with τave = 1.10 ns. Similar excited-state lifetimes can be obtained for samples in other solvents, without a clear trend dependency on solvent polarity. This finding, combined with the lower PLQY observed for Si(O1NO2)2 in MeOH and CH3CN, points towards the fact that a specific emitter–solvent interaction, such as H-bonding, might be at play, providing efficient quenching channels (see below).
These data allow us to estimate the radiative (kr) and non-radiative (knr) rate constants characterising the emissive excited state by using the following equations (eqn (1) and (2)):
 | (1) |
 | (2) |
At a later stage, the chiroptical properties of the two enantioenriched fractions (ee > 99.5%) were investigated in CH2Cl2 solution. The ECD spectra obtained for the two eluted fractions provided definitive confirmation of the resolution of the two enantiomers, as evidenced by the mirror-image shape of the spectra (Fig. 3a). As far as the first eluted fraction is concerned, the ECD spectrum displays several Cotton effects with bands possessing moderate intensity, observed at λmax = 354 nm (Δε = +6.6 M−1 cm−1), 333 nm (−16.1), 313 nm (+8.1), 287 nm (+12.3), and 259 nm (−18.2) (Table 2). A very similar, mirror-image ECD spectrum was found for the second enantiomer. Additional ECD spectra recorded in CH3CN can be found in the ESI (Fig. S10 and Table S3†), exhibiting nearly identical spectral features in both intensity and wavelength.
 | ||
| Fig. 3 (a) Experimental ECD spectra in dilute CH2Cl2 at a concentration of 2 × 10−4 M and (b) computed Boltzmann-weighted ECD spectra for Λ-Si(O1NO2)2 (first eluted, blue trace) and Δ-Si(O1NO2)2 (second eluted, red trace). | ||
| Isomer | λ max (Δε) [nm, (M−1 cm−1)] |
|---|---|
| Λ | 259 (−18.15), 278 (12.31), 313 (8.14), 333 (−16.07), 354 (6.60) |
| Δ | 259 (18.48), 276 (−11.82), 313 (−8.01), 334 (15.98), 354 (−6.64) |
Regrettably, CH2Cl2 samples of the two enantiomers exhibit only negligible circularly polarized luminescence (CPL) with very low dissymmetry factors |gLum| on the order of 10−4 at λem = 400–450 nm (Fig. S12 of the ESI†).
Remarkably, the complex Si(O1NO2)2 exhibits interesting photophysical properties in the solid state. The spectra recorded for the neat powder as well as thin-film samples at a 10% doping level in 120k poly(methyl-methacrylate) (PMMA) and 35k polystyrene (PS) matrices are displayed in Fig. 4. The most meaningful photophysical data are compiled in Table 3.
 | ||
| Fig. 4 Photoluminescence emission spectra for Si(O1NO2)2 as crystalline powder (grey trace), 10 wt% doped thin film in PMMA (green trace) and 10 wt% doped thin film in PS (aqua trace). Spectra were recorded upon excitation at λexc = 320 nm. | ||
| Compound | Doping % polymer | λ em [nm] | PLQY (%) | CIE (x, y) | τ obs [ns] |
|
|---|---|---|---|---|---|---|
| Si(O1NO2)2 | 10% PMMA 120k | 403 | 26 | 0.17, 0.07 | 1.79 (51%), 4.43 (62%) | 5.03 |
| 10% PS 35k | 420 | 32 | 0.17, 0.11 | 2.15 (92%) | — |

As far as the neat powder sample is concerned, the compound Si(O1NO2)2 exhibits a broad and featureless emission profile with a maximum at λem = 420 nm that is bathochromically shifted by 345 cm−1 compared to the CH2Cl2 sample, yet with a comparable PLQY value (11%) and τave = 2.36 ns. On the other hand, dispersion of the compound in doped polymer thin films yields emission profiles seemingly independent of the polarity of the polymer matrix, with maxima peaking at λem = 402 and 403 nm and with a remarkable enhancement of the PLQY values up to 29% and 26% for PS and PMMA samples, respectively. Overall, these findings point towards an emissive singlet-manifold excited state with admixed 1ILCT/1LC nature.
Upon lowering the temperature to 77 K, samples of racemic and enantiopure HPLC-purified Si(O1NO2)2 in a toluene glassy matrix exhibit a structured steady-state emission profile that allows us to identify two coexisting emissive phenomena (Fig. 5). The first one is located at higher energy and it is characterised by a short-lived emission peaking at λem = 392 nm with a fast two-exponential decay in the range of a few nanoseconds (τave = 6.43 ns at λem = 400 nm). The second one is found at lower energy and it is characterized by a green structured emission instead, with maxima at λem = 446, 478, 510, and 550 nm and a long-lived excited state. This latter emission can be fitted by using a two-exponential decay model that provides an average lifetime value as long as τave = 262 ms at λem = 480 nm. Both the structured profile and emission decay time allowed us to ascribe the short- and long-lived radiative processes to two excited states that are electronically decoupled with strong 1ILCT and 3LC character, respectively.
 | ||
| Fig. 5 Low-temperature photoluminescence emission spectra in a toluene glassy matrix at 77 K of Λ-Si(O1NO2)2 (first eluted, blue trace) and Δ-Si(O1NO2)2 (second eluted, red trace). Spectra were recorded upon excitation at λexc = 320 nm. | ||
It should be noted that since this emission profile is observed in both the racemate and the HPLC-enriched enantiomers, it is intrinsic to the complex and not due to trace impurities.
Computational investigation
First, the structures of both the ligand (O1NO2)H2 and the complex Si(O1NO2)2 were optimized starting from the coordinates obtained by single-crystal X-ray diffraction. For the complex, the conformational analysis was carried out on the stereoisomer and yielded four different structures associated with the possible helical twists adopted by the non-planar O1NO2 chelates. Indeed, it can be noticed that the phenol rings of the coordinated O1NO2 ligands are not coplanar, as shown by the ∠(C–C–C–C) dihedral angle values of −24.9 to −25.8° versus −25.8 to −29.5° for experimental and theoretical structures, respectively.
stereoisomer and yielded four different structures associated with the possible helical twists adopted by the non-planar O1NO2 chelates. Indeed, it can be noticed that the phenol rings of the coordinated O1NO2 ligands are not coplanar, as shown by the ∠(C–C–C–C) dihedral angle values of −24.9 to −25.8° versus −25.8 to −29.5° for experimental and theoretical structures, respectively.
Some selected geometrical parameters and energetics are listed in Table 4. The stereoisomer Λ(δ,δ) was shown to be at the absolute minimum, named GSa, while the Λ(λ,λ) stereoisomer, obtained by complete inversion of the O1NO2 twist, corresponds to a second energy-minimum structure, named GSb. These two minima (Fig. 6) are computed to be almost degenerate, with GSa lying only 0.1 kcal mol−1 above GSb, and its geometrical parameters excellently agree with those observed experimentally in the single-crystal X-ray structure.
 | ||
| Fig. 6 DFT-optimized structures of GS (left) and GSc (right). | ||
 complex. Distances, dihedral angles and Gibbs free energy differences are given in [Å], [°], and [kcal mol−1], respectively. The Boltzmann population of the different conformers is computed at 300 K
complex. Distances, dihedral angles and Gibbs free energy differences are given in [Å], [°], and [kcal mol−1], respectively. The Boltzmann population of the different conformers is computed at 300 K
| RX Λ(λ,λ) | GSaΛ(λ,λ) | GSi1Λ(λ,δ) | GSbΛ(δ,δ) | GSi2Λ(δ,λ) | |
|---|---|---|---|---|---|
| Si–O1 | 1.747 | 1.772 | 1.770 | 1.773 | 1.769 |
| Si–N1 | 1.874 | 1.887 | 1.891 | 1.887 | 1.891 |
| Si–O2 | 1.764 | 1.784 | 1.780 | 1.783 | 1.782 |
| Si–O3 | 1.755 | 1.772 | 1.769 | 1.773 | 1.770 |
| Si–N2 | 1.872 | 1.887 | 1.891 | 1.887 | 1.891 |
| Si–O4 | 1.758 | 1.784 | 1.781 | 1.783 | 1.780 |
| C1–C2–C3–C4 | −24.9 | −29.3 | −25.8 | 29.5 | 25.9 |
| C5–C6–C7–C8 | −25.8 | −29.3 | 25.8 | 29.3 | −25.7 |
| ΔG | 0.1 | 2.5 | 0.0 | 2.7 | |
| Relative Boltzmann population | 0.451 | 0.008 | 0.534 | 0.006 |
Two other energy minima, named GSi1 and GSi2, were obtained for the conformers with an alternate helical twist of one of the two O1NO2 chelates, Λ(λ,δ) and Λ(δ,λ), respectively. These conformers lie energetically slightly above the GSa geometry. The associated transition state between the Λ(δ,δ) and Λ(λ,δ) energy minima is low in energy, with the barrier being as small as ΔG‡ = 4.9 kcal mol−1, providing the possibility for fast interconversion in solution at room temperature between the GSa and GSb structures through the GSi1 and GSi2 intermediates (see Fig. S13† and Table 4).
Given the low interconversion barriers between the different conformers, the optical properties of the Si(O1NO2)2 complex in solution, whether enantiopure or as a racemate, should be the result of the contributions arising from multiple different conformers. To model the observed absorption spectrum, the electronic transitions for GSa, GSi1, GSi2 and GSb structures of the complex Si(O1NO2)2 as well as for the proligand (O1NO2)H2 were computed within the framework of time-dependent density functional theory (TD-DFT) and the corresponding data are listed in Tables S4–S8.†
The calculated absorption spectrum of the proligand (O1NO2)H2 agrees well with the experimental one (Fig. 7). The absorption band between 270 and 320 nm is the convolution of three electronic transitions computed at 286 (S0 → S1), 297 (S0 → S2) and 310 nm (S0 → S3). The computed transition at higher energy can be described as a pure π–π* localized on the phenol-triazole-phenol core (Fig. S14 and Table S4†). The two lower-energy transitions are very similar in nature and can be described as a mixture of π–π* excitation with some degrees of charge transfer character from the phenol to the triazole-phenyl moiety.
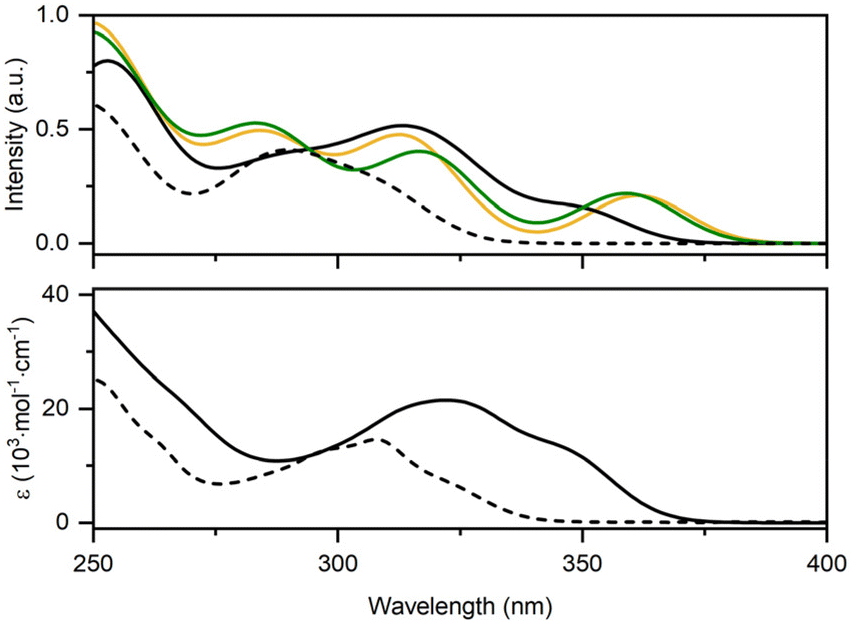 | ||
| Fig. 7 Top: Computed electronic absorption spectra of the proligand (O1NO2)H2 (dashed trace) and of the different conformers of Si(O1NO2)2: GSa (yellow), GSi1 (black), GSi2 (purple), and GSb (green), where GSi1 and GSi2 are superimposed. Bottom: Experimental electronic absorption spectra in dilute CH2Cl2 of (O1NO2)H2 (dashed trace) and Si(O1NO2)2 (black solid line). | ||
Experimentally, upon complexation of the proligand (O1NO2)H2 onto the Si(IV) centre, a bathochromic shift of the absorption spectrum of Si(O1NO2)2 is observed, yet none of the computed absorption spectra of the four conformers displayed in Fig. 7 represent the experimental profile satisfactorily. Regardless, it is evident from the computed data how the conformation of the complex has a significant impact on the shape and intensity of the absorption spectra: the computed absorption spectra of GSa and GSb appear to be relatively similar, whereas the absorption spectra of GSi1 and GSi2 are superimposable, as expected, and resemble the experimental one more closely.
As far as the conformer GSi1 is concerned, a large absorption band is present at λabs = 270–370 nm. The shoulder at 350 nm agrees well with the experimental one observed at λabs = 345 nm. This less energetic band is the superposition of two electronic transitions with mixed 1ILCT/1LC character corresponding to S0 → S1 and S0 → S2 transitions (Fig. 8) computed at λabs = 350 and 349 nm, respectively. The more intense band computed at around λabs = 315 nm is also in good agreement with the experimental peak measured at λabs = 322 nm and is due to the convolution of many electronic transitions, with the main contribution arising from the predominantly 1LC S0 → S9 transition computed at 310 nm. At higher energy, the experimental shoulder located around λabs = 290 nm is mainly due to 1LC S0 → S11 and S0 → S12 transitions computed at 294 and 293 nm, respectively.
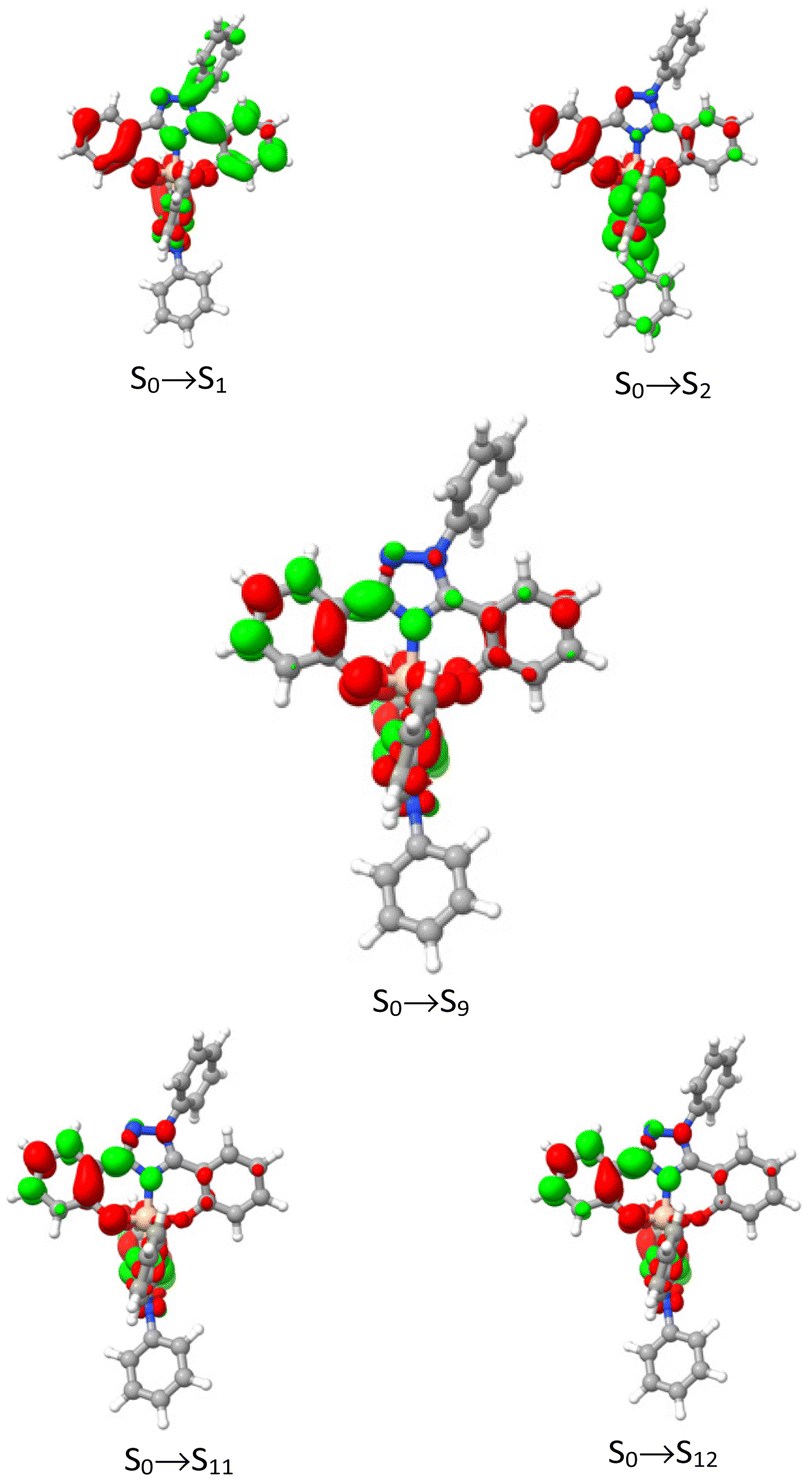 | ||
| Fig. 8 Electronic density difference maps (EDDMs) computed for complex Si(O1NO2)2 between S0 → S1 (top left), S0 → S2 (top right), S0 → S9 (center), S0 → S11 (bottom left) and S0 → S12 (bottom right) at Franck–Condon geometry. Electronically enriched and depleted areas are colored in green and red, respectively. | ||
The high conformational flexibility of the complex makes the simulation of the ECD spectra even more challenging, as it is highly sensitive to geometric parameters, or small variations thereof. For example, Fig. S15 and S16 of the ESI† clearly exemplify the variations in the computed ECD spectra for the different conformers of Λ and Δ enantiomers, respectively. At this stage, it is possible to derive the Boltzmann-weighted populations for the different conformers by employing the relative energies for the different geometries listed in Table 4, and to further derive the computed Boltzmann-weighted ECD spectra for both enantiomers, shown in Fig. 3b along the experimental spectra. Indeed, the so-obtained Boltzmann-weighted ECD spectra agree satisfactorily with the experimental data, yet with an overall bathochromic shift of about 30–40 nm compared to the experimental data. Additionally, according to these results, it is possible to assign with confidence the absolute configuration of the resolved enantiomers, with Λ being the first and Δ being the second eluted enantiomer.
The excited-state properties of the pro-ligand (O1NO2)H2 were investigated by considering both structures with and without taking into account the possible keto–enol tautomerism.18 Optimization of the S1 potential energy surface (PES) leads to several minimum-energy conformations with computed emissions at λem,theo = 415 nm (non-ESIPT process) to 1617 and 808 nm (K* form), as listed in Table 4 and Fig. 9. All these values are far from the experimental emission centred at λem,exp = 519 nm. On the other hand, optimisation of the S2 state of the lowest-energy K* structure leads to a computed emission at λem,theo = 516 nm, in good agreement with the experimental value (Table 1). It is reasonable to think that the weak experimental emission of the proligand arises from the S2 tautomeric K* form and that the structures with computed emissions in the IR domain would act as close-energy non-radiative deactivation channels, in agreement with the low PLQY in solution and the relatively large knr values observed experimentally (Table 1).
 | ||
| Fig. 9 Structure, emission wavelength (λem,theo) and relative Gibbs free energies (ΔG) of different minima on the S1 PES of the (O1NO2)H2 ligand. | ||
Upon complexation, the absence of the acidic protons of the phenolates in the complex Si(O1NO2)2 leads to a sharply different picture. Upon S1 geometry relaxation from the admixed 1LC/1LLCT at the Franck–Condon (FC) point, two minima are found on the PES during optimization (Fig. 10). The first state has a pure 1LLCT character and a computed emission wavelength at λem,theo,1 = 528 nm, while the second state possesses 1ILCT character with a computed emission at λem,theo,2 = 492 nm; however, both of these values are far from the experimental emission at λem,exp = 414 nm.
 | ||
| Fig. 10 Electronic density difference maps (EDDMs) for the complex Si(O1NO2)2 between S1 → S0 state, computed on the S1 PES for both minima and transition state structures along with emission wavelength (λem,theo) and relative Gibbs free energies (ΔG). Electronically enriched and depleted areas are colored in green and red, respectively. | ||
Taking into account the relative energies, the 1ILCT state lies only 1.1 kcal mol−1 above the 1LLCT state, and both minima are linked by a transition state (TS) located 2.3 kcal mol−1 above the 1LLCT state, which has a corresponding emission wavelength λem,theo,TS = 453 nm. These barriers associated with the TS are low enough to allow fast hopping between the different electron density distributions. Additionally, given the overall C2-symmetric nature of Si(O1NO2)2, both minima are doubly degenerate. It should be noted that the computed energy minima of S1 are probably not representative of the average structure present in solution.
Experimental section
General considerations
All reagents and solvents were purchased from commercial chemical suppliers (Acros, Alfa Aesar, Sigma-Aldrich and TCI Europe) and used without further purification. NMR spectra were recorded with a 500 MHz apparatus in deuterated solvent at 25 °C. All 13C NMR spectra are decoupled 1H (13C{1H}). The chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz, respectively. The following abbreviations are used: s: singlet; d: doublet; t triplet; q: quadruplet; quint: quintuplet; sext: sextet; sept: septet; m: multiplet; and br: broad signal. Thermogravimetric analyses were carried out by using a Q50 system from TA Instruments under air with a thermal scanning rate of 5 °C min−1.Synthesis
To a CH2Cl2 solution (6 mL) of the ligand (O1NO2)H2 (250 mg, 0.76 mmol, 2 equiv.) in a Schlenk tube under argon at room temperature was added a 1 M CH2Cl2 solution of SiCl4 (380 μL, 0.38 mmol, 1 equiv.). After 15 minutes of stirring, Et3N (250 μL, 1.8 mmol, 4.7 equiv.) was added dropwise, and the mixture was stirred for 6 hours. After evaporation of the CH2Cl2 solvent, toluene was added to the crude product and the mixture was filtered to eliminate Et3NHCl. The filtered solution was concentrated and the crude product was purified by column chromatography on silica gel using a pentane/CH2Cl2 (50/50 up to 0/100 gradient). The desired product was obtained in 86% yield (223 mg, 0.327 mmol).1 H NMR (500 MHz, CDCl3) δ 7.96 (dd, J = 7.8, 1.7 Hz, 2H), 7.71–7.67 (m, 4H), 7.66–7.61 (m, 6H), 6.96 (ddd, J = 8.4, 7.2, 1.8 Hz, 2H), 6.93–6.85 (m, 4H), 6.81 (ddd, J = 8.2, 7.3, 1.1 Hz, 2H), 6.50 (ddd, J = 8.2, 7.2, 1.1 Hz, 2H), 6.20 (ddd, J = 8.4, 5.0, 0.9 Hz, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ 160.7 (2C), 159.1 (2C), 154.9 (2C), 149.1 (2C), 137.7 (2C), 133.5 (2C), 132.3 (2C), 130.8 (2C), 130.3 (4C), 126.6 (4C), 125.8 (4C), 125.3 (2C), 122.1 (2C), 121.0 (2C), 119.0 (2C), 118.2 (2C), 113.5 (2C), 110.5 (2C). 29Si NMR (99 MHz, CDCl3) δ −194.1. HRMS (ESI+, m/z) [M + H]+ calc. for C40H27N6O428Si 683.1857 found 683.1846.
Photophysical techniques
Steady-state emission spectra were recorded on a Horiba Jobin–Yvon IBH FL-322 Fluorolog 3 spectrometer equipped with a 450 W xenon arc lamp, double-grating excitation, and emission monochromators (2.1 nm mm−1 dispersion; 1200 grooves per mm) and a Hamamatsu R13456 red-sensitive Peltier-cooled PMT detector. Phosphorescence emission spectra were recorded on a Horiba PPD-850 red-sensitive Peltier-cooled PMT detector. Emission and excitation spectra were corrected for source intensity (lamp and grating) and emission spectral response (detector and grating) by standard correction curves.
Time-resolved measurements were performed using either the time-correlated single-photon counting (TCSPC) or the Multi-Channel Scaling (MCS) electronics option of the TimeHarp 260 board installed on a PicoQuant FluoTime 300 fluorimeter (PicoQuant GmbH, Germany), equipped with a PDL 820 laser pulse driver. A pulsed laser diode LDH-P-C-375 (λ = 375 nm, pulse full width at half maximum FWHM < 40 ps driven at a repetition rate in the range of 50 kHz–40 MHz) was used to excite the samples either with either single pulse or burst mode. The excitation source was mounted directly on the sample chamber at 90°. The photons were collected using a PMA Hybrid-07 single photon counting detector. The data were acquired by using the commercially available software EasyTau II (PicoQuant GmbH, Germany), while data analysis was performed using the built-in software FluoFit (PicoQuant GmbH, Germany).
All the PLQYs of the samples were recorded at a fixed excitation wavelength by using a Hamamatsu Photonics absolute PLQY measurement system Quantaurus QY equipped with a CW Xenon light source (150 W), mO1NO2chromator, integrating sphere, C7473 photonics multi-channel analyser and employing the commercially available U6039-05 PLQY measurement software (Hamamatsu Photonics Ltd, Shizuoka, Japan). All measurements were repeated five times at the excitation wavelength λexc = 320 nm and their average values are reported in this article, unless otherwise stated.
ECD spectra were recorded on a JASCO J-815 spectrophotometer equipped with a JASCO Peltier cell holder PTC-423 to maintain the temperature at 25.0 ± 0.2 °C. A CD quartz cell of 1 mm of optical path length was used. The CD spectrometer was purged with nitrogen before recording each spectrum, which was baseline subtracted. The baseline was always measured for the same solvent and in the same cell as the samples. Acquisition parameters: intervals of 0.1 nm, a scanning speed of 50 nm min−1, a bandwidth of 2 nm, and 5 accumulations per sample.
 | (3) |
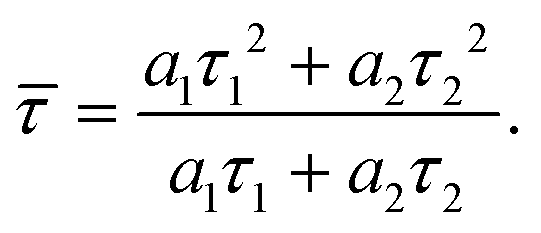 | (4) |
All solvents employed were Merck Uvasol® or Carlo Erba Spectrosol® spectrophotometric grade. Deaerated samples were prepared by the freeze–pump–thaw technique by using a custom quartz cuvette equipped with a Rotaflo® stopcock.
Computational details
All calculations have been performed with GAUSSIAN 16 version C0119 at the DFT level of theory employing the B3LYP functional20 and including Grimme's dispersion corrections.21 All atoms were described by the 6-31+G** basis set.22 The solvent (CH2Cl2) was taken into account through the polarizable continuum model (PCM) formalism.23 All structures were fully optimized and the nature of the encountered stationary points was determined by frequency analysis. Energy minima were characterized by a full set of real frequencies and transition states were characterised by one imaginary frequency. Gibbs free energies were extracted from the frequency calculation. Absorption and ECD spectra were computed by means of TD-DFT on the basis of the optimized structures. The emission wavelength was computed after geometry optimization of the lowest singlet state using the same protocol.
Electronic density difference maps (EDDMs) have been computed using the Dgrid package24 on the basis of the GAUSSIAN checkpoint file.
All calculations were performed starting from the Λ(λ,λ) enantiomer directly extracted from the experimental data. The other structures of the Λ family, Λ(λ,δ), Λ(δ,λ) and Λ(δ,δ), were obtained through the computed reaction path of ligand rotation. The structures of the Δ family, Δ(λ,λ), Δ(λ,δ), Δ(δ,λ) and Δ(δ,δ) were obtained by mirroring those of the Λ family.
Conclusions
In conclusion, we have synthesized and characterized a racemic neutral hexacoordinate Si(IV) complex containing two Deferasirox-inspired tridentate O^N^O ligands and separated its two helical enantiomers via chiral HPLC resolution. The homoleptic complex exhibits remarkable blue/near-UV photoluminescence, particularly in doped polymer thin films and very long-lived green phosphorescence at low temperatures. Our studies, including X-ray crystallography, (chiro)optical steady-state time-resolved spectroscopy and subsequent TD-DFT calculations, allowed us to model the ECD spectra and assign the absolute configuration of the resolved enantiomers.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data of Si(O1NO2)2 have been deposited at the Cambridge Crystallographic Data Centre (CCDC) 2422992.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Université de Strasbourg and the CNRS. This research was financed in part by the French National Research Agency (Agence Nationale de la Recherche – ANR) under the projects ANR-22-CE07-0049-01, ANR-21-CE29-0015 “ChirON”, ANR-22-CE07-0049-02 “BoostOLED” and the ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002). Dr J. Crassous (ISCR, Université de Rennes 1) is kindly acknowledged for the preliminary measurements of CPL. N. Kyritsakas of the Service de Radiocristallographie, Fédération de chimie Le Bel – FR2010, Université de Strasbourg & CNRS, is kindly acknowledged for the help in solving the X-ray structure. Dr Benoît Heinrich of the Institut de Physique et Chimie des Matériaux de Strasbourg is kindly acknowledged for the thermogravimetric studies.References
- T. J. Barton and P. Boudjouk, in Silicon-Based Polymer Science, American Chemical Society, 1989, vol. 224, pp. 3–46 Search PubMed.
- M. A. Brook, Silicon in organic, organometallic, and polymer chemistry, Wiley, New York Weinheim, 2000 Search PubMed.
- W. A. Chalifoux, S. K. Reznik and J. L. Leighton, Nature, 2012, 487, 86–89 CAS.
- S. Rendler and M. Oestreich, Synthesis, 2005, 1727–1747 CAS.
- Organosilicon Chemistry III: From Molecules to Materials, ed. N. Auner, N. Auner and J. Weis, Wiley-VCH, Weinheim, 1. Auflage., 2008 Search PubMed.
- M. Kocherga, J. Castaneda, M. G. Walter, Y. Zhang, N.-A. Saleh, L. Wang, D. S. Jones, J. Merkert, B. Donovan-Merkert, Y. Li, T. Hofmann and T. A. Schmedake, Chem. Commun., 2018, 54, 14073–14076 RSC.
- M. Kocherga, K. M. Boyle, J. Merkert, T. A. Schmedake and M. G. Walter, Mater. Adv., 2022, 3, 2373–2379 RSC.
- T. Thierry, V. Giuso, F. Polo, P. Mercandelli, Y.-T. Chen, C.-H. Chang, M. Mauro and S. Bellemin-Laponnaz, Dalton Trans., 2024, 53, 6445–6450 RSC.
- U. Heinz, K. Hegetschweiler, P. Acklin, B. Faller, R. Lattmann and H. P. Schnebli, Angew. Chem., Int. Ed., 1999, 38, 2568–2570 CrossRef CAS.
- S. Tury, F. Assayag, F. Bonin, S. Chateau-Joubert, J.-L. Servely, S. Vacher, V. Becette, M. Caly, A. Rapinat, D. Gentien, P. de la Grange, A. Schnitzler, F. Lallemand, E. Marangoni, I. Bièche and C. Callens, J. Pathol., 2018, 246, 103–114 CrossRef CAS.
- S. Puri, R. Kumar, I. G. Rojas, O. Salvatori and M. Edgerton, Antimicrob. Agents Chemother., 2019, 63, e02152-18 CrossRef PubMed.
- J.-H. Moon, C. Kim, H.-S. Lee, S.-W. Kim and J.-Y. Lee, J. Med. Microbiol., 2013, 62, 1307–1316 CrossRef CAS PubMed.
- S. Moreau-Marquis, G. A. O'Toole and B. A. Stanton, Am. J. Respir. Cell Mol. Biol., 2009, 41, 305–313 CrossRef CAS.
- S. J. Post, J. A. Shapiro and W. M. Wuest, MedChemComm, 2019, 10, 505–512 RSC.
- A. C. Sedgwick, K.-C. Yan, D. N. Mangel, Y. Shang, A. Steinbrueck, H.-H. Han, J. T. I. Brewster, X.-L. Hu, D. W. Snelson, V. M. Lynch, H. Tian, X.-P. He and J. L. Sessler, J. Am. Chem. Soc., 2021, 143, 1278–1283 CrossRef CAS.
- X.-L. Hu, A. C. Sedgwick, D. N. Mangel, Y. Shang, A. Steinbrueck, K.-C. Yan, L. Zhu, D. W. Snelson, S. Sen, C. V. Chau, G. Juarez, V. M. Lynch, X.-P. He and J. L. Sessler, J. Am. Chem. Soc., 2022, 144, 7382–7390 CrossRef CAS.
- G. Dahm, E. Borré, C. Fu, S. Bellemin-Laponnaz and M. Mauro, Chem. – Asian J., 2015, 10, 2368–2379 CrossRef CAS.
- A. Nina-Diogo, B. Bertrand, S. Thorimbert, G. Gontard, S. Nassem-Kahn, A. Echeverri, J. Contreras-García, C. Allain, G. Lemercier, E. Luppi and C. Botuha, Adv. Opt. Mater., 2023, 11, 2300336 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- M. D. Kohout, DGRID, Max Planck Society, Radebeul, Germany, 2011 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2422992. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00392j |
| ‡ These authors have equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |